
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of cubic spherosilicates for self-assembled organic–inorganic biohybrids based on functionalized methacrylates†
Łukasz
John
 *,
Mateusz
Janeta
and
Sławomir
Szafert
*,
Mateusz
Janeta
and
Sławomir
Szafert
Faculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: lukasz.john@chem.uni.wroc.pl
First published on 23rd November 2017
Abstract
The aim of this work is to develop an efficient synthetic approach to hexahedral cage-like organic–inorganic siloxane core biohybrids containing side chains fully functionalized by methacrylate groups derived from monomers such as 2-hydroxyethyl methacrylate (HEMA) or ethylene glycol dimethacrylate (EGDMA). The resulting hybrids were characterized using spectroscopic methods (FTIR, 1H, 13C, 29Si NMR), thermogravimetric and DSC analyses, and high resolution mass spectrometry (HR-MS). The obtained compounds, after polycondensation/polymerization reactions, were utilized in constructing 3D macroporous scaffolds which were examined using a scanning electron microscope (SEM). Covalent networks created by fully functionalized cubic spherosilicates can mimic certain biomaterials and constitute sophisticated, highly organized building blocks of complex systems.
1. Introduction
Functional compounds with well-defined structures are some of the most promising materials in many areas of chemistry and related fields. In this case, polyhedral oligomeric silsesquioxanes (POSSs) constitute a perfect example taking part in a powerful strategy towards designing next-generation functional species for, inter alia, bioapplications. POSSs, also named spherosilicates, belong to a group of organosilicon compounds with the general formula (RSiO3/2)n (where: R = H, alkyl, aryl, alkenyl groups and derivatives thereof; n = 6, 8, 10, 12, 14, 16, 18). They are prepared by the hydrolysis and subsequent condensation of trifunctional silanes, RSiX3 (where: X = Cl, OR, OAc, NH2, and other), at a slightly elevated or room temperature using acidic or basic catalysts.1 As the alkyl carbon–silicon bond is inert towards hydrolysis, the pendant silyl group readily forms a siloxane core. Such silanes are easy to hydrolyse giving silanol![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH groups which further undergo polycondensation forming cage-like structures. Among fully functionalized POSSs such as T8, T10, or T12 containing 8, 10, or 12 silicon atoms, respectively, most interest has been focused on cubic spherosilicates (hexahedral octafunctionalized silsesquioxanes, T8).2–4 Their attractiveness derives from the fact that they are highly soluble in common organic solvents making them useful compounds for the construction of numerous well-defined hybrid materials. Such compounds can effectively anchor various reactive substituents, R, to the siloxane core and, in fact, the structural and physicochemical properties as well as the potential applications of the POSSs are directly related to the precisely designed side chains. Here, covalently bonded organic arms make labile functionalities suitable for polymerization and grafting, making it possible to design the building of networks through polymerization, condensation, addition, substitution reactions, and even the sol–gel methodology.
Si–OH groups which further undergo polycondensation forming cage-like structures. Among fully functionalized POSSs such as T8, T10, or T12 containing 8, 10, or 12 silicon atoms, respectively, most interest has been focused on cubic spherosilicates (hexahedral octafunctionalized silsesquioxanes, T8).2–4 Their attractiveness derives from the fact that they are highly soluble in common organic solvents making them useful compounds for the construction of numerous well-defined hybrid materials. Such compounds can effectively anchor various reactive substituents, R, to the siloxane core and, in fact, the structural and physicochemical properties as well as the potential applications of the POSSs are directly related to the precisely designed side chains. Here, covalently bonded organic arms make labile functionalities suitable for polymerization and grafting, making it possible to design the building of networks through polymerization, condensation, addition, substitution reactions, and even the sol–gel methodology.
Depending on the nature of the organic arms, spherosilicates can easily create self-assembled structures. The introduction of covalent or Lewis acid–base bonds at the molecular level between the inorganic siloxane core and the organic arms seems to be an irresistibly attractive structural variation that can be incorporated into these materials. Such systems are called class II hybrids and can be prepared by carrying out conventional condensation, polymerization, or sol–gel reactions, allowing for the attachment of organic groups onto inorganic materials.5–7 Many successful examples that illustrate countless possibilities of POSS modification have already been described. For instance, Nischang studied tailor-made hybrid organic–inorganic materials based on POSSs by means of thiolene ‘click’ chemistry.8,9 Also, Yang, van Santen, Li, and co-workers described an efficient methodology for constructing hybrid materials using POSS-made molecular blocks containing vinyl side chains.10 Schwab et al. reported on a synthetic strategy for the modification of POSS cages by their incorporation into thermoplastic resin structures with much better properties compared with relative conventional organic systems.11 Modified POSS species containing tetrakis(4-azidophenyl)methane terminals, in turn, form self-assembling materials.12 Also, Kataoka and Endo et al. reported on the synthesis of ordered coordination polymers based on POSSs possessing eight carboxylic side chains forming polymeric networks.13 The construction of methacrylate- and acrylate-functionalized POSSs and the characterization of the relationship between chemical structures and physical properties were studied by Ervithayasuporn et al.14 It should be emphasized that interest in methacrylate-based materials has recently increased again, as they are interesting to many research groups in the field of biomaterials.6,7,14–18 In this work, we have developed an efficient synthetic approach to hexahedral cage-like organic–inorganic siloxane core biohybrids containing side chains fully functionalized by methacrylate groups derived from monomers such as 2-hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA). Covalent networks created by modified cubic spherosilicates can mimic potential macroporous hybrid biomaterials. Because of this, the obtained compounds were utilized in constructing 3D macroporous scaffolds. These aspects of studies on tailor-made organic–inorganic porous hybrids are shown in this paper.
2. Results and discussion
To date, a wide range of synthetic biomaterials for improving on the current methods of bone repair have been discovered. Such materials can be classified into three major groups: (i) bioactive glasses,19–21 (ii) composites based on glasses embedded in polymer matrices,22,23 and (iii) natural polymers, e.g. chitosan, gelatin, etc.24,25 Despite several advantages of the above-mentioned systems, they also possess some disadvantages, such as fragility in the case of bioactive glass scaffolds or different degradation rates during biodegradation in the case of polymer-based composites. Such drawbacks can be ‘cleverly’ omitted using organic–inorganic hybrids in the construction of biomaterials.26 Hybrids containing polymers attached to silica networks via covalent bonds constitute promising materials for bone substitution. An additional advantage of such materials is the molecular level co-network formed between the silica core and the flexible polymer which creates covalent bonding between various components giving unprecedented control of the mechanical properties, degradation rates, and enables osteogenic cells to grow on an amorphous silica surface.27–292.1. Synthesis and characterization of the starting materials
As a starting material, we used the ionic salt of octakis(3-aminopropyl)octasilsesquioxane, containing a CF3COO− anion, with the general formula [OAS-POSS-NH3]CF3COO (1). It can be obtained in hydrolytic condensation using (3-aminopropyl)trimethoxysilane (APTMS) and 1.5 equivalents of trifluoroacetic acid, CF3COOH, (Scheme 1). The composition of 1 was unambiguously confirmed using multinuclear (1H, 13C, and 29Si) NMR spectroscopy and high-resolution mass spectrometry (HRMS). In the FT-IR spectra of 1, the siloxane Si–O–Si moieties gave narrow strong absorption at 1132 cm−1, confirming a high symmetry of the resulting salt, characteristic of the hexahedral T8 structure. Moreover, the silanol ν(Si–OH) vibration (usually observed around 990 cm−1) was not observed, supporting the lack of cage-opening reactions. Furthermore, the structure of 1 was confirmed by nuclear magnetic resonance. In 29Si NMR, only one signal assigned to the silicon nucleus at approximately δ = −66.6 ppm was observed, confirming the formation of an alkyl-substituted cubic POSS. Also, the formation of a hexahedral structure possessing terminal CH2CH2CH2NH3+ organic arms was indicated in 1H NMR (chemical shifts at δ = 7.97, 2.79, 1.61, and 0.62 ppm). The mass spectrum {HRMS (ESI+, TOF/CH3OH)}: m/z 881.2862 {calcd for [M + H-8CF3COOH]+ 881.2871}, 441.1475 {calcd for [M + 2H-8CF3COOH]2+ 441.1469} confirmed the formation of a closed cage-like species possessing eight Si atoms with terminal aminopropyl side arms. Because pure, liquid octakis(3-aminopropyl)octasilsesquioxane [OAS-POSS-NH2] (2) constantly evolves in solution, applying 1 allowed us to obtain molecular 2in situ, avoiding drawbacks such as cage-opening rearrangements and other side reactions such as the formation of bigger Tn (where n is 10 and/or 12) spherosilicates and random silanes.3 Because of the above-mentioned arguments, it is much more convenient from a synthetic point of view to generate 2 in an immediate one-pot transformation starting from ionic salt 1 directly before further investigations.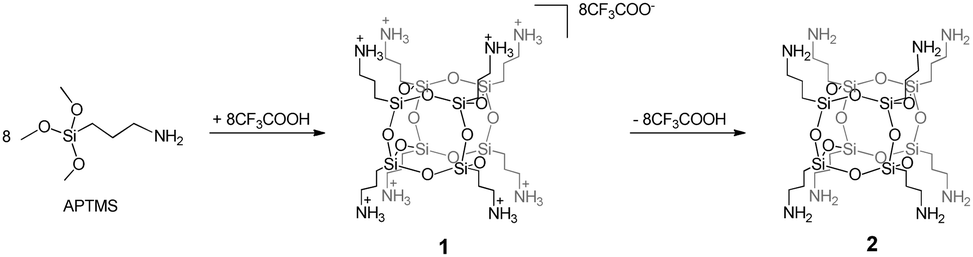 | ||
| Scheme 1 Synthesis of 1 and 2. | ||
The composition of 2 was confirmed using common spectroscopic methods and mass spectrometry (see Experimental, Section 4.3.2).
2.2. Synthesis and characterization of fully functionalized methacrylate cubic POSS derivatives
Copolymers based on methacrylate POSS are some of the first examples of hybrids30 synthesized using various methods.31–33 Recently, we have reported on the synthesis of biohybrids based on the 3-(trimethoxysilyl)propyl methacrylate-POSS compound.34 Inspired by those studies, spherosilicate 2 was functionalized in this work by (2-hydroxyethyl)methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) monomers giving [OAS-POSS-NH]HEMA (3) and [OAS-POSS-NH]EGDMA (4), respectively. These organic monomers were used because they constitute commonly applied agents in medical biomaterials. HEMA is, among others, a precursor for the production of contact lenses, soft tissues, and vascular prostheses and can be used as a drug carrier.35 EGDMA, in turn, is used to prepare bioinspired composites with interesting properties, for instance photocurable dental materials,36 systems directed towards reduced oxidative stress and co-stimulatory factor expression in human monocytic cells,37 porous hydrogel devices for the implantable delivery of insulin,38etc.Both materials, as polymers and copolymers, possess various advantages such as biocompatibility, hydrophilicity, and hardness similar to that observed for human bones and can be used as the filling of bone or tooth defects.36,39 From the chemical perspective, HEMA and EGDMA constitute valuable agents for cross-linking connections, because they contain hydroxyl –OH and terminal vinyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups. Such moieties are able to form not only linear chain copolymers but also more sophisticated structures. In the case of attaching them to the siloxane core, they can easily create self-assembled terminated and cross-linked dendrimers.40
C groups. Such moieties are able to form not only linear chain copolymers but also more sophisticated structures. In the case of attaching them to the siloxane core, they can easily create self-assembled terminated and cross-linked dendrimers.40
Compound 2 can be used as an ideal starting building block for the controlled synthesis of well-defined hexahedral spherosilicates 3 and 4 fully functionalized by HEMA and EGDMA, respectively, in quantitative yields (Scheme 2). During the synthesis, an excess of the organic monomers was used in order to achieve the octafunctionalization of the siloxane core. After the reaction, unreacted monomers can be simply evaporated under vacuum. The 29Si NMR spectra of the resulting hybrids 3 and 4 exhibited one symmetrical peak at around δ = −66 ppm, as expected for an alkyl-substituted hexahedral Si8O12 cage. The presence of other Si nuclei that would be assigned to structures with lower symmetries such as T10 and T12 was excluded. This strongly confirmed the formation of a T8 cube-like structure of 3 and 4 organic–inorganic hybrids which is strictly composed of three siloxane units. Furthermore, no silanol groups were observed, which would suggest the formation of open-like cages, e.g. T8(OH)2, or random structures containing Si–OH moieties.
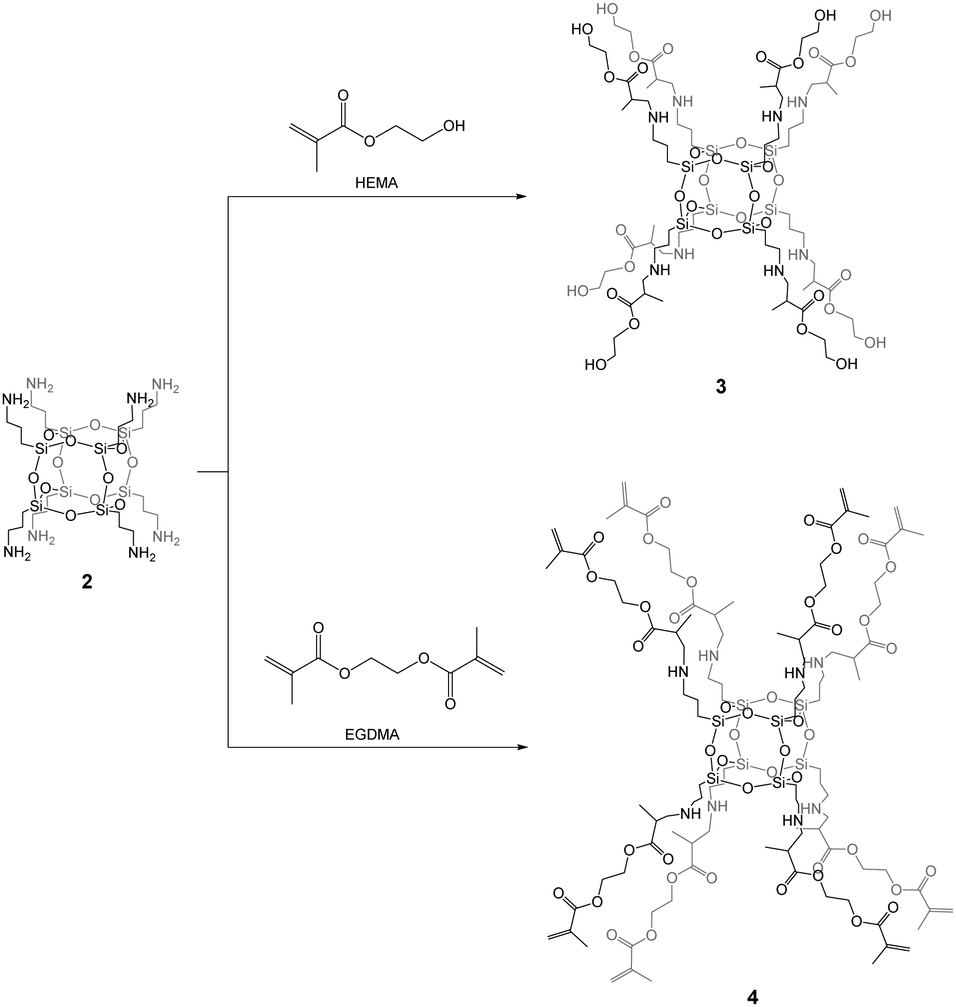 | ||
| Scheme 2 Synthesis of 3 and 4. | ||
Closed-frame structures constructed by eight silicon atoms were also confirmed by the mass spectrum for 3: {HRMS (ESI+, TOF/CH3OH) m/z: 1920.8 {calcd for [M + H]+ 1920.78}}, and 4: {HRMS (ESI+, TOF/CH3OH) m/z: 1790.72 {calcd for [M + H]+ 1790.72}}. The inherence of the side chains of both 3 and 4 was examined by NMR (see Experimental, paragraphs 4.3.3 and 4.3.4 and ESI,† Fig. S13–S24). The structural analysis of 3 and 4 was also performed by FTIR (Fig. 1a and b). The spectrum of 3 gives a characteristic CO peak at 1724 cm−1, while a peak at 1636 cm−1, due to absorption, indicates CC methacrylate vibrations. Moreover, the siloxane Si–O–Si stretching gave strong absorption around 1155 cm−1. The above-mentioned vibration bands are also confirmed for 4 and located at 1724, 1638, and 1155 cm−1 for CO, CC, and Si–O–Si, respectively. In the case of the polymerized hybrid 3, the characteristic vibrational band of CC disappeared (Fig. 1c), which may suggest that all terminal vinyl groups are polymerized.
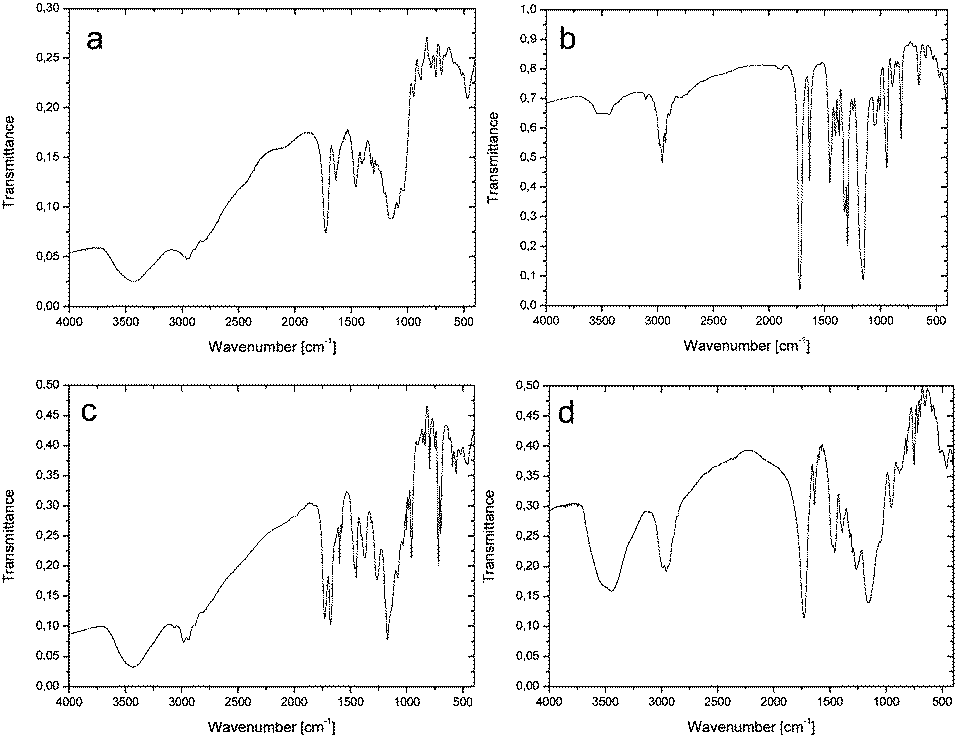 | ||
| Fig. 1 FTIR spectra of (a) 3 and (b) 4, and (c) polymerized 3 and (d) polymerized 4. | ||
For polymerized 4 (Fig. 1d), the situation is slightly different, because a weak peak at 1637 cm−1, matched to the CC band, is still present. This suggests that not all the vinyl groups were polymerized.2,3,41–43 Increasing the polymerization time does not cause this band to disappear.
2.3. Thermal analysis
The thermal behaviour of synthesized non-polymerized and polymerized hybrids 3 and 4 was evaluated using TGA and DSC to determine their thermal stability. A DSC analysis was performed in a nitrogen atmosphere from 30 to 300 °C, whereas a thermogravimetric analysis was conducted from 30 to 1000 °C under the same conditions. The residues after thermal decomposition were analysed using elemental analysis. All the thermograms are collected in the ESI† (see Fig. S5–S12). The TGA thermogram of 3 (Fig. S5, ESI†) shows degradation in five different stages. In the first two phases, a mass loss of about 13% occurs, which can be attributed to the evaporation of solvent molecules. In the next phase, two mass losses of about 17.5% are observed in the region between 280 and 400 °C, which may be correlated with the gradual decomposition of the organic arms attached to the siloxane core. The final decomposition stage with a curve ranging from 420 to 630 °C, with a mass loss of about another 17.5%, leads to a SiO2 residue; the overall mass loss during thermal decomposition is about 67%. The DSC thermogram of 3 (Fig. S9, ESI†) shows the first endothermic peak at ca. 40 °C, attributed to the initial loss of water/methanol molecules, and the second one at around 185 °C, the result of the evaporation process. No exothermic peaks are observed.The TGA curve of 4 (Fig. S6, ESI†) shows that the degradation process starts at 148 °C and also continues in five steps, where solvent evaporation and organic arm degradation are observed. The biggest mass loss attributed to side chain decomposition is noticed ranging from 390 to 580 °C, where thermolysis ends, with an overall mass loss of about 84% leading to a silicon dioxide residue. The DSC thermogram, in turn, manifests one clear endothermic peak that starts at ca. 200 °C. This endothermic peak is connected with the following evaporation of decomposed ‘by-products’. No exothermic peaks are observed.
Polymerized hybrids 3 and 4 decompose in three major stages with mass losses of about 79 and 84%, respectively. Polymerized 3 starts to decompose at ca. 150 °C and stops decomposing at 640 °C (Fig. S7, ESI†), whereas polymerized 4 at 160 and 605 °C (Fig. S8, ESI†), respectively. In the case of polymerized 3, no exothermic peaks on the DSC curve are observed. The DSC thermograms of both materials show three endothermic peaks relating to solvent and thermolysed product evaporation (Fig. S11 and S12, ESI†). The DSC curve of polymerized 4 also has one exothermic peak in the 132–154 °C region, which may be attributed to further polymerization reactions. Based on the FTIR of polymerized 4, the material contains some unreacted terminal vinyl groups (characteristic ν(CC) at 1637 cm−1), which occur along the polymeric network and probably start to further react in this temperature region. Unfortunately, to deeply understand this phenomenon, additional analyses are needed which were not the aim of this study. This high temperature degradation depicts the high stability behaviour of the obtained biomaterials, for hybrids both before and after the polymerization reaction. The thermal analysis of these hybrids indicates a strong and stable network for potential biomaterial construction.44,45
2.4. Macroporous scaffold preparation
Covalent networks created by fully functionalized cubic POSSs can mimic certain biomaterials and without a doubt constitute sophisticated highly organized building blocks of complex systems. Numerous applications of these compounds are conceivable, particularly in mimicking the functions of large constructions, e.g. similar to those observed in bone-tissue engineering. This new branch of ‘supramolecular chemistry’ should spark new developments in self-assembled biosystems based on well-defined functionalized covalently-bound building blocks.The resulting POSS-based hybrids 3 and 4, functionalized by methacrylate derivatives, constitute a perfect network for the construction of biomimetic 3D macroporous scaffolds for tissue engineering. To that aim, the obtained sols of both hybrids were transferred into plastic moulds filled with the porogen (3 was added after polycondensation as an oil-like liquid, whereas 4 was added with the concentration similar to that of 3, and then, after the gentle saturation of the sugar template, it was polymerized according to the procedure described in Experimental, Section 4.3.6). After the saturation of the porogen, the materials were aged for three days at 40 °C. After this time, when a flexible foam-like structure appeared, the template was washed out using de-ionized water at 60 °C and the presence of sugar was monitored by the Molisch test. After sugar removal, the resulting scaffolds were thermally condensed at 65–70 °C for 2–3 days to obtain reproducible and hard platforms.
A macroporous structure created by white granulated sugar guarantees a pore size in the range of 150–350 μm (Fig. 2). It fulfils the requirements of tissue ingrowth, mass transport (ions, nutrition, etc.), osteoblast attachment, cell movement through the scaffold, the permeability of the material, etc.46 The open porosity and roughness of the pore surface were assured by the addition of ammonium carbonate. This inorganic salt decomposes at ca. 60 °C, during porogen wash-out, to carbon dioxide, which opens closed pores, and ammonia, which affects the surface. The roughness of the surface constitutes a very important factor that forces cell adhesion abilities. It was reported that the bonding strength between the artificial tissue and the natural bone is greater for rough materials due to favourable osteoblast adhesion.47 The surface roughness was significantly high and could not be captured with AFM. The resulting structures of 3 and 4 are sisterly similar.
 | ||
| Fig. 2 SEM images of 3 (a and b) and 4 (c and d) scaffolds. | ||
2.5. POSS-based biomaterials containing methacrylate-derived side arms
POSS-based materials functionalized by organic groups able to polymerize/polycondensate can be considered unusual monomers grafted on the siloxane core.During our investigations regarding methacrylate-derivative compounds, we obtained three unique hybrid systems based on a hexahedral siloxane core and fully functionalized side arms which can be bound together via polycondensation (3 containing HEMA-based arms), polymerization (3 and 4 possessing HEMA- and EGDMA-based side chains, respectively), or sol–gel processes (5 containing TMSPMA-based organic arms; TMSPMA is 3-(trimethoxysilyl)propyl methacrylate) (Fig. 3).34 These species seem to be promising from the potential hard-tissue-engineering point of view. In vitro studies carried out on POSSs constitute strong evidence for their low cytotoxicity.47,48 To date, our recent studies, reported in 5, have indicated that hexahedral organic–inorganic hybrids based on a siloxane cage-like core with eight 3-(trimethoxysilyl)propyl methacrylate side arms can be considered unique three-dimensional scaffolds that fulfil potential hard-tissue-engineering requirements realized by multiple factors such as the chemical composition, structural dimensions, topography, and microstructural properties.34 Our investigations also prove that the methacrylate derivatives used, not only those attached to the Si8O12 cage, are safe components and constitute efficient reservoirs for osteoblasts.42 For this reason, they may be used in future experiments, as they do not have any toxic influence on cells. However, for the full characterization of the resulting 3 and 4 biohybrids towards their application as bone artificial scaffolds, the performance of both biological and mechanical studies is necessary. Investigations along this line are currently underway.
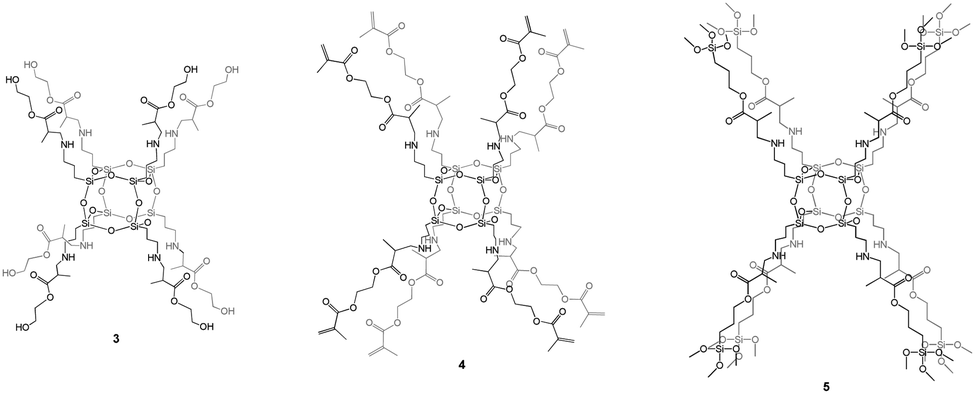 | ||
| Fig. 3 Examples of spherosilicates containing various methacrylate-based organic side arms. | ||
2.6. A brief comment on the biological assessment of methacrylate-based hybrid/composite systems
Based on our previous studies, the aim of this paper was to report on an efficient synthetic approach to organic–inorganic biohybrids serving as attractive materials from the bone-tissue-engineering point of view. Here, we would like to summarize the general potential of methacrylate derivatives, such as 2-hydroxyethyl methacrylate, ethylene glycol dimethacrylate, and 3-(trimethoxysilyl)propyl methacrylate, which prove promising 3D platforms for the hard-tissue architecture (Fig. 4).34,41–43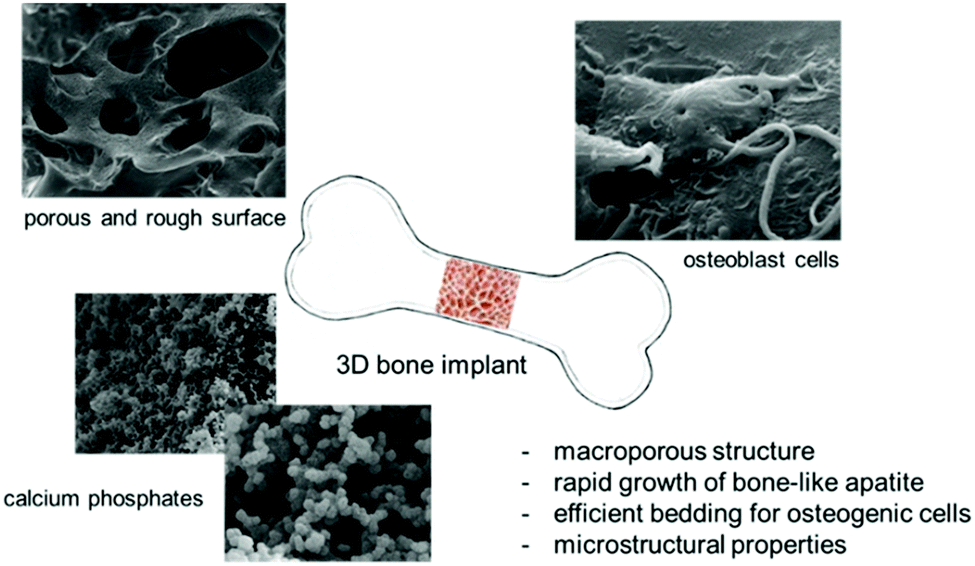 | ||
| Fig. 4 General potential of the organic–inorganic biohybrids based on functionalized methacrylate systems. | ||
Silicon-based systems can create a perfect environment for biomineralization leading to the formation of bone-like apatites and other calcium phosphates. We showed that methacrylate-derived materials after exposition in biological fluids (Dulbecco's Modified Eagle's Medium) force the crystallization of hydroxyapatite on their surface after merely 0.5 h of incubation.41
This result was confirmed for all the studied composites containing HEMA, EGDMA, and TMSPMA derivatives. It is worth noting that, during the static evaluation of in vitro bioactivity, we observed, inter alia, continuous silicon release from the polymeric matrix, which is considered a positive signal from the hybrid material because these ions are essential nutrients for bone metabolism. Moreover, Si affects the reduction of the activity of osteoclast cells and an increase in the number of osteoblasts which effectively rebuild healthy bones.49
During the design stage of complex biohybrids/biocomposites, surface modification should be considered, always aimed at creating favourable conditions for ion exchange, mass transport, attaching growth factors, drugs and other factors crucial from the osteogenesis perspective, and setting up a perfect bedding for the rapid growth of calcium phosphates, cell differentiation and then proliferation. This can be performed at different biomaterial formation steps. The surface of the resulting organic–inorganic hybrids/composites can be easily modified using simple approaches. We showed that the surface of the three-dimensional scaffold can be modified using ammonium carbonate. The addition of this salt directly to a porogen matrix affects surface roughness, while it decomposes to ammonia and carbon dioxide. Ammonia is an agent that influences roughness, and CO2 increases open porosity inside the hybrid network.41,42 Also, the size, ranging from 1 to 800 μm, and the geometry of macropores, which may be spherical, hexagonal, or cubic, can be modulated using various sorts of porogens such as sodium chloride, ammonium chloride, or the above-mentioned granulated white sugar. Another method of creating a rough surface, cheap and simple to scale up, includes the formation of synthetic hydroxyapatite or carbonate hydroxyapatite on the scaffold surface before biological investigations.43
Biomaterials intended for bone-tissue engineering should have special mechanical properties. We proved that composites containing functionalized methacrylates attached to a cage-like siloxane core show excellent mechanical properties compared to other types of tissue-engineering materials, such as pure polymeric and bioactive glass systems,34e.g. for scaffold-like organic–inorganic hybrid 5, Young's modulus, hardness, and uniaxial compressive strength of 4.36 ± 0.57 GPa, 0.57 ± 0.13 GPa, and 0.21 ± 0.03 MPa, respectively, comparable with values observed for natural bones and much better compared with some synthetic materials, for instance composite scaffolds based on gelatin and partially hydrolysed α-tricalcium phosphate.50,51
Furthermore, a 2-hydroxyethyl methacrylate-based composite doped with calcium and phosphate ions constitutes a non-cytotoxic material on mouse fibroblasts (BALB/3T3 clone A31).41 Also, a similar system was tested on the human osteoblast cell culture.42 The evaluation of the cytotoxicity of hybrid biocomposites on the NHOst cell line indicated that scaffold hybrids containing Ca2+, Sr2+, and PO43− ions do not have any negative impact on the survival of osteoblasts and, moreover, cells are able to attach to and grow on these materials.
The above arguments indicate that organic–inorganic hybrids based on methacrylate derivatives constitute promising systems with prospects to become novel and competitive biomaterials. It is necessary, however, to verify parameters like biomineralization, cytotoxicity, differentiation and proliferation of osteoblasts, and mechanical properties by means of laboratory investigations on the macro scale. The results suggest that well-defined, tailor-made, and fully functionalized spherosilicates, whose synthesis is described in this article and our recent papers, seem to be the most valuable. In our opinion, these biomaterials point the way to novel implants with huge application potential.
3. Conclusions
We have presented a synthetic approach to fully substituted spherosilicates containing functionalized methacrylate derivatives as side arms, yielding [OAS-POSS-NH]HEMA and [OAS-POSS-NH]EGDMA. These compounds were obtained upon the treatment of [OAS-POSS-NH2] with methacrylate-based monomers: 2-hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) in the case of [OAS-POSS-NH]HEMA and [OAS-POSS-NH]EGDMA, respectively. The reactions were efficient (yields of ca. 100%), and no other undesirable processes like cage-opening reactions or cage rearrangements leading to higher forms of POSSs like T10 or T12 were observed. The performed thermal studies show the high-stability behaviour of the obtained non-polymerized and polymerized functionalized materials and indicate a strong and thermally stable network for the construction of porous materials. The hybrids [OAS-POSS-NH]HEMA and [OAS-POSS-NH]EGDMA were used for the construction of three-dimensional bioscaffolds templated by granulated white sugar doped with ammonium carbonate (ca. 2 wt%), and the resulting 3D networks possess open interconnected rough pores that force, among others, cell adhesion abilities.The described studies point the way to novel organic–inorganic hybrids for potential applications in hard-tissue engineering. In the near future, we intend to report analyses of the mechanical properties and cytotoxicity, proliferation, and degradability studies conducted on [OAS-POSS-NH]HEMA and [OAS-POSS-NH]EGDMA species. Intensive investigations along these lines are currently underway.
4. Experimental
4.1. General procedures and chemicals
All reactions were carried out under nitrogen gas flow with the use of a standard Schlenk line. The glassware was pre-dried before use at 120 °C. The empty syringes were purged with nitrogen gas before being used for the injection of the solvents and reagents. The progress of the reaction was monitored by thin layer chromatography (TLC) performed on Merck silica gel 60 F254 plates. Chromatograms were visualized using UV light (254 nm). Amberlite IRA-400 ion exchange resin (40.0 g) was prepared by washing with water (4 × 200 mL), next with 1 M NaOH (3 × 200 mL), again with water (6 × 200 mL), and finally with MeOH (elution solvent, 6 × 200 mL). The resin was suspended in MeOH (200 mL) and chilled (−18 °C, 5 h) before use. Preparative separations were performed on a Sepacore® chromatography system (Büchi Labortechnik) consisting of two C-605 pump modules, a C-620 control unit, a C-640 UV-VIS detector, and a C-660 fraction collector. The system was controlled by the SepacoreControl 1.3 software. Flash chromatography separations were performed on pre-packed modified silica gel C18 (40–63 mm) polypropylene cartridges (21.0 × 129 mm, Büchi) at a flow rate of 25 mL min−1. The samples were dissolved in methanol and injected through a six-way valve with a 20 mL loop.(3-Aminopropyl)trimethoxysilane (97%, Sigma-Aldrich), trifluoroacetic acid (99%, Aldrich), granulated sugar (Diamant, Pfeifer & Langen Marketing, Poland), the photoinitiator 2-hydroxy-2-methylpropiophenone (97%, Aldrich), 2-hydroxyethyl methacrylate (97%, Aldrich), ethylene glycol dimethacrylate (98%, Aldrich), and (NH4)2CO3 (reagent-grade, Avantor) were of reagent-grade quality and were used without further purification.
4.2. Methods
1H and 13C NMR spectra were recorded using a Bruker Avance 500 or a Bruker Avance III 600 spectrometer equipped with broadband inverse gradient probe heads. 1H NMR spectra were collected at 500.13 MHz with a relaxation delay of 1.0 s and a pulse width of 7°. Spectra were referenced to the residual solvent signals (MeOD 3.34, DMSO-d6 2.50, CDCl3 7.26, D2O 4.79 ppm) as an internal reference. 13C NMR spectra were collected at 125.77 MHz with a relaxation delay of 2.0 s and a pulse width of 15° and referenced to solvent signals ((13CH3)2SO 39.52, 13CDCl3 77.16 ppm). 29Si NMR spectra were recorded on a Bruker AMX-300 spectrometer using Wildmad PTFE-FEP (polytetrafluoroethylene/fluorinated ethylene polypropylene copolymer) 5 mm tube liners and collected at 59.62 MHz with a relaxation delay of 10.0 s and a pulse width of 13°. Cr(acac)3 was added at a concentration of ∼10–2 mol L−1 as a shiftless relaxation agent. Chemical shifts were referenced to tetramethylsilane (TMS) (δ = 0.00 ppm). For proton and carbon assignments, COSY, HMBC, and HMQC experiments were performed on a Bruker Avance 500 spectrometer. Two-dimensional NMR spectra were recorded with 2048 data points in the t2 domain and up to 2048 points in the t1 domain with a 1.0 s recovery delay. All 2D spectra were recorded with gradient selection. FTIR spectra were recorded on a Bruker Vertex 70 FTIR spectrometer in the transmission mode in the 4000–400 cm−1 range. The sample chamber was continuously flushed with N2. The spectra were recorded using KBr pellets. Optical grade, random cuttings of KBr were ground with 1.0 wt% of the sample to be analysed and pressed as KBr pellets. HRMSs were carried out on a Bruker microTOF-Q spectrometer equipped with an ESI source. The samples were dissolved in chloroform or methanol. The experimental parameters were as follows: scan range: 200–2500 m/z; drying gas: nitrogen; temperature: 200 °C; ion source voltage: 4500 V; in-source collision energy: 10 eV. The instrument was operated in the positive ion mode and was calibrated externally with the Tunemix mixture (Bruker Daltonics). The analysed solutions were introduced at a flow rate of 4.0 L min−1. The Compass Data Analysis software was used to determine the formulae of the compounds. The distance between the isotopic peaks allowed the calculation of the charge of the analysed ions. Elemental analyses (C, H, and N) were performed using a Vario EL III element analyser. TGA analyses were recorded using a Mettler Toledo TGA-2 instrument. Samples for thermogravimetric characterization were placed in open alumina crucibles (70 μL volume) in a N2 atmosphere. A heating rate of 10 °C min−1 was applied, and all samples were studied between 30 and 1000 °C. DSC analyses were recorded using a Mettler Toledo DSC-3 instrument. Samples for DSC characterization were placed in open alumina crucibles (40 μL volume) in a N2 atmosphere. A heating rate of 5 °C min−1 was applied, and all samples were studied between 30 and 300 °C. SEM images were collected on a Hitachi S-3400N-II variable-pressure scanning electron microscope. Samples were sputter-coated with 7 nm Au to facilitate viewing by SEM. Energy dispersive X-ray (EDX) spectra were obtained using a EDS Thermo Scientific Ultra Dry system.4.3. Syntheses
Note. Compound 2 should be freshly prepared before use and stored as a methanol or ethanol solution at −18 °C.
This compound was prepared following the described procedure.2 Amberlite IRA-400 ion-exchange resin was prepared by washing with water (4 × 200 mL), 1 M NaOH (3 × 200 mL), water (6 × 200 mL), and MeOH (elution solvent, 6 × 200 mL). The resin was suspended in MeOH (200 mL) and chilled (−18 °C, 5 h) before use. Half of the resin beads were loaded onto a column (3.0 cm outside diameter) and the other half was used to prepare a suspension of 1 (0.764 g, 0.426 mmol) in a minimum amount of the eluent (ca. 5 mL) at −18 °C. The solvent was removed with nitrogen flow at 0 °C and then evaporated (0 °C, 0.5 mbar) to afford 2 (50%, 0.200 g) as a yellowish resin. 1H NMR (500 MHz, MeOD, 20 °C): δ = 2.60 (t, 3J(H,H) = 7.9 Hz, 16H; CH2NH2), 1.54 (m, 16H; SiCH2CH2CH2NH2), 0.63 ppm (t, 3J(H,H) = 8.6 Hz, 16H; SiCH2); 13C{1H} NMR (126 MHz, DMSO-d6, 20 °C): δ = 44.4 (s; SiCH2CH2CH2NH2), 26.6 (s; SiCH2CH2CH2NH2), 9.8 ppm (s; SiCH2CH2CH2NH2); 29Si{1H} NMR (59.6 MHz, DMSO-d6, 20 °C): δ = −66.5 ppm (s); FTIR (KBr pellets): ν = 3041 (s, νN–H), 2935 (m, νC–H), 2872 (m, νC–H), 1615 (m, δNH2), 1500 (m, νC–N), 1237 (m, νSi–C), 1116 (s, νring-asym![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Si–O–Si), 941 (m, νring-symSi–O–Si), 798 (w, δSi–C), 701 (m, δO–Si–O) cm−1; HRMS (ESI+, TOF/CH3OH): m/z: 881.2916 {calcd for [M + H]+ 881.2871}, 441.1635 {calcd for [M + 2H]2+ 441.1469}, 294.4442 {calcd for [M + 3H]3+ 294.4339}; elemental analysis calcd (%) for C24H64N8O12Si8 (881.50): C 32.70, H 7.32, N 12.71; found: C 32.69, H 7.35, N 12.69.
Si–O–Si), 941 (m, νring-symSi–O–Si), 798 (w, δSi–C), 701 (m, δO–Si–O) cm−1; HRMS (ESI+, TOF/CH3OH): m/z: 881.2916 {calcd for [M + H]+ 881.2871}, 441.1635 {calcd for [M + 2H]2+ 441.1469}, 294.4442 {calcd for [M + 3H]3+ 294.4339}; elemental analysis calcd (%) for C24H64N8O12Si8 (881.50): C 32.70, H 7.32, N 12.71; found: C 32.69, H 7.35, N 12.69.
O), 1636 (m, νCC), 1457 (m, νC–N), 1405 (br, νCH3 and νCOO), 1240 (m, νSi–C), 1155 (br, νring-asymSi–O–Si and δC–O–C), 992 (w, νCC), 947 (m, νring-symSi–O–Si), 883 (m, νCC), 793 (w, δSi–C), 699 (m, δO–Si–O) cm−1; HRMS (ESI+, TOF/CH3OH): m/z: 1920.8 {calcd for [M + H]+ 1920.78}; elemental analysis calcd (%) for C72H168N8O36Si8: C 44.42, H 8.70, N 5.76; found: C 44.58 H 8.60, N 5.28; FTIR of polymerized 3 (KBr, pellets): 3430 (br, νO–H), 3153 (vw, νN–H), 2979 (w, νC–H), 1728 (s, νCO), 1697 (s, νCO), 1598 (m, δSi–C), 1447 (m, νC–N), 1376 (m, νCOO), 1261 (m, νSi–C), 1171 (br, νring-asymSi–O–Si and δC–O–C), 855 (w, νring-symSi–O–Si), 796 (m, δSi–C), 697 (s, δO–Si–O).
O), 1638 (s, νCC), 1454 (m, νC–N and δCOO), 1247 (m, νSi–C), 1155 (vs, νring-asymSi–O–Si), 1006 (w, νCC), 944 (m, νring-symSi–O–Si), 815 (m, δSi–C), 654 (m, δO–Si–O) cm−1; HRMS (ESI+, TOF/CH3OH): m/z: 1790.72 {calcd for [M + H]+ 1790.72}; elemental analysis calcd (%) for C66H134N8O33Si8: C 44.22, H 7.53, N 6.25; found: C 44.26, H 7.42, N 6.69; FTIR of polymerized 4 (KBr, pellets): 3441 (br, νN–H), 2957 (s, νC–H), 1730 (vs, νCO), 1637 (w, νCC), 1457 (s, br, νC–N and δCOO), 1265 (νSi–C), 1153 (s, br, νring-asymSi–O–Si and δC–O–C), 954 (m, νring-asymSi–O–Si), 881 (vw, br νCC), 814 (w, νSi–C), 697 (w, δO–Si–O).
Note. HEMA and EGDMA contain monomethyl ether hydroquinone as an inhibitor. Therefore, these compounds should be purified by double extraction of the monomer using a 10% aqueous solution of sodium hydroxide. This approach takes advantage of the fact that phenols are soluble in the aqueous basic environment forming phenolates. After phenol removal, the mixture can be extracted using distilled water to eliminate phenolates and excess NaOH. This method is also useful in removing other contaminants from the commercial product. The inhibitor can also be removed by vacuum distillation (60–80 °C; 30 Torr), but a viscous polymer liquid remains at the bottom of the flask in this method.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland (Grant No. 2016/23/B/ST5/01480 and 2016/21/N/ST5/03293).References
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed.
- M. Janeta, Ł. John, J. Ejfler and S. Szafert, Chem. – Eur. J., 2014, 20, 15966 CrossRef CAS PubMed.
- M. Janeta, Ł. John, J. Ejfler and S. Szafert, RSC Adv., 2015, 5, 72340 RSC.
- T. Jaroentomeechai, P. Yingsukkamol, C. Phurat, E. Somsook, T. Osotchan and V. Ervithayasuporn, Inorg. Chem., 2012, 51, 12266 Search PubMed.
- B. M. Novak, Adv. Mater., 1993, 5, 422 CrossRef CAS.
- A. L. B. Maçon, T. Kasuga, R. Becer and J. R. Jones, Polym. Chem., 2017, 8, 3603 RSC.
- A. L. B. Maçon, S. Li, J. J. Chung, A. Nomm–Nommeots, A. K. Solanki, M. M. Stevens and J. R. Jones, J. Mater. Chem. B, 2016, 4, 6032 RSC.
- J. G. Croissant, X. Cattoën, J.-O. Durand, M. W. C. Man and N. M. Khashab, Nanoscale, 2016, 8, 19945 RSC.
- F. Alves and I. Nischang, Chem. – Eur. J., 2013, 19, 17310 CrossRef CAS PubMed.
- L. Zhang, H. C. L. Abbenhuis, Q. Yang, Y.-M. Wang, P. C. M. M. Magusin, B. Mezari, R. A. van Santen and C. Li, Angew. Chem., Int. Ed., 2007, 46, 5003 CrossRef CAS PubMed.
- J. J. Schwab and J. D. Lichtenhan, Appl. Organomet. Chem., 1998, 12, 707 CrossRef CAS.
- M. J. Huang, C. H. Hsu, J. Wang, S. Mei, X. H. Dong, Y. W. Li, M. X. Li, H. Liu, W. Zhang, T. Z. Aida, W. B. Zhang, K. Yue and S. Z. D. Cheng, Science, 2015, 348, 424 CrossRef CAS PubMed.
- S. Banerjee, S. Kataoka, T. Takahashi, Y. Kamimura, K. Suzuki, K. Sato and A. Endo, Dalton Trans., 2016, 45, 17082 RSC.
- V. Ervithayasuporn and S. Chimjarn, Inorg. Chem., 2013, 52, 13108 CrossRef CAS PubMed.
- E. A. Elizalde-Peña, I. A. Quintero-Ortega, D. G. Zárate-Triviño, A. Nuño-Licona, J. Gough, I. C. Sanchez, D. I. Medina and G. Luna-Barcenas, Mater. Sci. Eng., C, 2017, 78, 892 CrossRef PubMed.
- S. Hu, W. Hu, Z. Li, L. Ren, Y. Zhao and X. Yuan, Mater. Sci. Eng., C, 2017, 76, 1112 CrossRef CAS PubMed.
- X. Fan, M. Cao, X. Zhang and Z. Li, Mater. Sci. Eng., C, 2017, 76, 211 CrossRef CAS PubMed.
- Y. Ju, C. Xing, D. Wu, Y. Wu, L. Wang and H. Zhao, Chem. – Eur. J., 2017, 23, 3366 CrossRef CAS PubMed.
- J. R. Jones, L. M. Ehrenfried and L. L. Hench, Biomaterials, 2006, 27, 964 CrossRef CAS PubMed.
- Q. A. Fu, E. Saiz and A. P. Tomsia, Adv. Funct. Mater., 2011, 21, 1058 CrossRef CAS PubMed.
- E. Jallot, J. Lao, Ł. John, J. Soulié, Ph. Moretto and J.-M. Nedelec, ACS Appl. Mater. Interfaces, 2010, 2, 1737 CAS.
- J. R. Jones, Acta Biomater., 2013, 9, 4457 CrossRef CAS PubMed.
- J. J. Blaker, A. Bismarck, A. R. Boccaccini, A. M. Young and S. N. Nazhat, Acta Biomater., 2010, 6, 756 CrossRef CAS PubMed.
- D. M. Wang, F. Romer, L. Connell, C. Walter, E. Saiz, S. Yue, P. D. Lee, D. S. McPhail, J. V. Hanna and J. R. Jones, J. Mater. Chem. B, 2015, 3, 7560 RSC.
- O. Mahony, S. Yue, C. Turdean–Ionescu, J. V. Hanna, M. E. Smith, P. D. Lee and J. R. Jones, J. Sol-Gel Sci. Technol., 2014, 69, 288 CrossRef CAS.
- J. J. Chung, B. S. T. Sum, S. Li, M. M. Stevens, T. K. Georgiou and J. R. Jones, Macromol. Rapid Commun., 2017, 38, 1700168 CrossRef PubMed.
- C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC.
- F. Mammeri, E. Le Bourhis, L. Rozes and C. Sanchez, J. Mater. Chem., 2005, 15, 3787 RSC.
- L. Nicole, C. Boissiere, D. Grosso, A. Quach and C. Sanchez, J. Mater. Chem., 2005, 15, 3598 RSC.
- J. D. Lichtenhan, Y. A. Otonari and M. J. Carr, Macromolecules, 1995, 28, 8435 CrossRef CAS.
- E. T. Kopesky, T. S. Haddad, R. E. Cohen and G. H. McKinley, Macromolecules, 2004, 37, 8992 CrossRef CAS.
- J. Pyun and K. Matyjaszewski, Macromolecules, 2000, 33, 217 CrossRef CAS.
- J. Pyun and K. Matyjaszewski, Chem. Mater., 2001, 13, 3436 CrossRef CAS.
- Ł. John, M. Janeta, M. Rajczakowska, J. Ejfler, D. Łydżba and S. Szafert, RSC Adv., 2016, 6, 66037 RSC.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Biomaterials Science: An Evolving, Multidisciplinary Endeavorw, Biomaterials Science, An Introduction to Materials in Medicine, Academic Press, 2013, 3rd edn, ISBN: 978-0-12-374626-9 Search PubMed.
- S. Shin, Y.-J. Kim, M. Toan, J.-G. Kim, T. Nguyen and J. Ku Cho, Eur. Polym. J., 2017, 92, 338 CrossRef CAS.
- A. Tamura, I. Fukumoto, N. Yui, M. Matsumura and H. Miura, J. Biomed. Mater. Res., Part A, 2015, 103, 1060 CrossRef PubMed.
- A. Kumar, P. Tyagi, H. Singh, Y. Kumar and S. S. Lahiri, J. Appl. Polym. Sci., 2012, 126, 894 CrossRef CAS.
- R. Filmon, F. Grizon, M. F. Basle and D. Chappard, Biomaterials, 2002, 23, 3053 CrossRef CAS PubMed.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard III, Angew. Chem., Int. Ed. Engl., 1990, 29, 138 CrossRef.
- Ł. John, M. Bałtrukiewicz, P. Sobota, R. Brykner, Ł. Cwynar-Zając and P. Dzięgiel, Mater. Sci. Eng., C, 2012, 32, 1849 CrossRef.
- Ł. John, M. Podgórska, J.-M. Nedelec, Ł. Cwynar-Zając and P. Dzięgiel, Mater. Sci. Eng., C, 2016, 68, 117 CrossRef PubMed.
- Ł. John, M. Janeta and S. Szafert, Mater. Sci. Eng., C, 2017, 78, 901 CrossRef PubMed.
- H. Rashid, M. Ahmad, M. U. Minhas, M. Sohail and M. F. Aamir, J. Chem. Soc. Pak., 2015, 37, 999 CAS.
- L. Ning, N. Xu, C. Xiao, R. Wang and Y. Liu, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 1017 CrossRef CAS.
- G. Wang, L. Zheng, H. Zhao, J. Miao, C. Sun, H. Liu, Z. Huang, X. Yu, J. Wang and X. Tao, ACS Appl. Mater. Interfaces, 2011, 3, 1692 CAS.
- C. Larsson, P. Thomsen, J. Lausmaa, M. Rodahl, B. Kasemo and L. E. Ericson, Biomaterials, 1994, 15, 1062 CrossRef CAS PubMed.
- A. Janaszewska, K. Gradzińska, M. Marcinkowska, B. Klajnert-Maculewicz and W. A. Stańczyk, Materials, 2015, 8, 6062 CrossRef PubMed.
- C. D. Seaborn and F. H. Nielsen, Biol. Trace Elem. Res., 2002, 89, 251 CrossRef CAS PubMed.
- S.-H. Lee and H. Shin, Adv. Drug Delivery Rev., 2007, 59, 339 CrossRef CAS PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra, TGA, DSC, NMR spectra (1H, 13C, 29Si, COSY, NOESY, HMQC, HMBC). See DOI: 10.1039/c7nj02533e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |