
The molecular design of cage metal complexes for biological applications: pathways of the synthesis, and X-ray structures of a series of new N2-, S2- and O2-alicyclic iron(II) di- and tetrachloroclathrochelates†
Genrikh E.
Zelinskii
a,
Alexander S.
Belov
a,
Irina G.
Belaya
a,
Anna V.
Vologzhanina
 a,
Valentin V.
Novikov
a,
Oleg A.
Varzatskii
b and
Yan Z.
Voloshin
*ac
a,
Valentin V.
Novikov
a,
Oleg A.
Varzatskii
b and
Yan Z.
Voloshin
*ac
aNesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, 119991 Moscow, Russia. E-mail: voloshin@ineos.ac.ru
bVernadskii Institute of General and Inorganic Chemistry NASU, 03680, Kiev, Ukraine
cKurnakov Institute of General and Inorganic Chemistry of the Russian Academy of Sciences, 119991, Moscow, Russia
First published on 27th November 2017
Abstract
The synthesis of new metal(II) di- and tetrahalogenoclathrochelates with apical functionalizing substituents as reactive macrobicyclic precursors is a key stage of the molecular design of cage metal complexes – prospective biological effectors. We found that the most convenient multistep synthetic pathway for their preparation includes (i) direct template condensation of a dihalogeno-α-dioxime with an appropriately functionalized boronic acid on the corresponding metal ion as a matrix, giving an apically functionalized metal hexahalogenoclathrochelate in a high yield; and (ii) its stepwise nucleophilic substitution with S2-, N2- or O2-bis-nucleophiles, forming stable six-membered alicyclic ribbed fragments, thus allowing obtaining the corresponding apically functionalized di- and tetrahalogenoclathrochelates. The latter reaction of an iron(II) hexachloroclathrochelate with different N2-, S2- and O2-bis-nucleophilic agents afforded chloroclathrochelate complexes with equivalent and non-equivalent alicyclic ribbed substituents, such as N2-, S2 or O2-containing six-membered cycles. In the case of anionic forms of pyrocatechol and 1,2-ethanedithiol as O2- and S2-bis-nucleophiles, generated in situ in the presence of triethylamine, such substitution proceeds easily and in a high yield. In the case of anionic derivatives of ethylenediamine as N2-bis-nucleophiles, only a mono-N2-alicyclic iron(II) tetrachloroclathrochelate was obtained in a moderate yield. The S2-alicyclic iron(II) tetrachloroclathrochelate underwent a further nucleophilic substitution of one of the two dichloroglyoximate fragments, giving its N2, S2-alicyclic dichloroclathrochelate derivative with three non-equivalent ribbed chelate fragments. The complexes obtained were characterized using elemental analysis, MALDI-TOF mass spectrometry, and IR, UV-vis, 1H and 13C{1H} NMR spectroscopies, and by single crystal X-ray diffraction (XRD). As follows from XRD data for four O2-, S2- and N2-ribbed-functionalized iron(II) clathrochelates, the geometry of their FeN6-coordination polyhedra is intermediate between a trigonal prism and a trigonal antiprism. UV-vis spectra of these cage complexes are indicative of a dramatic redistribution of the electron density in a quasiaromatic clathrochelate framework caused by its ribbed functionalization with six-membered O2-, S2- and/or N2-alicyclic substituent(s).
Introduction
Several examples of efficient transcription inhibition by designed iron(II) cage and bis-cage complexes1,2 have been previously reported3–6 for a model in vitro system based on a T7 RNA polymerase (T7 RNAP). This small single-subunit polymerase is a convenient and reliable model in the search for transcription inhibitors7 and these ribbed-functionalized clathrochelates and bis-clathrochelates have demonstrated structure- and concentration-dependent inhibition in its transcription assay. Their inhibition mode has been deduced4 using experimental preincubation data and molecular docking calculations: the rigid and bulky three-dimensional inhibitor molecule is located in the same region of a supramolecular complex, formed by the macromolecules of T7 RNAP, matrix DNA and RNA synthesized – in the transcriptional “bubble” – and is involved in the intermolecular contacts with protein residues, as well as with DNA and RNA. The surfaces of DNA and RNA form the framework of the pocket for the inhibitor's binding, while the two structural elements of T7 RNAP interact with the ribbed phenyl substituents of a clathrochelate molecule, thus completing the formation of the above pocket. As a result, trapping of this macrobicyclic inhibitor occurred. So, the activity of such cage inhibitors is affected not only by a geometric complementarity between these clathrochelate guests and the above pockets as hosts, but also by the ability of the peripheral and terminal groups in their apical and ribbed substituents to form stable supramolecular bonds with hydrophobic and hydrophilic (polar) fragments of the biomacromolecules forming a hosting site. Moreover, such host–guest supramolecular binding depends not only on the hydrophilic–hydrophobic balance in an inhibitor cage molecule but also on the chemical nature of its peripheral and terminal groups: it is also affected by a spatial orientation of these groups within this pocket and, therefore, by their positions at a rigid quasiaromatic clathrochelate framework. Among the iron(II) clathrochelates and bis-clathrochelates, which have been tested in this model system to date, monoribbed-functionalized cage complexes (Scheme 1) with hydrophobic ribbed substituents in two of their three chelate fragments, with the third fragment of this type containing the functionalizing substituents with terminal biorelevant polar and H-bond donors (in particular carboxyl) terminal groups, have been previously recognized as the most efficient submicromolar inhibitors.4,6 The same iron(II) cage complexes have recently been recognized8 as efficient transcription inhibitors in the system of DNA polymerase. These leader cage and bis-cage inhibitors have been studied9 for their binding to bovine serum albumin (BSA), β-lactoglobulin, lysozyme, and insulin using steady-state and time-resolved fluorescence spectroscopies. Quenching of the protein's fluorescence and a decrease in its excited state lifetime have been observed only in the case of BSA, thus suggesting the formation of strong supramolecular assemblies by these mono- and bis-clathrochelates with serum albumins, but not with other globular proteins.9 These compounds have been also tested10 as prospective antifibrillogenic agents in the in vitro system with insulin. They were found to substantially affect the kinetics of insulin fibrillization, thus reducing the amount of fibrils formed, decreasing their diameter and preventing the lateral aggregation of mature fibrils and the formation of superfibrillar clusters.10 Very recently, based on in vitro studies, we found11 that an iron(II) hexachloroclathrochelate exhibits substantial cytotoxicity and a high selectivity (as compared with normal cells) in human promyelocytic leukemia cells. Such toxicity is explained11 by the ability of this electrophilic complex to increase the intracellular oxidative stress, caused by alkylation of glutathione, thus leading to the inhibition of the cellular antioxidative system, as well as by the catalytic generation of reactive oxygen species with partially alkylated clathrochelate products.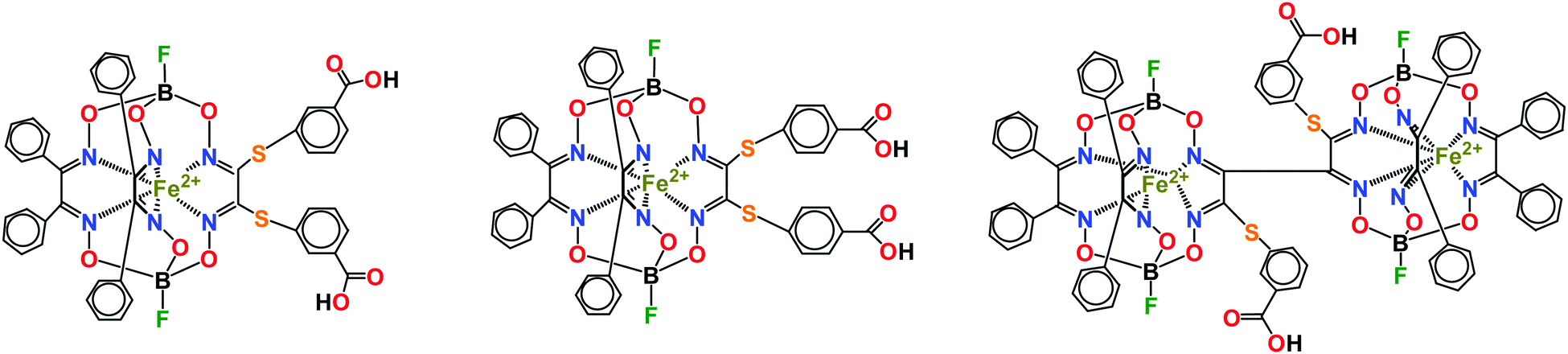 | ||
| Scheme 1 The most efficient clathrochelate and bis-clathrochelate inhibitors in the transcription system of T7 RNAP. | ||
The previously elaborated most convenient pathway for the synthesis of monoribbed-functionalized iron and cobalt mono- and dihalogenoclathrochelates12–16 is based on the condensation of the corresponding mono- or dihalogeno-α-dioxime with a macrocyclic metal bis-dioximate, the derivative of an aliphatic or aromatic α-dioxime, followed by chemical transformations of thus obtained mono- or dihalogenoclathrochelate precursors. However, this synthetic approach can be performed under vigorous reaction conditions only for several types of α-dioximes and only for fluoroboron-capped cage complexes. The bis-α-benzildioximate mono- and dihalogenoclathrochelates of this type have been recognized as the most suitable reactive macrobicyclic precursors of the above biological effectors: they easily undergo well-known classical organic reactions, such as N,O,S,C,P-nucleophilic substitution12,13,17–22 (including cadmium-promoted reactions with low-active nucleophiles21,22), free-radical substitution,23–28 copper-promoted cyanation,29 electrophilic addition (for their amine derivatives30,31), and copper(0)- and copper(I)-promoted reactions of halogen exchange,32 reductive homocoupling,6,33 and hydrodehalogenation.29,33 However, the apical functionalization of these fluoroboron-capped halogenoclathrochelate precursors, allowing tuning the physical properties of their macrobicyclic derivatives (in particular their solubility in water or biological media), and performing their efficient binding to a given biological target, is hardly possible (or even impossible). In this work, we aimed to elaborate the efficient synthetic strategies and procedures for a gram-scale synthesis of new iron(II) di- and tetrahalogenoclathrochelates as suitable macrobicyclic precursors for the molecular design of cage metal complexes (prospective biological effectors) and to study their structure and reactivity.
Experimental
The reagents used, triethylamine, pyrocatechol (Prch), 1,2-ethylenediamine, 1,2-ethanedithiol, sorbents and organic solvents were obtained commercially (SAF). The hexachloroclathrochelate Fe(Cl2Gm)3(Bn-C4H9)2 (where Cl2Gm2− is a dichloroglyoxime dianion) was prepared as described in ref. 17.Analytical data (C, H, N contents) were obtained with a Carlo Erba model 1106 microanalyzer.
MALDI-TOF mass spectra were recorded with and without a matrix using a MALDI-TOF-MS Bruker Autoflex II (Bruker Daltonics) mass spectrometer in reflecto-mol mode. The ionization was induced by a UV-laser with a wavelength of 337 nm. The samples were applied to a nickel plate, and 2,5-dihydroxybenzoic acid was used as the matrix. The accuracy of measurements was 0.1%.
1H and 13C NMR spectra were recorded from solutions in CD2Cl2, CDCl3 and CD3OD with Bruker AMX-400 and Avance 600 spectrometers. The measurements were performed using the residual signals of these deuterated solvents.
UV-vis spectra of solutions in dichloromethane were recorded in the range 230–800 nm with a Varian Cary 50 spectrophotometer. The individual Gaussian components of these spectra were calculated using the Fityk program.34
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture). Then the reaction mixture was evaporated to dryness and the solid residue was extracted with benzene (5 ml). The extract was filtered, evaporated to dryness and the solid residue was extracted with a chloroform–hexane 3:1 mixture. The extract was flash-chromatographically separated on silica gel (30 mm layer; first eluent: hexane; second eluent: hexane–chloroform 2:1 mixture). The first elute was discarded; the second elute was filtered and evaporated to dryness and the solid residue was dried in vacuo. Yield: 0.072 g (70%). Anal. calc. (%) for C16Cl4B2O6S2H22FeN6: C, 28.35; H, 3.27; N, 12.40. Found (%): C, 28.49; H, 3.45; N, 12.34. 1H NMR (CD2Cl2, δ, ppm): 0.65 (t, 4H, CH2B), 0.93 (t, 6H, CH3), 1.40 (m, 8H, (CH2)2 (Bu)), 3.43 (s, 4H, SCH2). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.53 (s, CH3), 16.97 (br. s, CH2B), 26.29, 26.69 (two s, (CH2)2 (Bu)), 28.94 (s, SCH2), 129.40 (s, ClC
:1 mixture). Then the reaction mixture was evaporated to dryness and the solid residue was extracted with benzene (5 ml). The extract was filtered, evaporated to dryness and the solid residue was extracted with a chloroform–hexane 3:1 mixture. The extract was flash-chromatographically separated on silica gel (30 mm layer; first eluent: hexane; second eluent: hexane–chloroform 2:1 mixture). The first elute was discarded; the second elute was filtered and evaporated to dryness and the solid residue was dried in vacuo. Yield: 0.072 g (70%). Anal. calc. (%) for C16Cl4B2O6S2H22FeN6: C, 28.35; H, 3.27; N, 12.40. Found (%): C, 28.49; H, 3.45; N, 12.34. 1H NMR (CD2Cl2, δ, ppm): 0.65 (t, 4H, CH2B), 0.93 (t, 6H, CH3), 1.40 (m, 8H, (CH2)2 (Bu)), 3.43 (s, 4H, SCH2). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.53 (s, CH3), 16.97 (br. s, CH2B), 26.29, 26.69 (two s, (CH2)2 (Bu)), 28.94 (s, SCH2), 129.40 (s, ClC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 144.89 (s, SCN). MS (MALDI-TOF): m/z: 678 [M]+˙. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 256 (5.5), 288 (3.0), 294 (5.8), 355 (1.6), 452 (10), 457 (6.9).
:1 mixture). Then the reaction mixture was evaporated to dryness and the solid residue was extracted with benzene (5 ml). The extract was filtered, evaporated to dryness and the product was extracted with a hexane–dichloromethane 2:1 mixture. The extract was flash-chromatographically separated on silica gel (70 mm layer, eluent: hexane–dichloromethane 2:1 mixture). The first elute was discarded; the second elute was filtered and evaporated to dryness and the solid residue was dried in vacuo. Yield: 0.068 g (33%). Anal. calc. (%) for C18Cl2B2O6S4H26FeN6: C, 30.92; H, 3.75; N, 12.02. Found (%): C, 30.75; H, 3.78; N, 11.85. 1H NMR (CD2Cl2, δ, ppm): 0.64 (t, 4H, CH2B), 0.96 (t, 6H, CH3), 1.42 (m, 8H, (CH2)2 (Bu)), 3.43 (s, 8H, SCH2). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.55 (s, CH3), 17.24 (br. s, CH2B), 26.36, 26.92 (two s, (CH2)2 (Bu)), 28.93 (s, SCH2), 127.67 (s, ClCN), 143.16 (s, SCN). MS (MALDI-TOF): m/z: 699 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 256 (13), 293 (11), 350 (3.6), 467 (7.5), 470 (18).
N), 143.96 (s, NCN). MS (MALDI-TOF): m/z: 644 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 256 (2.7), 277 (10), 360 (6.7), 418 (4.1), 487 (10), 597 (0.7).
:1 mixture). Then ethylenediamine (0.103 ml, 1.5 mmol) was added to the reaction mixture (in three portions, during 3 h). Then the reaction mixture was evaporated to dryness, and the solid residue was extracted with benzene (5 ml). The extract was filtered, evaporated to dryness and the product was extracted with dichloromethane (2 ml). The extract was flash-chromatographically separated on silica gel (30 mm layer, first eluent: hexane–dichloromethane 1:1 mixture; second eluent: hexane–dichloromethane 1:2 mixture). The first elute was discarded; the second elute was filtered, evaporated to dryness and the solid residue was dried in vacuo. Yield: 0.41 g (81%). Anal. calc. (%) for C18Cl2B2O6S2H26FeN8: C, 32.61; H, 3.95; N, 16.90. Found (%): C, 32.48; H, 4.11; N, 16.70. 1H NMR (CD2Cl2, δ, ppm): 0.50 (t, 4H, CH2B), 0.91 (t, 6H, CH3), 1.35 (m, 8H, (CH2)2 (Bu)), 3.39 (s, 4H, SCH2), 3.49 (s, 4H, NCH2), 5.63 (s, 2H, NH). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.57 (s, CH3), 17.79 (br. s, CH2B), 26.55, 27.19 (two s, (CH2)2 (Bu)), 28.99 (s, SCH2), 40.87 (s, NCH2), 125.47 (s, ClCN), 141.24, 141.48 (two s, SCN + NCN). MS (MALDI-TOF): m/z: 665 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 236 (13), 289 (4.2), 378 (3.6), 411 (0.3), 426 (1.9), 447 (2.4), 484 (4.2), 521 (3.8), 535 (1.0), 577 (0.4).
:1 mixture). Then the reaction mixture was evaporated to dryness, the solid residue was extracted with dichloromethane (2 ml) and the extract was flash-chromatographically separated on silica gel (70 mm layer, eluent: dichloromethane–hexane 1:1 mixture). First two elutes were discarded; the third elute was filtered and evaporated to dryness. The solid residue was washed with ethanol and hexane, and dried in vacuo. Yield: 0.02 g (6%). Anal. calc. (%) for C32H30B2FeN6O12: C, 50.04; H, 3.94; N, 10.94. Found (%): C, 50.18; H, 3.80; N, 10.90. 1H NMR (CDCl3, δ, ppm): 0.63 (t, 4H, CH2B), 0.95 (t, 6H, CH3), 1.42 (m, 8H, (CH2)2 (Bu)), 7.15 (m, 4H, HC(Prch)), 7.28 (m, 4H, HC(Prch)). 13C{1H} NMR (CDCl3, δ, ppm): 14.34 (s, CH3), 26.27, 26.44 (two s, (CH2)2 (Bu)), 117.71, 125.98 (two s, HC(Prch)), 135.17 (s, OCC), 138.67 (s, CN). MS (MALDI-TOF): m/z: 768 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 268 (7.2), 282 (12), 294 (1.1), 429 (4.4), 432 (3.5), 464 (6.0), 466 (6.9).
:1 mixture). Then the reaction mixture was evaporated to dryness, the solid residue was extracted with dichloromethane (2 ml) and flash-chromatographically separated on silica gel (70 mm layer, eluent: dichloromethane–hexane 1:1 mixture). The first orange elute was filtered and evaporated to dryness. The solid residue was washed with hexane and dried in vacuo. Yield: 0.145 g (69%). Anal. calc. (%) for C20H22B2Cl4FeN6O8: C, 34.63; H, 3.20; N, 12.12. Found (%): C, 34.48; H, 3.15; N, 12.17. 1H NMR (CD2Cl2, δ, ppm): 0.66 (t, 4H, CH2B), 0.97 (t, 6H, CH3), 1.43 (m, 8H, (CH2)2), 7.27 (m, 2H, HC(Prch)), 7.35 (m, 2H, HC(Prch)). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.49 (s, CH3), 16.93 (br. s, CH2B), 26.36, 26.58 (two s, (CH2)2), 118.09, 126.97 (two s, HC(Prch)), 130.00 (s, ClCN), 136.40 (s, OCC), 138.63 (s, OCN). MS (MALDI-TOF): m/z: 694 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 239 (13), 273 (13), 283 (1.0), 343 (1.5), 422 (0.9), 436 (5.4), 450 (9.8).
:1 mixture). Then the reaction mixture was evaporated to dryness, the solid residue was extracted with dichloromethane (2 ml) and the extract was flash-chromatographically separated on silica gel (70 mm layer, eluent: dichloromethane–hexane 2:3 mixture). The first elute was discarded; the second elute was filtered and evaporated to dryness. The solid residue was washed with ethanol and hexane, and dried in vacuo. Yield: 0.28 g (61%). Anal. calc. (%) for C26H26B2Cl2FeN6O10: C, 42.73; H, 3.59; N, 11.50. Found (%): C, 42.83; H, 3.64; N, 11.60. 1H NMR (CDCl3, δ, ppm): 0.66 (t, 4H, CH2B), 0.94 (t, 6H, CH3), 1.42 (m, 8H, (CH2)2), 7.18 (m, 4H, HC(Prch)), 7.28 (m, 4H, HC(Prch)). 13C{1H} NMR (CDCl3, δ, ppm): 14.32 (s, CH3), 16.81 (br. s, CH2B), 26.08, 26.29 (two s, (CH2)2), 117.71, 126.22 (two s, HC(Prch)), 128.56 (s, ClCN), 135.51 (s, OCC), 138.41 (s, OCN). MS (MALDI-TOF): m/z: 731 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 253 (5.4), 273 (13), 302 (3.7), 423 (3.3), 447 (9.6), 461 (6.5).
N), 144.89 (s, SCN). MS (MALDI-TOF): m/z: 678 [M]+˙. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 256 (5.5), 288 (3.0), 294 (5.8), 355 (1.6), 452 (10), 457 (6.9).
:1 mixture). Then the reaction mixture was evaporated to dryness and the solid residue was extracted with benzene (5 ml). The extract was filtered, evaporated to dryness and the product was extracted with a hexane–dichloromethane 2:1 mixture. The extract was flash-chromatographically separated on silica gel (70 mm layer, eluent: hexane–dichloromethane 2:1 mixture). The first elute was discarded; the second elute was filtered and evaporated to dryness and the solid residue was dried in vacuo. Yield: 0.068 g (33%). Anal. calc. (%) for C18Cl2B2O6S4H26FeN6: C, 30.92; H, 3.75; N, 12.02. Found (%): C, 30.75; H, 3.78; N, 11.85. 1H NMR (CD2Cl2, δ, ppm): 0.64 (t, 4H, CH2B), 0.96 (t, 6H, CH3), 1.42 (m, 8H, (CH2)2 (Bu)), 3.43 (s, 8H, SCH2). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.55 (s, CH3), 17.24 (br. s, CH2B), 26.36, 26.92 (two s, (CH2)2 (Bu)), 28.93 (s, SCH2), 127.67 (s, ClCN), 143.16 (s, SCN). MS (MALDI-TOF): m/z: 699 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 256 (13), 293 (11), 350 (3.6), 467 (7.5), 470 (18).
N), 143.96 (s, NCN). MS (MALDI-TOF): m/z: 644 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 256 (2.7), 277 (10), 360 (6.7), 418 (4.1), 487 (10), 597 (0.7).
:1 mixture). Then ethylenediamine (0.103 ml, 1.5 mmol) was added to the reaction mixture (in three portions, during 3 h). Then the reaction mixture was evaporated to dryness, and the solid residue was extracted with benzene (5 ml). The extract was filtered, evaporated to dryness and the product was extracted with dichloromethane (2 ml). The extract was flash-chromatographically separated on silica gel (30 mm layer, first eluent: hexane–dichloromethane 1:1 mixture; second eluent: hexane–dichloromethane 1:2 mixture). The first elute was discarded; the second elute was filtered, evaporated to dryness and the solid residue was dried in vacuo. Yield: 0.41 g (81%). Anal. calc. (%) for C18Cl2B2O6S2H26FeN8: C, 32.61; H, 3.95; N, 16.90. Found (%): C, 32.48; H, 4.11; N, 16.70. 1H NMR (CD2Cl2, δ, ppm): 0.50 (t, 4H, CH2B), 0.91 (t, 6H, CH3), 1.35 (m, 8H, (CH2)2 (Bu)), 3.39 (s, 4H, SCH2), 3.49 (s, 4H, NCH2), 5.63 (s, 2H, NH). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.57 (s, CH3), 17.79 (br. s, CH2B), 26.55, 27.19 (two s, (CH2)2 (Bu)), 28.99 (s, SCH2), 40.87 (s, NCH2), 125.47 (s, ClCN), 141.24, 141.48 (two s, SCN + NCN). MS (MALDI-TOF): m/z: 665 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 236 (13), 289 (4.2), 378 (3.6), 411 (0.3), 426 (1.9), 447 (2.4), 484 (4.2), 521 (3.8), 535 (1.0), 577 (0.4).
:1 mixture). Then the reaction mixture was evaporated to dryness, the solid residue was extracted with dichloromethane (2 ml) and the extract was flash-chromatographically separated on silica gel (70 mm layer, eluent: dichloromethane–hexane 1:1 mixture). First two elutes were discarded; the third elute was filtered and evaporated to dryness. The solid residue was washed with ethanol and hexane, and dried in vacuo. Yield: 0.02 g (6%). Anal. calc. (%) for C32H30B2FeN6O12: C, 50.04; H, 3.94; N, 10.94. Found (%): C, 50.18; H, 3.80; N, 10.90. 1H NMR (CDCl3, δ, ppm): 0.63 (t, 4H, CH2B), 0.95 (t, 6H, CH3), 1.42 (m, 8H, (CH2)2 (Bu)), 7.15 (m, 4H, HC(Prch)), 7.28 (m, 4H, HC(Prch)). 13C{1H} NMR (CDCl3, δ, ppm): 14.34 (s, CH3), 26.27, 26.44 (two s, (CH2)2 (Bu)), 117.71, 125.98 (two s, HC(Prch)), 135.17 (s, OCC), 138.67 (s, CN). MS (MALDI-TOF): m/z: 768 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 268 (7.2), 282 (12), 294 (1.1), 429 (4.4), 432 (3.5), 464 (6.0), 466 (6.9).
:1 mixture). Then the reaction mixture was evaporated to dryness, the solid residue was extracted with dichloromethane (2 ml) and flash-chromatographically separated on silica gel (70 mm layer, eluent: dichloromethane–hexane 1:1 mixture). The first orange elute was filtered and evaporated to dryness. The solid residue was washed with hexane and dried in vacuo. Yield: 0.145 g (69%). Anal. calc. (%) for C20H22B2Cl4FeN6O8: C, 34.63; H, 3.20; N, 12.12. Found (%): C, 34.48; H, 3.15; N, 12.17. 1H NMR (CD2Cl2, δ, ppm): 0.66 (t, 4H, CH2B), 0.97 (t, 6H, CH3), 1.43 (m, 8H, (CH2)2), 7.27 (m, 2H, HC(Prch)), 7.35 (m, 2H, HC(Prch)). 13C{1H} NMR (CD2Cl2, δ, ppm): 14.49 (s, CH3), 16.93 (br. s, CH2B), 26.36, 26.58 (two s, (CH2)2), 118.09, 126.97 (two s, HC(Prch)), 130.00 (s, ClCN), 136.40 (s, OCC), 138.63 (s, OCN). MS (MALDI-TOF): m/z: 694 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 239 (13), 273 (13), 283 (1.0), 343 (1.5), 422 (0.9), 436 (5.4), 450 (9.8).
:1 mixture). Then the reaction mixture was evaporated to dryness, the solid residue was extracted with dichloromethane (2 ml) and the extract was flash-chromatographically separated on silica gel (70 mm layer, eluent: dichloromethane–hexane 2:3 mixture). The first elute was discarded; the second elute was filtered and evaporated to dryness. The solid residue was washed with ethanol and hexane, and dried in vacuo. Yield: 0.28 g (61%). Anal. calc. (%) for C26H26B2Cl2FeN6O10: C, 42.73; H, 3.59; N, 11.50. Found (%): C, 42.83; H, 3.64; N, 11.60. 1H NMR (CDCl3, δ, ppm): 0.66 (t, 4H, CH2B), 0.94 (t, 6H, CH3), 1.42 (m, 8H, (CH2)2), 7.18 (m, 4H, HC(Prch)), 7.28 (m, 4H, HC(Prch)). 13C{1H} NMR (CDCl3, δ, ppm): 14.32 (s, CH3), 16.81 (br. s, CH2B), 26.08, 26.29 (two s, (CH2)2), 117.71, 126.22 (two s, HC(Prch)), 128.56 (s, ClCN), 135.51 (s, OCC), 138.41 (s, OCN). MS (MALDI-TOF): m/z: 731 [M]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 253 (5.4), 273 (13), 302 (3.7), 423 (3.3), 447 (9.6), 461 (6.5).
X-ray crystallography
Single crystals of the complexes Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2, Fe(PrchGm)3(Bn-C4H9)2 and 1.5Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2·0.5Fe(S2-Nx)3(Bn-C4H9)2 were grown at room temperature from dichloromethane–hexane mixtures. In the latter case, a mixture of the clathrochelate products was used for crystallization. The intensities of reflections were measured with a Bruker Apex II CCD diffractometer with multilayer optics using Cu-Kα radiation (λ = 1.54178 Å) for the crystals 1.5Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2·0.5Fe(S2-Nx)3(Bn-C4H9)2 and Fe(PrchGm)3(Bn-C4H9)2 and Mo-Kα radiation (λ = 0.71073 Å, graphite monochromator) for the complex Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2. The structures were solved using the direct method and refined by full-matrix least squares against F2. Non-hydrogen atoms were refined in anisotropic approximation except the disordered sulfur and chlorine atoms in the crystal 1.5Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2·0.5Fe(S2-Nx)3(Bn-C4H9)2. The asymmetric unit of the crystal 1.5Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2·0.5Fe(S2-Nx)3(Bn-C4H9)2 contains two independent clathrochelate molecules. One of them is the molecule Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2, while a substantial residual density near the ribbed chlorine atoms was observed for the second macrobicyclic entity. These two peaks with high electron density values were refined as carbon atoms with partial occupancies. Free refinement of their occupancies gave values equal to 0.5; thus, an occupation of carbon atoms was fixed at 0.5. The positions of these ribbed substituents were split onto sulfur and chlorine atoms, and the S–C distances for this chelate fragment were constrained to be 1.75 Å. As a result, the second crystallographic position was found to be equiprobably occupied by the molecules Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2 and Fe(S2-Nx)3(Bn-C4H9)2. The best available single crystal of the clathrochelate Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2 had very poor quality that resulted in high convergence factors for its X-ray structure. H(C) atoms were included in the refinement by the riding model with Uiso(H) = nUeq(C), where n = 1.5 for the methyl groups and 1.2 for the other atoms. All calculations were performed using the SHELXL35 and OLEX236 program packages. The crystallographic data and experimental details are collected in Table S1 {see the ESI†}. CCDC 1433324–1433326 contain the supplementary crystallographic data for this paper.†Results and discussion
Feasible pathways for the synthesis of monoribbed-functionalized iron(II) clathrochelates with apical functionalizing substituents include (i) the direct template condensation using a mixture of the corresponding α-dioximes; (ii) the condensation of a suitable macrocyclic precursor with the corresponding α-dioxime; (iii) the halogenation of a suitable glyoximate clathrochelate precursor; and (iv) the partial and stepwise nucleophilic substitution of the apically functionalized metal hexachloroclathrochelates. It should be noted that the dihalogeno-α-dioximate ligands are substantially weaker donors than their aliphatic and aromatic analogs. As a result, the use of a mixture of the corresponding α-dioximes in the direct template condensation leads mainly to C3-symmetric clathrochelate products with equivalent ribbed fragments, but not the target cage complexes with non-equivalent chelate moieties. Condensation of macrocyclic bis-α-dioximates with these weakly donor dihalogenodioximes typically proceeds under vigorous reaction conditions, also giving C3-symmetric boron-capped aliphatic or aromatic tris-dioximates as the clathrochelate products of a symmetrization reaction. Moreover, the preparation of the above macrocyclic bis-dioximates, the derivatives of the corresponding functionalized boronic acids, as their precursors, seems to be a real synthetic challenge. Halogenation of apically functionalized glyoximate macrobicyclic complexes is complicated by side reactions of their apical and ribbed substituents, as well as by the complete destruction of their cage frameworks under these reaction conditions. Therefore, the latter pathway (iv) seems to be the most convenient: metal(II) hexahalogenoclathrochelates can be obtained in high yields using the direct template condensation on the metal(II) ion as a matrix.13,17,18 Their nucleophilic substitution with several bis-nucleophiles, forming stable six-membered alicyclic ribbed fragments,37,38 can be performed stepwise, thus giving the corresponding di- and tetrahalogenoclathrochelate precursors. Moreover, it was shown39 that nucleophilic substitution of vic-dichloroclathrochelates with secondary aliphatic amines as N-nucleophiles gives the monoamine macrobicyclic products of such substitution of only one of the two chlorine atoms in the vic-position of the same ribbed fragment. This partial functionalization allows obtaining the corresponding monohalogenoclathrochelates, which can undergo further nucleophilic substitution with more active nucleophiles, such as primary aliphatic amines and thiolate anions.Analogous synthetic pathways can also be used for the synthesis of tetrahalogenoclathrochelates. In this case, the preparation of their macrocyclic tetrahalogeno-bis-α-dioximate precursors seems to be especially questionable. Stepwise nucleophilic substitution using different bis-nucleophilic agents allows obtaining dichloroclathrochelate complexes with non-equivalent alicyclic ribbed substituents, such as N2- and S2-containing six-membered cycles. It should be noted that the remaining halogen atoms of the tetraamine iron(II) dihalogenoclathrochelates are unreactive under standard conditions of this reaction.39 In general, the inherent electron-donating amine groups at a quasiaromatic polyazomethine cage framework deactivate the remaining halogen substituents and, as a result, they do not undergo nucleophilic substitution even at high temperatures and with a large excess of a nucleophilic agent as well. In the case of anionic forms of ethylenediamine and 1,2-ethanedithiol as N2- and S2-bis-nucleophiles, generated in situ in the presence of triethylamine, such substitution proceeds easily and in a high yield due to the formation of stable six-membered alicyclic ribbed substituents at a macrobicyclic framework. Indeed, six-membered alicycles are most less strained,40 and therefore, cyclohexane-based metal tris-dioximate clathrochelates are substantially more stable than their cycloheptane-containing analogs,1 whereas the chelate complexes of cyclopentanedione-1,2 dioxime have not been reported in the literature to date. Previously, 1,2-ethanedithiol and ethylenediamine have been successfully used for the preparation of N2- and S2-alicyclic mono- and triribbed-functionalized iron, ruthenium(II), and cobalt(II,III) clathrochelates.37,38 They have been prepared in relatively high yields using intramolecular nucleophilic substitution (cyclization) and pseudo-high dilution reaction conditions, thus limiting the formation of various macrobicyclic by-products, which are known to be usually formed in the case of such polynucleophilic agents.
At the same time, nucleophilic substitution with anionic derivatives of aliphatic diols as O2-bis-nucleophiles proceeds without the destruction of a tris-dioximate clathrochelate framework only at very low temperatures in the presence of cadmium(II) amides as promoters, giving the target O2-alicyclic cage products in low yields.22 In contrast, the same reactions with phenolate anions or pyrocatechol-based podands as O-nucleophiles gave di- and triribbed-functionalized macrobicyclic tetra- and hexaphenolates, and bis- and tris-crown ether iron and ruthenium(II) clathrochelates, respectively, in moderate yields.17,18 So, we used nucleophilic substitution with anionic derivatives of pyrocathechol, generated in situ in the presence of triethylamine, for the preparation of iron(II) di- and tetrachloroclathrochelate precursors with O2-containing six-membered cyclic ribbed substituent(s).
In the case of anionic derivatives of ethylenediamine as N2-bis-nucleophiles, only mono-N2-alicyclic iron(II) tetrachloroclathrochelate Fe(N2-Nx)(Cl2Gm)2(Bn-C4H9)2 was obtained in a moderate yield. We failed to isolate its bis-N2-alicyclic dichloroclathrochelate analog even in a low yield: the use of an excess of ethylenediamine led to complete destruction of a cage framework. At the same time, the tetrachloroclathrochelate Fe(S2-Nx)(Cl2Gm)2(Bn-C4H9)2 undergoes further nucleophilic substitution of one of its two dichloroglyoximate fragments as shown in Scheme 2, thus giving N2, S2-alicyclic dichloroclathrochelate Fe(N2-Nx)(S2-Nx)(Cl2Gm)2(Bn-C4H9)2 with three non-equivalent ribbed chelate fragments. However, this complex cannot be regarded as a dichloroclathrochelate precursor: its two remaining chlorine atoms do not undergo nucleophilic substitution under standard reaction conditions. Their low reactivity is caused by the electron-donating effect of the inherent alicyclic diamine substituent at a quasiaromatic cage framework of Fe(N2-Nx)(S2-Nx)(Cl2Gm)2(Bn-C4H9)2. The same effect has been previously observed17,18,39 in a series of amine iron and ruthenium(II) tris-dioximate clathrochelates.
 | ||
| Scheme 2 Synthesis of S2- and N2-alicyclic di- and tetrachloroclathrochelates. | ||
Stepwise nucleophilic substitution with anionic derivatives of 1,2-ethanedithiol allowed us to obtain both mono- and diribbed-substituted S2-alicyclic tetra- and dichloroclathrochelates Fe(S2-Nx)(Cl2Gm)2(Bn-C4H9)2 and Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2 (Scheme 2). These cage complexes were obtained using a classical approach of macrocyclic chemistry41–44 under pseudo-high dilution conditions by a simultaneous dropwise addition of the reagents (i.e. the hexadichloroclathrochelate Fe(Cl2Gm)3(Bn-C4H9)2 and 1,2-ethanedithiol) in their molar ratios 1:1 and 1:2, respectively, to the intensively stirring solvent. However, in both these cases, the target clathrochelates were contaminated by their triribbed-functionalized analog Fe(S2-Nx)3(Bn-C4H9)2 and these mixtures are poorly separable even by column chromatography.
We also performed the study of a stepwise nucleophilic substitution of the hexachloroclathrochelate precursor Fe(Cl2Gm)3(Bn-C4H9)2 with the pyrocatecholate dianion. Mono- and bis-pyrocatecholate macrobicyclic products Fe(Cl2Gm)2(PrchGm)(Bn-C4H9)2 and Fe(Cl2Gm)(PrchGm)2(Bn-C4H9)2 were obtained in relatively high yields using clathrochelate precursor-to-nucleophile molar ratios 1:1 and 1:2, respectively (Scheme 3). At the same time, we did not detect the corresponding tris-pyrocatecholate cage complex with equivalent ribbed fragments among the clathrochelate products of nucleophilic substitution under standard conditions of this reaction. Such a C3-symmetric complex was obtained in a low yield (approximately 7%) only under vigorous reaction conditions with a large excess of pyrocatechol at high temperature using a boiling nitromethane as a solvent. In contrast, aiming to obtain the monoribbed-functionalized tetrachloroclathrochelate of this type, the complex Fe(Cl2Gm)2(PrchGm)(Bn-C4H9)2, and trying to avoid the formation of the bis-pyrocatecholate iron(II) macrobicyclic complex, we performed nucleophilic substitution at low temperature (−50 °C) in dichloromethane medium, thus allowing an increase in its yield of up to approximately 70%. At the same time, the complex Fe(Cl2Gm)(PrchGm)2(Bn-C4H9)2 was obtained in a relatively high yield in carbon tetrachloride as a solvent, allowing performing this reaction at approximately 75 °C.
 | ||
| Scheme 3 Synthesis of pyrocatechol cage complexes. | ||
The complexes obtained were characterized using elemental analysis, MALDI-TOF mass spectrometry, and IR, UV-vis, 1H and 13C{1H} NMR spectroscopies, and by single crystal X-ray diffraction.
The numbers and positions of the signals in the solution 1H and 13C{1H} NMR spectra of the diamagnetic iron(II) di- and tetrachloroclathrochelates and their derivatives (see the ESI,† Fig. S1–S16), and the ratios of the integral intensities of protons of the apical n-butyl groups, the aliphatic and aromatic chelate fragments and those of the functionalizing ribbed substituents in their 1H NMR spectra confirmed the compositions and symmetries of the macrobicyclic molecules obtained, thus allowing deducing their chemical constitution. In particular, the 13C{1H} NMR spectra of monoribbed-functionalized iron(II) tetrachloroclathrochelates Fe(N2-Nx)(Cl2Gm)2(Bn-C4H9)2 and Fe(S2-Nx)(Cl2Gm)2(Bn-C4H9)2, as well as those of the diribbed-functionalized dichloroclathrochelate Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2, the derivative of 1,2-ethanedithiol, whose molecules have a symmetry plane passing through their B⋯Fe⋯B axes, contain the signals of azomethine carbon atoms of two types. On the other hand, that of their analog Fe(N2-Nx)(S2-Nx)(Cl2Gm)(Bn-C4H9)2 with all the non-equivalent chelate fragment contains three signals of this type in the range 120–145 ppm: its molecule does not have the molecular C3 symmetry pseudoaxis, passing through the capping boron atoms, but it has a symmetry plane, passing through the middles of the chelate C–C bonds and this caged ion as well.
The molecular structures of the ribbed-functionalized iron(II) clathrochelates Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2, Fe(PrchGm)3(Bn-C4H9)2, Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2 and·Fe(S2-Nx)3(Bn-C4H9)2 are shown in Fig. 1–3; the main geometrical parameters of their macrobicyclic molecules are listed in Table 1. The Fe–N distances in these molecules fall within a narrow range (1.91–1.93 Å) and are characteristic of iron(II) tris-dioximate clathrochelates.1,2 A cage framework of the iron(II) complex Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2 is similar to those of the amine clathrochelates Fe((C6H11NH)2Gm)2(Cl2Gm)(BC6H5)239 and Fe((n-C4H9NH)2Gm)2(Cl2Gm)(BC6H5)2 with non-equivalent ribbed fragments.17 The encapsulated iron(II) ion is slightly shifted from the geometrical centers of their macrobicyclic frameworks in the direction of the α-dichloroglyoximate chelate fragment. The C–C bond lengths in these fragments (1.41–1.45 Å) are shorter than those in the amine chelate ribbed fragment (1.47 Å).
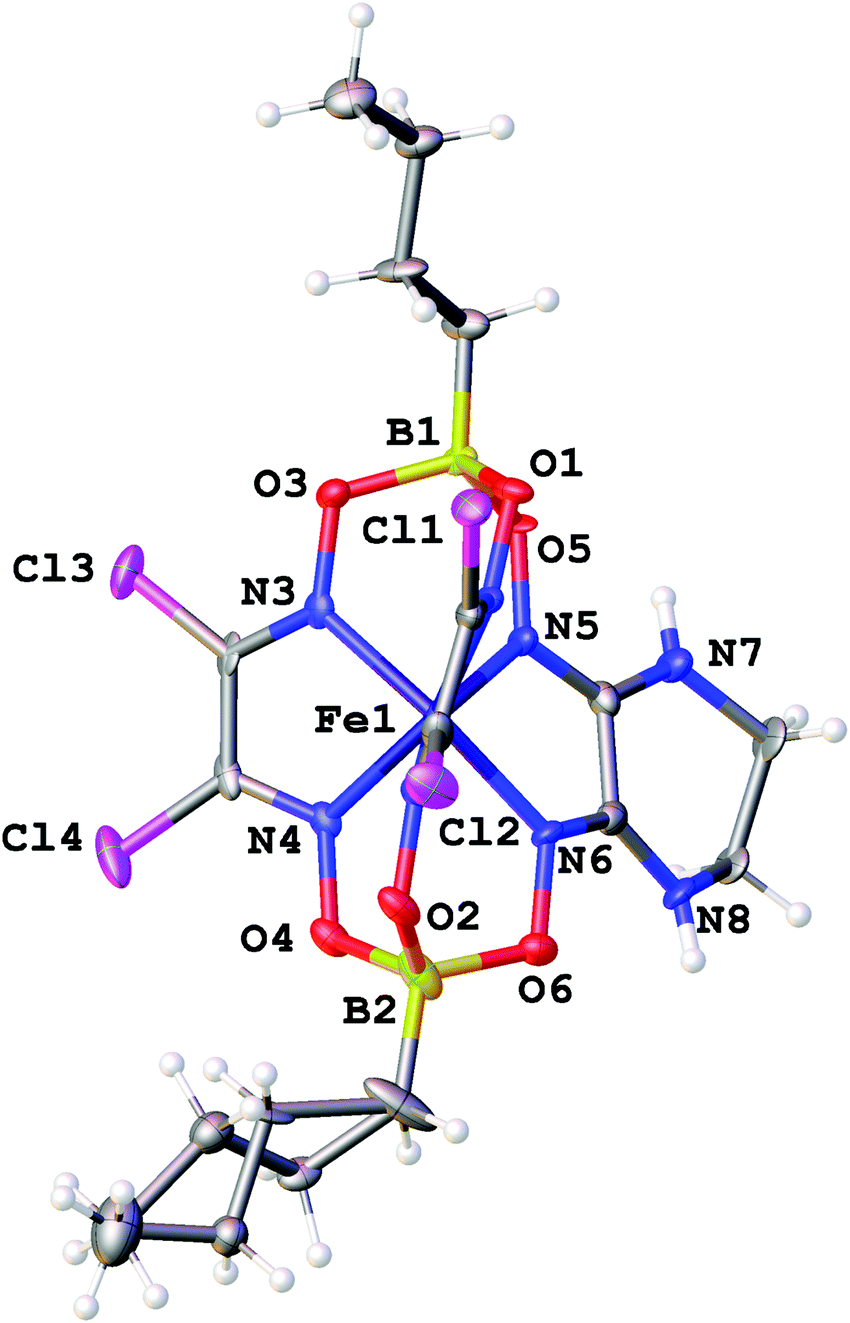 | ||
| Fig. 1 General view of the molecule Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2 in representation of atoms with thermal ellipsoids given with p = 50%. One of its apical n-butyl substituents is equiprobably disordered over two sites. | ||
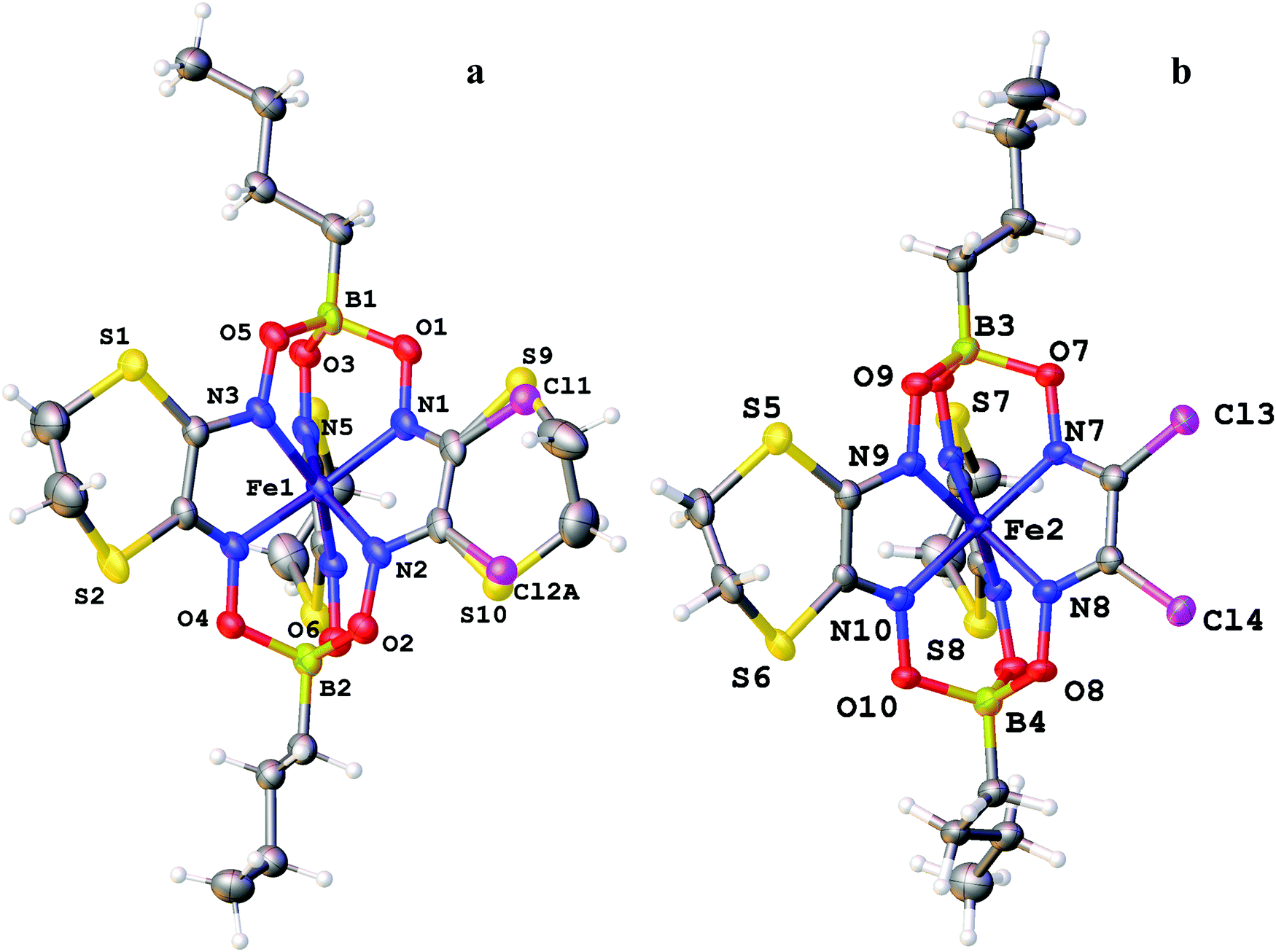 | ||
| Fig. 2 General views of the disordered (a) and ordered (b) molecules of the clathrochelate Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2 in representation of atoms with thermal ellipsoids given with p = 50%. The disordered crystallographic site is equiprobably occupied by the macrobicycles Fe(S2-Nx)3(Bn-C4H9)2 and Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2. | ||
 | ||
| Fig. 3 General view of the clathrochelate Fe(PrchGm)3(Bn-C4H9)2 in representation of atoms with thermal ellipsoids given with p = 50%. Only half of its molecule is symmetrically independent. | ||
| Parameter | Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2 | Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2 | Fe(PrchGm)3(Bn-C4H9)2 | |
|---|---|---|---|---|
| Type A | Type B | |||
| a The distance for the functionalized ribbed fragment. b Only half of this molecule is symmetrically independent. | ||||
| Fe–N(1) (Å) | 1.905(6) | 1.912(4)a | 1.909(4)a | 1.928(1) |
| Fe–N(2) (Å) | 1.918(6) | 1.904(4)a | 1.906(3)a | 1.935(1) |
| Fe–N(3) (Å) | 1.914(6) | 1.905(4) | 1.911(3) | 1.928(1) |
| Fe–N(4) (Å) | 1.913(6) | 1.910(4) | 1.903(3) | |
| Fe–N(5) (Å) | 1.933(6)a | 1.909(4) | 1.903(3) | |
| Fe–N(6) (Å) | 1.934(6)a | 1.906(4) | 1.907(4) | |
| B–O (Å) |
1.497(10)–1.538(11)
av. 1.519 |
1.499(6)–1.512(6)
av. 1.506 |
1.495(6)–1.524(5)
av. 1.509 |
1.505(2)–1.517(2)
av. 1.510 |
| N–O (Å) |
1.353(8)–1.415(7)
av. 1.379 |
1.364(5)–1.382(5)
av. 1.372 |
1.355(4)–1.373(4)
av. 1.366 |
1.369(1)–1.376(1)
av. 1.374 |
| CN (Å) |
1.283(9)–1.325(9)
av. 1.308 |
1.293(6)–1.313(5)
av. 1.303 |
1.300(5)–1.317(5)
av. 1.307 |
1.291(2)–1.300(2)
av. 1.295 |
| C–C (Å) |
1.417(11)–1.469(10)
av. 1.443 |
1.430(7)–1.449(6)
av. 1.439 |
1.429(6)–1.449(6)
av. 1.440 |
1.438(3)–1.444(2)
av. 1.442 |
| B–C (Å) |
1.570(11)–1.580(11)
av. 1.575 |
1.575(7)–1.587(7)
av. 1.365 |
1.570(6)–1.576(6)
av. 1.573 |
1.594(2) |
| NC–CN (°) |
9.0(11)–10.5(7)
av. 10.0 |
7.8(6)–10.8(7)
av. 9.3 |
4.1(5)–10.7(4)
av. 8.5 |
8.5(2)–9.2(2)
av. 9.0 |
| φ (°) | 26.5 | 25.3 | 24.0 | 23.4 |
| α (°) | 79.6 | 79.4 | 79.0 | 80.0 |
| h (Å) | 2.36 | 2.34 | 2.34 | 2.41 |
The Fe–N distances in the di- and triribbed-functionalized macrobicyclic molecules of the crystal 1.5Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2·0.5Fe(S2-Nx)3(Bn-C4H9)2 vary from 1.903(3) to 1.912(4) Å, thus being slightly smaller than those for their tris-pyrocatecholate clathrochelate analog (1.928(1)–1.935(1) Å). Although in all the cases the geometry of their FeN6-coordination polyhedra is intermediate between a trigonal prism (TP, the distortion angle φ = 0°) and a trigonal antiprism (TAP, φ = 60°), passing to the molecule Fe(PrchGm)3(Bn-C4H9)2 is accompanied by elongation of the Fe–N bonds and by an increase in φ from 23.4 to 26.5° due to a rotational–translational elongation of its cage framework. The height h of the corresponding coordination polyhedron is slightly greater (2.41 Å) as compared to those in the range 2.34–2.36 Å which have been observed for its n-butylboron-capped tetra- and hexafunctionalized clathrochelate analogs.1,2 Other geometrical parameters of their macrobicyclic frameworks are also characteristic of the boron-capped clathrochelate iron(II) tris-dioximates.
Strong intermolecular bonding is observed only in the crystal Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2 that contains the intermolecular N–H⋯O connected chains with a distance of ri(N⋯O) = 3.309(8) Å and with an N⋯H⋯O angle of approximately 157°, which are parallel to its crystallographic axis b (Fig. 4). In contrast, only weak intermolecular C–H⋯π and C–H⋯O interactions are observed in the crystals 1.5Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2·0.5Fe(S2-Nx)3(Bn-C4H9)2 and Fe(PrchGm)3(Bn-C4H9)2. The absence of strong intermolecular bonding in the case of the clathrochelates Fe(S2-Nx)2(Cl2Gm)(Bn-C4H9)2 and Fe(S2-Nx)3(Bn-C4H9)2 resulted in their co-crystallization, thus giving the corresponding co-crystals.
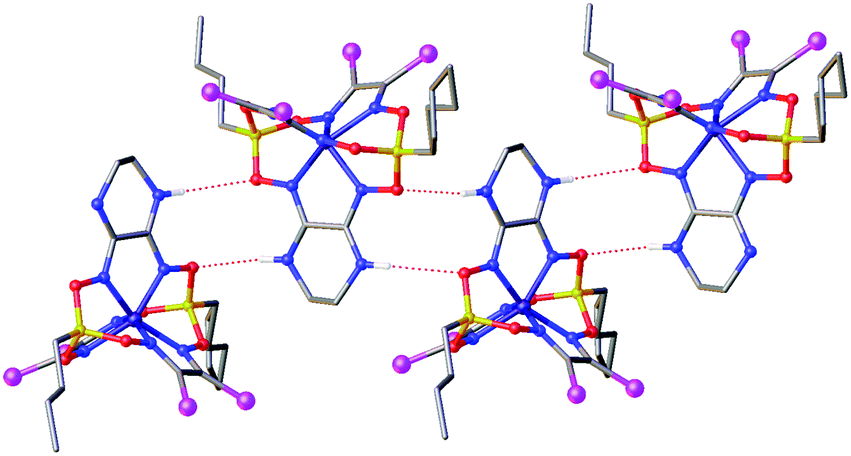 | ||
| Fig. 4 Hydrogen bonding between the clathrochelate molecules in the crystal Fe(Cl2Gm)2(N2-Nx)(Bn-C4H9)2. Hydrogen atoms, which do not form such supramolecular bonds, are omitted for clarity. | ||
Similar co-crystallization of the different clathrochelates has been previously observed15 for the two constitutional isomers of the complex Fe(CH3ClGm)(CH3(n-C4H9NH)Gm)2(BC6H5)2: 10% impurity of the chloroform solvate of its mer,fac,fac-isomer in the crystal of a mer,mer-isomer of this complex was detected. The co-crystal FeBd2((CF3)2Gm)(BF)2·FeBd2((CF3)GmI)(BF)2·2C6H14 that is isostructural to the complex FeBd2((CF3)2Gm)(BF)2·C6H14 is described;45 it should be noted that the attempted crystallization of a pure clathrochelate FeBd2((CF3)GmI)(BF)2 met with failure.45
Deconvolution of UV-vis spectra of the O2-, S2-, N2- and N2, S2-alicyclic iron(II) di- and tetrachloroclathrochelates under study (see the ESI,† Fig. S17–S23) into their Gaussian components gave in the visible range from two to six intense metal-to-ligand charge transfer (MLCT) Fed → Lπ* bands, respectively, with maxima in the range 410–600 nm; the bands of π–π* transitions in their polyazomethine macrobicyclic ligands were observed in the range 240–380 nm (see the ESI,† Table S2). The spectrum of the hexachloroclathrochelate Fe(Cl2Gm)3(Bn-C4H9)2 in its visible range contains two MLCT bands with maxima at approximately 420 and 450 nm. Thus, passing from two chlorine atoms in this dichloroclathrohelate molecule to its derivatives with O2-, S2-, N2- and N2, S2-alicyclic ribbed fragments led to a substantial (up to 40 nm) shift of the longwave band of this type and to the appearance of up to four new bands with maxima in the range 580–600 nm. This fact is indicative of a dramatic redistribution of the electron density in a quasiaromatic clathrochelate framework caused by its ribbed functionalization with six-membered O2-, S2- and N2-alicyclic substituent(s).
Conclusion
Thus, we developed straightforward synthetic strategies and convenient procedures allowing a gram-scale synthesis of various types of di- and tetrachloroclathrochelates, which seem to be suitable macrobicyclic precursors for the preparation of a wide range of cage complexes of given symmetry and functionality for their further biological testing in different in vitro, ex vivo and in vivo systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthesis of cage complexes was supported by the Russian Science Foundation (grant 16-13-10475). The spectral and X-ray diffraction studies were performed with the financial support of the EU Research and Innovation Staff Exchange (RISE) (H2020-MSCA-RISE-2017, Project 778245 ‘CLATHROPROBES’). The contribution of the Center for molecule composition studies of INEOS RAS is gratefully acknowledged. A. S. B., G. E. Z. and I. G. B. also thank the RFBR (grants 17-03-00587, 16-33-00146 and 16-33-60196) for the financial support.References
- Y. Z. Voloshin, N. A. Kostromina and R. Krämer, Clathrochelates: Synthesis, Structure and Properties, Elsevier, Amsterdam, 2002 Search PubMed.
- Y. Z. Voloshin, I. G. Belaya and R. Krämer, Cage metal complexes: clathrochelates revisited, Springer, Heidelberg, 2017 Search PubMed.
- Y. Z. Voloshin, V. V. Novikov and Y. V. Nelyubina, RSC Adv., 2015, 5, 72621–72637 RSC.
- V. V. Novikov, O. A. Varzatskii, V. V. Negrutska, Y. N. Bubnov, L. G. Palchykovska, I. Y. Dubey and Y. Z. Voloshin, J. Inorg. Biochem., 2013, 124, 42–45 CrossRef CAS PubMed.
- A. S. Belov, A. V. Vologzhanina, V. V. Novikov, V. V. Negrutska, I. Y. Dubey, Z. A. Mikhailova, E. G. Lebed and Y. Z. Voloshin, Inorg. Chim. Acta, 2014, 421, 300–306 CrossRef CAS.
- O. A. Varzatskii, V. V. Novikov, S. V. Shulga, A. S. Belov, A. V. Vologzhanina, V. V. Negrutska, I. Y. Dubey, Y. N. Bubnov and Y. Z. Voloshin, Chem. Commun., 2014, 50, 3166–3168 RSC.
- A. Oleksy, A. G. Blanco, R. Boer, I. Uson, J. Aymami, A. Rodger, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2006, 45, 1227–1231 CrossRef PubMed.
- Y. Z. Voloshin, K. Y. Zhizhin, V. V. Novikov, O. A. Varzatskii and N. T. Kuznetsov, 16th International Seminar on Inclusion Compounds (ISIC-16), Kazan (Russia), June 26–30, 2017, p. 36.
- M. Y. Losytskyy, V. B. Kovalska, O. A. Varzatskii, A. M. Sergeev, S. M. Yarmoluk and Y. Z. Voloshin, J. Fluoresc., 2013, 23, 889–895 CrossRef CAS PubMed.
- V. B. Kovalska, M. Y. Losytskyy, O. A. Varzatskii, V. V. Cherepanov, Y. Z. Voloshin, A. A. Mokhir, S. M. Yarmoluk and S. V. Volkov, Bioorg. Med. Chem., 2014, 22, 1883–1888 CrossRef CAS PubMed.
- J. Blechinger, O. A. Varzackii, V. Kovalska, G. E. Zelinskii, Y. Z. Voloshin, E. Kinski and A. Mokhir, Bioorg. Med. Chem. Lett., 2016, 26, 626–629 CrossRef CAS PubMed.
- Y. Z. Voloshin, V. E. Zavodnik, O. A. Varzatskii, V. K. Belsky, A. V. Palchik, N. G. Strizhakova and M. Y. Antipin, Dalton Trans., 2002, 1193 RSC.
- Y. Z. Voloshin, O. A. Varzatskii, V. V. Novikov, N. G. Strizhakova, I. I. Vorontsov, A. V. Vologzhanina, K. A. Lyssenko, G. V. Romanenko, M. V. Fedin, V. I. Ovcharenko and Y. N. Bubnov, Eur. J. Inorg. Chem., 2010, 5401–5415 CrossRef CAS.
- Y. Z. Voloshin, O. A. Varzatskii, A. V. Palchik, Z. A. Starikova, M. Yu. Antipin, E. G. Lebed and Y. N. Bubnov, Inorg. Chim. Acta, 2006, 359, 553–569 CrossRef CAS.
- Y. Z. Voloshin, O. A. Varzatskii, A. V. Palchik, I. I. Vorontsov, M. Y. Antipin and E. G. Lebed, Inorg. Chim. Acta, 2005, 358, 131–146 CrossRef CAS.
- Y. Z. Voloshin, A. S. Belov, A. V. Vologzhanina, G. G. Aleksandrov, A. V. Dolganov, V. V. Novikov, O. A. Varzatskii and Y. N. Bubnov, Dalton Trans., 2012, 41, 6078–6093 RSC.
- Y. Z. Voloshin, O. A. Varzatskii, T. E. Kron, V. K. Belsky, V. E. Zavodnik, N. G. Strizhakova and A. V. Palchik, Inorg. Chem., 2000, 39, 1907–1918 CrossRef CAS PubMed.
- Y. Z. Voloshin, O. A. Varzatskii, T. E. Kron, V. K. Belsky, V. E. Zavodnik, N. G. Strizhakova, V. A. Nadtochenko and V. A. Smirnov, Dalton Trans., 2002, 1203–1211 RSC.
- O. I. Artyushin, I. L. Odinets, E. V. Matveeva, A. V. Vologzhanina, O. A. Varzatskii, S. E. Lubimov and Y. Z. Voloshin, Dalton Trans., 2014, 43, 9677–9689 RSC.
- O. I. Artyushin, E. V. Matveeva, A. V. Vologzhanina and Y. Z. Voloshin, Dalton Trans., 2016, 45, 5328–5333 RSC.
- Y. Z. Voloshin, O. A. Varzatskii, P. A. Stuzhin, S. V. Shul'ga, S. V. Volkov, A. V. Vologzhanina, E. G. Lebed and Y. N. Bubnov, Inorg. Chem. Commun., 2011, 14, 1504–1507 CrossRef.
- O. A. Varzatskii, Y. Z. Voloshin, S. V. Korobko, S. V. Shulga, R. Krämer, A. S. Belov, A. V. Vologzhanina and Y. N. Bubnov, Polyhedron, 2009, 28, 3431–3438 CrossRef CAS.
- M. A. Vershinin, A. B. Burdukov, I. V. Eltsov, V. A. Reznikov, E. G. Boguslavsky and Y. Z. Voloshin, Polyhedron, 2011, 30, 1233–1237 CrossRef CAS.
- A. B. Burdukov, M. A. Vershinin, N. V. Pervukhina, S. G. Kozlova, I. V. Eltsov and Y. Z. Voloshin, Russ. Chem. Bull., 2011, 60, 2504–2509 CrossRef CAS.
- M. A. Vershinin, A. B. Burdukov, N. V. Pervukhina, I. V. Eltsov and Y. Z. Voloshin, Macroheterocycles, 2012, 5, 11–16 CrossRef CAS.
- M. A. Vershinin, A. B. Burdukov, N. V. Pervukhina, I. V. Eltsov and Y. Z. Voloshin, Macroheterocycles, 2015, 8, 71–74 CrossRef CAS.
- M. A. Vershinin, A. B. Burdukov, N. V. Pervukhina, I. V. Eltsov and Y. Z. Voloshin, Inorg. Chem. Commun., 2013, 30, 159–162 CrossRef CAS.
- M. A. Vershinin, A. B. Burdukov, N. V. Pervukhina and I. V. Eltsov, J. Struct. Chem., 2015, 56, 379–381 CrossRef CAS.
- Y. Z. Voloshin, A. S. Belov, O. A. Varzatskii, S. V. Shul’ga, P. A. Stuzhin, Z. A. Starikova, E. G. Lebed and Y. N. Bubnov, Dalton Trans., 2012, 41, 921–928 RSC.
- M. A. Vershinin, A. B. Burdukov, E. G. Boguslavskii, N. V. Pervukhina, N. V. Kuratieva, I. V. Eltsov, V. A. Reznikov, O. A. Varzatskii, Y. Z. Voloshin and Y. N. Bubnov, Inorg. Chim. Acta, 2011, 366, 91–97 CrossRef CAS.
- A. B. Burdukov, R. Šipoš, M. A. Vershinin, N. V. Pervukhina, N. V. Kuratieva, P. E. Plyusnin, I. V. Eltsov and Y. Z. Voloshin, J. Coord. Chem., 2015, 68, 3894–3902 CrossRef CAS.
- Y. Z. Voloshin, O. A. Varzatskii, S. V. Shul’ga, I. N. Denisenko, A. V. Vologzhanina and Y. N. Bubnov, Inorg. Chem. Commun., 2012, 17, 128–131 CrossRef CAS.
- O. A. Varzatskii, S. V. Shul'ga, A. S. Belov, V. V. Novikov, A. V. Dolganov, A. V. Vologzhanina and Y. Z. Voloshin, Dalton Trans., 2014, 43, 17934–17948 RSC.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. Z. Voloshin, O. A. Varzatskii, A. S. Belov, Z. A. Starikova, N. G. Strizhakova, A. V. Dolganov, D. I. Kochubey and Y. N. Bubnov, Inorg. Chim. Acta, 2010, 363, 134–146 CrossRef CAS.
- Y. Z. Voloshin, O. A. Varzatskii, A. S. Belov, Z. A. Starikova, A. V. Dolganov, V. V. Novikov and Y. N. Bubnov, Inorg. Chim. Acta, 2011, 370, 322–332 CrossRef CAS.
- V. E. Zavodnik, O. A. Varzatskii, V. K. Belsky, I. I. Vorontsov and M. Y. Antipin, Inorg. Chim. Acta, 2001, 321, 116–134 CrossRef.
- D. Barton and W. D. Ollis, Comprehensive organic chemistry, Pergamon, Oxford, 1979 Search PubMed.
- R. M. Izatt and J. J. Christensen, Synthesis of macrocycles, The design of selective complexing agents, Wiley-Interscience, New York, 1987 Search PubMed.
- D. Parker, Macrocycle synthesis: a practical approach, Oxford University Press, Oxford, 1996 Search PubMed.
- N. V. Gerbeleu, V. B. Arion and F. J. Burgess, Template synthesis of macrocyclic compounds, Wiley-VCH, Weinheim, 2000 Search PubMed.
- F. Diederich, P. J. Stang and R. R. Tykwinski, Modern supramolecular chemistry: strategies for macrocycle synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- O. A. Varzatskii, I. N. Denisenko, S. V. Volkov, A. V. Dolganov, A. V. Vologzhanina, Y. N. Bubnov and Y. Z. Voloshin, Inorg. Chem. Commun., 2013, 33, 147–150 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1433324–1433326. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj03051g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |