
Artificial macrocycles as IL-17A/IL-17RA antagonists†
Wenjia
Wang
a,
Matthew R.
Groves
 b and
Alexander
Dömling
*c
b and
Alexander
Dömling
*c
aDepartment of Drug Design, University of Groningen, A. Deusinglaan 1, Groningen, The Netherlands. E-mail: w.wang@rug.nl
bDepartment of Drug Design, University of Groningen, A. Deusinglaan 1, Groningen, The Netherlands. E-mail: m.r.groves@rug.nl
cDepartment of Drug Design, University of Groningen, A. Deusinglaan 1, Groningen, The Netherlands. E-mail: a.s.s.domling@rug.nl
First published on 25th October 2017
Abstract
Interleukin 17(A) is a pro-inflammatory cytokine involved in several auto-immune and inflammatory diseases. Current antagonists against IL-17(A) or its receptor (IL-17RA) that show efficacy in clinical trials are monoclonal antibodies (mAbs). However, recently designed artificial macrocycles are potent IL-17A/IL-17RA antagonists. Based on co-crystal structures, a better understanding of the biological activity and SAR of the macrocycles has been gained, demonstrating that they can compete with mAbs for difficult targets such as PPIs.
Inflammation is a complex biological protective response of body tissue against harmful stimuli (either acute or chronic) in which monocytes and lymphocytes play a role in the later stages. In recent decades, antagonists of inflammatory factors have been developed for the treatment of various inflammatory diseases, such as Crohn's disease, rheumatoid arthritis, ankylosing spondylitis and psoriasis, among which antagonists against tumor necrosis factor (TNF) are the most promising. Currently, while TNF inhibitors are widely used in different diseases, they behave differently in patients, and some side effects appear after long-term treatment. Thus, a major priority is the investigation of new approaches to treat TNF related inflammatory diseases. As interleukin-17 (IL-17A) and TNF share similar effector functions, IL-17A could be targeted in patients with inflammation who are not responsive to TNF inhibitors. Therefore, much attention is being devoted to drugs targeting IL-17A.
Interleukin-17, synonymous with IL-17A (the archetype protein in the IL-17 family consisting of IL-17A-F), was first discovered in 1993 as a pro-inflammatory cytokine predominantly produced by a subset of CD4+ cells (T helper cells (Th-17)), although it has been found that other cell types such as mast cells and neutrophils produce IL-17A as well.1 For several decades, IL-17A has been well known to participate in various acute inflammation reactions e.g. the release of pro-inflammatory cytokines IL-6 and IL-8 from mesenchymal cells leading to fever, and the accumulation of neutrophils in blood and tissue.2 IL-17A also contributes to chronic inflammation associated with matrix destruction,3 resulting in joint damage and defective tissue repair. Additionally, IL-17A increases the expression of the receptor activator of NF-κB ligand (RANKL) on osteoblasts, increasing the RANK signal in osteoclasts, as shown in the bone destruction of rheumatoid arthritis.4 When acting on endothelial cells, IL-17A stimulates inflammation and pro-coagulant activity.5 IL-17A also promotes endothelial cells and dendritic cells to release cytokines and enzymes.6,7 In monocytes and dendritic cells, IL-17A is involved in inflammation by modulating the production of pro-inflammatory cytokines.
The first receptor identified for IL-17A was IL-17A receptor A, and soon thereafter, other components required for the IL-17 pathway were found.8,9 Since IL-17F shares 50% sequence homology with IL-17A, a heterodimer consisting of IL-17A and IL-17F can also interact with the IL-17 receptor complex. The receptor complex contains IL-17RA and IL-17RC ligands.9 After the release of IL-17, two IL-17RA complex related intracellular signal pathways are activated. In the first pathway, IL-17RA can recruit adapter NF-κB activator 1 (Act-1) to form a complex with its conserved cytoplasmic domain SEFIR, which is common to all IL-17R family members.10,11 Subsequently, Act-1 binds to TNF receptor associated factor 6 (TRAF6) and acts as an E3 ubiquitin ligase on TRAF6, which recruits transforming growth factor activated kinases (TAK) 1 to mediate nuclear transportation of transcription factors such as nuclear factor-κB (NF-κB), activator protein 1 (AP1) and CCAAT/enhancer binding proteins (C/EBP).12 It is well established that mitogen-activated protein (MAP) kinase located downstream of TRAF6 is also required for AP1 activation.13
The second pathway is activated by IKKi-dependent phosphorylation of Act-1 at three Ser sites, which in turn suppresses the recruitment of TRAF6, thereby blocking the NF-rB pathway. In addition, Act-1 can modulate the mRNA stability of alternative splicing factor 1 (ASF) and ELAV-like protein 1 (ELAV1) through the ubiquitination of TRAF5 and its binding efficiency to TRAF2. At the same time, Act-1 can interact with and ubiquitinate ELAV1 through TRAF2 and TRAF5.14
Based on these observations, several agents have been designed to block the IL-17 pathway. These can be broadly divided into antibody inhibitors and small molecule inhibitors. In the first approach, several mAbs have been generated and are now undergoing clinical trials (Table 1). Rheumatoid arthritis and multiple sclerosis are diseases that have been widely studied using mouse models, and preclinical studies in humans related to IL-17A and trials of IL-17A mAb inhibitors to treat rheumatoid arthritis and multiple sclerosis have been reported.15
| mAba | Target | Indication | Development phase |
|---|---|---|---|
| a Clinical trial information: https://clinicaltrials.gov/. | |||
| Secukinumab (Novartis) | IL-17A | Plaque psoriasis | On the market |
| Ankylosing spondylitis | On the market | ||
| Rheumatoid arthritis | Phase III | ||
| Uveitis | Phase III | ||
| Ixekizumab (Lilly) | IL-17A | Plaque psoriasis | On the market |
| Rheumatoid arthritis | Phase III | ||
| Brodalumab (Amgen) | IL-17RA | Plaque psoriasis, rheumatoid arthritis | Phase III |
| ABT-122 (AbbVie) | IL-17A TNF | Plaque psoriasis, rheumatoid arthritis | Phase II |
| KHK4827 (Kyowa Hakko Kirin) | IL-17RA | Plaque psoriasis | Phase III |
| Perakizumab (Roche) | IL-17A | Psoriatic arthritis | Discontinued |
| RG 7624 (Roche) | IL-17A | Autoimmune diseases | Discontinued |
| IL-17F | |||
| ANB004 (AnaptysBio) | IL-17 | Autoimmune, inflammatory diseases | No information available |
| COVA322 (Covagen AG) | IL-17A TNF | Inflammatory diseases | No information available |
IL-17A signals through the formation of a heterodimeric receptor complex involving IL-17RA and IL-17RC and the compounds described here act by disrupting the interaction of IL-17A with IL-17RA, which is believed to be the first step in receptor activation. There are several ways to block IL-17A signalling by targeting IL-17A proteins or receptors. The priority option is direct action against IL-17A or IL-17RA. Two monoclonal antibodies directed against IL-17A are FDA approved: secukinumab (AIN457), a fully human IL-17A specific monoclonal antibody derived from human lgG1 kappa isotype and ixekizumab (LY2439821), a humanized lgG4 antibody. Other IL-17A targeted antibodies that have completed phase II are shown in Table 1. Secukinumab and ixekizumab are both specific for IL-17A homodimers and IL-17A but not effective on IL-17F homodimers.15,16 CJM112 is another IgG1 against IL-17A that is being tested for potential treatment of hidradenitis suppurativa and psoriasis.17 CNT06785 is a fully human Ig IL-17A antibody in phase II trials for rheumatoid arthritis and moderate-to-severe chronic obstructive pulmonary disease (COPD). However, there are no published data or further plans for development.18,19 Unlike secukinumab and ixekizumab, bimekizumab (previously UCB 4940) is a mAb targeting both IL-17A and IL-17F. A phase II trial for the add-on use of bimekizumab to certolizumab pegol in patients with rheumatoid arthritis has been completed with failure.20 Clinical trials of ABT-122, a dual-variable-domain immunoglobulin targeting both TNF and IL-17A for amplifying efficiency which was designed to treat moderate–severe psoriatic arthritis and rheumatoid arthritis, have currently completed phase II.21,22 The other targets for potential intervention are members of the IL-17RA complex, of which IL-17RA inhibition is the broadest way to regulate the IL-17A signal pathway. Brodalumab (AMG 827) is a human mAb neutralizing IL-17RA with high affinity that can block the biological activity of IL-17, IL-17F, a heterodimer composed of 17A/17F, or 17E. During phase II trials, brodalumab showed efficiency but with a greater risk of adverse events compared to the placebo group.23
In addition to biological therapy, small-molecule inhibitors that target IL-17A signaling by binding to the soluble ligand have been developed and are being tested in various clinical trials. Recently, some studies have suggested that some macrocycles might interfere with the interaction between IL-17A and IL-17RA with efficiency comparable to that of mAbs.
The crystal structure of the human IL-17A–IL-17RA interaction reveals an IL-17A homodimer which forms two symmetrical interactions with IL-17RA24 (Fig. 1). Both chains (A and B) of the IL-17A homodimer interact with the IL-17RA D1 or D2 domain. Compared with the apo-structure, the entire N-terminus of chain A bends away from IL-17RA, thus the N-terminus of the IL-17A homodimer is slightly separated in a buried cavity of the complex. The buried surface area of IL-17A–IL-17RA is ∼2000 Å2. The overall interaction comprises a large, flat and featureless binding interface, resulting in numerous additive but weak polar and hydrophobic interactions.
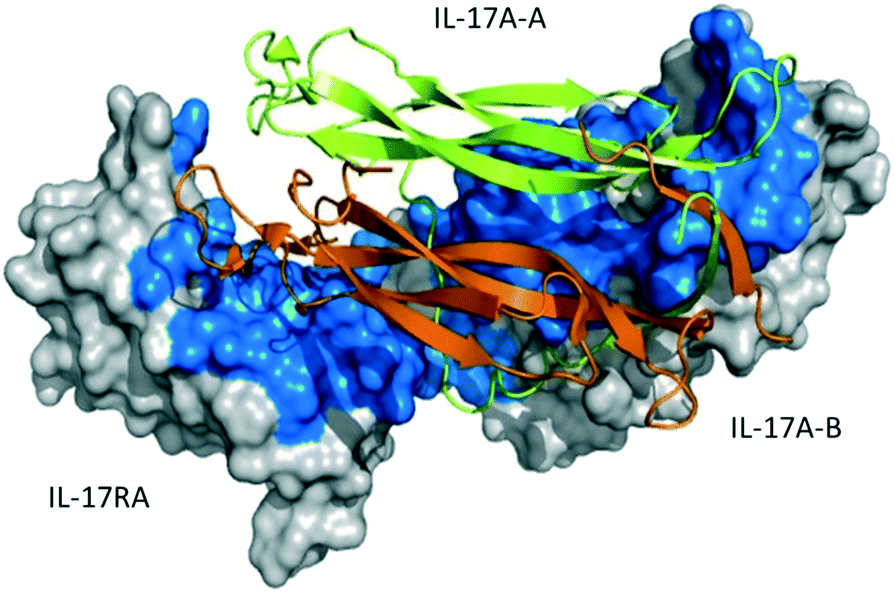 | ||
| Fig. 1 Complex between the IL17A dimer and its receptor (PDB ID 4HSA). The IL17A dimer is shown in orange and green cartoon and IL-17RA as a surface representation. The footprint of IL17A on IL-17RA is shown in blue. | ||
Recently, several groups have discovered artificial macrocycles that efficiently antagonize the IL-17A/IL-17RA interactions (Scheme 1). The biotech company Ensemble Therapeutics disclosed macrocycles binding to IL-17A.25 The discovery of compound 1 involved synthesis of large libraries of macrocycles using DNA-encoded library (DEL) synthesis technology. Recently, H/D exchange MS was used to determine the binding region of macrocycle 1 on IL-17A, which is predicted to bind to the β-hairpin pocket.26 Another group from Pfizer designed potent derivative macrocycles 2 and 3 and elucidated their high-resolution co-crystal structure with IL-17A.27,28 Both compounds bind into the homodimeric interface. Compound 3 shows multiple interactions with its receptor including hydrogen bonds, hydrophobic interactions and stacking interactions of the aromatic components of the macrocycle (Fig. 2).
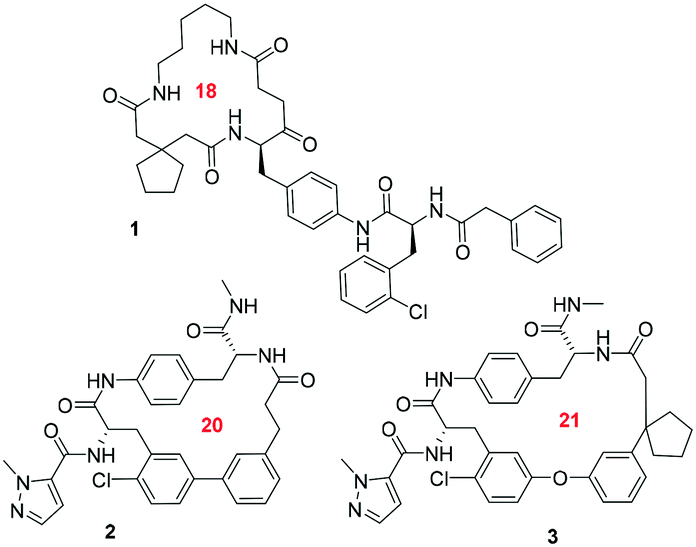 | ||
| Scheme 1 Structures of macrocycles interacting with IL17A. The ring size is indicated in red. | ||
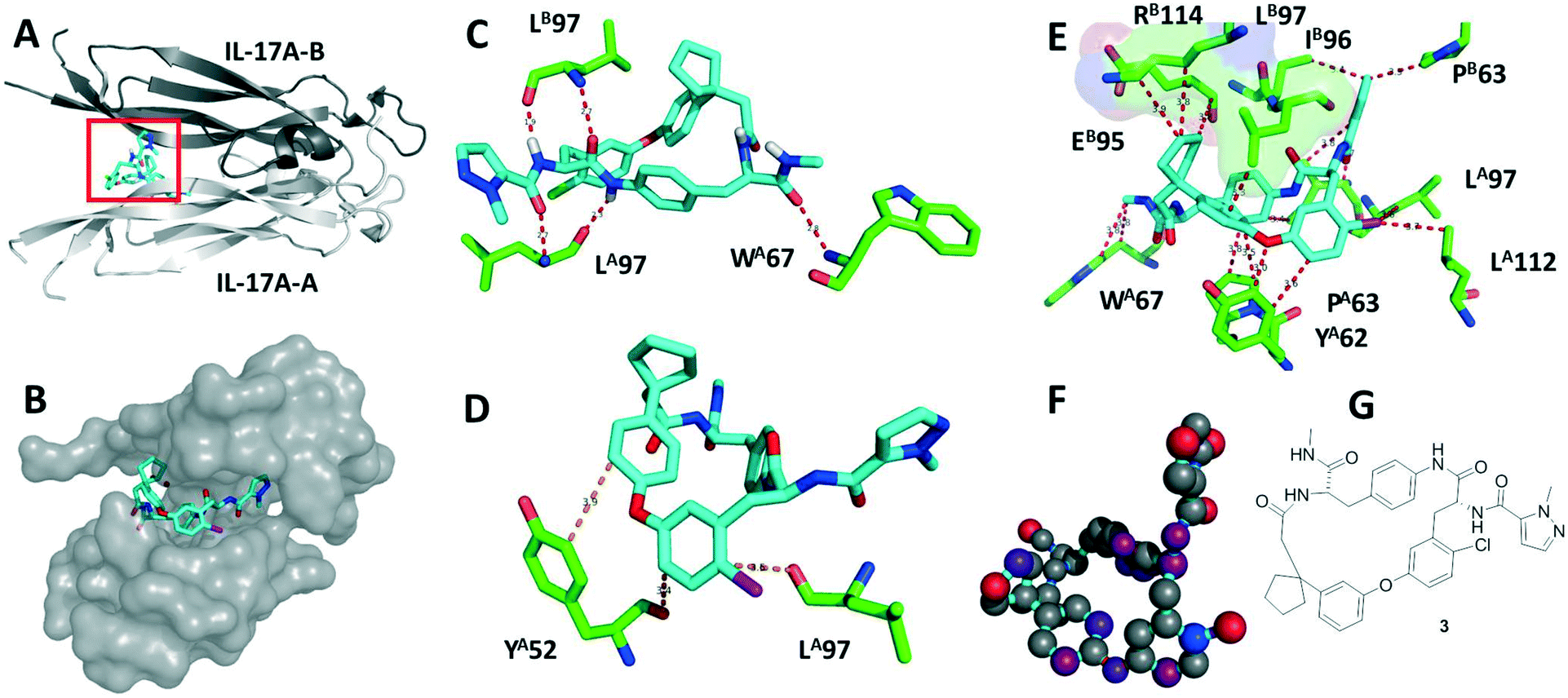 | ||
| Fig. 2 Macrocycle 3 bound to the IL17A dimer (PDB ID 5HI4). A: Secondary structure elements of IL17A chain A (light grey) and B (dark grey) and boxed macrocycle 3 (cyan sticks); B: surface representation of IL17A with 3; C: the hydrogen bonding network involves L97 from the IL17A and B monomers and W67 of IL17A A monomers; D: pi-stacking interactions with Y52 and L97; E: van der Waals interactions involving P63 B, L97 A, L112 A, Y62 A, P63 A, and W67 A; the subpocket (surface representation) formed by R114 B, E95 B and L97 B harbours the cyclopentenyl moiety which is of great importance for the SAR; F: the relative contribution of the heavy atoms of 3 to the binding to IL17A from low to high (grey to red) calculated using SCORPION;33 G: 2D structure of 3. | ||
Co-crystallisation of the macrocycles with IL17A was achieved using antibody antigen-binding fragment (Fab) co-crystallization chaperones – a technique known to facilitate the crystallization of difficult targets (Fig. 2). Macrocycle 3 shows multiple interactions with its dimeric receptor, including hydrogen bonds, hydrophobic interactions and stacking interactions of the aromatic components of the macrocycle (Fig. 2C–E). Notably, the atoms of the spiro-cyclopentyl moiety are important activity elements in all active macrocycle series, which can be rationalized by the shape and electrostatic complementarity with the Lys114, Leu97 pocket (Fig. 2E).
Recently, design guidelines for macrocycles were formulated based on the analysis of orally available cycles to obtain drug-like compounds. Thus, macrocycle 3 with a ring size of 21, a MW of 682, clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P ∼ 5, tPSA ∼ 140, HBD = 4, HBA = 6, four substituents, an overall polar/nonpolar atom balance of ∼0.3 and an N/O ratio of 6/5 fits well into these guidelines. Macrocycle 3 exhibits five hot-spot interacting areas (Fig. 2G). Besides, it shows measurable activity in the keratinocyte-based bioassay for IL-17A inhibition, indicating reasonable membrane permeation.
P ∼ 5, tPSA ∼ 140, HBD = 4, HBA = 6, four substituents, an overall polar/nonpolar atom balance of ∼0.3 and an N/O ratio of 6/5 fits well into these guidelines. Macrocycle 3 exhibits five hot-spot interacting areas (Fig. 2G). Besides, it shows measurable activity in the keratinocyte-based bioassay for IL-17A inhibition, indicating reasonable membrane permeation.
Protein–protein interactions (PPIs) on membranes are promising drug targets, and many mAb-based drugs are currently marketed. Traditionally, PPI inhibitors are designed as antibodies rather than small molecules, such as herceptin (anti-Her2), secukinumab (anti-IL17A) or atezolizumab (anti-PDL1). mAbs efficiently target large featureless protein surfaces and can be developed in a straightforward process to the market. Moreover, the attrition rate of mAbs during clinical development appears to be much lower than that of small molecules. Hence, some traditional small molecule-oriented pharma companies have recently announced a strong focus on biologics or their complete exit from the field of small molecule drug research and development. However, mAbs have a number of disadvantages as well, such as high cost-of-good, non-oral applications, poor tissue penetration, often long half-life times and most importantly being applicable only to extracellular targets. Recent findings from several groups demonstrate that several potent macrocycles show strong affinity to IL-17A and may lead to the discovery of lower cost and more effective treatments for IL-17A related inflammatory diseases. In nature, macrocycles are not uncommon and frequently exhibit useful biological activities29 and have major advantages over open chain analogues: including higher affinity and selectivity,30 preferable entropic signature, better oral bioavailability or higher stability.31,32
In summary, IL-17A/IL-17RA antagonists derived from medicinal chemistry might offer better options by reinvestigating beyond r-o-5 small compound classes such as artificial macrocycles, natural products, or peptidomimetics to treat inflammatory diseases. These compounds will also potentially enable research into novel targets such as large receptor ligand interactions, DNA and RNA. The application of synthetic macrocycles to challenging PPI targets represents an important emerging area, with potential implications for the future balance of effort between biological and small molecule drug discovery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NIH (2R01GM097082-05) and by Innovative Medicines Initiative (grant agreement no 115489). Moreover, funding has also been received from the European Union's Horizon 2020 research and innovation programme under MSC ITN “Accelerated Early stage drug dIScovery” (AEGIS), grant agreement No 675555.Notes and references
- L. E. Harrington, R. D. Hatton, P. R. Mangan, H. Turner, T. L. Murphy, K. M. Murphy and C. T. Weaver, Nat. Immunol., 2005, 6, 1123–1132 CrossRef PubMed.
- F. Fossiez, O. Djossou, P. Chomarat, L. Flores-Romo, S. Ait-Yahia, C. Maat, J.-J. Pin, P. Garrone, E. Garcia and S. Saeland, J. Exp. Med., 1996, 183, 2593–2603 CrossRef PubMed.
- P. Miossec, Arthritis Rheum., 2003, 48, 594–601 CrossRef PubMed.
- M. Chabaud and P. Miossec, Arthritis Rheum., 2001, 44, 1293–1303 CrossRef PubMed.
- A. Hot, V. Lenief and P. Miossec, Ann. Rheum. Dis., 2012, 71, 768–776 CrossRef PubMed.
- Z. Yao, W. C. Fanslow, M. F. Seldin, A.-M. Rousseau, S. L. Painter, M. R. Comeau, J. I. Cohen and M. K. Spriggs, Immunity, 1995, 3, 811–821 CrossRef PubMed.
- Z. Yao, M. K. Spriggs, J. M. Derry, L. Strockbine, L. S. Park, T. VandenBos, J. Zappone, S. L. Painter and R. J. Armitage, Cytokine, 1997, 9, 794–800 CrossRef PubMed.
- D. Toy, D. Kugler, M. Wolfson, T. V. Bos, J. Gurgel, J. Derry, J. Tocker and J. Peschon, J. Immunol., 2006, 177, 36–39 CrossRef.
- L. K. Ely, S. Fischer and K. C. Garcia, Nat. Immunol., 2009, 10, 1245–1251 CrossRef PubMed.
- S. H. Chang, H. Park and C. Dong, J. Biol. Chem., 2006, 281, 35603–35607 CrossRef PubMed.
- Y. Qian, C. Liu, J. Hartupee, C. Z. Altuntas, M. F. Gulen, D. Jane-Wit, J. Xiao, Y. Lu, N. Giltiay and J. Liu, Nat. Immunol., 2007, 8, 247–256 CrossRef PubMed.
- F. Shen, Z. Hu, J. Goswami and S. L. Gaffen, J. Biol. Chem., 2006, 281, 24138–24148 CrossRef PubMed.
- J. K. Kolls and A. Lindén, Immunity, 2004, 21, 467–476 CrossRef PubMed.
- S. Zhu and Y. Qian, Clin. Sci., 2012, 122, 487–511 CrossRef PubMed.
- R. G. Langley, B. E. Elewski, M. Lebwohl, K. Reich, C. E. Griffiths, K. Papp, L. Puig, H. Nakagawa, L. Spelman and B. Sigurgeirsson, N. Engl. J. Med., 2014, 371, 326–338 CrossRef PubMed.
- C. E. Griffiths, K. Reich, M. Lebwohl, P. van de Kerkhof, C. Paul, A. Menter, G. S. Cameron, J. Erickson, L. Zhang and R. J. Secrest, Lancet, 2015, 386, 541–551 CrossRef.
- Single and Multiple Dose Escalation Study to Assess the Safety and Tolerability of CJM112 in Psoriasis. https://clinicaltrials.gov/ct2/show/NCT01828086 (accessed May 20, 2017).
- An Efficacy And Safety Study of CNTO 6785 In Participants With Active Rheumatoid Arthritis Despite Methotrexate Therapy. https://clinicaltrials.gov/ct2/show/NCT01909427 (accessed May 20, 2017).
- A Study to Evaluate the Effectiveness and Safety of CNTO6785 in Patients With Moderate to Severe Chronic Obstructive Pulmonary Disease. https://clinicaltrials.gov/ct2/show/NCT01966549 (accessed May 20, 2017).
- Multiple Dose Study of UCB4940 as add-on to Certolizumab Pegol in Subjects With Rheumatoid Arthritis. https://clinicaltrials.gov/ct2/show/NCT02430909?term=bimekizumab&rank=6 (accessed May 21, 2017).
- A Phase 2 Study to Investigate the Safety, Tolerability and Efficacy of ABT-122 in Subjects With Active Psoriatic Arthritis Who Have an Inadequate Response to Methotrexate. https://clinicaltrials.gov/ct2/show/NCT02349451?term=ABT-122&rank=1 (accessed May 22, 2017).
- Phase 2, Multicenter, Open-Label Extension (OLE) Study With ABT-122 in Rheumatoid Arthritis Subjects Who Have Completed the Preceding M12–963 Study. https://clinicaltrials.gov/ct2/show/NCT02433340?term=ABT-122&rank=2 (accessed May 22, 2017).
- Study of Efficacy, Safety, and Withdrawal and Retreatment With Brodalumab (AMG 827) in Moderate to Severe Plaque Psoriasis Subjects (AMAGINE-1). https://clinicaltrials.gov/ct2/show/NCT01708590?term=Brodalumab&rank=3 (accessed May 20, 2017).
- S. Liu, X. Song, B. A. Chrunyk, S. Shanker, L. R. Hoth, E. S. Marr and M. C. Griffor, Nat. Commun., 2013, 4, 1888 CrossRef PubMed.
- M. Taylor, N. K. Terrett, W. H. Connors, K. C. Shortsleeves, B. A. Seigal, C. Snedeker, S. P. Hale, T. F. Briggs, F. G. Favaloro and T. J. Cipriani, Int. Pat. Appl., WO2013116682A1, 2013 Search PubMed.
- A. Espada, H. Broughton, S. Jones, M. J. Chalmers and J. A. Dodge, J. Med. Chem., 2016, 59, 2255–2260 CrossRef PubMed.
- S. Liu, J. Desharnais, P. V. Sahasrabudhe, P. Jin, W. Li, B. D. Oates, S. Shanker, M. E. Banker, B. A. Chrunyk, X. Song, X. Feng, M. Griffor, J. Jimenez, G. Chen, D. Tumelty, A. Bhat, C. W. Bradshaw, G. Woodnutt, R. W. Lappe, A. Thorarensen, X. Qiu, J. M. Withka and L. D. Wood, Sci. Rep., 2016, 6, 26071 CrossRef PubMed.
- S. Liu, L. A. Dakin, L. Xing, J. M. Withka, P. V. Sahasrabudhe, W. Li, M. E. Banker, P. Balbo, S. Shanker, B. A. Chrunyk, Z. Guo, J. M. Chen, J. A. Young, G. Bai, J. T. Starr, S. W. Wright, J. Bussenius, S. Tan, A. Gopalsamy, B. A. Lefker, F. Vincent, L. H. Jones, H. Xu, L. R. Hoth, K. F. Geoghegan, X. Qiu, M. E. Bunnage and A. Thorarensen, Sci. Rep., 2016, 6, 30859 CrossRef PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef PubMed.
- F. Giordanetto and J. Kihlberg, J. Med. Chem., 2013, 57, 278–295 CrossRef PubMed.
- J. Mallinson and I. Collins, Future Med. Chem., 2012, 4, 1409–1438 CrossRef CAS PubMed.
- D. H. Williams, C. T. Calderone, D. P. O'Brien and R. Zerella, Chem. Commun., 2002, 1266–1267 RSC.
- B. Kuhn, J. E. Fuchs, M. Reutlinger, M. Stahl and N. R. Taylor, J. Chem. Inf. Model., 2011, 51, 3180–3198 CrossRef CAS PubMed.
Footnote |
| † The authors declare no competing interests. |
| This journal is © The Royal Society of Chemistry 2018 |