
Light driven epoxidation of olefins using a graphene oxide/g-C3N4 supported Mo (salen) complex†
Gang
Bian
 a,
Pingping
Jiang
*a,
Fang
Wang
a,
Yirui
Shen
a,
Kelei
Jiang
b,
Lin
Liu
a and
Weijie
Zhang
c
a,
Pingping
Jiang
*a,
Fang
Wang
a,
Yirui
Shen
a,
Kelei
Jiang
b,
Lin
Liu
a and
Weijie
Zhang
c
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: ppjiang@jiangnan.edu.cn
bWuxi WeiFu Lida Catalytic Converter Co. Ltd, Wuxi 214177, China
cState Key Laboratory for Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, and Engineering Research Center for Nano-Preparation Technology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China
First published on 3rd November 2017
Abstract
A novel Schiff (Mo) base complex supported on GO surface was successfully prepared, which was further combined with graphitic carbon nitride (g-C3N4) polymer (Mo-GO/g-C3N4) to make it photo-active. The photocatalytic activity of the resulting nano-composite was tested under simulated sunlight (AM 1.5) for the epoxidation of various olefin substrates. In addition, surface photovoltage (SPV) measurements were applied to explore the electron acceleration effect caused by GO and molybdenum. It was demonstrated that both GO and the metal active centers can efficiently accelerate electron transfer, which would finally contribute to the superior catalytic performance.
Introduction
Owing to the increasingly severe global energy crisis and the depletion of fossil fuels, numerous scientists have started to pay attention to the development of new energy sources. Remarkably, the utilization of solar energy is the most attractive as not only is sunlight renewable and the simplest energy source available but also solar energy is clean and environmentally-friendly.1–3 Thus far, solar energy has been applied in various reactions, such as overall water splitting,4,5 degradation of organic contaminants,6 organic synthesis7 and hydrogen (oxygen) evolution.8–10 Recently, great interest has been shown by chemists to carry out various photocatalytic oxidation reactions. Since Inoue et al. first applied tetraphenylporphyrinantimony(V) as a sensitizer in the photochemical epoxidation of alkenes,11 several photocatalytic epoxidation systems have been developed. Jafarpour et al.12 prepared a cobalt Schiff base complex immobilized onto TiO2 nanoparticles, which exhibited a synergistic effect under the visible-light irradiation. After a series of single factor tests, the authors observed that the composite exhibited high photocatalytic activity in epoxidation reactions. Chen et al.13 developed a manganese(V) nitrido complex as an efficient catalyst for the visible-light induced oxidation of both alkenes and alcohols. They demonstrated that MnV (N) is a useful platform for the construction of highly efficient photochemical oxidation catalysts.As mentioned above, the sensitizer is one of the most important components of a photocatalytic system. Recently, graphitic carbon nitride (g-C3N4) polymer, as a metal-free sensitizer, has attracted increasing interest due to its high quantum yield, strong stability, good biocompatibility and non-toxicity.14–17 Chen et al.18 found that g-C3N4 with a semiconductor absorption around 460 nm can be used to oxidize benzene to phenol. With the doping of transition metals to modify the semiconductor properties, they discovered that 10 wt% Fe-modified g-C3N4 supported on SBA-15 could achieve 6.7% phenol in the dark and 11.9% upon irradiation with visible-light. From this, it can be observed that metal doping is a simple and efficient way to enhance the photocatalytic performance of the catalysts. Moreover, the most widely used transition metals such as Fe, Co, V, Cu, Mn, Ti and Mo can be applied in the epoxidation reactions performed in our study.
Graphene oxide (GO), a new class of carbon nanomaterial with specific 2D structure and unique properties,19–21 has emerged as an ideal carrier for a variety of catalytic species.22–25 Besides its rich surface oxygen-containing functionalities, which could be beneficial for the catalytic reactions, the 2D structure not only exhibits a large surface area for excellent dispersion of the catalytic species but also facilitates the charge transfer during the reaction process. The active sites can be easily accessed by the substrates with limited mass transfer resistance through the deficient sites in GO, which significantly enhances the catalytic performance of graphene oxide-based composite catalysts.
Herein, we successfully immobilized a molybdenum (Schiff base) complex onto the GO surface and combined them with graphitic carbon nitride (g-C3N4) polymer (Mo-GO/g-C3N4). As shown in Scheme 1, the Schiff base ligand protrudes from the GO surface and can firmly grab the active metal centres, similar to the pliers of a crab, which can grip the Mo atoms tightly onto the GO surface. With the further incorporation of g-C3N4, the composite could be successfully applied to photocatalytic epoxidation using several olefin substrates.
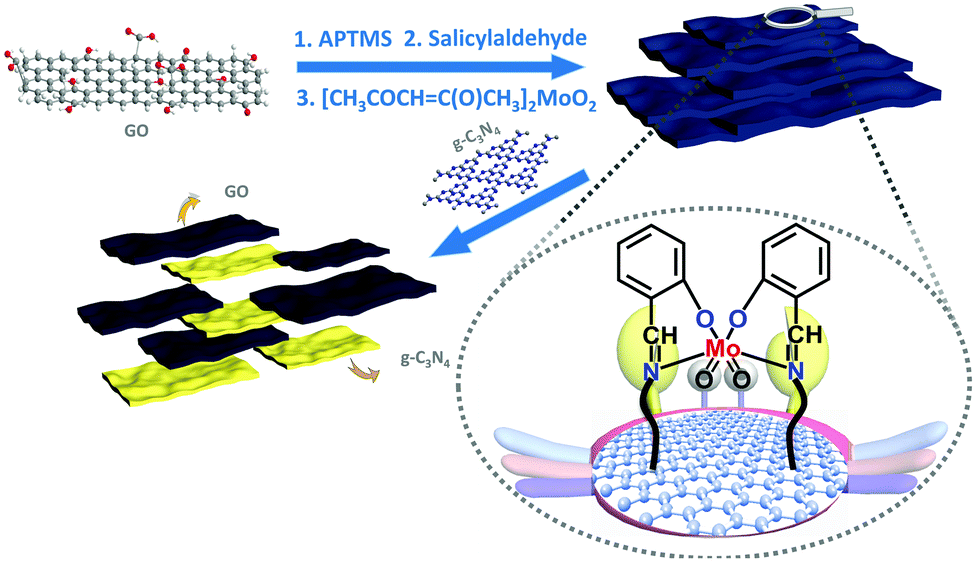 | ||
| Scheme 1 Synthetic route to prepare Mo-GO/g-C3N4 composite. | ||
Results and discussion
The FT-IR spectra are displayed in Fig. 1 and reveal the chemical nature of Mo-GO/g-C3N4. Two major characteristic peaks at 1322 and 1573 cm−1 from Mo-GO/g-C3N4 indicate the distinctive stretching modes of the aromatic C–N heterocycles.26 The sharp peak at 809 cm−1 verified the presence of triazine units in the catalyst, which confirmed the doping of the g-C3N4 nanosheets. In addition, the typical bond absorptions at 3406 cm−1 (–OH stretching vibration) and 1622 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in Mo-GO/g-C3N4 provided strong evidence for the existence of GO in the composite. In addition, Mo-GO/g-C3N4 showed a characteristic IR absorption in the region of 890–940 cm−1, which were assigned to the cis-MoO2 fragment.27,28 These results confirm the successful anchoring of the Mo complexes onto the GO surface.
C) in Mo-GO/g-C3N4 provided strong evidence for the existence of GO in the composite. In addition, Mo-GO/g-C3N4 showed a characteristic IR absorption in the region of 890–940 cm−1, which were assigned to the cis-MoO2 fragment.27,28 These results confirm the successful anchoring of the Mo complexes onto the GO surface.
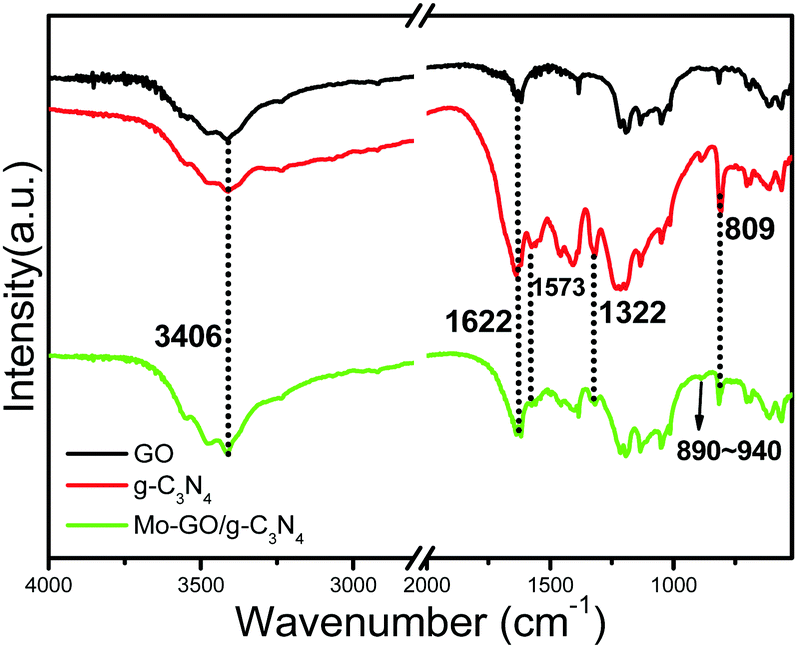 | ||
| Fig. 1 FT-IR spectra of GO, g-C3N4 and Mo-GO/g-C3N4. | ||
The chemical states of the individual elements in Mo-GO/g-C3N4 were investigated using X-ray photoelectron spectroscopy (XPS, Fig. 2). Fig. 2a shows peaks at binding energies of 284.2, 398.9, 532.3 and 231.8 eV for C 1s, N 1s, O 1s and Mo 3d, respectively. The high-resolution spectrum of C1s (Fig. 2b) shows two distinct peaks at 284.8 and 287.0 eV, which can be attributed to the non-oxygenated carbon (C–C/CC) peak from graphene oxide29 and N-sp2 carbon (C–N) in g-C3N4,30 respectively. The N 1s spectrum (Fig. 2c) can be fitted into two main peaks. One at 399.1 eV was assigned to the tertiary nitrogen bonded to the carbon atoms (N–(C)3) in g-C3N4;31 the other at 416.5 eV was due to the resonances of the N 2pxy orbitals.32 The O 1s spectrum (Fig. 2d) was distributed into two peaks. The major peak at 531.0 eV is the characteristic of Mo–O,33 indicating a strong coordination between the two elements. The shoulder peak at 532.0 eV indicated the existence of residual O2− species bonded to the C atoms in graphene oxide.34 As shown in Fig. 2e, two major peaks with binding energies at 233.2 and 236.4 eV correspond to Mo 3d5/2 and Mo 3d3/2, respectively, which revealed an oxidation state of Mo to be (6+).35,36
 | ||
| Fig. 2 Survey scan XPS spectra of Mo-GO/g-C3N4 (a) and high-resolution spectra of C 1s (b), N 1s (c), O 1s (d) and Mo 3d (e). | ||
Field-emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM) and scanning electron microscope-energy dispersive spectrometry (SEM-EDS) images are shown in Fig. 3 to present the morphology and elemental distribution of the samples. When compared with pristine g-C3N4 (Fig. 3a and c), Mo-GO/g-C3N4 not only maintained its original structure but also obtained more crumpling features due to the combination of the GO nanosheets. Similar characteristics are exhibited in the TEM images (Fig. 3b and d); Mo-GO/g-C3N4 contained both the plicated structure from g-C3N4 and the layered features of GO, which verified that our preparation process did not destroy the microstructure of the raw materials. In the SEM-EDS image of Mo-GO/g-C3N4 (Fig. 3f), it could be observed that Mo is homogeneously distributed throughout the composite, implying the uniform immobilization of the Mo (salen) complexes on the GO surface. Hence, it can be concluded that the immobilized catalyst would be fully extended and display a sheet-like morphology in the reaction solvent, which can serve as a versatile platform for access and removal with the surface catalytic active sites.37
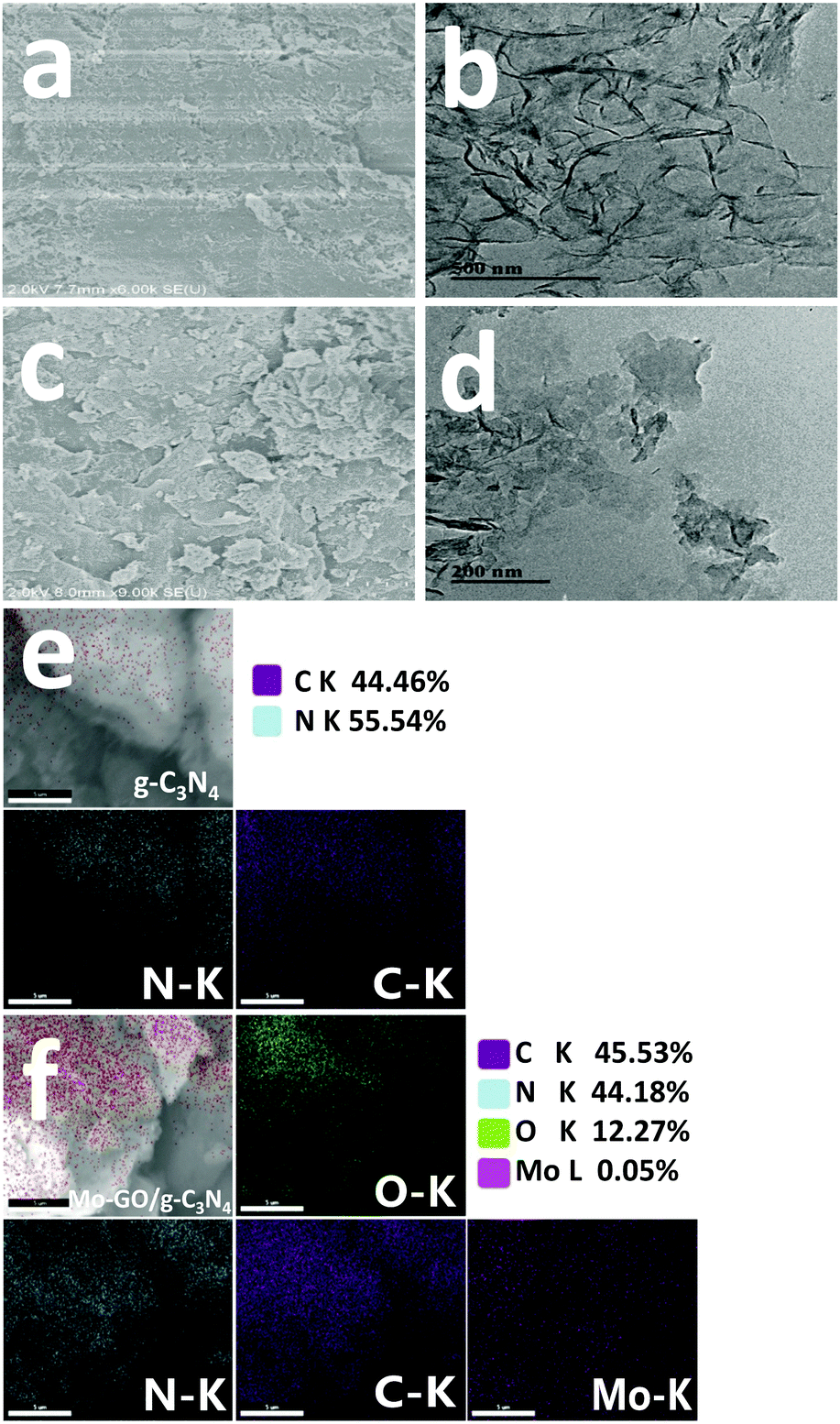 | ||
| Fig. 3 SEM (a) and TEM micrographs (b) and SEM-EDS element mapping images (e) of g-C3N4; SEM (c) and TEM micrographs (d) and SEM-EDS element mapping images (f) of Mo-GO/g-C3N4. | ||
After the successful preparation of Mo-GO/g-C3N4, its catalytic performance was evaluated through the epoxidation of olefins using O2 as the oxidant. First, the catalytic activity was evaluated in terms of the influence of the light source and solvent used in the epoxidation of cyclooctene. As shown in the left side of Fig. 4, three different light sources (visible-light, AM 1.5 and light lamp) were used to estimate the catalytic activity. Although the full wave band Xe Lamp exhibited the highest conversion, the overpowered intensity converted the epoxides into a variety of by-products such as aldehydes and alcohols, which suppressed the selectivity of the reaction to be as low as 64%. In addition, the catalytic performance under visible-light irradiation was even worse since the g-C3N4 material was excited the most in the ultra-violet light region (λ < 400 nm).38 Hence, to simultaneously optimize the conversion and selectivity, AM 1.5 was chosen as the light source. Remarkably, hardly any epoxide was detected when the reactor was placed under dark conditions, which implied that the reaction was driven specifically by the energy provided by the light source and not because of other factors. Moreover, the solvent effect was also investigated. As shown in the right side of Fig. 4, CH3CN provided the best catalytic activity. According to the solubility parameter close principle, CHCl3 should present the optimum catalytic performance in the epoxidation reaction since it has the highest solubility for O2. However, CH3CN, exhibiting the strongest polarity, showed the best reactivity. The N atoms in CH3CN can incorporate well with the –CN– groups in g-C3N4, which act as the catalytic carrier, thus the catalyst offered better contact with the substrates, which consequently enhanced the catalytic activity. The results obtained from the solvent contrast experiment are also in accordance with those obtained in an earlier report.39 Consequently, CH3CN was chosen to be the optimal reaction solvent. In addition, both the conversion and selectivity were obtained through the internal standard method according to the GC data, which was further confirmed by GC-MS. The method is presented in the section of “Photocatalytic tests” (ESI†) and the GC data of cyclooctene is provided in Fig. S1 (ESI†) as an example.
 | ||
| Fig. 4 Photocatalytic performance of Mo-GO/g-C3N4 during cyclooctene epoxidation assessed using different illuminantsa and solvents.b aReaction conditions: cyclooctene (10 mmol), CH3CN (10 mL, solvent), chlorobenzene (5 mmol), catalyst (10 mg) and O2. The mixture was continuously stirred under different illuminants for 6 h. b Reaction conditions: cyclooctene (10 mmol), solvent (10 mL), chlorobenzene (5 mmol), catalyst (10 mg) and O2. The mixture was continuously stirred under a Xe lamp (300 W) with an AM 1.5G filter for 6 h. | ||
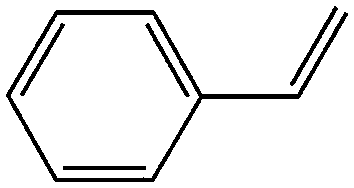
Further, a comprehensive assessment of the catalytic performance using Mo-GO/g-C3N4 was carried out using different olefin substrates and the results are presented in Table 1. When comparing the three different catalysts (g-C3N4, GO/g-C3N4 and Mo-GO/g-C3N4) during the epoxidation of cyclooctene (Table 1, entries 3, 5, 6), the addition of GO efficiently improved the selectivity, which was mainly attributed to the moderate oxygen-containing functionalities present on the GO surface.40 When the Mo active centers were introduced, both the conversion and selectivity were dramatically enhanced further. On one hand, Mo has a specific selectivity for the epoxidation products. On the other hand, the incorporation of metals ions can further accelerate the rate of electron transport, which also benefited the catalytic reactivity. Moreover, Mo-GO (Table 1, entry 4) without g-C3N4 had almost no photocatalytic activity in the epoxidation of cyclooctene. Due to the absence of a photosensitizer and the inability of molybdenum and GO to absorb light, the energy from illuminant cannot be supplied the reaction, thus inhibiting the epoxidation reaction. When compared with other photocatalysts used in epoxidation reactions,10,13,41 Mo-GO/g-C3N4 showed enhanced catalytic activity either in terms of the conversion or TOF value, indicating the superior activity of the photocatalytic system. Furthermore, it is well-known that the higher electron-donating ability of the olefin double bond always leads to increased epoxidation reactivity. Hence, cyclohexene and cyclooctene (Table 1, entries 2, 6) with internal double bonds exhibited higher reactivity when compared with 1-hexene and 1-octene (Table 1, entries 11 and 13), which contain terminal double bonds. Remarkably, Mo-GO/g-C3N4 revealed superior catalytic activity towards styrene (Table 1, entry 15). The π–π interactions between the sp2 hybridized C atoms in GO and benzene ring accelerated the electron transfer, which promoted the catalytic efficiency. Moreover, the –CN– groups in g-C3N4 would conjugate with the π-bonds in styrene, which further increased the activity.
| Entry | Substrate | Catalyst | Conversion (%) | Selectivity (%) | TOF (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: olefin (10 mmol), CH3CN (10 mL, solvent), chlorobenzene (5 mmol), catalyst (10 mg) and O2. The mixture was continuously stirred under a Xe lamp (300 W) equipped with an AM 1.5G filter for 6 h. | |||||
| 1 |

|
GO/g-C3N4 | 25 | 57 | 54 |
| 2 | Mo-GO/g-C3N4 | 40 | 98 | 149 | |
| 3 |

|
g-C3N4 | 28 | 27 | 29 |
| 4 | Mo-GO | 3 | — | — | |
| 5 | GO/g-C3N4 | 29 | 63 | 70 | |
| 6 | Mo-GO/g-C3N4 | 44 | 98 | 164 | |
| 7 | CoL2@TiO210 | 10 (yield) | — | — | |
| 8 | TiO2/AA/Fe41 | 23 (yield) | — | — | |
| 9 | [Mn(N)(CN)4]2–13 | — | — | 117 | |
| 10 |

|
GO/g-C3N4 | 20 | 43 | 33 |
| 11 | Mo-GO/g-C3N4 | 35 | 83 | 111 | |
| 12 |
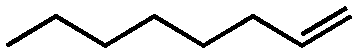
|
GO/g-C3N4 | 17 | 39 | 25 |
| 13 | Mo-GO/g-C3N4 | 30 | 79 | 90 | |
| 14 |
|
GO/g-C3N4 | 41 | 96 | 150 |
| 15 | Mo-GO/g-C3N4 | 81 | 98 | 302 | |
The stability of the Mo-GO/g-C3N4 composite was confirmed during the epoxidation of cyclooctene (Fig. 5). After the photocatalytic epoxidation, the catalyst was easily recovered from the reaction mixture using centrifugation and was ready for recycling after washing twice with water and acetonitrile, in sequence. As summarized in Fig. 6, the catalytic reactivity remained almost the same after recycling 6 times and the selectivity was still as high as 97%. A metal leaching test was also performed and the catalyst was screened by centrifugation and decantation of the mixture. No detectable molybdenum was detected based on the ICP-AES result, indicating its strong stability. Such superior stability primarily originated from the robust coordination between the metal active centers and the Schiff base, which formed covalent bonds and immobilized the Mo atoms firmly onto the GO surface. As proposed in Scheme 1, the Schiff-base ligand, which appeared like a large pincer of a crab, tightly vised the metal centers on the GO carrier, thus contributing to the outstanding stability.
 | ||
| Fig. 5 Recycle runs of Mo-GO/g-C3N4 during cyclooctene epoxidation.a aReaction conditions: cyclooctene (10 mmol), CH3CN (10 mL, solvent), chlorobenzene (5 mmol), catalyst (10 mg) and O2. The mixture was continuously stirred under a Xe lamp (300 W) equipped with an AM 1.5G filter for 6 h. | ||
 | ||
| Fig. 6 Proposed photocatalytic process of epoxidation catalyzed using Mo-GO/g-C3N4. | ||
To further confirm the excellent catalytic performance of the as-prepared catalyst and explore its mechanism, a conceivable catalytic process was proposed and surface photovoltage (SPV) measurements were applied to demonstrate this. As displayed in Fig. 6, the electrons and holes in g-C3N4 were both excited under light irradiation. In the presence of GO, the electrons and holes can be easily transferred, which prevents their recombination. Such hypothesis was consistent with the catalytic results; combining GO with the as-obtained composite efficiently improved the selectivity and produced more epoxides. Moreover, as the metal active centers (Mo) were introduced, the transport velocity of the electrons and holes was further accelerated because of the free electrons and unoccupied orbitals that exist in the outer-shell of the transition metal atoms. This accelerating effect has also been revealed in the epoxidation of cyclooctene (Table 1), in which the Mo active centers dramatically improved the activity. Due to the intrinsically high selectivity of molybdenum for epoxidation, the selectivity of cyclooctene was up to 98%. Finally, the holes were transferred through the metal sites and contributed to the generation of the epoxides.
From the SPV spectrum (Fig. 7), it can be easily identified that the intensities of the photoelectric signals were enhanced after the addition of both GO and molybdenum, which implies that the electron transfer was accelerated. In particular, the photoelectric signal was significantly increased when molybdenum was introduced. The SPV results confirmed our hypothesis that both GO and Mo can improve the catalytic activity, which may benefit our future study.
 | ||
| Fig. 7 Surface photovoltage (SPV) spectra of g-C3N4, GO/g-C3N4 and Mo-GO/g-C3N4 composites. | ||
Conclusions
In conclusion, we have demonstrated the synthesis of a Mo (Schiff base) complex immobilized on GO sheets. g-C3N4 was introduced as the sensitizer to harvest solar energy. The composite exhibited outstanding photocatalytic activity for epoxidation and was quite stable during the recycling tests. After six runs, the activity of the recycled catalyst was almost the same as the fresh catalyst and the selectivity was ∼97%. Moreover, the electron acceleration effect induced by GO and molybdenum was further demonstrated using surface photovoltage (SPV) measurements. Both of them can dramatically accelerate electron transfer. This type of designed strategy can also be readily extended to other catalytic reactions and more photocatalytic systems are anticipated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Financial Support from MOE & SAFEA, 111 Project (B13025), the Fundamental Research Funds for the Central Universities (JUSRP51507 and JUSRP51623A) and the Postgraduate Innovation Project of Jiangsu Province (KYLX15-1156).Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- L. Hammarström and S. Hammes-Schiffer, Acc. Chem. Res., 2009, 42, 1859 CrossRef PubMed.
- R. Eisenberg, Science, 2009, 324, 44 CrossRef CAS PubMed.
- Y. An, Y. Liu, P. An, J. Dong, B. Xu, Y. Dai, X. Qin, X. Zhang, W. Myung-Hwan and B. Huang, Angew. Chem., 2017, 129, 3082 CrossRef.
- B. Qiu, Q. Zhu, M. Du, L. Fan, M. Xing and J. Zhang, Angew. Chem., 2017, 129, 2728 CrossRef.
- X. Xiao, S. Tu, M. Lu, H. Zhong, C. Zheng, X. Zuo and J. Nan, Appl. Catal., B, 2016, 198, 124 CrossRef CAS.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188 RSC.
- J. Ran, G. Gao, F. T. Li, T. Y. Ma, A. Du and S. Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- C. Cui, S. Li, Y. Qiu, H. Hu, X. Li, C. Li, J. Gao and W. Tang, Appl. Catal., B, 2017, 200, 666 CrossRef CAS.
- M. Xing, B. Qiu, M. Du, Q. Zhu, L. Wang and J. Zhang, Adv. Funct. Mater., 2017, 1702624 CrossRef.
- H. Inoue, M. Sumitani, A. Sekita and M. Hida, Chem. Commun., 1987, 1681 RSC.
- M. Jafarpour, H. Kargar and A. Rezaeifard, RSC Adv., 2016, 6, 79085 RSC.
- G. Chen, L. Chen, L. Ma, H. K. Kwong and T. C. Lau, Chem. Commun., 2016, 52, 9271 RSC.
- C. Li, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, M. Craft, G. Chen and R. Xu, Adv. Funct. Mater., 2017, 27, 1604328 CrossRef.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336 CrossRef CAS PubMed.
- Y. Zhao and M. Antonietti, Angew. Chem., 2017, 56, 9336 CrossRef CAS PubMed.
- Z. Tong, D. Yang, Z. Li, Y. Nan, F. Ding, Y. Shen and Z. Jiang, ACS Nano, 2017, 11, 1103 CrossRef CAS PubMed.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed.
- A. A. Jeffery, S. R. Rao and M. Rajamathi, Carbon, 2017, 112, 8 CrossRef CAS.
- D. Wang, H. Duan, J. Lü and C. Lü, J. Mater. Chem. A, 2017, 5, 5088 CAS.
- S. Wang, R. Cazelles, W. C. Liao, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nano Lett., 2017, 17, 2043 CrossRef CAS PubMed.
- Q. Gu, G. Wen, Y. Ding, K. H. Wu, C. Chen and D. Su, Green Chem., 2017, 19, 1175 RSC.
- C. Zhang, Y. Lu, Q. Jiang and J. Hu, Nanotechnol., 2016, 27, 355402 CrossRef PubMed.
- Y. Li, X. Fu, B. Gong, X. Zou, X. Tu and J. Chen, J. Mol. Catal. A: Chem., 2010, 322, 55 CrossRef CAS.
- Y. Leng, J. Liu, C. Zhang and P. Jiang, Catal. Sci. Technol., 2014, 4, 997 CAS.
- K. S. Rao, J. Sentilnathan, H. W. Cho, J. J. Wu and M. Yoshimura, Adv. Funct. Mater., 2015, 25, 298 CrossRef CAS.
- H. Tao, C. Yan, A. W. Robertson, Y. Gao, J. Ding, Y. Zhang, T. Ma and Z. Sun, Chem. Commun., 2017, 53, 873 RSC.
- D. Peng, H. Wang, K. Yu, Y. Chang, X. Ma and S. Dong, RSC Adv., 2016, 6, 77760 RSC.
- N. Miyata, M. Yanagihara, M. Watanabe, Y. Harada and S. Shin, J. Phys. Soc. Jpn., 2002, 71, 1761 CrossRef CAS.
- D. M. Fernandes and C. Freire, J. Appl. Electrochem., 2014, 44, 655 CrossRef CAS.
- Y. Sun, X. Hu, W. Luo, F. Xia and Y. Huang, Adv. Funct. Mater., 2013, 23, 2436 CrossRef CAS.
- H. Cheng, X. Qian, Y. Kuwahara, K. Mori and H. Yamashita, Adv. Mater., 2015, 27, 4616 CrossRef CAS PubMed.
- S. Wang, B. Liu, Z. Yuan and Z. Zhang, J. Taiwan Inst. Chem. Eng., 2016, 58, 92 CrossRef CAS.
- J. Ji, G. Zhang, H. Chen, S. Wang, G. Zhang, F. Zhang and X. Fan, Chem. Sci., 2011, 2, 484 RSC.
- G. Dong, L. Yang, F. Wang, L. Zang and C. Wang, ACS Catal., 2016, 6, 6511 CrossRef CAS.
- M. Jafarpour, H. Kargar and A. Rezaeifard, RSC Adv., 2016, 6, 79085 RSC.
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., 2010, 122, 6965 CrossRef.
- M. Jafarpour, A. Rezaeifard and F. Feizpour, ChemistrySelect, 2017, 2, 2901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details include chemicals, preparation methods of GO, g-C3N4 and Mo-GO/g-C3N4, characterization and photocatalytic tests. See DOI: 10.1039/c7nj02894f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |