
Syntheses, crystal structures, DNA binding, DNA cleavage, molecular docking and DFT study of Cu(II) complexes involving N2O4 donor azo Schiff base ligands†
Saikat
Banerjee
 a,
Pravat
Ghorai
a,
Paula
Brandão
b,
Dipanjan
Ghosh
c,
Sutanwi
Bhuiya
a,
Dhrubajyoti
Chattopadhyay
d,
Suman
Das
a and
Amrita
Saha
*a
a,
Pravat
Ghorai
a,
Paula
Brandão
b,
Dipanjan
Ghosh
c,
Sutanwi
Bhuiya
a,
Dhrubajyoti
Chattopadhyay
d,
Suman
Das
a and
Amrita
Saha
*a
aDepartment of Chemistry, Jadavpur University, Kolkata-700032, India. E-mail: asaha@chemistry.jdvu.ac.in; amritasahachemju@gmail.com; Tel: +91-33-24572941
bDepartamento de Química, CICECO, Universidade de Aveiro, 3810-193 Aveiro, Portugal
cNIPER, Kolkata – 700032, India
dAmity University, Kolkata – 700135, India
First published on 9th November 2017
Abstract
Here, we have reported three novel copper(II) complexes (1–3) involving azo Schiff base ligands. All the complexes have been well characterized using different spectroscopic tools and single crystal X-ray diffraction analysis. Structural and electronic parameters of the complexes have been justified by DFT and TDDFT computation. All the complexes showed minor groove binding to the AT-rich sequence of DNA. The binding properties of the complexes have been extensively studied, and are further supported by a molecular docking analysis. These complexes also showed H2O2-mediated DNA cleavage properties involving a hydroxyl radical. MTT assay of the complexes was performed and they were found to be cytotoxic. The intrinsic binding constants (Kb) were calculated to be 7.11 × 105 M−1, 8.36 × 105 M−1 and 10.81 × 105 M−1 for complexes 1–3, respectively. The complexes show interesting supramolecular architectures in the solid state mainly supported by π–π stacking interactions.
Introduction
After the discovery of cis-platin, transition metal complexes have drawn considerable attention in the field of medicinal chemistry, since they target proteins and DNA.1 In the last few years, intensive efforts have been made to develop small molecule drugs using transition metal complexes of various metal ions like vanadium(V), cobalt(II), nickel(II), copper(II), zinc(II), palladium(II), platinum(II) etc.2 It has been reported that complexes with bulky ligands show DNA groove binding whereas complexes with a planar aromatic moiety bind DNA both through coordination and non-covalent interactions like π–π stacking, electrostatic binding, H-bonding etc. resulting in intercalative binding and partial intercalative binding.3 Therefore, ligand design is the key step for studying DNA or protein interactions.Obviously, Schiff base ligands demand utmost attention due to their easy synthetic procedure and wide applications in the field of both material and coordination chemistry. To date, numerous N2O4 donor Schiff base ligands have been synthesized and used for the development of transition metal complexes. Usually, these N2O4 donor Schiff base ligands behave as compartmental ligands. Chemists are mainly interested in developing heteronuclear transition metal complexes of different topologies using this type of ligand. Magnetism and catalysis are found to be the most focused area in this connection. Schiff base ligands with an azo arm are an interesting platform to incorporate different metal ions and they have applications in the field of pharmaceuticals, optical data storage, non-linear optics, photo-switching devices and dye-sensitized solar cells etc. Azo compounds generally exist in the stable trans form and show photo-induced cis–trans isomerism.4 In the last few decades, azo-derived transition metal complexes have been reported worldwide with a number of interesting photo physical and chemical properties. Even a number of ion and molecular sensors have been reported with azo-derived compounds in the literature.5
Among transition metals, copper attracts the utmost attention for its oxidative nature, bio essential activity etc.6–11 Copper(II) ion is present in different metalloproteins like plastocyanin, hemocyanin, cerluplasmin etc.12 The copper(II) ion also acts as a cofactor in a number of enzymes like cytochrome C oxidase, carbon monoxide dehydrogenase, CA1-3 DNAzymes, laccase, nitrite reductase, nitrous-oxide reductase etc. Sigman et al. first reported that the cationic part of a copper ion coordinated 1,10-phenanthroline complex can interact with and is able to cleave a strand of DNA.13
It has been found that copper complexes interact noncovalently with the DNA double helix rather than creating coordinated covalent adducts with DNA.14 Furthermore, copper complexes are usually less toxic and they have important cellular effects such as neurotransmission, cellular respiration etc. It has also been found that among transition metal complexes, copper complexes have comparatively strong Lewis acidity and high nucleobase affinity to induce efficient DNA cleavage activity. This may be the reason why much attention has been given by scientists to explore the properties of coordination complexes of copper(II) as DNA/protein targeting agents or as pharmaceutical molecules, for foot-printing and folding studies etc.15 This lays the foundation of our study of DNA interaction, which explores the structural and electronic properties of azo-derived Schiff base copper(II) complexes.
In this study, we are for the first time developing simple Cu(II) square planar complexes (1–3) involving azo-appended Schiff base ligands to study DNA binding, oxidative DNA cleavage and anticancer properties. In the solid state, all the complexes result in an interesting supramolecular architecture mainly through π–π stacking interactions. Other unconventional interactions like CH–π and H-bonding interactions are also present. The mode of the DNA binding study has been further justified by a molecular docking study. The structural and electronic parameters of the complexes have been supported by DFT and TDDFT analyses.
Experimental
Materials and physical measurements
All reagents were of analytical grade and the solvents were purchased from commercial sources and used without further purification.Elemental analysis for C, H and N was carried out using a Perkin-Elmer 240C elemental analyzer. Infrared spectra (400–4000 cm−1) were recorded from KBr pellets on a Nicolet Magna IR 750 series-II FTIR spectrophotometer. Absorption spectra were measured using a Cary 60 spectrophotometer (Agilent) with a 1 cm-path-length quartz cell. Electron spray ionization mass (ESI-MS positive) spectra were recorded on a MICROMASS Q-TOF mass spectrometer. Emission spectra were collected using a Fluromax-4 spectrofluorimeter at room temperature (298 K). Measurements of 1H NMR spectra were conducted using a Bruker 300 spectrometer in CDCl3 and DMSO-d6 solvent. The CD study was carried out using a JASCO J815 spectropolarimeter (Jasco International Co., JAPAN) attached with a PFD-425L/15 thermal programmer interfaced in a quartz cuvette having a 1 cm path length.
Synthesis of azoaldehydes, ligands and complexes
 | ||
| Scheme 1 The route to the syntheses of azoaldehyde1. | ||
 | ||
| Scheme 2 The route to the syntheses of azoaldehyde2. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The resulting deep red colored Schiff base ligand (H2L1) was used for a further complexation reaction.
:1, v/v). The resulting deep red colored Schiff base ligand (H2L1) was used for a further complexation reaction.
Yield: 0.5173 g (87%). Anal. calc. for C33H34N6O5: C 66.65%; H 5.76%; N 14.13%. Found: C 66.49%; H 5.62%; N 14.07%. IR (cm−1, KBr): ν(Ar–OH) 3460b; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) 1650 s; ν(NN) 1455 s; ν(C–H) 765 s. ESI-MS (positive) in MeOH: The base peak was detected at m/z = 595.11, corresponding to [H2L + 1]+. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 270 (12120) and 362 (17990).
N) 1650 s; ν(NN) 1455 s; ν(C–H) 765 s. ESI-MS (positive) in MeOH: The base peak was detected at m/z = 595.11, corresponding to [H2L + 1]+. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 270 (12120) and 362 (17990).
1H NMR (DMSO-d6, 300 MHz) δ ppm: 1.12–1.25 (–CH) (m, 1H), 1.74 (–OH) (bs, 1H), 3.40–3.57 (–CH2) (s, 2H), 4.00 (–OCH3) (s, 3H), 7.25–7.27 (Ar–H) (m, 2H), 7.57 (Ar–H) (s, 2H), 7.75–7.78 (Ar–H) (m, 2H), 8.36 (–CHN) (s, 1H).
:1, v/v). The resulting deep red colored Schiff base ligand (H2L2) was used for a further complexation reaction.
Yield: 0.4263 g (86%). Anal. calc. for C33H34N6O4: C 68.49%; H 5.92%; N 14.52%. Found: C 68.39%; H 5.78%; N 14.42%. IR (cm−1, KBr): ν(Ar–OH) 3460b; ν(CN) 1650 s; ν(NN) 1455 s; ν(C–H) 765 s. ESI-MS (positive) in MeOH: the base peak was detected at m/z = 579.21, corresponding to [H2L + 1]+. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 270 (12120) and 362 (17990).
1H NMR (CDCl3, 300 MHz) δ ppm: 1.15 (–CH3) (s, 3H), 3.57 (–CH2) (s, 2H), 4.00 (–OCH3) (s, 3H), 7.26–7.29 (Ar–H) (m, 3 H), 7.57 (Ar–H) (s, 2 H), 7.75–7.78 (Ar–H) (m, 2 H), 8.36 (–CHN) (s, 1H).
:1, v/v). The resulting deep red colored Schiff base ligand (H2L3) was used for a further complexation reaction.
Yield: 0.5282 g (96%). Anal. calc. for C31H30N6O4: C 67.62%; H 5.49%; N 15.26%. Found: C 67.51%; H 5.38%; N 15.19%. IR (cm−1, KBr): ν(CN) 1648s; ν(NN) 1455 s; ν(C–H) 764 s. ESI-MS (positive) in MeOH: the base peak was detected at m/z = 551.28, corresponding to [M + 1]+. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 290 (4533) and 388 (5736).
1H NMR (CDCl3, 300 MHz) δ ppm: 2.25 (–CH2) (m, 1H), 3.82–3.84 (–CH2) (bs, 2H), 4.01 (–OCH3) (s, 3H), 7.42–7.90 (Ar–H) (m, 7 H), 8.43 (–CHN) (s, 1H).
Complex 1. Yield: 0.5587 g (83%). Anal. calc. for C33H33CuN6O6: C 55.88%; H 4.94%; N 12.48%. Found: C 55.78%; H 4.81%; N 12.37%. IR (cm−1, KBr): ν(–OH) 3082b; ν(C
N) 1624 s; ν(NN) 1442 s; ν(C–H) 775 s. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 279 (34209), 392 (49476) and 605 (721).
Complex 2. Yield: 0.5883 g (87%). Anal. calc. for C33H36CuN6O6: C 58.61%; H 5.37%; N 12.43%. Found: C 58.52%; H 5.27%; N 12.39%. IR (cm−1, KBr): ν(C
N) 1615 s; ν(NN) 1466 s; ν(C–H) 764 s. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 280 (38365), 394 (56827) and 612 (825).
Complex 3. Yield: 0.5898 g (91%). Anal. calc. for C31H32CuN6O6: C 57.44%; H 4.98%; N 12.97%. Found: C 57.40%; H 4.91%; N 12.93%. IR (cm−1, KBr): ν(–OH) 3481b; ν(C
N) 1621 s; ν(NN) 1456 s; ν(C–H) 768 s. UV-Vis, λmax (nm), (ε (dm3 mol−1 cm−1)) in methanol: 280 (14164), 376 (28640) and 635 (645).
X-ray crystallography
Single crystal X-ray data of complexes 1–3 were collected on a Bruker SMART APEX-II CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 150(2) K. Data processing, structure solution, and refinement were performed using the Bruker Apex-II program suite. All available reflections in the 2θmax range were harvested and corrected for Lorentz and polarization factors with Bruker SAINT plus.18 Reflections were then corrected for absorption, inter-frame scaling and other systematic errors with SADABS.19 The structures were solved by direct methods and refined by means of a full matrix least-square technique based on F2 with the SHELX-2016/6 software package.20 All the non-hydrogen atoms were refined with anisotropic thermal parameters. C–H hydrogen atoms were inserted at geometrical positions with Uiso = 1/2Ueq to those they are attached to. Crystal data and details of data collection and refinement for 1–3 are summarized in Table 1.| Complex | 1 | 2 | 3 |
|---|---|---|---|
| Empirical formula | C33H33CuN6O6 | C33H36CuN6O6 | C31H32CuN6O6 |
| Formula weight | 673.19 | 676.22 | 648.16 |
| Temperature (K) | 150(2) | 150(2) | 150(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
| a (Å) | 9.5695(11) | 12.0130(14) | 11.0747(3) |
| b (Å) | 10.6070(10) | 12.2684(15) | 10.6797(3) |
| c (Å) | 16.3461(15) | 12.3980(15) | 25.2355(7) |
| α (°) | 71.986(4) | 70.845(4) | 90 |
| β (°) | 83.215(5) | 80.210(4) | 91.650(2) |
| γ (°) | 85.146(5) | 72.219(4) | 90 |
| Volume (Å3) | 1564.8(3) | 1638.6(3) | 2983.48(14) |
| Z | 2 | 2 | 4 |
| D calc (g cm−3) | 1.429 | 1.371 | 1.443 |
| Absorption coefficient (mm−1) | 0.753 | 0.719 | 0.787 |
| F(000) | 700 | 706 | 1348 |
| θ Range for data collection (°) | 2.98–25.00 | 2.98–25.00 | 2.94–25.06 |
| Reflections collected | 5952 | 20489 |
38353 |
| Independent reflections/Rint | 5952 | 5566/0.0893 | 6624/0.0419 |
| Observed reflections [I > 2σ(I)] | 4234 | 3399 | 4002 |
| Data/restraints/parameters | 5952/2/440 | 5566/0/428 | 6624/0/419 |
| Goodness-of-fit on F2 | 1.052 | 0.997 | 1.033 |
| Final indices [I > 2σ(I)] | R 1 = 0.0582; wR2 = 0.1351 | R 1 = 0.0674; wR2 = 0.1640 | R 1 = 0.0789; wR2 = 0.2192 |
| R indices (all data) | R 1 = 0.1027; wR2 = 0.1572 | R 1 = 0.1338; wR2 = 0.1908 | R 1 = 0.1278; wR2 = 0.2660 |
| Largest diff. peak/hole (e Å−3) | 0.694/−0.855 | 1.235/−0.646 | 0.996/−0.726 |
Spectroscopic studies on CT-DNA binding ability
The stock solution of CT-DNA was prepared following the standard procedure. Briefly, a requisite amount of DNA (∼10 mg per mL buffer solution) was dissolved in Tris–HCl buffer medium (5 mM Tris–HCl/50 mM NaCl, pH = 7.4). The ratio of UV absorbance at 260 nm and 280 nm of the solution is ca. 1.85, which indicates that the CT-DNA sample does not have any protein contamination. The concentration of the stock solution was measured by considering the molar extinction coefficient of the CT-DNA at 260 nm to be 6600 M−1 cm−1. The stock solution was stored at 4 °C and used within four days. The stock solutions of the complexes were prepared in the DMSO–buffer mixture (v/v = 1:1000). All the data were recorded at room temperature. First, an absorption spectrum of 20 μM of each complex was taken followed by the gradual addition of the CT-DNA solution (0–6 eq.). A change in the absorption band was observed upon each addition of DNA to the complex. In order to eliminate the absorbance of CT-DNA itself, an equal amount of CT-DNA was added to a reference cuvette. Then, a steady-state fluorescence study was carried out to have a better insight into the experiment.
Binding stoichiometry (Job's plot)
Job's continuation method21 was employed to find the binding stoichiometry of the complexes with CT-DNA using absorption spectroscopy. At a constant temperature (25 °C), the absorbance was noted for solutions where the concentrations of both the complex and DNA were varied but the sum of their concentrations was kept constant at 50 μM. The relative change in absorbance (ΔA/A0) was plotted as a function of the mole fraction of the complex. The break point in the resulting plot corresponds to the mole fraction of the complex in the DNA bound + complex form. From the break point, the stoichiometry was estimated. The results reported are averages of at least three experiments.Circular dichroism (CD) study
Circular dichroism (CD) is a very useful tool for conformational analysis of bio-macromolecules like proteins and nucleic acids at the molecular level.22 Here, CD measurements were carried out on a JASCO J815 spectropolarimeter (Jasco International Co., JAPAN) attached with a PFD-425L/15 thermal programmer interfaced in a quartz cuvette having a 1 cm path length. All CD spectra were measured in the wavelength range of 200–400 nm at a scan speed of 100 nm min−1. Each spectrum was an average of five readings.DNA cleavage study
Since all the complexes bind with DNA, we were also interested in studying their oxidative cleavage properties. We incubated each of the complexes 1–3 (20 μM) separately with supercoiled plasmid DNA (pUC19) (60 μM) in the presence of H2O2 (250 μM) in 5 mM Tris–HCl/50 mM NaCl buffer (pH 7.4) in 20 μL at 37 °C for 4 h and the result was analyzed by electrophoresis of the DNA in 1% agarose gel in buffer (10 mM Tris–HCl, pH 7.2) under physiologically relevant conditions. The DNA bands were visualized under a UV tranluminator and band pictures were captured using chemidoc (Chemidoc™ XRS+, BioRad). The control reactions were done using H2O2 alone or a mixture of H2O2 and CuCl2 with plasmid DNA.A time course experiment was done for oxidative cleavage of plasmid DNA by the individual complexes keeping the same reaction conditions (i.e. complexes 1–3 (20 μM), plasmid DNA (pUC19) (60 μM), H2O2 (250 μM) in 5 mM Tris–HCl/50 mM NaCl buffer (pH 7.4) at 37 °C and a reaction volume of 100 μL). Twenty μL samples were collected at 1 h, 2 h, 3 h and 4 h time intervals from the same reaction mixtures and stored at −80 °C before running the agarose gel.
Computational method
All computations were performed using the GAUSSIAN09 (G09)23 software package. Coordinates obtained from single crystal X-ray data were used for optimization of the structures of complexes 1–3. For optimization, we used the density functional theory method at the B3LYP level24 for complexes 1–3. The lanL2DZ effective potential (ECP) basis set of Hay and Wadt for copper atoms and the standard 6-31+G(d) basis set for C, H, N and O atoms were chosen for optimization.25–27TDDFT calculation was performed with the optimized geometry to ensure only positive eigen values. Time-dependent density functional theory (TDDFT)28–31 was performed using the conductor-like polarizable continuum model (CPCM)32–35 and the same B3LYP level and basis sets in a methanol solvent system. GAUSSSUM36 was used to calculate the fractional contributions of various groups to each molecular orbital.
Molecular docking simulation study
A molecular docking study was performed to investigate the probable binding site and mode of binding of the three complexes (1–3) with CT-DNA using the docking program AutoDock (version 4.2). The structure of the DNA duplex of the dodecamer sequence d(CGCGAATTCGCG)2 obtained from the RCSB Protein Data Bank (PDB ID: 1BNA) was used for the docking study. The three-dimensional structures of the complexes were created in Chem3D Ultra 8.0 and modified accordingly using two well-known software packages, Gaussian 09W and AutoDock 4.2. Gasteiger partial charges were added to the ligand atoms. Hydrogen atoms were united and rotatable bonds were identified. The rigid residue of DNA was considered for this docking simulation. Grid maps of 120 × 120 × 120 Å grid points and 0.375 Å grid spacing were generated using the Auto Grid program. Other Auto Dock parameters were set to the default values. The Lamarckian genetic algorithm (LGA) was used to carry out the docking calculations. Each run of the docking simulation was set to conclude after a maximum of 250000 energy evaluations. The best optimized docked model with the lowest docking energy was chosen for further analysis of the docking simulations, which was best viewed in PyMOL software.
In vitro cytotoxicity assay
Use of cultured cell lines in vitro to evaluate the cytotoxicity effects of the complexes is a common practice. We used A549, adenocarcinomic human alveolar basal epithelial cells, for the in vitro cytotoxicity assay. The cells were cultured in DMEM high glucose media (200 μL mL−1) with 10% FBS and Pen/Strep and maintained in 5% CO2 and 37 °C humidified incubators. In a 96-well plate, in each well about 1 × 104 cultured cells were added. Stock solutions of 100× of the complexes were prepared in DMSO and 2 μL of the complexes was added in each well so that the final concentrations of the complexes were 1, 5, 10, 25, 50, 100 and 150 μM. For the control wells, 2 μL of DMSO was added. An MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed 48 hours after the addition of the complexes. Briefly, 20 μL of MTT solution (5 mg mL−1) was added to each well for 3 hours and then the solution was removed carefully leaving the formazan crystals in the well and 200 μL of DMSO was added to solubilize the formazan crystals. The absorbance was taken at 490 nm. All data points are the average of triplicate results and the relative cell viability (%) was expressed as a percentage relative to the DMSO control cells.Results and discussion
Preparation of ligands H2L1, H2L2, and H2L3 and complexes 1–3
The ligands H2L1–3 were synthesized by the condensation reaction between 1,3-diaminopropane or substituted 1,3-diaminopropane and azo appended aldehyde in a 1:2 molar ratio in a methanol–chloroform solvent mixture (Scheme 3).
 | ||
| Scheme 3 The route to the syntheses of H2L1–3. | ||
The ligand possesses six potential donor sites; two azomethine nitrogen atoms, two phenolic oxygen atoms and two oxygen atoms of methoxy groups. The methanolic solution of Cu(NO3)2·3H2O was subjected to reaction with H2L1–3 in the presence of Et3N in a 1:1:1 molar ratio under stirring. Upon slow evaporation of the solvent, deep brown colored crystals of the complexes were obtained.
Crystal structure description of complexes 1–3
The structures of complexes 1–3 with the atomic numbering scheme are shown in Fig. 1–3. Selected bond distances and bond angles are given in Table 2. | ||
| Fig. 1 Ortep view of complex 1. Atoms are shown as 30% thermal ellipsoids. H atoms and solvent molecule are omitted for clarity. | ||
 | ||
| Fig. 2 Ortep view of complex 2. Atoms are shown as 30% thermal ellipsoids. H atoms and solvent molecules are omitted for clarity. | ||
 | ||
| Fig. 3 Ortep view of complex 3. Atoms are shown as 30% thermal ellipsoids. H atoms and solvent molecules are omitted for clarity. | ||
| Complex 1 | Complex 2 | Complex 3 | ||||
|---|---|---|---|---|---|---|
| X-ray | Calculated | X-ray | Calculated | X-ray | Calculated | |
| Cu–O1 | 1.901(3) | 1.92663 | 1.892(4) | 1.92782 | 1.918(4) | 1.92775 |
| Cu–O2 | 1.900(4) | 1.92766 | 1.914(4) | 1.92781 | 1.931(4) | 1.92773 |
| Cu–N3 | 1.950(4) | 1.99940 | 1.945(5) | 1.99410 | 1.959(6) | 1.99521 |
| Cu–N4 | 1.946(4) | 1.99105 | 1.944(4) | 1.99415 | 1.958(6) | 1.99073 |
| N1–N2 | 1.266(5) | 1.26643 | 1.269(6) | 1.26612 | 1.235(8) | 1.26615 |
| N5–N6 | 1.250(7) | 1.26644 | 1.264(9) | 1.26611 | 1.268(7) | 1.26616 |
| C14–C15–C16 | 114.5(5) | 114.01379 | 111.4(5) | 111.64925 | 119.6(9) | 119.8623 |
| O1–Cu–N3 | 94.0(1) | 92.50288 | 93.5(2) | 92.46997 | 93.4(2) | 92.75531 |
| O1–Cu–N4 | 158.5(1) | 155.32958 | 158.2(2) | 155.62890 | 172.1(2) | 169.97583 |
| O1–Cu–O2 | 87.6(1) | 94.46403 | 89.7(2) | 94.22463 | 86.0(2) | 94.57123 |
| N3–Cu–N4 | 92.3(2) | 91.21777 | 90.5(2) | 91.00757 | 89.0(2) | 91.27836 |
| N3–Cu–O2 | 158.5(1) | 155.06048 | 160.6(2) | 155.63275 | 172.3(2) | 162.55942 |
| N4–Cu–O2 | 94.0(1) | 92.34482 | 93.6(2) | 92.46547 | 92.6(2) | 92.43576 |
The structures of complexes 1–2 were solved in the triclinic space group P. In contrast, the structure of complex 3 was solved in the monoclinic space group P21/c. In the molecule, crystallographic imposed symmetry was absent and the asymmetric unit consists of a discrete copper(II) complex having a quadridentate azo-azomethine Schiff base ligand. The deprotonated ligand, L2−, forms three adjacent six-membered rings (6-6-6) during complexation. The terminal two rings consist of o-vanillin fragments with an azo tail and the starting diamine forms the middle one. The Cu–O bonds are in the cis configuration and the Cu–O distances are shorter than the Cu–N distances. The azomethine bond distances of the complexes are 1.473(6), 1.465(8) Å for complex 1 (N3–C21 and N4–C25), 1.292(4), 1.286(5) Å for complex 2 (N3–C13 and N4–C17), and 1.292(4), 1.286(5) Å for complex 3 (N3–C13 and N4–C17), respectively. The azo bonds (N1–N2 and N5–N6) exhibit an NN double bond character with distances of 1.266(5) and 1.250(7) Å for 1, 1.269(6) and 1.264(9) Å for 2, and 1.235(8) and 1.268(7) Å for 3, respectively. The central Cu(II) atom has distorted square planar coordination with a significant tetrahedral distortion. The coordinating atoms N3, N4, O1 and O2 deviate from the least-square mean plane by 0.342, 0.342, 0.369 and 0.369 Å for 1, 0.337, 0.340, 0.348 and 0.351 Å for 2, and 0.126, 0.125, 0.132 and 0.131 Å for 3, respectively. For complexes 1 and 3, the Cu atom is located perfectly within the same plane resulting in 0.00 Å deviations, whereas the Cu atom of complex 2 is located 0.019 Å above the same. Although there is no crystallographically imposed molecular symmetry, two phenolate rings are close to planar with a dihedral angle of 38.96° (C2/C7 and C28/C33 for 1), 31.40° (C2/C7 and C27/C32 for 2), and 5.66° (C2/C7 and C25/C30 for 3), respectively. The two outer azo coordinated benzene rings are twisted with respect to the phenolate rings. The dihedral angles between C12/C17 and C39/C44 (for 1), C12/C17 and C38/C43 (for 2), and C12/C17 and C36/C41 (for 3) are 79.08°, 3.51° and 10.41°, respectively. The bite angles of all the complexes are very comparable and range from 86.0° to 94.0°, and they significantly deviate from the ideal angle of 90°. The larger bite angle [N4–Cu–O2, 94.0(1)° for 1, N3–Cu–O1, 93.59° for 2 and N3–Cu–O1, 93.39° for 3] arises from the six-membered chelate ring, which is somewhat puckered due to the sp3 hybridization of the saturated portion of the chelating ligand. Again, the sums of the equatorial angles N3–Cu–N4, N3–Cu–O1, O1–Cu–O2 and O2–Cu–N4 for complexes 1–3 are 367.87°, 367.32° and 361.01°, respectively, and slightly differ from the ideal value of 360.00°, which is consistent with the distorted geometry.
The encapsulated water molecule of complex 1 forms strong hydrogen bonding interactions with both the phenolic and methoxy oxygen atoms. Here, dimerization occurs through H-bonding interactions. These dimers further assemble to form a 1D supramolecular architecture via CH–π interactions where the H-atom of the methyl group of a dimer interacts with the π electron cloud of the azobenzene of another dimer. The distances of H-bonding interactions are 2.199 Å and 2.208 Å, respectively. The distance of CH–π interaction is 3.621 Å (Fig. 4).
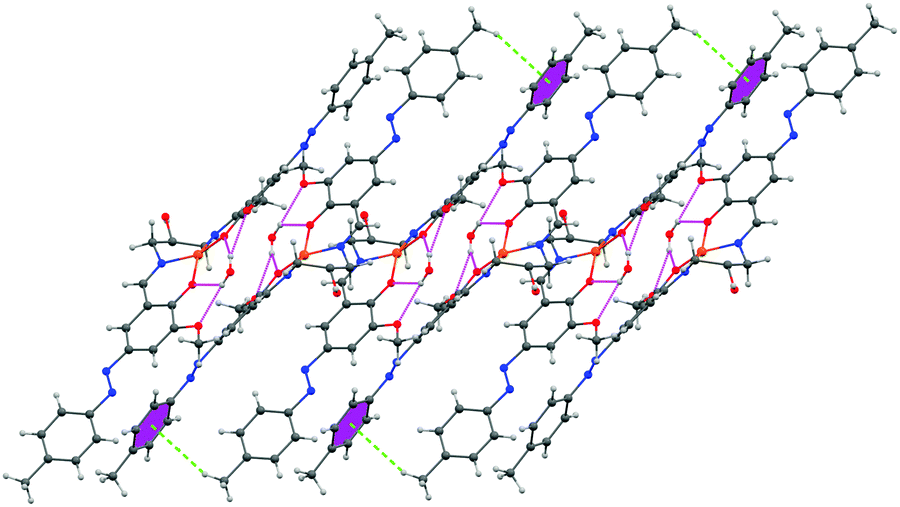 | ||
| Fig. 4 1D supramolecular architecture of complex 1 propagating along the b axis showing H-bonding interaction and CH–π interaction. Hydrogen atoms of least interest are omitted for clarity. | ||
Complex 2 forms a 1D supramolecular architecture through π–π interactions. There are edge to edge π–π interactions between NN and the π bond of the benzene ring. The distances of π–π interactions are 3.724 Å and 3.990 Å, respectively (Fig. S1, ESI†).
Complex 3 dimerizes through π–π interactions. These dimers further assemble through π–π interactions to form a 1D network. The distances of the π–π interactions are 4.203 Å, 3.685 Å, 3.613 Å and 3.577 Å, respectively (Fig. S2 and S3, ESI†).
Spectroscopic characterization of the ligands (H2L1–3) and complexes (1–3)
The ligands (H2L1–3) and complexes 1–3 have been characterized using IR, UV-Vis, NMR (only Ligands), and CHN analysis. In the IR spectra of the ligands, a broad band appeared around 3460 cm−1 attributed to the phenolic OH stretching frequency. For complexes 1–3, bands that appeared around 1624, 1615 and 1621 cm−1, respectively, are assigned to the >CHN stretching frequencies. In contrast, the characteristic azo NN stretching frequencies of complexes 1–3 are obtained around 1442, 1466 and 1456 cm−1, respectively. In the 1H NMR spectra of H2L1–3 (CDCl3), imine protons appeared around 8.36 ppm. In contrast, aromatic protons appeared around 7.26–7.90 ppm. Methoxy protons of H2L1–3 appeared around 4.0 ppm. For H2L1, aliphatic protons and –OH protons appeared around 3.40–3.57, 1.12–1.25 and 1.74 ppm, respectively. In the case of H2L2, aliphatic protons appeared around 3.57 and 1.15 ppm. In contrast, for H2L3, aliphatic protons appeared around 3.82–3.84 and 2.25 ppm (Fig. S4–S6, ESI†). The ESI mass spectrum of H2L3 is depicted in Fig. S7 (m/z = 551.28) (ESI†). UV-Vis spectra of the ligands and complexes 1–3 were recorded in methanol solvent at room temperature. The ligands show absorption bands around 410 nm and 260 nm attributed to the n → π* and π → π* transitions, respectively. Each complex shows a weak absorption band around 605 nm assigned as spin-allowed d–d transition of the Cu(II) metal ion.37
DNA binding study of complexes 1–3
DNA binding studies are one of the important key steps in developing a new drug. The elucidation of the mode of binding of a drug with nucleic acids is essential to understand the structural as well as probable functional aspects of the drug. So, here, we have studied the interaction of the complexes with naturally occurring CT-DNA. DNA binding of the complexes was studied using different spectroscopic tools, viz. UV-vis absorption and fluorescence measurement. The binding studies were carried out in Tis–HCl buffer medium (5 mM Tris–HCl/50 mM NaCl, pH = 7.4). Each complex shows two absorption bands around ∼280 nm and ∼390 nm, respectively (Fig. 5). | ||
| Fig. 5 UV-VIS spectrum of complexes 1–3 in methanol. | ||
Since the absorption band of CT-DNA at 260 nm coincides with the absorption bands (∼280 nm) of the complexes, the change in absorption intensity has been studied for λmax = 370 nm. 20 μM of the complex was titrated with a gradually increasing concentration of CT-DNA (0–120 μM). When any complex strongly binds with the base pairs of the DNA, it results in hypochromism and bathochromism of the absorption intensity. This is due to stacking between the aromatic part of the intercalator and the DNA resulting in a conformational change of the nucleic acid helix along with changes in the environment polarity. On the other hand, minor groove binding of the complex to DNA results in hyperchromism with no red/blue shift due to changes in the secondary structure of the DNA. The absorption spectra obtained here indicate that the complexes do not intercalate in the DNA base pairs.38 A gradual decrease in the absorption band has been observed for each complex upon the addition of DNA to the complex. Complexes 1–3 show analogous changes in the absorption band pattern (Fig. 6 and Fig. S8, S9, ESI†).
 | ||
| Fig. 6 UV-Vis spectra of 2 × 10−5 (M) DNA with incremental addition of complex 1 (0–120 μM). | ||
Analysis of the absorption spectroscopic data was carried out using the Benesi Hildebrand equation (eqn (1))39 for (1:1) complexation.
 | (1) |
To further support the interaction mode of the complexes with DNA obtained from the absorption titration study, a steady-state competitive binding experiment has been performed. EB is an excellent intercalator of B-DNA structures and gives strong fluorescence at around 590 nm when excited at ca. 510 nm. 20 μM EB saturated CT-DNA was fluorimetrically titrated in Tris–HCl buffer medium (5 mM Tris–HCl/50 mM NaCl, pH = 7.4) against each of the complexes. Upon addition of 20 μM complex each time, there is a gradual decrease in fluorescence intensity. This is owing to either replacement of the EB from DNA-bound EB or the complex is accepting an electron in an excited state or it is deforming the secondary structure of the DNA. Upon incremental addition of the complex, an abnormal hump at around 642 nm has been observed for the complexes.40 This is due to the fluorescence properties of the complex itself and probably the excitation wavelength is overlapping with that of the d–d transition of the complex. Upon exciting the complex in buffer medium at 510 nm, the same emission band was observed in the absence of CT-DNA. With an increase in concentration of the complex, the fluorescence intensity at 642 nm increases. Spectra of EB saturated DNA in the presence and the absence of the complexes are depicted in Fig. 7 and Fig. S13, S14 (ESI†).
To quantify the binding affinity, the Stern–Volmer equation41 has been employed;
 | (2) |
 | ||
| Fig. 7 Fluorescence spectra of (a) 20 μM EB bound DNA with incremental addition of complex 1 (0–120 μM). (b) Stern–Volmer plot for the quenching of the fluorescence of the ethidium bromide (EB)–DNA complex caused by complex 1. | ||
The apparent binding constant (Kapp) of each of the complexes with CT-DNA was calculated by employing the equation,42
| KEB[EB] = Kapp[Complex] | (3) |
| Complex | K b | K app |
|---|---|---|
| 1 | 7.11 × 105 | 7.5 × 105 |
| 2 | 8.36 × 105 | 8.33 × 105 |
| 3 | 10.81 × 105 | 10.71 × 105 |
These data suggest minor groove binding to the DNA bases.44 To evaluate the binding mode, we carried out viscosity measurements for the DNA solution in the absence and in the presence of complexes 1–3. Our data reveal that there is no enhancement of the viscosity of the DNA solution in the presence of the said complexes. This experiment rules out the chance of intercalation of compounds 1–3 inside the base pairs. Details of these studies along with the results are given in the ESI† (Fig. S15).
Binding stoichiometry (Job's plot)
The binding stoichiometry of the complexes with CT-DNA was determined by employing the continuation variation method known as Job's plot (Fig. S16–S18, ESI†) using UV-Vis absorption spectroscopy. In our study, the inflection point was found to be at χcomplex = 0.5. From the inflection point, the stoichiometry of association was found to be 1:1.
Circular dichroism study
We have also made an endeavor to explore the binding mode of the complexes 1–3 with CT-DNA employing the circular dichroism technique. When a small molecule binds to DNA by the mechanism of intercalation, the secondary structure of DNA is perturbed markedly and there is a signature change in the intrinsic CD spectra of DNA.45 On the other hand, groove binding does not have so much impact on the CD signal. Fig. S19 (ESI†) depicts the effect of the complexes 1–3 on the CD spectrum of CT-DNA. Fig. S19 (ESI†) shows the characteristics of the right-handed B form. Gradual addition of the complexes to the DNA does not cause any significant change in the intrinsic CD spectrum of the DNA. This observation predicts that none of the complexes intercalate into the DNA helix, rather they associate with DNA by the groove binding mode.DNA cleavage studies of complexes 1–3
The stability of DNA is determined by the phosphodiester backbone. The relative potential of copper(II) complexes to cleave pUC19 plasmid DNA (60 μM) in the presence of an activator i.e. hydrogen peroxide (250 μM) (H2O2) was studied by the agarose gel electrophoresis method in 5 mM Tris–HCl/50 mM NaCl buffer (pH 7.4) at 37 °C. The nuclease competence of the complexes (1–3) was measured by their ability to convert the SC DNA (form I) into nicked circular (NC) DNA (form II) followed by conversion into a linear form (form III) with prominent DNA cleavage. It is known that SC DNA (form I) migrates at a relatively faster rate compared to nicked circular (NC) DNA (form II) and the linear form (form III).The cleavage competence increased with time, and the quantity of pUC19 plasmid DNA decreased gradually due to the degradation of SC DNA (form I) to the nicked circular (form II) and linear DNA (form III), respectively. In Fig. 8, lane 1 i.e. the control shows no cleavage of the SC form of pUC19 plasmid DNA without any added complex. It has also been found that plasmid DNA incubated with either the copper(II) chloride and hydrogen peroxide or complex alone does not show any change in the supercoiled form (Fig. 8 lanes 2 and 4). In the presence of hydrogen peroxide (H2O2), the copper(II) complexes can cleave supercoiled plasmid DNA (Lane 3). This advocates that these complexes mediated DNA cleavage via an oxidative mechanism. The observed distribution of supercoiled, nicked circular and linear forms of DNA in the agarose gels gives an account of the degree of DNA cleavage by these complexes. Complexes 1–3 showed an identical result. Only complex 1 is represented in Fig. 8.
 | ||
| Fig. 8 DNA cleavage of pUC19 plasmid DNA. Lane 1, DNA + H2O2; lane 2, DNA + CuCl2 + H2O2; lane-3, DNA + complex 1 + H2O2; lane 4, DNA + complex 1. | ||
Time-dependent DNA cleavage study
A time-dependent assay was performed to determine the ability of DNA cleavage of every individual copper(II) complex. In the presence of H2O2, complexes 1–3 show significant DNA cleavage activity. At a 250 μM concentration of H2O2, all the complexes convert SC DNA (form I) into nicked circular (NC) DNA (form II) followed by conversion into the linear form (form III) with prominent DNA cleavage. Complex 1 shows around 40% cleavage (NC) (form II), whereas complexes 2 and 3 show around 55% and 50% cleavage (NC) (form II), respectively, upon 4 h of incubation under identical conditions. The concentration dependence of the complexes of this oxidative DNA cleavage study has been studied keeping the concentration of supercoiled pUC19 DNA (60 μM) fixed. It has been found that complex 3 shows maximum DNA cleavage at 50 μM DNA concentration. With an increase in the concentration of complexes 2 and 3, the percentage of NC DNA (form II) increases and complete conversion occurs at nearly 20 μM complex concentration. After 4 h of incubation, NC DNA (form II) converts into the linear form (III). After 4 h of incubation, the order of extent of DNA cleavage has been found to be 3 > 2 > 1. Under similar conditions, a lower amount of the linear form of DNA has been found for complex 1, whereas almost equal proportions of NC and linear forms of DNA have been found for complexes 2 and 3, respectively.The plausible mechanism of oxidative cleavage of SC pUC19 plasmid DNA by 1–3 was studied by adding various radical scavengers and singlet oxygen quenchers like DMSO, KI, NaN3, SOD and L-histidine to the reaction mixtures. The extent of oxidative cleavage of SC pUC19 plasmid DNA drastically falls upon addition of DMSO, which implies that the cleavage occurs due to the generation of ˙OH radicals. Sodium azide resulted in smearing at higher concentration. Other scavengers do not show any role in DNA cleavage. Therefore, only ˙OH radicals generated from copper–oxygen species are responsible for DNA cleavage (Table 4 and Fig. 9–12).
| Serial no | Reaction conditions | % Form | ||
|---|---|---|---|---|
| SC | NC | Linear | ||
| 1 | DNA control | 95 | 5 | 0 |
| 2 | DNA + H2O2 | 95 | 5 | 0 |
| 3 | DNA + CuCl2 + H2O2 | 95 | 5 | 0 |
| 4 | DNA + H2O2 + 1 | 40 | 45 | 15 |
| 5 | DNA + H2O2 + 2 | 0 | 55 | 45 |
| 6 | DNA + H2O2 + 3 | 0 | 50 | 50 |
 | ||
| Fig. 9 Time-dependent oxidative DNA cleavage of pUC19 plasmid DNA (60 μM) in the presence of 150 μM of H2O2 and 20 μM of complex 1 at 37 °C after incubation for 1–4 h; sampling was done at 1 h, 2 h, 3 h and 4 h and a 1% agarose gel was run. | ||
 | ||
| Fig. 10 Time-dependent oxidative DNA cleavage of pUC19 plasmid DNA (60 μM) in the presence of 150 μM of H2O2 and 20 μM of complex 2 at 37 °C after incubation for 1–4 h; sampling was done at 1 h, 2 h, 3 h and 4 h and a 1% agarose gel was run. | ||
 | ||
| Fig. 11 Time-dependent oxidative DNA cleavage of pUC19 plasmid DNA (60 μM) in the presence of 150 μM of H2O2 and 20 μM of complex 3 at 37 °C after incubation for 1–4 h; sampling was done at 1 h, 2 h, 3 h and 4 h and a 1% agarose gel was run. | ||
 | ||
| Fig. 12 Role of various radical scavengers and singlet oxygen quenchers on oxidative DNA cleavage. The scavengers were used with an appropriate concentration along with pUC19 plasmid DNA (60 μM) in the presence of 150 μM of H2O2 and 20 μM of complex 1 at 37 °C. Lane 1, DNA + complex 1; lane 2, DNA + complex 1 + L-histidine + H2O2; lane 3, DNA + complex 1 + NaN3 + H2O2; lane 4, DNA + complex 1 + KI + H2O2; lane 5, DNA + complex 1 + SOD + H2O2; lane 6, DNA + complex 1 + DMSO + H2O2. | ||
DFT study
DFT computation of the optimized structures of complexes 1–3 was performed in order to have a better understanding of structural information and electronic transitions. The calculated bond lengths and angles show good agreement with the X-ray structure (Table 2). The Mulliken charge distribution shows that the positive charge on the metal atoms is 0.641000, 0.644754 and 0.634335, respectively. The Mulliken charge distributions of complexes 1–3 are depicted in Table S1 (ESI†). The energy (eV) and composition of some selected molecular orbitals of complexes 1–3 and key electronic transitions are presented in Tables S2–S4, ESI† and Table 5, respectively.| E excitation (eV) | λ excitation (nm) | Osc. strength (f) | Key transition | Character | |
|---|---|---|---|---|---|
| Contour plots of some selected M.O.s are given in Fig. S20–S22 (ESI). | |||||
| Complex-1 | 2.05 | 603.53 | 0.0036 | HOMO−3(α) → LUMO(α) (40%), HOMO(β) → LUMO(β) (46%) | d–d |
| 3.15 | 394.18 | 0.2895 | HOMO(α) → LUMO(α) (13%), HOMO(β) → LUMO(β) (15%) | π → π* | |
| 3.29 | 376 | 0.762 | HOMO(α) → LUMO(α) (33%), HOMO(β) → LUMO+1(β) (43%) | π → π* | |
| 3.30 | 375.39 | 0.0323 | HOMO−4(α) → LUMO+2(α) (18%), HOMO−3(β) → LUMO(β) (23%) | π → π* | |
| 3.34 | 371.22 | 0.0015 | HOMO−2(β) → LUMO(β) (94%) | π → π* | |
| Complex-2 | 2.13 | 620.38 | 0.1817 | HOMO−3(α) → LUMO(α) (44%), HOMO−3(β) → LUMO+2(β) (33%) | d–d |
| 3.48 | 387.56 | 0.0052 | HOMO−1(α) → LUMO+2(α) (43%), HOMO−3(β) → LUMO+2(β) (38%) | π → π* | |
| 3.55 | 375.49 | 0.0746 | HOMO(α) → LUMO(α) (43%), HOMO(β) → LUMO(β) (43%) | π → π* | |
| Complex-3 | 2.36 | 609.53 | 0.0782 | HOMO(α) → LUMO+3(α) (36%), HOMO−1(β) → LUMO+1(β) (47%) | d–d |
| 3.44 | 385.43 | 0.0043 | HOMO−1(α) → LUMO(α) (41%), HOMO−2(β) → LUMO+1(β) (23%) | π → π* | |
| 3.79 | 373.45 | 0.0157 | HOMO−1(α) → LUMO+1(α) (44%), HOMO(β) → LUMO(β) (35%) | π → π* | |
TDDFT study
For a better understanding of the electronic transition, TDDFT calculations were performed using the B3LYP/CPCM method using the same basis sets in methanol. The calculated electronic transitions along with the calculated oscillator strengths (f) are given in Table 5. Complexes 1–3 show three intense absorption bands around 260 nm, 410 nm and 605 nm for ligand based n–π and π–π* transitions and the spin-allowed d–d transition of the Cu(II) metal ion, respectively. The π(L) → π*(L) transitions of complexes 1–3 are well characterized theoretically, including oscillation strength and key electronic transitions with their individual contribution. They are also depicted in Table 5. The spin-allowed d–d transition of complexes 1–3 is obtained around 605 nm, which is theoretically associated with 2.05 eV (λ = 603 nm, f = 0.0036), 2.13 eV (λ = 620 nm, f = 0.0817) and 2.23 eV (λ = 609 nm, f = 0.0043) amount of energy.Molecular docking studies with DNA
The results obtained from spectroscopic titration proposed that the three synthesized complexes do not interact with DNA via intercalation. The binding constant values were in the range of 105 M−1 for all three DNA-bound complexes. This diminishes the possibility of association through the mode of intercalation of these complexes between the DNA base pairs primarily due to less effective stacking forces between the aromatic nucleus and DNA bases.46 Molecular docking simulation was used to gain an insight into the preferential docking location and orientation of the complexes within the DNA groove. Fig. 13 represents the docked complexes of the three complexes with DNA. | ||
| Fig. 13 Stereo view of the docked conformation of complexes 1, 2 and 3 with DNA (panels A, C and E, respectively). The site of the interaction of the three complexes is magnified in the right panel (B, D and F) in the near vicinity (within 4.0 Å) of the DNA at the binding site. | ||
The right panel of each figure marks the DNA bases in the near vicinity (within 4 Å) of the complexes. It is observed that the minor groove of DNA is the most suitable position for the binding in the case of all three complexes. The minimized free energies of the docked structures of complexes 1, 2 and 3 with DNA were found to be −9.13, −9.47 and −9.44 kcal mol−1, respectively, as evidenced from molecular docking analyses. Energy minimized docked structures evidenced that the best optimized geometry of the metal complexes was bound to the AT-rich sequence of the minor groove as shown in Fig. 13.
Interactions between the complexes and DNA are primarily non-covalent interactions, which comprise hydrophobic as well as electrostatic interactions. The interactions take place either through the phosphate backbone atoms of the DNA or via the DNA bases.47 However, the interaction between the complexes with DNA cannot be accepted to be exclusively hydrophobic or electrostatic in nature as the prospect of hydrogen bonding cannot be ruled out. Inside the DNA groove, hydrogen bonding occurs between electronegative nitrogen or oxygen atoms of the three complexes and the NH of thiamine. The hydrogen bonded interactions of the three complexes within the DNA groove are presented in Fig. 14.
 | ||
| Fig. 14 Molecular modeling of the hydrogen bonded interaction of complexes 1 (panel A), 2 (panel B) and 3 (panel C) with DNA. Yellow dotted line represents the hydrogen bonding in each case. | ||
Cytotoxicity assay
The cytotoxicity of the three Cu(II) complexes has been studied through MTT assay in A549 lung cancer cell lines. Cytotoxicity and IC50 values have been determined for all complexes using the standard procedure. In the in vitro cytotoxicity assay with A549 cells, all three complexes did not show cytotoxicity at concentrations below 10 μM. But above the concentration of 10 μM, all three complexes exhibited cytotoxicity. The IC50 values of these complexes are given below (Fig. 15 and Fig. S23, S24). | ||
| Fig. 15 Represents % cell viability of A549 cells treated with different concentrations (0–150 μM) of complex 1 for 12 h determined by MTT assay. Results are expressed as the mean of three independent experiments. | ||
Since the Cu(II) complexes are found to have cytotoxicity at higher concentrations, they cannot be used as anticancer drugs. In this work we have established that depending upon the ligand system, simple Cu(II) complexes show different IC50 values and further studies can provide better insight for the development of anticancer drugs in the future.
Conclusion
Here, we have reported three novel Cu(II) complexes involving three new N2O4 donor azo Schiff base ligands. All the complexes have been well characterized by different spectroscopic tools and single crystal X-ray diffraction analysis. All structural and electronic parameters have been further justified by DFT and TDDFT computation. Here, the ligands are designed and synthesized in such a way that an azo arm is being appended to the para position in the phenyl ring with respect to the phenolic –OH group. The purpose of introducing the azo arm in the phenyl ring of o-vanillin is to have extended π–π stacking interactions within the molecular architecture/structure. As evidenced from the solid state architecture of all the complexes, there are nice and significant π–π stacking interactions present in every molecule apart from other CH-π and H-bonding interactions, which resulted in the formation of a 1D network. The azo arm is placed away from the donor centre of the ligand and it cannot directly take part in the coordination to the metal centre. Here, we expected that our complexes might exhibit intercalating properties with DNA through this long azo arm. Our purpose was to utilize π–π stacking interactions in DNA binding to get this type of intercalation. But, probably the overall bulkiness of the complexes is barring them from undergoing intercalation. The intrinsic binding constants (Kb) were calculated to be 7.11 × 105 M−1, 8.36 × 105 M−1 and 10.81 × 105 M−1 for complexes 1–3, respectively. The order of ability of oxidative DNA cleavage of the complexes has been found to be 3 > 2 > 1. This resulted in only groove binding to the AT-rich sequence of DNA primarily through hydrophobic or electrostatic interactions. Besides, hydrogen bonding interactions between electronegative nitrogen or oxygen atoms of the complexes and the NH of thiamine play a significant role. It has been confirmed that the overall hydrophobicity of the ligand system governed by the substituent present in the ligands of the Cu(II) complexes plays a crucial role in DNA binding, cleavage and anticancer properties. From the MTT assay it has been found that complexes show different IC50 values depending upon the ligand system. So, rational modification of the ligand system may be useful to develop DNA intercalators and anticancer drugs as well in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. gratefully acknowledges the financial support of this work by the DST, India (Sanction No. SB/FT/CS-102/2014, dated-18.07.2015). D. G. gratefully acknowledges the financial support of this work by DST, India, vide sanction no. SB/FT/LS-444/2012.Notes and references
- (a) E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS PubMed; (b) G. Sava, G. Jaouen, E. A. Hillard and A. Bergamo, Dalton Trans., 2012, 41, 8226–8234 RSC; (c) C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS; (d) N. P. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- (a) S. Packianathan, T. Arun and N. Raman, J. Photochem. Photobiol., B, 2015, 148, 160–167 CrossRef CAS PubMed; (b) C. Shiju, D. Arish, N. Bhuvanesh and S. Kumaresan, Spectrochim. Acta, Part A, 2015, 145, 213–222 CrossRef CAS PubMed.
- S. Delaney, M. Pascaly, P. K. Bhattacharya, K. Han and J. K. Barton, Inorg. Chem., 2002, 41, 1966–1974 CrossRef CAS PubMed.
- (a) A. Kumar, R. Pandey, R. K. Gupta, V. Mishra, S. M. Mobinb and D. S. Pandey, Dalton Trans., 2014, 43, 6365–6376 RSC; (b) T. Eren, M. Kose, N. Kurtoglu, G. Ceyhan, V. McKee and M. Kurtoglu, Inorg. Chim. Acta, 2015, 430, 268–279 CrossRef CAS; (c) M. Sarigul, P. Deveci, M. Kose, U. Arslan, H. T. Dagi and M. Kurtoglu, J. Mol. Struct., 2015, 1096, 64–73 CrossRef CAS; (d) G. Ozkan, M. Kose, H. Zengin, V. McKee and M. Kurtoglu, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2015, 150, 966–973 CrossRef CAS PubMed.
- (a) A. Kumar, A. Kumar and D. S. Pandey, Dalton Trans., 2016, 45, 8475–8484 RSC; (b) A. K. Mahapatra, S. K. Manna, K. Maiti, R. Maji, C. Das Mukhopadhyay, D. Sarkar and T. K. Mondal, RSC Adv., 2014, 4, 36615–36622 RSC; (c) Y. Zhang, Q. Li, J. Guo, Y. Li, Q. Yang and J. Du, RSC Adv., 2015, 5, 66674–66680 RSC; (d) C. J. Ward, P. Patel and T. D. James, J. Chem. Soc., Perkin Trans. 1, 2002, 462–470 RSC.
- D. R. Green and J. C. Reed, Science, 1998, 281, 1309–1312 CrossRef CAS PubMed.
- Y. Ma, L. Cao, T. Kawabata, T. Yoshino, B. B. Yang and S. Okada, Free Radical Biol. Med., 1998, 25, 568–575 CrossRef CAS PubMed.
- F. Liang, C. Wu, H. Lin, T. Li, D. Gao, Z. Li, J. Wei, C. Zheng and M. Sun, Bioorg. Med. Chem. Lett., 2003, 13, 2469–2472 CrossRef CAS PubMed.
- J. Easmon, G. Pürstinger, G. Heinisch, T. Roth, H. H. Fiebig, W. Holzer, W. Jäger, M. Jenny and J. Hofmann, J. Med. Chem., 2001, 44, 2164–2171 CrossRef CAS PubMed.
- S. Tabassum, S. Amir, F. Arjmand, C. Pettinari, F. Marchetti, N. Masciocchi, G. Lupidi and R. Pettinari, Eur. J. Med. Chem., 2013, 60, 216–229 CrossRef CAS PubMed.
- V. M. Manikandamathavan, V. Rajapandian, A. J. Freddy, T. Weyhermuller, V. Subramanian and B. U. Nair, Eur. J. Med. Chem., 2012, 57, 449–454 CrossRef CAS PubMed.
- (a) E. T. Adman, Adv. Protein Chem., 1991, 42, 145 CrossRef CAS PubMed; (b) M. Choi, N. Sukumar, A. Liu and V. L. Davidson, Biochemistry, 2009, 48, 9174–9184 CrossRef CAS PubMed.
- (a) D. S. Sigman, Acc. Chem. Res., 1986, 19, 180–186 CrossRef CAS; (b) D. S. Sigman, A. Mazumder and D. M. Perrin, Chem. Rev., 1993, 93, 2295–2316 CrossRef CAS; (c) D. Sigman, D. Graham, V. D’Aurora and A. Stern, J. Biol. Chem., 1979, 254, 12269–12272 CAS.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- (a) T. Deb, P. K. Gopal, D. Ganguly, P. Das, M. Paul, M. B. Saha, S. Paul and S. Das, RSC Adv., 2014, 4, 18419–18430 RSC; (b) K. Ghosh, P. Kumar, N. Tyagi, U. P. Singh, N. Goel, A. Chakraborty, P. Roy and M. C. Baratto, Polyhedron, 2011, 30, 2667–2677 CrossRef CAS; (c) J. Chen, X. Wang, Y. Shao, J. Zhu, Y. Zhu, Y. Li, Q. Xu and Z. Guo, Inorg. Chem., 2007, 46, 3306–3312 CrossRef CAS PubMed; (d) A. G. Papavassiliou, Biochem. J., 1995, 305, 345–357 CrossRef CAS PubMed.
- M. Odabaşoğlu, Ç. Albayrak, B. Koşar and O. Büyükgüngör, Spectrochim. Acta, Part A, 2012, 92, 357–364 CrossRef PubMed.
- S. Banerjee, P. Brandão and A. Saha, RSC Adv., 2016, 6, 101924 RSC.
- G. M. Sheldrick, SAINT, Version 6.02, SADABS, Version 2.03, Bruker AXS Inc., Madison, Wisconsin, 2002 Search PubMed.
- G. M. Sheldrick, SADABS: Software for Empirical Absorption Correction, University of Gottingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1999–2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- (a) P. Job, Ann. Chim. Appl., 1928, 9, 113–203 CAS; (b) C. Y. Huang, in Methods Enzymol., ed. S. P. C. Colowick, N. O. Kaplan, Academic Press, 1982, vol. 87, p. 509 Search PubMed.
- (a) A. Ray, G. Suresh Kumar, S. Das and M. Maiti, Biochemistry, 1999, 38, 6239–6247 CrossRef CAS PubMed; (b) K. Bhadra, G. Suresh Kumar, S. Das, Md. M. Islam and M. Maiti, Bioorg. Med. Chem., 2005, 13, 4851–4863 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- N. M. O’Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- M. S. Ray, R. Bhattacharya, S. Chaudhuri, L. Righi, G. Bocelli, G. Mukhopadhyay and A. Ghosh, Polyhedron, 2003, 22, 617–624 CrossRef CAS.
- I. S. Haworth, A. H. Elcock, J. Freemann, A. Rodger and W. G. J. Richards, J. Biomol. Struct. Dyn., 1991, 9, 23–43 CAS.
- (a) F. J. Meyer-Almes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS PubMed; (b) B. C. Baguley and M. LeBret, Biochemistry, 1984, 23, 937–943 CrossRef CAS PubMed; (c) R. F. Pasternack, M. Caccam, B. Keogh, T. A. Stephenson, A. P. Williams and E. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS; (d) M. Cory, D. D. McKee, J. Kagan, D. W. Henry and J. A. Miller, J. Am. Chem. Soc., 1985, 107, 2528–2536 CrossRef CAS; (e) J. R. Johnson, in Circular Dichroism: Principles and Applications, ed. K. Nakanishi, N. Berova and R. W. Woody, VCH, New York, 1994, pp. 523–540 Search PubMed; (f) X. Sheng, X.-M. Lu, Y.-T. Chen, G.-Y. Lu, J.-J. Zhang, Y. Shao, F. Liu and Q. Xu, Chem. – Eur. J., 2007, 13, 9703–9712 CrossRef CAS PubMed.
- M. G. Jelić, N. Boukos, M. M. Lalović, N. Ž. Romčević, V. M. Leovac, B. B. Hadžić, S. S. Baloš, L. S. Jovanović, M. P. Slankamenac, M. B. Živanov and L. S. Vojinović-Ješić, Opt. Mater., 2013, 35(12), 2728–2735 CrossRef.
- C. V. Kumar and E. H. Asuncion, J. Chem. Soc., Chem. Commun., 1992, 6, 470–472 RSC.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forror and J. A. Hartley, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed.
- (a) V. A. Bloomfield, D. M. Crothers and I. Tinoco, Physical Chemistry of Nucleic Acids, Harper and Row, New York, 1974, p. 432 Search PubMed; (b) F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Chem. Commun., 2005, 2540–2548 RSC; (c) J. Bernadou, G. Pratviel, F. Bennis, M. Girardet and B. Meunier, Biochemistry, 1989, 28, 7268–7275 CrossRef CAS PubMed.
- (a) A. Wolfe, G. H. Shimer Jr. and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed; (b) N. Chitrapriya, J. H. Shin, I. H. Hwang, Y. Kim, C. Kim and S. K. Kim, RSC Adv., 2015, 5, 68067–68075 RSC.
- (a) S. S. Jain, M. Polak and N. V. Hud, Nucleic Acids Res., 2003, 31, 4608–4615 CrossRef CAS PubMed; (b) J. L. Mergny, G. Duval-Valentin, C. H. Nguyen, L. Perrouault, B. Faucon, M. Rougee, T. Montenay-Garestier, E. Bisagni and C. Helene, Science, 1992, 256, 1681–1684 CAS.
- N. Raman, S. Sobha and A. Thamaraichelvan, Spectrochim. Acta, Part A, 2011, 78, 888–898 CrossRef CAS PubMed.
- F. Arjmand and I. Yousuf, J. Organomet. Chem., 2013, 743, 55–62 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1470725–1470727 (1–3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj03293e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |