
Cross-linked chitosan with a dicationic ionic liquid as a recyclable biopolymer-supported catalyst for cycloaddition of carbon dioxide with epoxides into cyclic carbonates†
Masoud
Taheri
ab,
Mehran
Ghiaci
 *a and
Andrey
Shchukarev
c
*a and
Andrey
Shchukarev
c
aDepartment of Chemistry, Isfahan University of Technology, Isfahan, 8415683111, Iran. E-mail: mghiaci@cc.iut.ac.ir
bCollege of Pardis, Chemistry Section, Isfahan University of Technology, Isfahan, 8415683111, Iran
cDepartment of Chemistry, Umeå University, Umeå SE-90187, Sweden
First published on 23rd November 2017
Abstract
Development of eco-friendly, desirable and reusable catalysts particularly for reactions in regard to carbon dioxide fixation is still attractive in academic and industrial sectors. In this study, a dicationic ionic liquid was anchored to a cheap biopolymer, i.e., chitosan and the prepared hybrid composite was used in the cycloaddition reaction of CO2 with various epoxides for preparing five-membered cyclic carbonates under solvent-free conditions in high yield and selectivity without the addition any metal co-catalyst. The catalyst was characterized by 1H and 13C NMR, FTIR, UV-vis, XPS, TG, FESEM, and BET techniques. The catalyst was recovered and recycled at least five times without losing its activity and selectivity. Moreover, it has been demonstrated that chitosan through hydrogen bonding, coordination of amine groups with CO2 and also loosely bonded bromide ion to imidazolium ion have synergistic effects on the yield and selectivity of cyclic carbonates under optimum conditions. On the basis of the obtained results, a feasible mechanism was proposed for the reaction.
1. Introduction
Carbon dioxide is probably the cheapest, nontoxic, abundant, non-flammable and renewable carbon resource and its production, particularly by human beings, is responsible for the greenhouse effect.1–3 Conversion of carbon dioxide into useful and desirable products is one of the most interesting challenges in academic and industrial sectors.4 Actually, carbon dioxide is a useful and environment-friendly gas, which can be compared in chemical usefulness with highly toxic chemical reagents, including phosgene, isocyanates and carbon monoxide, and can replace them in various conventional applications.5–8 In general CO2 is used in the synthesis of urea, inorganic carbonate compounds, salicylic acid and as additives for the synthesis of methanol from carbon monoxide.9 One challenge in utilization of carbon dioxide is its inherent kinetic and thermodynamic stability, due to which its activation needs high pressure and temperature or high energy reactants.10–12 Reaction of ethylene oxide as a cheap starting material with carbon dioxide, which affords five-membered ring carbonates, is one of the most cost-effective reactions.13 Production of cyclic carbonates as valuable intermediates for the synthesis of fine chemicals, green polar aprotic solvents, antifoam additives, and monomers of polycarbonates and electrolytes is an atom economic process for overcoming the inherent stability of CO2.14,15 In this regard, a broad range of catalysts, particularly in homogeneous form including complexes of transition metals, ammonium salts, ionic liquids, and phosphines, have been used for cycloaddition of CO2 with epoxides, in which isolation of the product from the reaction mixture needs complicated processes. Therefore, performing the reactions via more eco-friendly processes with various heterogeneous catalysts have been reported and developed in the last few decades.16–26In this context, different heterogeneous catalysts with desirable selectivity have been developed for cycloaddition of carbon dioxide with epoxides.15,27–30 The heterogeneous catalysts have been prepared on the basis of transition metal complexes, metal oxides,21,31,32 immobilized catalysts,16,28,33 carbon materials,34 and metal–organic frameworks.12 In an interesting study, D'Elia et al. tested the activity of transition-metal complexes as Lewis acids under homogeneous and heterogeneous conditions for cycloaddition reaction of CO2 with epoxides.23 To build eco-friendly catalysts, the focus of research groups should be to develop catalysts based on metal-free resources.2,8,28 Today, for the cycloaddition of CO2 to epoxides, the focus is on using organic bases, carbon base materials, ionic liquids, organic copolymers and eco-friendly materials.2,30,35–37
The cycloaddition reaction of CO2 with epoxides has been investigated mechanistically and theoretically.8,38,39 In this context, it is known that the ring opening of epoxides is the rate-determining step, where the produced alkoxide species is necessary for reaction with carbon dioxide. In addition, for performing the reaction in a milder condition, one should catalyze the ring opening of the epoxide with abundant hydrogen bonding donors that could accelerate the ring opening and subsequent carbon dioxide insertion.7,35,39
Today, metal free catalysts, due to their eco-friendly nature, have attracted much attention in the field of carbon dioxide fixation.28,30 In this respect, ionic liquids (ILs), which are known as green solvents and catalysts with special features such as low vapor pressure, non-flammability, satisfying thermal stability, chemical stability and conductivity and specific solubility, are the center of attention.40–42 Ionic liquids can be designed according to a specific application by different methods, such as ion-exchange.43 Wang et al. designed a metal-free mesoporous polyionic liquid44 for fixation of CO2, which provides a matrix with desirable performance and tunable porosity in heterogeneous catalysis. Furthermore dicationic ionic liquids show higher thermal stability, flexibility, viscosity, melting point, lower volatility and more exchangeable sites versus mono-cationic ionic liquids; for these reasons, ionic liquids have been attracting a growing interest.45–49
Along with the abovementioned specific features, ionic liquids have some drawbacks, including high viscosity, which decreases the diffusion of substrates when the reaction is carried out in an ionic liquid medium. Apart from this, it is difficult to separate and purify the applied ionic liquid from the reaction in both cases, i.e., when it is used as a catalyst or reaction media.50–55 From the economic point of view and because of their high cost, perhaps immobilization of ionic liquids on solid supports could address these problems. In this respect, the proposed methods for immobilization and heterogenization of oligomeric ionic liquid have attracted many research groups all over the world.56,57 Immobilized ionic liquids not only show the advantages of both ionic liquids and supports but also show new features because of their synergetic effects.58 Various solid supports for immobilization of ionic liquids have been reported, including zeolites, polymers, and carbon materials.59–65
Chitosan, as the second abundant polysaccharide, which is derived from chitin, is a cheap biopolymer with interesting features such as biodegradability, biocompatibility, non-toxicity and can be physically or chemically modified with various materials from its amine or hydroxyl groups of β-(1–4)-linked D-glucosamine and N-acetyl-D-glucosamine units. Due to these features, chitosan has been considered as a suitable support for heterogenization of catalysts.66–70 Moreover, the existence of hydroxyl and amine groups has been evidenced to promote accelerated cycloaddition of CO2 with epoxides; also, because chitosan bears a large number of both hydroxyl and amine groups, it is considered as a suitable support for this process.33,71,72
In this study, we synthesized a stable composite of chitosan and a dicationic ionic liquid by cross-linking the two components through a nucleophilic reaction between mostly free amines groups of chitosan with the –CH2Br groups designed on the dicationic ionic liquid (Scheme 2). The as prepared Ch–ILBr catalyst was used to obtain optimized settings under solvent free conditions and as a metal free catalyst for the synthesis of cyclic carbonates from cycloaddition of CO2 and epoxides. Under the obtained optimized conditions, other Ch–ILX (Ch–ILX; X = Br−, Cl−, CO32−) catalysts were prepared via anion exchange and their catalytic activities were compared.
2. Experimental
Materials
1,4-Dibromobutane, styrene oxide, epichlorohydrin, 1,2-propylene oxide, 1,2-epoxybutane, 1,2-epoxycyclohexane and imidazole were purchased from Merck Co. Chitosan was purchased from Acros Co. (medium molecular weight, 75–85% deacetylated). Sodium hydride and phosphorus pentoxide were obtained from Sigma-Aldrich Co. Acetonitrile was purchased from Dae Jung Co. Acetonitrile was dried with phosphorus pentoxide. All chemicals were used without further purification.Characterization techniques
FTIR spectra were recorded using KBr pellets by Jasco 680-plus FTIR spectrophotometer. The product yields of the reaction were determined by Agilent 7890 gas chromatograph (GC) equipped with a FID detector. The synthesized ionic liquid and its intermediate were determined and identified by a Bruker AMX nuclear magnetic resonance (NMR) 300 MHz 1H in CD3OD and D2O, respectively, and the 1H NMR of the styrene carbonate, as the only obtained product, and 13C NMR for the synthesized ionic liquid was obtained by a Bruker 400 MHz (13C 125 MHz) in CDCl3 and D2O respectively. Tetramethylsilane was used as the internal reference. Field emission scanning electron microscopy (FESEM) by TESCAN Mira-3 at an operating voltage of 15 kV was used to study the surface morphology of Ch–ILBr after coating with a gold film. Energy dispersive spectrometry (EDS) was conducted using TESCAN Mira-3, at an operating voltage of 15 kV for the elemental analysis of Ch–ILBr. Ultraviolet-visible (UV-vis) spectra of the samples were recorded using a Jasco V60 spectrophotometer. For studying the elemental states of Ch–ILBr, a KRATOS AXIS Ultra DLD X-ray photoelectron spectroscopy (XPS) system with Al Kα as the X-ray radiation source was used. The nitrogen adsorption–desorption isotherms for calculating the surface area and determining the structural type by BET and pores size with BJH method were acquired at 77 K using a PHSCHINA PHS-1020 surface area and porosity analyzer. The thermal analysis of Chitosan and Ch–ILBr was conducted with a BAHR-Thermoanalyse STA/TGA-503. Identification of the reaction products was carried out by an Agilent 5975C gas chromatograph coupled with a mass spectrometer (GC-MS).Preparation of the catalyst
Ch–ILBr was synthesized using the following procedure: in the first step, 1,1′-(butane-1,4-diyl)bis(3-(4-bromobutyl)-1H-imidazol-3-ium)bromide (ILBr) was synthesized by dissolving 44 mmol imidazole in 30 mL acetonitrile; the solution was stirred in an ice bath and 60 mmol sodium hydride was added to this solution under N2 atmosphere and stirred for 30 min. Then, the ice bath was removed and the reaction mixture was stirred at room temperature for 2 h. Then, 22 mmol 1,4-dibromobutane was added and the mixture was refluxed for 7 h, followed by filtration of the mixture and purification of the synthesized 1,4-di(1H-imidazol-1-yl)butane (A in Scheme 1) using column chromatography on silica gel (EtOAc–CH3OH). 1H NMR (400 MHz, D2O): δ 1.68 (4H, CH2), 3.99 (4H, CH2), 6.99 (2H, CH), 7.11 (2H, CH), 7.70 (2H, CH) ppm, (Fig. S1, ESI†). | ||
| Scheme 1 Preparation of 1,1′-(butane-1,4-diyl)bis(3-(4-bromobutyl)-1H-imidazol-3-ium)bromide. | ||
In the next step, 21 mmol of the resultant dried product (A in Scheme 1) was dispersed in acetonitrile and 81 mmol of 1,4-dibromobutane (excess) was added to the reaction mixture and refluxed at 70 °C under N2 atmosphere for 24 h. Then, acetonitrile was removed under vacuum and the crude product was purified using column chromatography on silica gel (solvent: EtOAc–CH3OH) (B in Scheme 1). 1H NMR (400 MHz, CD3OD): δ 1.93 (4H, CH2), 2.01 (8H, CH2), 4.31 (4H, CH2), 4.36 (8H, CH2), 7.76 (4H, CH), 9.29 (2H, CH) ppm, (Fig. S2, ESI†). 13C NMR (100 MHz, D2O): δ 26.50, 28.19, 28.90, 33.79, 49.07, 122.60, 122.85, 136.50 ppm, (Fig. S3, ESI†).
In the last step, 2.5 g of chitosan was dispersed in acetonitrile (10 mL) and 5 mmol of the synthesized ionic liquid was added to it and the mixture was stirred under nitrogen atmosphere at 70 °C for 72 h (Scheme 2). Then, the solid was filtrated and soxhleted with methanol for 15 h to remove the non-connected ionic liquid from the composite. The obtained solid, Ch–ILBr, was dried at 70 °C.
 | ||
| Scheme 2 Synthetic procedure for preparation of Ch–ILBr catalyst. | ||
General procedure for cycloaddition reaction of CO2 with epoxides
For investigation of the catalytic activity of the synthesized Ch–ILBr, 8.73 mmol of epoxide and a given amount of the catalyst were added to a 50 mL stainless steel autoclave reactor and charged with desired CO2 pressure. Then, the reactor was placed in an oil bath and after heating to a preset temperature, the timer was started. After the desired time duration, the autoclave was cooled to room temperature and the additional CO2 was released. Next, 5 mL ethanol was added to the mixture to extract the unreacted epoxide and the product. By adding n-decane as internal standard, the obtained liquid was analyzed by GC and the products were identified by GC/MS. 1H NMR (400 MHz, CDCl3): δ 4.33–4.42 (1H, CH2), 4.71–4.83 (1H, CH2), 5.67–5.71 (1H, CH), 7.26–7.38 (3H, CH), 7.44–7.46 (m, 2H, CH) ppm, Fig. S4 (ESI†).For evaluation of the catalytic activity and investigating the effect of the anion counterpart, the Ch–ILX (X = Cl−, CO32−) were prepared by anion exchange of Ch–ILBr in NaCl (1 M) or K2CO3 (1 M) solution, respectively, for 24 h and dried at 70 °C. The activity of the newly prepared Ch–ILX catalysts was determined under optimized conditions obtained for the Ch–ILBr catalyst. The catalyst reusability was investigated with Ch–ILBr, where in each run, the separated catalyst was washed with ethanol and ethyl acetate and used for the next run after drying at 70 °C.
3. Results and discussion
Characterization of synthesized catalyst
The 1H NMR spectrum of 1,4-di(1H-imidazol-1-yl)butane (Fig. S1, ESI†) shows peaks at δ 7.7 (2H), 7.1 (2H), 6.9 (2H), 3.9 (4H) and 1.6 (4H) ppm corresponding to C–H in the imidazole rings and the aliphatic CH2 groups in the butyl moiety. Synthesis of the desired ionic liquid (ILBr) was confirmed by 1H and 13C NMR spectroscopy. 1H NMR (400 MHz, CD3OD): δ 1.93 (4H, CH2), 2.01 (8H, CH2), 4.31 (4H, CH2), 4.36 (8H, CH2), 7.76 (4H, CH), 9.29 (2H, CH) ppm, (Fig. S2, ESI†). 13C NMR (100 MHz, D2O): δ 26.50, 28.19, 28.90, 33.79, 49.07, 122.60, 122.85, 136.50 ppm, (Fig. S3, ESI†).The FTIR spectra of ILBr, chitosan and Ch–ILBr are shown in Fig. 1. As shown in the spectrum of ILBr, the bands at 3069 cm−1 and 2945 cm−1 are attributed to the C–H stretching vibrations in the imidazolium rings and aliphatic C–H in the butyl moieties, respectively. The bands at 1640 cm−1 and 1453 cm−1 are attributed to aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations and the band appearing at 1564 cm−1 is related to the CN stretching vibration in the imidazolium rings. The observed band at 1162 cm−1 could be assigned to the C–N stretching vibration.14,29,73–75 The obtained FTIR spectrum of chitosan shows a broadband for O–H and N–H in the 3200–3600 cm−1 region and the bands at 2875 cm−1 and 2925 cm−1 are related to the C–H stretching vibrations of aliphatic moieties. The bands appearing at 1659 cm−1 and 1596 cm−1 are related to the carbonyl group of residual acetyl amides and the bending vibration of the N–H bond, respectively. The stretching vibrations of C–O bonds in chitosan appear at 1159, 1079 and 1030 cm−1.76–78 The FTIR spectra of the Ch–ILBr was not really informative, except if we believe that the shoulder appearing at 3084 cm−1 could be related to the aromatic C–H of the imidazolium rings. Therefore, to be more certain, we decided using the XPS technique for confirming the presence of the ionic liquid in the Ch–ILBr composite.
C stretching vibrations and the band appearing at 1564 cm−1 is related to the CN stretching vibration in the imidazolium rings. The observed band at 1162 cm−1 could be assigned to the C–N stretching vibration.14,29,73–75 The obtained FTIR spectrum of chitosan shows a broadband for O–H and N–H in the 3200–3600 cm−1 region and the bands at 2875 cm−1 and 2925 cm−1 are related to the C–H stretching vibrations of aliphatic moieties. The bands appearing at 1659 cm−1 and 1596 cm−1 are related to the carbonyl group of residual acetyl amides and the bending vibration of the N–H bond, respectively. The stretching vibrations of C–O bonds in chitosan appear at 1159, 1079 and 1030 cm−1.76–78 The FTIR spectra of the Ch–ILBr was not really informative, except if we believe that the shoulder appearing at 3084 cm−1 could be related to the aromatic C–H of the imidazolium rings. Therefore, to be more certain, we decided using the XPS technique for confirming the presence of the ionic liquid in the Ch–ILBr composite.
 | ||
| Fig. 1 FTIR spectra of ILBr, chitosan and Ch–ILBr. | ||
Fig. 2 shows the UV-vis DRS of chitosan, ILBr, and Ch–ILBr, where the chitosan shows adsorption in the range of 250–350 nm, which could be related to the residual protein impurities remaining in the chitosan. The ILBr ionic liquid shows a strong absorption band (in aqueous solution) in the range of 200–230 nm with a shoulder peak extended to 400 nm, which could be related to the π–π stacking of the imidazolium rings in a perpendicular arrangement relative to each other. In the DR spectrum of Ch–ILBr, the adsorption band at 220 nm was attributed to the ionic liquid moieties of the prepared cross-linked chitosan–ionic liquid composite.79–84 The formation of the composite was confirmed with other techniques such as XPS and TGA analysis.
 | ||
| Fig. 2 UV-vis spectra of (a) chitosan, Ch–ILBr and (b) ILBr. | ||
The X-ray photoelectron spectroscopy (XPS) spectra of the Ch–ILBr composite were used for investigating the electronic and chemical state of the elements; the results are shown in Fig. 3. As shown in Fig. 3, the Ch–ILBr composite contains C, N, O, and Br as the main constructing elements along with a trace amount of sodium, which was attributed to sodium bromide (Fig. 3a). In the Fig. 3b, the C 1s high resolution spectrum is deconvoluted to four peaks: the peaks at 285 eV (C–C, C–H, 13.7%), 286.3 eV (C–N, C–O, 43.84%), 287.7 eV (C–O–C, 7.97%) and 288.8 eV due to the amide and carboxyl residues (1.35%) of chitosan.85–89 In the high resolution of N 1s spectrum, three peaks at 399.1, 400 and 401 eV are attributed to C–N (3.88%), CON (1.15%), and C–N in the imidazolium moiety (7.94%), respectively (Fig. 3c).90–92 In the deconvoluted high resolution spectrum of O 1s, two peaks at 531 and 532.7 eV (Fig. 3d) can be fitted to CO and C–O, respectively.85 In the high resolution spectrum of Br 3d (Fig. 3e), there are two sets of doublets. The doublet at 67.5 and 68.8 eV is ascribed to 3d5/2 and 3d3/2, respectively, of the bromide ion incorporated within the ionic liquid (2.98%).93–95 The second doublet at binding energies of 68.4 and 69.5 eV id attributed to 3d5/2 and 3d3/2, respectively, of the bromide ion of sodium bromide existing in a trace amount in the sample.96–98
 | ||
| Fig. 3 (a) Global XPS of Ch–ILBr; (b) C 1s, (c) N 1s, (d) O 1s and (e) Br 3d high resolution spectra. | ||
The thermo-analysis results of chitosan before and after modification with the ionic liquid under argon atmosphere on increasing the temperature at the rate of 10 °C min−1 are presented in Fig. 4. In the thermogram of chitosan, an initial weight loss of about 7% up to 150 °C was attributed to the loss of water trapped or hydrogen bonded to chitosan. However, the Ch–ILBr composite in the same condition (up to 150 °C) loses at least a few percent lesser water than chitosan. Of course, we could expect that the Ch–ILBr composite, because of its more hydrophilic nature, should absorb more water and hence lose more water. Is there any inconsistency in our data? The answer is NO because the Ch–ILBr composite is more porous (BET surface area = 57 m2 g−1) than chitosan (BET surface area = 30 m2 g−1), which lowers its heat capacity and for this reason the composite loses less water in comparison with chitosan. The second step of weight loss for the chitosan starts at 150 °C, losing only 5 wt% upto 300 °C. However, the Ch–ILBr composite at this temperature range was stable. The main weight loss for both samples occurs at about 300 °C and at an even lower temperature for the Ch–ILBr composite probably because of the presence of bromide ions. Finally, from the carbonaceous residue of both samples, we estimated that the amount of grafted ionic liquid was 8–10 wt%.
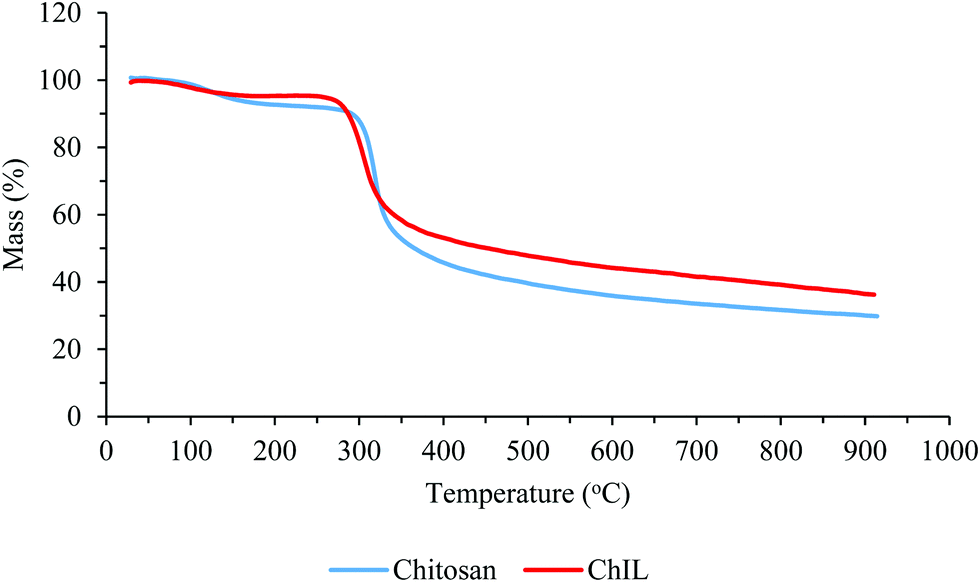 | ||
| Fig. 4 Thermogravimetric analysis of chitosan and Ch–ILBr. | ||
The field emission-scanning electron microscopy (FE-SEM) images of chitosan after modification with the ionic liquid are shown in Fig. 5. Chitosan is a macroporous solid with a BET specific surface area of 30 m2 g−1. The Ch–ILBr composite is mesoporous with a surface area of 57 m2 g−1, pore volume of 0.16 cm3 g−1 and an average pore diameter of 2.48 nm (Fig. S5, ESI†). Furthermore, the obtained EDS data for the Ch–ILBr composite confirm the existence of bromide as a structural component of the synthesized catalyst (Fig. 5c).
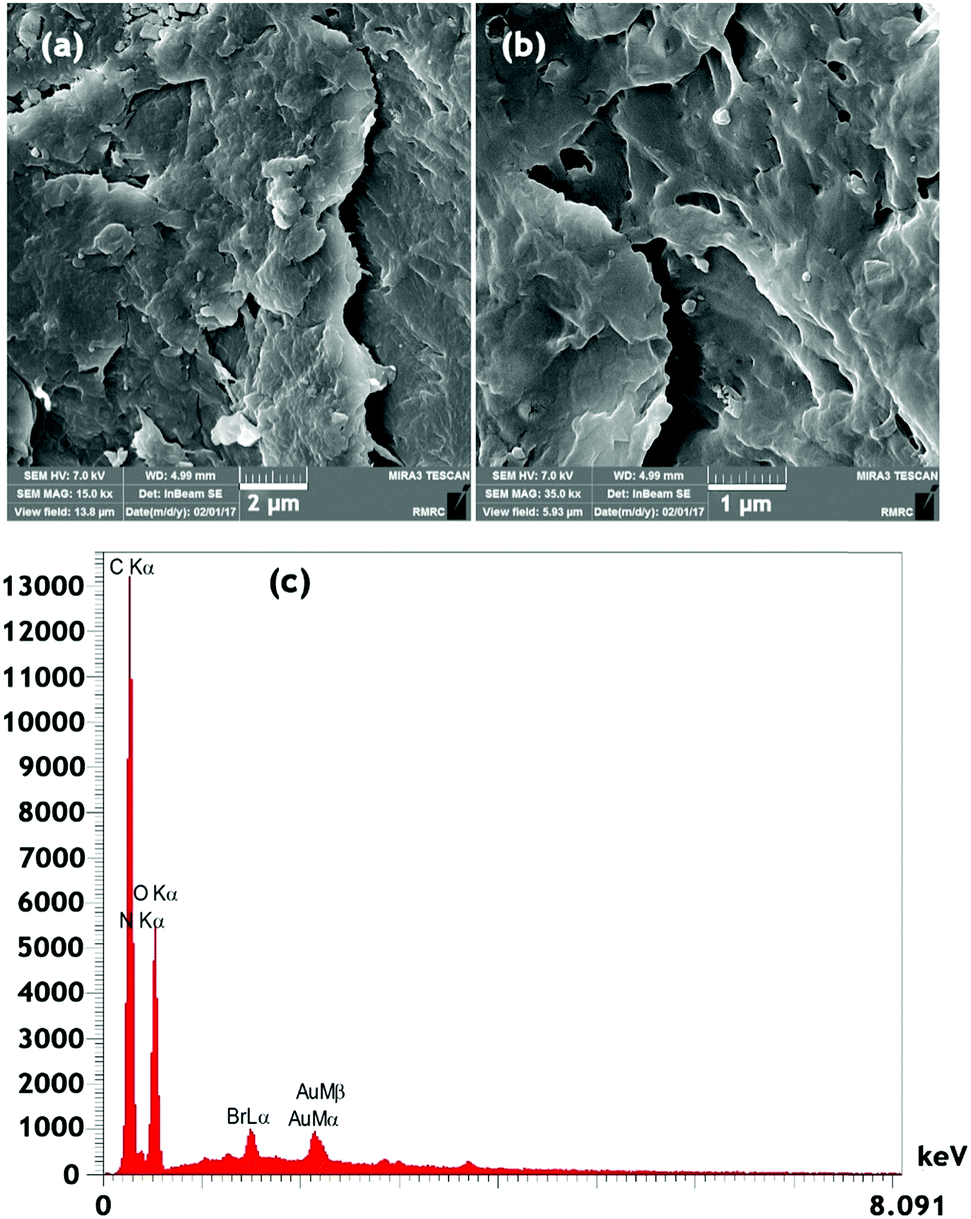 | ||
| Fig. 5 (a and b) FESEM images, (c) EDS of Ch–ILBr. | ||
Catalytic activity
In the interesting studies reported by Sakakura et al.,99 Zhang et al.65 and the recently published data from our laboratory,100 the synergistic effect of the support and the ionic liquids on the yield of cyclic carbonate has been investigated. In this respect, cycloaddition of CO2 and styrene oxide for producing the corresponding styrene carbonate (Scheme 3) was selected as the model reaction to investigate catalytic activity of the synthesized Ch–ILBr as a more greenish catalyst for the synthesis of cyclic carbonates under solvent free conditions. In this regard, the effects of various parameters including temperature, catalyst amount, CO2 pressure and reaction time were studied and optimized.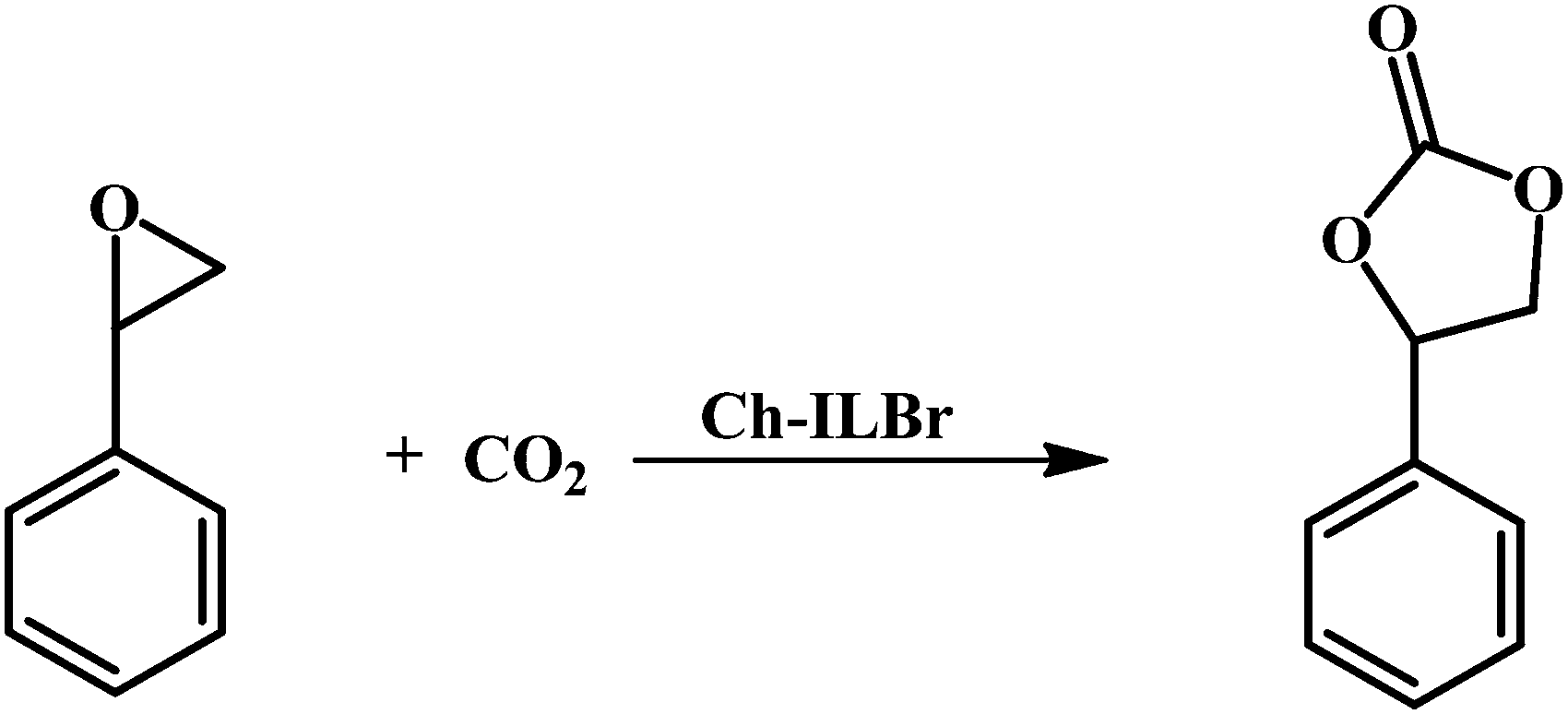 | ||
| Scheme 3 Cycloaddition of CO2 and styrene oxide to synthesize styrene carbonate. | ||
The effect of reaction temperature was investigated and the results are summarized in Table 1. As demonstrated, by increasing the temperature in the range of 90–130 °C, the yield of styrene carbonate increased up to 98% with 100% selectivity. If we consider the economics of the process, perhaps choosing 120 °C as the optimum temperature, which would lead to 2% lowering in the conversion, but saving energy could be reasonable! We did perform the reaction in the presence of non-modified chitosan, and as expected (entry 6, Table 1), the yield was not satisfactory; therefore, 120 °C was chosen as the optimum temperature.
| Entry | Temperature (°C) | Conversionb (%) | TONa |
|---|---|---|---|
| Reaction conditions: 8.73 mmol styrene oxide, 200 mg Ch–ILBr, 5 h, 20 bar CO2.a TON: converted mole of styrene oxide/mole of Br− (0.3 mol%, 1.43 wt% obtained from EDS).b Selectivity 100%.c 200 mg chitosan. | |||
| 1 | 90 | 48 | 117.3 |
| 2 | 100 | 86 | 210.3 |
| 3 | 110 | 91 | 222.5 |
| 4 | 120 | 96 | 223.4 |
| 5 | 130 | 98 | 228.1 |
| 6c | 120 | 11 | — |
Table 2 shows the influence of the amount of catalyst, carbon dioxide pressure, time of reaction, and exchanging the counter anion of the ionic liquid on the yield of styrene carbonate. As shown, the amount of catalyst used in the reaction was changed in the range of 100–200 mg, where 150 mg catalyst was chosen as optimum (Table 2, entries 1–4). Moreover, by increasing PCO2, the yield of styrene carbonate increased and reached a maximum of 96% when the pressure was 20 bar (Table 2; entry 5). In addition, by exchanging the anion accompanied with the imidazolium cation with chloride and carbonate anion and applying them as the alternative catalysts, the result showed that by decreasing the nucleophilicity of the anion, which influences the rate of epoxide ring opening, the yield of the product decreased as expected (Table 2, entries 2, 8, and 9).38,55,65,101,102 Finally, in the optimized conditions, during 5 h of reaction, the conversion reached to 96% with 100% selectivity (Table 2, entries 6 and 7).
| Entry | Conversionb (%) | TONa |
|---|---|---|
| Reaction conditions: 8.73 mmol styrene oxide, 120 °C, 5 h, 20 bar CO2,a TON: converted mole of styrene oxide/mole of Br− (0.3 mol%, 1.43 wt% obtained from EDS).b Selectivity 100%.c 200 mg Ch–ILBr.d 150 mg Ch–ILBr.e 125 mg Ch–ILBr.f 100 mg Ch–ILBr.g 10 bar CO2.h 4 h.i 6 h.j Needed amount of Ch–ILCl.k Needed amount of Ch–ILCO3. | ||
| 1c | 96 | 223.4 |
| 2d | 96 | 312.7 |
| 3e | 92 | 360.16 |
| 4f | 77 | 377.64 |
| 5d,g | 88 | 286.6 |
| 6d,h | 83 | 270.3 |
| 7d,i | 98 | 319.2 |
| 8j | 79 | 257.3 |
| 9k | 68 | 221.5 |
Cycloaddition reaction of CO2 with other epoxides catalyzed with Ch–ILBr
To explore the applicability of the proposed green catalyst, various epoxides were reacted with carbon dioxide under the optimized conditions obtained for the synthesis of styrene carbonate and the results are presented in Table 3. As expected, cyclohexene oxide, because of steric hindrance, did not show a desired yield of the corresponding product even after 17 h (Table 3, entry 5).28| Entry | Epoxide | Product | Conversionb (%) | TONa |
|---|---|---|---|---|
| Reaction conditions: 8.73 mmol epoxide, 120 °C, 5 h, 150 mg Ch–ILBr, 20 bar CO2.a TON: converted mole of epoxide/mole of Br− (0.3 mol%, 1.43 wt% obtained from EDS).b Selectivity 100%.c Reaction time was 17 h. | ||||
| 1 |

|

|
96 | 312.7 |
| 2 |
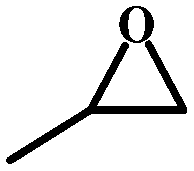
|
|
99 | 322.4 |
| 3 |

|
|
98.5 | 320.8 |
| 4 |

|

|
98 | 319.3 |
| 5c |
|

|
27 | 87.9 |
Reusability of Ch–ILBr catalyst
To investigate the reusability of the prepared catalyst, after each run, the catalyst was separated from the reaction mixture and washed thoroughly by ethanol and ethyl acetate and then dried in an oven (70 °C). The entire catalyst was recovered without any color change, and used for the next run (Fig. 6). As shown in Fig. 6, in the 5th run, the yield decreased by about 10%. However, when the used catalyst was stirred in a dilute solution of potassium bromide overnight and then washed until the filtrate did not show any trace of bromide anion by drying and using it in a new run, the reactivity retrieved to 96%. | ||
| Fig. 6 Reusability of Ch–ILBr in cycloaddition of CO2 and styrene oxide (reaction conditions: 8.73 mmol styrene oxide, 120 °C, 5 h, 150 mg Ch–ILBr, 20 bar CO2). | ||
In Table 4, we have compared the result obtained with previously reported catalysts for cycloaddition of CO2 with styrene oxide. Copolymer of phosphonium salts and triphenylphosphane,30 polystyrene–triethanolammonium iodide,36 polystyrene–3,3′-[1,4-phenylenebis(methylene)]bis(1-vinyl-1H-imidazol-3-ium) dibromide,15 polystyrene–1-(2-hydroxyl-ethyl)-imidazolium,27 polystyrene–N,N-dimethylglycine,102 chitosan–trimethyl chloride,20 copolymer of glycidyl-3-vinylimidazolium bromide and divinylbenzene,39 copolymer of 3-aminoethyl-1-vinylimidazolium hydroxide and divinylbenzene,14 and graphene oxide modified by hexamethylenetetramine methyl iodide102 are examples of catalysts which have been used for cycloaddition of CO2 with styrene oxide. The catalyst used in the present study is more eco-friendly, and also shows desired activity. Furthermore, in comparison to other catalysts, Ch–ILBr has a biopolymer support with lowest mol% of bromide anion, which is beneficial from the viewpoint of environment and economy.
| Entry | Catalyst | Catalyst amount (mol%) | Time (h) | Temp. (°C) | P (bar) | Con. (%) | Sel. (%) | TON | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Copolymer of phosphonium salts and triphenylphosphane. b Polystyrene–triethanolammonium iodide. c Polystyrene–3,3′-[1,4-phenylenebis(methylene)]bis(1-vinyl-1H-imidazol-3-ium)dibromide. d Polystyrene–1-(2-hydroxyl-ethyl)-imidazolium. e Polystyrene–N,N-dimethylglycine. f Chitosan–trimethyl chloride. g Copolymer of glycidyl-3-vinylimidazolium bromide and divinylbenzene. h Copolymer of 3-aminoethyl-1-vinylimidazolium hydroxide and divinylbenzene. i Graphene oxide modified by hexamethylenetetramine methyl iodide. | |||||||||
| 1 | ZnBr2/polymer (A2 + A6)a | 0.5 | 12 | 130 | 10 | 96 | 96 | 192 | 30 |
| 2 | Ps–N+(EtOH)3I−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
2 | 4 | 90 | 10 | 100 | 98 | 50 | 36 |
| 3 | PS–Brc | 1 | 3 | 150 | 100 | 33 | 99 | 33 | 15 |
| 4 | PS–HEIMBrd | 1.6 | 6 | 120 | 25 | 99 | 99 | 61.87 | 27 |
| 5 | PS–CQNSe | 1.3 | 5 | 150 | 20 | 93.1 | 98.2 | 71.61 | 103 |
| 6 | CS–N+Me3Cl−f |
1.7 | 6 | 160 | 40 | 88 | 88 | 51.76 | 20 |
| 7 | PGDBr–5-2OHg | 1.9 | 16 | 70 | 40 | 93 | 99 | 48.94 | 39 |
| 8 | PAD-3h | 1.9 | 8 | 110 | 1 | 92 | 98 | 48.42 | 14 |
| 9 | GO-H-Mei | 0.46 | 4 | 90 | 20 | 71.3 | 98.6 | 155 | 104 |
| 10 | Ch–ILBr | 0.3 | 5 | 120 | 20 | 96 | 100 | 312.7 | This work |
Reaction mechanism for cycloaddition of CO2 with epoxides in the presence of Ch–ILBr
According to previously reported results,8,14,15,27,33,65 we have proposed the following mechanism for cycloaddition reaction of CO2 with epoxide when catalyzed by the Ch–ILBr catalyst. As shown in Scheme 4, in the first step hydrogen bonding between the hydroxyl groups of the chitosan with oxygen of epoxide, the ring opening is facilitated when the bromide anion attacks the nucleophile. In addition, simultaneous coordination of CO2 with the nitrogen of the amine group in chitosan could result in a carbamic acid moiety, leading to intermediate 1 that finally, through an intermolecular nucleophilic ring closure, results in the formation of the cyclic carbonate and regeneration of the used catalyst.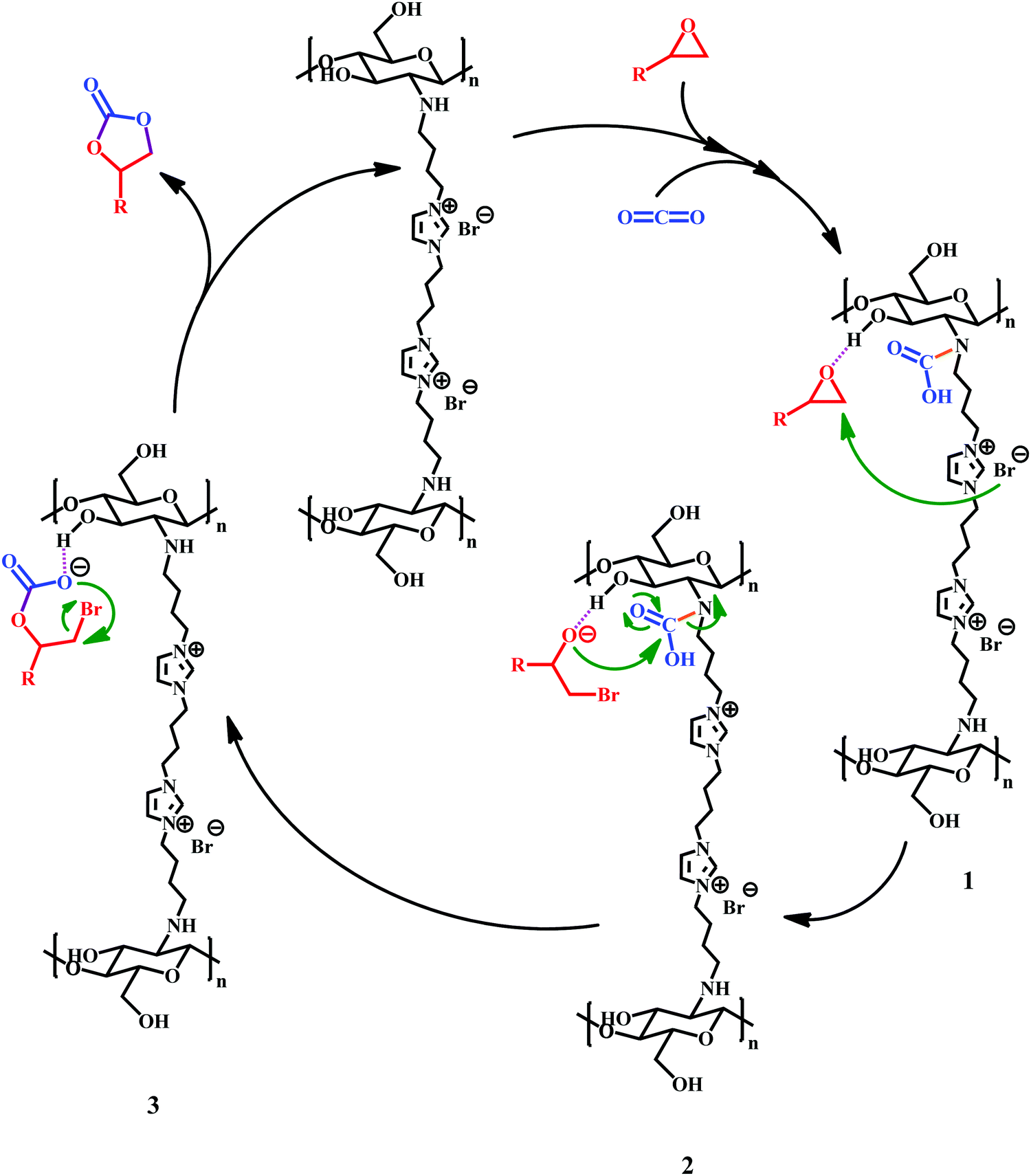 | ||
| Scheme 4 Proposed mechanism for cycloaddition reaction of CO2 with epoxide catalyzed by Ch–ILBr. | ||
4. Conclusion
In summary, ionic liquid cross-linked-immobilized on chitosan was designed and synthesized successfully for the synthesis of cyclic carbonate from cycloaddition of CO2 with epoxides as metal free catalysts under solvent free conditions. The synthesized Ch–ILBr was thoroughly characterized by different techniques such as NMR, FTIR, UV-vis, XPS, TG, FESEM and BET. The obtained results confirm the synthesis of the desired catalyst, which shows high catalytic activity for variously epoxides with exclusive selectivity. The catalyst reusability was investigated and the results showed that the catalyst can be easily separated and reused.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support from the Research Council of the Isfahan University of Technology (IUT), Iran.References
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vaginc and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 CAS.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- Z. Z. Yang, L. N. He, J. Gao, A. H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- S. Iksi, A. Aghmiz, R. Rivas, M. D. González, L. Cuesta Aluja, J. Castilla, A. Orejón, F. El Guemmout and A. M. Masdeu Bultó, J. Mol. Catal. A: Chem., 2014, 383–384, 143–152 CrossRef CAS.
- B. H. Xu, J. Q. Wang, J. Sun, Y. Huang, J. P. Zhang, X. P. Zhang and S. J. Zhang, Green Chem., 2015, 17, 108–122 RSC.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F. Q. Bai and J. y. Hasegawa, Catal. Sci. Technol., 2015, 5, 2314–2321 CAS.
- F. a. Al Qaisi, N. Genjang, M. Nieger and T. Repo, Inorg. Chim. Acta, 2016, 442, 81–85 CrossRef CAS.
- X. Sheng, L. Qiao, Y. Qin, X. Wang and F. Wang, Polyhedron, 2014, 74, 129–133 CrossRef CAS.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 CAS.
- L. Song, C. Chen, X. Chen and N. Zhang, New J. Chem., 2016, 40, 2904–2909 RSC.
- Y. N. Li, R. Ma, L. N. He and Z. F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 CAS.
- Z. Guo, X. Cai, J. Xie, X. Wang, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 12812–12821 CAS.
- C. Aprile, F. Giacalone, P. Agrigento, L. F. Liotta, J. A. Martens, P. P. Pescarmona and M. Gruttadauria, ChemSusChem, 2011, 4, 1830–1837 CrossRef CAS PubMed.
- S. Udayakumar, M. K. Lee, H. L. Shim and D. W. Park, Appl. Catal., A, 2009, 365, 88–95 CrossRef CAS.
- L. Han, H. J. Choi, D. K. Kim, S. W. Park, B. Liu and D. W. Park, J. Mol. Catal. A: Chem., 2011, 338, 58–64 CAS.
- J. Xu, F. Wu, Q. Jiang, J. K. Shang and Y. X. Li, J. Mol. Catal. A: Chem., 2015, 403, 77–83 CrossRef CAS.
- S. Duan, X. Jing, D. Li and H. Jing, J. Mol. Catal. A: Chem., 2016, 411, 34–39 CrossRef CAS.
- Y. Zhao, J. S. Tian, X. H. Qi, Z. N. Han, Y. Y. Zhuang and L. N. He, J. Mol. Catal. A: Chem., 2007, 271, 284–289 CrossRef CAS.
- P. Yan and H. Jing, Adv. Synth. Catal., 2009, 351, 1325–1332 CrossRef CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- V. D'Elia, J. D. A. Pelletier and J. M. Basset, ChemCatChem, 2015, 7, 1906–1917 CrossRef.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- W. Wang, Y. Wang, C. Li, L. Yan, M. Jiang and Y. Ding, ACS Sustainable Chem. Eng., 2017, 5, 4523–4528 CrossRef CAS.
- C. Li, W. Wang, L. Yan, Y. Wang, M. Jianga and Y. Ding, J. Mater. Chem. A, 2016, 4, 16017–16027 CAS.
- J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- T. Sakai, Y. Tsutsumi and T. Ema, Green Chem., 2008, 10, 337–341 RSC.
- S. Udayakumar, M. Lee, H. Shim, S. Park and D. Park, Catal. Commun., 2009, 10, 659–664 CrossRef CAS.
- J. Wang, J. G. W. Yang, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 15708–15711 RSC.
- M. Aresta, J. Mol. Catal. A: Chem., 2003, 204–205, 245–252 CrossRef CAS.
- Y. Xie, R. X. Yang, N. Y. Huang, H. J. Luo and W. Q. Deng, J. Energy Chem., 2014, 23, 22–28 CrossRef CAS.
- L. Han, H. J. Choi, S. J. Choi, B. Liu and D. W. Park, Green Chem., 2011, 13, 1023–1028 RSC.
- M. Galvan, M. Selva, A. Perosa and M. Noè, Asian J. Org. Chem., 2014, 3, 504–513 CrossRef CAS.
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D’Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS.
- C. Kohrt and T. Werner, ChemSusChem, 2015, 8, 2031–2034 CrossRef CAS PubMed.
- B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5673–5678 CrossRef CAS.
- C. Carvalho Rocha, T. Onfroy, J. Pilmé, A. Denicourt Nowicki, A. Roucoux and F. Launay, J. Catal., 2016, 333, 29–39 CrossRef CAS.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- X. Lu, J. Hu, X. Yao, Z. Wang and J. Li, Biomacromolecules, 2006, 7, 975–980 CrossRef CAS PubMed.
- X. Sun, C. Huang, Z. Xue and T. Mu, Energy Fuels, 2015, 29, 1923–1930 CrossRef CAS.
- X. Zhai, L. Li, H. Gao, C. Si and C. Yue, Microchim. Acta, 2012, 177, 373–380 CrossRef CAS.
- H. Olivier Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- X. Wang, Y. Zhou, Z. Guo, G. Chen, J. Li, Y. Shi, Y. Liu and J. Wang, Chem. Sci., 2015, 6, 6916–6924 RSC.
- X. Chen, J. Zhao, J. Zhang, L. Qiu, D. Xu, H. Zhang, X. Han, B. Sun, G. Fu, Y. Zhang and F. Yan, J. Mater. Chem., 2012, 22, 18018–18024 RSC.
- J. L. Anderson, R. Ding, A. Ellern and D. W. Armstrong, J. Am. Chem. Soc., 2005, 127, 593–604 CrossRef CAS PubMed.
- H. Shirota, T. Mandai, H. Fukazawa and T. Kato, J. Chem. Eng. Data, 2011, 56, 2453–2459 CrossRef CAS.
- T. Payagala, J. Huang, Z. S. Breitbach, P. S. Sharma and D. W. Armstron, Chem. Mater., 2007, 19, 5848–5850 CrossRef CAS.
- V. H. Jadhav, J. G. Kim, H. J. Jeong and D. W. Kim, J. Org. Chem., 2015, 80, 7275–7280 CrossRef CAS PubMed.
- D. W. Kim and D. Y. Chi, Angew. Chem., Int. Ed. Engl., 2004, 43, 483–485 CrossRef CAS PubMed.
- C. P. Mehnert, Chemistry, 2004, 11, 50–56 CrossRef CAS PubMed.
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chemistry, 2010, 16, 8047–8053 CrossRef CAS PubMed.
- W. Xie, L. Hu and X. Yang, Ind. Eng. Chem. Res., 2015, 54, 1505–1512 CrossRef CAS.
- K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530–531 CrossRef CAS PubMed.
- R. Yao, H. Wang and J. Han, Front. Chem. Sci. Eng., 2012, 6, 239–245 CrossRef CAS.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afework, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS PubMed.
- J. F. Wei, J. Jiao, J. J. Feng, J. Lv, X. R. Zhang, X. Y. Shi and Z. G. Chen, J. Org. Chem., 2009, 74, 6283–6286 CrossRef CAS PubMed.
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC.
- J. Li, X. Y. Shi, Y. Y. Bi, J. F. Wei and Z. G. Chen, ACS Catal., 2011, 1, 657–664 CrossRef CAS.
- M. J. Park, J. K. Lee, B. S. Lee, Y. W. Lee, I. S. Choi and S. g. Lee, Chem. Mater., 2006, 18, 1546–1551 CrossRef CAS.
- B. Yu, F. Zhou, G. Liu, Y. Liang, W. T. Huck and W. Liu, Chem. Commun., 2006, 2356–2358 RSC.
- F. Adam, J. N. Appaturi and E. P. Ng, J. Mol. Catal. A: Chem., 2014, 386, 42–48 CrossRef CAS.
- N. Fattori, C. M. Maroneze, L. P. da Costa, M. Strauss, F. A. Sigoli, I. O. Mazali and Y. Gushikem, Langmuir, 2012, 28, 10281–10288 CrossRef CAS PubMed.
- X. Wang, P. Hu, F. Xue and Y. Wei, Carbohydr. Polym., 2014, 114, 476–483 CrossRef CAS PubMed.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654–660 RSC.
- J. Tharun, Y. Hwang, R. Roshan, S. Ahn, A. C. Kathalikkattil and D. W. Park, Catal. Sci. Technol., 2012, 2, 1674–1680 CAS.
- M. Zeng, X. Zhang, L. Shao, C. Qi and X. M. Zhang, J. Organomet. Chem., 2012, 704, 29–37 CrossRef CAS.
- N. V. Kramareva, A. Y. Stakheev, O. P. Tkachenko, K. V. Klementiev, W. Grünert, E. D. Finashina and L. M. Kustov, J. Mol. Catal. A: Chem., 2004, 209, 97–106 CrossRef CAS.
- S. Puvaneswary, S. Talebian, H. B. Raghavendran, M. R. Murali, M. Mehrali, A. M. Afifi, N. H. Kasim and T. Kamarul, Carbohydr. Polym., 2015, 134, 799–807 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513–1528 CAS.
- Y. Xie, K. Ding, Z. Liu, J. Li, G. An, R. Tao, Z. Sun and Z. Yang, Chem. – Eur. J., 2010, 16, 6687–6692 CrossRef CAS PubMed.
- R. Kurt, R. Sanjines and F. L. A. Karimi, Diamond Relat. Mater., 2000, 9, 566–572 CrossRef CAS.
- C. Cai, H. Wang and J. Han, Appl. Surf. Sci., 2011, 257, 9802–9808 CrossRef CAS.
- M. Ghiaci, M. Zarghani, F. Moeinpour and A. Khojastehnezhad, Appl. Organomet. Chem., 2014, 28, 589–594 CrossRef CAS.
- M. Ghiaci, N. Dorostkar, M. V. Martínez Huerta, J. L. G. Fierro and P. Moshiri, J. Mol. Catal. A: Chem., 2013, 379, 340–349 CrossRef CAS.
- J. Brugnerotto, J. Lizardi, F. M. Goycoolea, W. Argüelles Monal, J. Desbrières and M. Rinaudo, Polymer, 2001, 42, 3569–3580 CrossRef CAS.
- O. Kuzmina, T. Heinze and D. Wawro, ISRN Polym. Sci., 2012, 2012, 1–9 CrossRef.
- C. N. Babu, P. Suresh, K. Srinivas, A. Sathyanarayana, N. Sampath and G. Prabusankar, Dalton Trans., 2016, 45, 8164–8173 RSC.
- R. Liu, S. Zhu, H. Shi, J. Hu, M. Shu, J. Liu and H. Zhu, Inorg. Chem. Commun., 2016, 74, 26–30 CrossRef CAS.
- K. Chen and M. Schmittel, Chem. Commun., 2014, 50, 5756–5759 RSC.
- K. Chauhan, R. Sharma, R. Dharela, G. S. Chauhan and R. K. Singhal, RSC Adv., 2016, 6, 75453–75464 RSC.
- A. L. Andrady, A. Torikai and T. Kobatake, J. Appl. Polym. Sci., 1996, 62, 1465–1471 CrossRef CAS.
- S. Sabar and M. A. Nawi, Desalin. Water Treat., 2015, 57, 10312–10323 CrossRef.
- Z. Geng, H. Zhang, Q. Xiong, Y. Zhang, H. Zhao and G. Wang, J. Mater. Chem. A, 2015, 3, 19455–19460 CAS.
- S. Shahabuddin, N. M. Sarih, F. H. Ismail, M. M. Shahid and N. M. Huang, RSC Adv., 2015, 5, 83857–83867 RSC.
- E. A. K. Nivethaa, S. Dhanavel, V. Narayanan, C. A. Vasu and A. Stephen, RSC Adv., 2015, 5, 1024–1032 RSC.
- A. E. G. Karakeçili, C. Satriano, M. E. Gümüşderelioğlu and G. Marletta, J. Appl. Polym. Sci., 2007, 106, 3884–3888 CrossRef.
- H. Maachou, M. J. Genet, D. Aliouche, C. C. Dupont Gillain and P. G. Rouxhet, Surf. Interface Anal., 2013, 45, 1088–1097 CrossRef CAS.
- Y. Wang, B. Li, Y. Zhou, D. Jia and Y. Song, Polym. Adv. Technol., 2011, 22, 1681–1684 CrossRef CAS.
- P. Yu, Y. Lin, L. Xiang, L. Su, J. P. Zhang and L. Mao, Langmuir, 2005, 21, 9000–9006 CrossRef CAS PubMed.
- G. Lawrie, I. Keen, B. Drew, A. C. Temple, L. Rintoul, P. Fredericks and L. Grøndahl, Biomacromolecules, 2007, 8, 2533–2541 CrossRef CAS PubMed.
- Y. S. Chi, S. Hwang, B. S. Lee, J. Kwak, I. S. Choi and S. g. Lee, Langmuir, 2005, 21, 4268–4271 CrossRef CAS PubMed.
- B. S. Lee, Y. S. Chi, J. K. Lee, I. S. Choi, C. E. Song, S. K. Namgoong and S. G. Lee, J. Am. Chem. Soc., 2004, 126, 480–481 CrossRef CAS PubMed.
- S. Men, D. S. Mitchell, K. R. Lovelock and P. Licence, ChemPhysChem, 2015, 16, 2211–2218 CrossRef CAS PubMed.
- S. Ghosal, A. Shbeeb and J. C. Hemminger, Geophys. Res. Lett., 2000, 27, 1879–1882 CrossRef CAS.
- S. Gao, N. Shang, C. Feng, C. Wang and Z. Wang, RSC Adv., 2014, 4, 39242–39247 RSC.
- H. Huang, X. Li, X. Han, N. Tian, Y. Zhang and T. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 3673–3679 RSC.
- T. Takahashi, T. Watahiki, S. Kitazume, H. Yasudaa and T. Sakakura, Chem. Commun., 2006, 1664–1666 RSC.
- Z. Akbari and M. Ghiaci, Ind. Eng. Chem. Res., 2017, 56, 9045–9053 CrossRef CAS.
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- D. H. Lan, L. Chen, C. T. Au and S. F. Yin, Carbon, 2015, 93, 22–31 CrossRef CAS.
- Y. Y. Zhang, L. Chen, S. F. Yin, S. L. Luo and C. T. Au, Catal. Lett., 2012, 142, 1376–1381 CrossRef CAS.
- D. H. Lan, Y. X. Gong, N. Y. Tan, S. S. Wu, J. Shen, K. C. Yao, B. Yi, C. T. Au and S. F. Yin, Carbon, 2018, 127, 245–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj03665e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |