
Effect of the nature of the nucleophile and solvent on an SNAr reaction†
Marcela
Gazitúa
 *a,
Ricardo A.
Tapia
b,
Renato
Contreras
c and
Paola R.
Campodónico
a
*a,
Ricardo A.
Tapia
b,
Renato
Contreras
c and
Paola R.
Campodónico
a
aCentro de Química Médica, Facultad de Medicina, Clínica Alemana Universidad del Desarrollo, Código Postal 7710162, Santiago, Chile. E-mail: migazitu@uc.cl; pcampodonico@udd.cl; Fax: +56 2 23279639; Tel: +56 2 23279682
bFacultad de Química, Pontificia Universidad Católica de Chile, Código Postal 7820436, Santiago, Chile. E-mail: rtapia@uc.cl; Tel: +56 2 23544429
cDepartamento de Química, Facultad de Ciencias, Universidad de Chile, Casilla 653, Santiago, Chile. E-mail: rcontrer@uchile.cl; Tel: +56 2 29787272
First published on 15th November 2017
Abstract
The reaction of 2,4-dinitrobenzenesulfonyl chloride toward propylamine was kinetically evaluated in 19 organic solvents and 10 ionic liquids as reaction media. This study was compared with a previous study to experimentally show that solvent effects and the nature of the reacting pair drastically affect the reaction rate and the reaction mechanism. While the reaction of the reference electrophile 2,4-dinitrobenzenesulfonyl chloride with piperidine is favored in polar solvents with the ability to donate or accept hydrogen bonds, the reaction with propylamine is favored in solvents with the ability to accept hydrogen bonds.
Introduction
The mechanisms and reactivities in nucleophilic aromatic substitution (SNAr) reactions have been extensively investigated1–18 and continue to attract attention. In general, the SNAr mechanism proceeds stepwise,5–11,13–15 with a first step where the nucleophile attacks the substrate to provide an intermediate, commonly called a Meisenheimer complex (MC); then, the leaving group (LG) departs in a second step through catalyzed or non-catalyzed pathways.4,10–13,15 The general mechanism for the reaction involving amine as the nucleophile is shown in Scheme 1.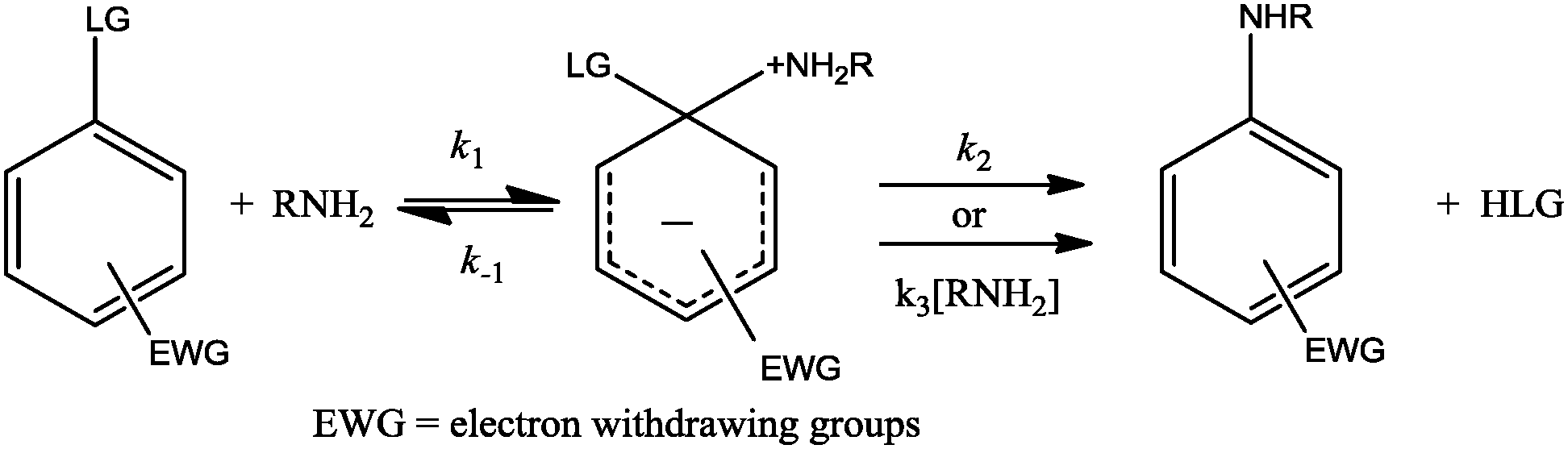 | ||
| Scheme 1 General mechanism for an SNAr reaction. | ||
To date, in few studies, systems that involve the possible existence of competing reactions between traditional nucleophilic substitution (SN) and SNAr have been considered. For example, Bunnet et al.17 studied a number of mono and dinitrophenyl esters of p-toluenesulfonic and mesitylenesulfonic acid. The authors have suggested that the attack at the carbon or sulphur centres depends on the reagent used, and the yield of SNAr or SN products is strongly influenced by the solvent. Currently, solvent effect in SNAr reactions has been studied in conventional organic solvents (COS)10,12,19–24 and room temperature ionic liquids (RTILs);12,17,18,20,25–28 this opens the discussion of the factors that determine selectivities, reaction rates, and mechanisms. For instance, D’anna et al.18 investigated the reaction of 2-substituted 5-nitrothiophenes with secondary amines. In their study, it was shown that the reactions were faster in all the RTILs used in comparison with those in methanol or benzene. This kinetic response was attributed to the stabilization of the MC through polarity effects together with a reduction of reactivity of the amine traced to the ability of the solvent to donate hydrogen bonds (HB). They suggest that In RTIL media, improvement in the reactivity is caused by the anion/cation pair of the RTILs. Habibi-Yangjeh et al.23 studied the reaction between 1-chloro-2,4-dinitrobenzene and aniline in mixtures of 1-ethyl-3-methylimidazolium ethylsulfate with methanol, chloroform, and dimethylsulfoxide. They have demonstrated through a multiparameter linear regression that the reaction rate constants increase with normalized polarity parameters (ENT), dipolarity/polarizability π*, and hydrogen bond basicity (HBB) and decrease with hydrogen bond acidity (HBA). Newington et al.24 investigated the reaction of p-fluorobenzene and p-anisidine in four COS and nine RTILs and under solvent-free conditions. They have found that the nature of the solvent exerts a dramatic effect on the reaction due to polarities and abilities of solvents to donate or accept HB.
Much of our recent effort has been focused in this area. For instance, in one of our previous studies, we reported the reaction of 2,4-dinitrophenylsulfonyl chloride (DNBSCl) with secondary alicyclic amines.13 In this reaction, the possibility of competitive SN/SNAr pathways was discarded; this suggested that this reaction occurred only via SNAr pathways and the solvent polarity affected the route of the SNAr reactions. The dual character of water and formamide as a HB donor and acceptor resulted in nucleophilic activation at the nitrogen atom of piperidine, and consequently, the reactivity of this N atom was increased. On the other hand, when RTILs were used as reaction media, EMIMDCN was found to be the best solvent as the reaction proceeded 5 times faster than that in an aqueous medium; this was probably due to the high polarizability of dicyanamide anion.13
In this study, we have considered this reaction again to answer three questions: first, is the solvent effect affected by a change in the amine structure, e.g. from secondary (piperidine in previous work) to primary amine (propylamine in this study)? Second, which reactions can be predicted to be accelerated or decelerated while using an RTIL rather than a COS? Third, is the anion or the cation responsible for the changes in reactivities when RTILs are used as solvents? Scheme 2 shows the acronyms of the RTILs, and the structures of the substrate and amines that have been used in this study.
 | ||
| Scheme 2 Acronyms of the RTILs and structures of the substrate and amines that have been used in this study. | ||
Results and discussion
Herein, we report a kinetic study on the reactions of DNBSCl with propylamine in a series with COS and RTILs as reaction media. The kinetic products were confirmed spectrophotometrically and corresponded to the product of the SNAr reaction for all the reaction media studied. The product analysis was performed using HPLC, and retention times and UV-visible spectra were compared with those of authentic samples obtained under the same experimental conditions (see Fig. S1–S8 in the ESI†). HPLC analyses at different reaction times show that the SN product is formed in minor proportion and reacts with excess amine to provide the SNAr product; this is in agreement with our previous results.13 The formation of a unique SNAr product discards the possibility of nucleophilic attack at the unsubstituted position on the ring.29Under amine excess, pseudo-first-order rate constants (kobs) were found for COS and RTILs. The values of kobs for all the reactions are in accordance with eqn (1), where k0 and kN are the rate coefficients for solvolysis and aminolysis of the substrate, respectively. We shall first analyse the solvent effects on the model reaction in COS as a reference system to discuss the solvent effects in the series of RTILs afterward. Linear plots of kobsvs. NT (total amine concentration) for a series of COS are shown (see Fig. 1).
 | ||
| Fig. 1 Linear plots of kobs against NT of the reactions of DNBSCl with propylamine at 25.0 °C in a series of COS. | ||
The values of kN were obtained as the slope of linear plots of eqn (1). These plots pass through the origin, and the reactions occur via a non-catalyzed route (k2 in Scheme 1 and Fig. S9–S19 and Tables S1–S11 in ESI†).
| kobs = k0 + kN[N]T | (1) |
Despite the small values of rate constants, if we compare the kN values of the reactions of DNBSCl with propylamine vs. piperidine13 quoted in Table 1 in a series of COS, we will note that the rate constants with piperidine are higher than those with propylamine in DMSO, FMA, and water as solvents. These data allow assessment of the effect of the nucleophile on the reaction. Note that the reactions of DNBSCl with piperidine are approximately 8, 20, and 88 times faster than the reactions of DNBSCl with propylamine in DMSO, FMA, and water, respectively. This response can be attributed to the more polar character of these solvents as compared to the case of remaining COS studied; they can interact more efficiently with the proton of piperidine. This result is probably due to two factors: (i) despite displaying similar pKa values (i.e. similar basicity), the structures of the amines are different. Propylamine is linear, and piperidine presents a chair conformation, which is most stable and produces lowest steric hindrance; this suggests that polar solvents would remove the proton from amine more easily. (ii) The double effect of hyperconjugation of –CH2 groups adjacent to the nitrogen atom of piperidine increases its electron density, thereby enhancing the nucleophilicity of N, which is favoured in polar solvents as compared to the case of propylamine.10,30 Furthermore, we can observe that DMSO, FMA, and water display different HB abilities. For instance, DMSO presents only HB accepting properties, whereas H2O and FMA both accept and donate HB. The high rate value in H2O (using piperidine as a nucleophile) suggests the presence of a HB between a hydrogen atom of H2O acting as a bridge between the nucleophile and the leaving group.13 This bridge facilitates the relay of electron density from the amine toward the electrophilic centre, thereby enhancing the nucleophilicity of the amine.13 This interaction is marginal when propylamine is used as a nucleophile if we consider the low value of its rate constant (see Table 1). This provides us an indication that the reaction with piperidine is favoured in more polar solvents with the ability to donate and accept HB, whereas the reaction with propylamine is more favoured in solvents that can accept HB and less favoured in the solvents that can donate HB. On the other hand, when alcohols are used as solvents, the reactions with propylamine are 3 times faster than those with piperidine. However, since the kN values are so small, we can say that the rate constants of both amines are practically the same. This is probably due to the low polarity of all alcohols and the better packing between the molecules of alcohol and propylamine in the bulk of solvent as compared to the case of piperidine.
| Solvent | 104kN propylamine | 104kN piperidinea |
|---|---|---|
| Acronyms of COS: N,N-DMF is N,N-dimethylformamide, MeCN is acetonitrile, DMSO is dimethyl sulfoxide and FMA is formamide. The value accompanying the kN constant corresponds to the error of the slope to obtain these values.a kN data were obtained from ref. 13. | ||
| N,N-DMF | 173 ± 6.0 | — |
| MeCN | 69.0 ± 3.3 | — |
| DMSO | 36.0 ± 2.8 | 278 ± 9 |
| FMA | 29.0 ± 1.6 | 577 ± 11 |
| tert-Butanol | 25.0 ± 0.68 | 16.4 ± 0.90 |
| 2-Propanol | 20.0 ± 0.66 | 6.40 ± 0.29 |
| Butanol | 14.0 ± 0.56 | 9.20 ± 0.43 |
| Methanol | 8.44 ± 0.32 | 4.80 ± 0.12 |
| Ethanol | 8.40 ± 0.31 | 7.20 ± 0.30 |
| H2O | 1.76 ± 0.040 | 156 ± 2.2 |
| Ethyl acetate | 30.9 ± 1.0 | — |
It seems that the solvent effects are implicated in the reactivity patterns in these systems; the nature of the amine is an additional factor on which the observed reactivity depends. This response is apparent, as shown in Fig. 1. Note that the best solvent for the reaction of DNBSCl with propylamine is N,N-DMF. The rate constant is 2.5 times faster in N,N-DMF as compared to that in MeCN, 20 times faster than that in ethanol, and 98 times faster than that in water. Moreover, note that the oxygen atom of N,N-DMF can accept a HB from propylamine, and the –CH3 groups in N,N-DMF generate an additional inductive effect on the nitrogen atom of the solvent; this suggests an enhancement of the reactivity of the system.10,30 In addition, N,N-DMF, MeCN, ethyl acetate, and DMSO only have the ability to accept HB, whereas water, FMA, and all alcohols can accept and donate HB which probably causes a decrease in the rate constants. This leads us to think that the rate constants for DNBSCl are enhanced in aprotic solvents for the reactions with propylamine. In summary, the reaction rate coefficients are solvent-dependent mainly in terms of the ability of the solvent to accept or donate HB; they are also affected by the solvent's intrinsic polarity and the structure of the nucleophile.
Now, to see if these medium effects are universal in any solvent, we have conducted a similar kinetic study in solvents of quite different nature, namely RTILs. We included a series of 10 RTILs for this purpose. Table 2 shows the kN values for the reactions of DNBSCl with propylamine and piperidine13 in RTILs, and Fig. 2 displays linear relationships between kobs and NT for propylamine to evaluate the solvent effects on the same reaction and similar conditions.
| RTILs | 104kN/s−1 M−1 propylamine | 104kN/s−1 M−1 piperidinea |
|---|---|---|
| The value accompanying the kN value corresponds to the error of the slope to obtain this value.a kN data were obtained from ref. 13. | ||
| BMIMBF4 | 59.1 ± 4.0 | 448 ± 42 |
| BMIMNTF2 | 58.2 ± 4.0 | 266 ± 21.3 |
| BMIMDCN | 48.2 ± 2.0 | 235 ± 5.30 |
| BMIMFAP | 17.9 ± 1.0 | 186 ± 5.60 |
| BMIMPF6 | 40.9 ± 2.0 | 206 ± 10.0 |
| BM2IMNTF2 | 104.3 ± 5.0 | 263 ± 15.0 |
| HMIMNTF2 | 29.3 ± 1.2 | 291 ± 20.0 |
| NEMMPNTF2 | 35.9 ± 1.6 | — |
| EMIMNTF2 | 27.3 ± 1.3 | 360 ± 19.0 |
| EMIMDCN | 38.2 ± 1.3 | 717 ± 32.0 |
 | ||
| Fig. 2 Linear plots of kobs against NT for the reactions of DNBSCl with propylamine at 25.0 °C in a series of RTILs. | ||
If we compare the kN values for the reaction of DNBSCl with propylamine vs. piperidine (see Table 2), it can be seen that the reactivity with piperidine is higher than that with propylamine in all the RTILs evaluated. The reaction of DNBSCl with piperidine in EMIMDCN is 19 times faster than the same reaction with propylamine. The slowest rate constant is in BMIMFAP for both amines, being 10 times faster for piperidine than that for propylamine. These responses are probably due to the fact that all RTILs used can donate and accept HB. This result is similar to that observed in the series of COS. This suggests that the reactivity follows the same pattern for both types of solvents. The reaction with piperidine is favoured in solvents that can donate and accept HB, whereas propylamine is favoured in solvents that can accept HB.
If we now analyse the kinetic responses among RTILs for propylamine, the reaction rate is 6 times faster in BM2IMNTF2 as compared to that in BMIMFAP, the slowest RTIL within the series. BM2IMNTF2 seems to be the best solvent and the fact that the acidic proton of imidazolium is blocked can be a key contribution to enhancement of reactivity. The presence of a –CH3 at position 2 on the imidazolium group prevents a HB with propylamine, thereby increasing the reactivity, a result which agrees with that observed for the COS series. The remaining RTILs in the series have an acidic proton available to form HB with propylamine, which decreases the reactivity (details in Fig. S20–S29 and Tables S12–S21 in ESI†).
To evaluate the performance of the RTILs as reaction media based on cation/anion effects, we compared different families of RTILs by fixing the cation (BMIM) and varying the anion (i.e. to evaluate the anion effect) and fixing the anion (NTF2) and varying the cation (i.e. to evaluate the cation effect) (see Fig. S30 and S31 in ESI†). When we fix the anion, no large differences in reaction rates are observed. EMIM presents the same kobs values as HMIM. Apparently, variation in the length of the alkyl chain of the imidazolium moiety from two to six carbons does not produce any change in the value of the rate constant, in accordance with previous work.13 BM2IM appears to be the most favourable reaction medium within the anions evaluated. Apparently, the steric hindrance effects from two to six carbon atoms in the alkyl chain in the cation are less important than the absence of the acidic proton of the imidazolium group.
Now, when we fix the cation, the results show the following anion-dependent order of decreasing quality: BF4 > NTF2 > DCN > PF6 > FAP. The rate constant in BF4 is twice that in FAP. As we can see, the size of the anion shows an inverse relationship with the reaction rate. At higher anion sizes, there is a lower rate constant probably due to steric hindrance between the nucleophile and the electrophile (see Fig. S31 in ESI†).
If we compare the performance of the best COS with the best RTIL for the reaction between DNBSCl and propylamine (Tables 1 and 2), N,N-DMF is 1,7 times faster than BM2IMNTF2. This result may probably be traced to the higher HB accepting capability of N,N-DMF as compared to the case of BM2IMNTF2. Note that the difference between them is their size, which can cause greater steric hindrance and consequently a decrease in reactivity.
In summary, the reactivity of the model substrate in RTILs is slightly more favourable with piperidine as a nucleophile. Note that EMIMDCN is the best solvent for the reaction of DNBDCl with piperidine, but not for the same reaction with propylamine. These results indicate that HB effects from the solvent to the nucleophile are qualitatively the same and that the nature of the nucleophile is also similar in both sets of molecular and ionic solvents. A definitive answer about differential solvent effects in mixtures of COS/RTILs can contribute an evidence on probable preferential solvation effects. This studyis in progress in our group.
Conclusions
The reaction of DNBSCl and propylamine has been examined for a series of conventional and non-conventional solvents and compared with that reported in a previous studywith piperidine. The analysis based on the kinetic study shows that DNBSCl is more reactive toward piperidine as a nucleophile in comparison with propylamine in polar solvents with the ability to donate and accept HB, whereas the reaction with propylamine is favoured only with solvents that can accept HB. The reactivity is marginally changed when alcohols are used as solvents probably due to their low polarity and the better packing between molecules of alcohol and propylamine in comparison with the case of piperidine. For the reaction between DNBSCl and propylamine in COS, the best solvent is N,N-DMF, and in the case of RTILs, the best solvent is BM2IMNTF2. The optimal solvent is that which is capable of assisting propylamine to enhance its nucleophilicity and has the ability to accept HB. Therefore, solvent effects and the nature of the reacting pair should be considered while analysing the reaction mechanism and its reactivities because both the reacting pair and the reaction medium are the determining factors in these processes.Experimental
Materials
Propylamine was purified before use. All organic solvents used were commercially available from Sigma-Aldrich and Merck with purity ≥99%. They were stored under anhydrous conditions and used as they were received. The certificate of analysis given by Merck S. A. for all RTILs show purity values between 99 and 100%, presence of halides ≤0.1% and content of water ≤1%. To ensure that they had no water, we put the RTILs into a vacuum drying oven LabTech Model LVO-2013 for 4 hours at a pressure of −0.06 MPa before use.Synthetic protocol of 2,4-dinitro-N-n-propylaniline
To a stirred solution of 1-fluoro-2,4-dinitrobenzene (200 mg, 1.07 mmol) in dry DMSO (2.0 mL) containing potassium carbonate (280 mg, 2.03 mmol), n-propylamine (72 mg, 1.22 mmol) was slowly added. Stirring was continued for 2 h at room temperature, and the reaction mixture was poured onto ice-water (20 g). The resulting solid was filtered, washed with water, dried, and recrystallized from dichloromethane–hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain 2,4-dinitro-N-n-propylaniline (170 mg, 70%); m.p. 98–100 °C (Lit. P. Gangopadhyay, 2000:100–102 °C); IR (KBr) νmax cm−1 3380, 3100, 1630, 1590, 1520, 1420, 1350, 1300; 1H (400 MHz, CDCl3): δ = 1.10 (t, J = 7.4 Hz, 3H), 1.83 (sextuplet, J = 7.1 Hz, 2H), 3.40 (q, J = 7.1 Hz, 2H), 6.93 (d, J = 9.5 Hz, 1H), 8.28 (dd, J = 9.5, 2.6 Hz, 1H), 8.59 (br s, 1H), 9.16 (d, J = 2.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 11.9, 22.5, 45.7, 114.3, 124.8, 130.7, 130.8, 136.3, 148.8.
:1) to obtain 2,4-dinitro-N-n-propylaniline (170 mg, 70%); m.p. 98–100 °C (Lit. P. Gangopadhyay, 2000:100–102 °C); IR (KBr) νmax cm−1 3380, 3100, 1630, 1590, 1520, 1420, 1350, 1300; 1H (400 MHz, CDCl3): δ = 1.10 (t, J = 7.4 Hz, 3H), 1.83 (sextuplet, J = 7.1 Hz, 2H), 3.40 (q, J = 7.1 Hz, 2H), 6.93 (d, J = 9.5 Hz, 1H), 8.28 (dd, J = 9.5, 2.6 Hz, 1H), 8.59 (br s, 1H), 9.16 (d, J = 2.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 11.9, 22.5, 45.7, 114.3, 124.8, 130.7, 130.8, 136.3, 148.8.
Synthetic protocol of 2,4-dinitro-N-n-propylbenzenesulfonamide31
A solution of 2,4-dinitrobenzenesulfonyl chloride (266.6, 1.0 mmol) in dry dichloromethane (20 mL) was slowly added to a mixture of n-propylamine (72 mg, 1.22 mmol) and pyridine (118.8 mg, 1.5 mmol) in dry dichloromethane (20 mL) at 0 °C. The reaction mixture was stirred at room temperature for 6.0 h. The solution was washed with 1 M HCl (5.0 mL) and water (5.0 mL), dried (Na2SO4), and concentrated. The residue was purified by column chromatography on silica gel using petroleum ether/chloroform (1:1) to obtain 2,4-dinitro-N-n-propylbenzenesulfonamide (165 mg, 57%); m.p. 81–82 °C; 1H NMR (400 MHz, CDCl3): δ 0.92 (t, J = 7.4 Hz, 3H), 1.55 (sextuplet, J = 7.4 Hz, 2H), 3.09 (q, J = 7.4 Hz, 2H), 5.36 (br.s, 1H), 8.37 (d, J = 8.7 Hz, 1H), 8.56 (dd, J = 8.7, 1.7 Hz, 1H), 8.66 (d, J = 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 10.9, 23.0, 45.6, 120.7, 127.0, 132.5, 139.3, 148.2, 149.7.
Kinetic measurements
The kinetics of 2,4-dinitrobenzenesulfonyl chloride in COS and RTILs were measured by a diode array spectrophotometer at 25.0 °C by monitoring the formation of the product between 350 and 400 nm depending on the solvent used. The initial substrate concentration was 5 × 10−5 M. Under excess amine, pseudo-first-order rate coefficients (kobs) were found throughout. The kobs values were obtained through the kinetic software (for first-order reactions) of the spectrophotometer.Chromatographic system and conditions
The HPLC system used for the analysis of the samples was a UV-DAD Elite Lachrom with quaternary pump L-2100, a UV-DAD detector L-2455, 8 μL injection loop, oven column L-2300 and autosampler L-2200 with cooling unit. The column attached was a Chromolith Fast Gradient RP 18 50-3 mm (Merck). The UV detector was set at 260 nm, which was found to be the most suitable wavelength for the detection of all the substrates, products, and internal standards. The flow-rate of the mobile phase was adjusted to 0.5 mL min−1 to keep the column pressure between 47 and 50 bar. The system was thermostated at 25 °C to maintain the same reactions conditions. Chromatograms were obtained in a computer system using the EZChrom Elite software from Agilent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Postdoctoral fellowships 3120060, Fondecyt grants 11140172, 1150759, project ICM-MINECON, RC-130006 – CILIS, granted by Fondo de Innovación para La Competitividad Del Ministerio de Economía, Fomento y Turismo, Chile and Instituto de Ciencias e Innovación en Medicina (ICIM-CAS UDD).References
- C. F. Bernasconi, MTP Int. Rev. Sci. Org. Chem. Ser. 1, 1973, 3, 33–63 CAS.
- F. Terrier, Nucleophilic Aromatic Displacement, New York, 1991 Search PubMed.
- M. R. Crampton, T. A. Emokpae, C. Isanbor, A. S. Batsanov, J. A. K. Howard and R. Mondal, Eur. J. Inorg. Chem., 2006, 1222–1230 CrossRef CAS.
- I. H. Um, S. W. Min and J. M. Dust, J. Org. Chem., 2007, 72, 8797–8803 CrossRef CAS PubMed.
- J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273–412 CrossRef CAS.
- J. H. Choi, B. C. Lee, H. W. Lee and I. Lee, J. Org. Chem., 2002, 1277–1281 CrossRef CAS.
- I. Um, J. Hong, J. Kim, O. Chae, S. Bae and X. However, J. Org. Chem., 2003, 5180–5185 CrossRef CAS PubMed.
- I. Um, S. Chun, O. Chae, M. Fujio and Y. Tsuno, J. Org. Chem., 2004, 3166–3172 CrossRef CAS PubMed.
- I. H. Um, J. Y. Hong and J. A. Seok, J. Org. Chem., 2005, 70, 1438–1444 CrossRef CAS PubMed.
- R. Ormazabal-Toledo, J. G. Santos, R. Paulina, E. A. Castro, P. R. Campodo and R. Contreras, J. Phys. Chem. B, 2013, 117, 5908–5915 CrossRef CAS PubMed.
- R. Ormazábal-Toledo, R. Contreras, R. A. Tapia and P. R. Campodónico, Org. Biomol. Chem., 2013, 11, 2302–2309 Search PubMed.
- R. Goumont, F. Terrier, D. Vichard, S. Lakhdar, J. M. Dust and E. Buncel, Tetrahedron Lett., 2005, 46, 8363–8367 CrossRef CAS.
- M. Gazitúa, R. A. Tapia, R. Contreras and P. R. Campodónico, New J. Chem., 2014, 38, 2611 RSC.
- R. Ormazábal-Toledo and P. R. C. Contreras, In Arene Chemistry: Reaction Mechanism and Methods for Aromatic Compounds, J. W. & Sons, New York, 2015 Search PubMed.
- F. Terrier, No Title, Germany, 2013 Search PubMed.
- M. R. Ashgar and B. H. M. Crampton, Org. Biomol. Chem., 2005, 3, 3971–3978 Search PubMed.
- J. F. Bunnett and J. Y. Bassett Jr., J. Am. Chem. Soc., 1959, 81, 2104–2109 CrossRef CAS.
- F. D’Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2006, 71, 5144–5150 CrossRef PubMed.
- J. Alarcón-Espósito, R. A. Tapia, R. Contreras and P. R. Campodónico, RSC Adv., 2015, 5, 99322–99328 RSC.
- X. Wang, E. J. Salaski, D. M. Berger and D. Powell, Org. Lett., 2009, 11, 5662–5664 CrossRef CAS PubMed.
- P. M. Mancini, G. Fortunato, C. Adam, L. R. Vottero and A. J. Terenzani, J. Phys. Org. Chem., 2002, 15, 258–269 CrossRef CAS.
- P. M. Mancini, G. G. Fortunate and L. R. Vottero, J. Phys. Org. Chem., 2005, 18, 336–346 CrossRef CAS.
- A. Izci and H. L. Hosgün, Turk. J. Chem., 2007, 31, 493–499 CAS.
- I. Newington, J. M. Perez-Arlandis and T. Welton, Org. Lett., 2007, 9, 5247–5250 CrossRef CAS PubMed.
- J. Alarcón-Espósito, R. Contreras, R. A. Tapia and P. R. Campodónico, Chem. – Eur. J., 2016, 22, 13347–13351 CrossRef PubMed.
- F. D’Anna, S. Marullo and R. Noto, J. Org. Chem., 2010, 75, 767–771 CrossRef PubMed.
- F. D’Anna, S. Marullo and R. Noto, J. Org. Chem., 2008, 73, 6224–6228 CrossRef PubMed.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Org. Biomol. Chem., 2013, 11, 2534–2542 CAS.
- E. Buncel, J. M. Dust and F. Terrier, Chem. Rev., 1995, 95, 2261–2280 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- L. Nondek and R. Ponce, J. Chromatogr., 1984, 294, 175–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C spectra, rate constant values and experimental conditions. See DOI: 10.1039/c7nj03212a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |