
Synthesis and biological evaluation of rapamycin-derived, next generation small molecules
Shiva Krishna Reddy
Guduru
 *ab and
Prabhat
Arya
c
*ab and
Prabhat
Arya
c
aCenter for Drug Discovery, Department of Pharmacology and Chemical Biology, Baylor College of Medicine, One Baylor Plaza, Houston, Texas 77030, USA. E-mail: gskreddy.chem@gmail.com; Shiva.Guduru@bcm.edu; Tel: +1 713 798 8794
bDepartment of Pharmacology and Chemical Biology, Baylor College of Medicine, One Baylor Plaza, Houston, Texas 77030, USA
cChemistry and Chemical Biology, Dr. Reddy's Institute of Life Sciences (DRILS), University of Hyderabad Campus, Hyderabad 500046, India
First published on 22nd November 2017
Abstract
Over the years, rapamycin has attracted serious attention due to its remarkable biological properties and as a potent inhibitor of the mammalian target of rapamycin (mTOR) protein through its binding with FKBP-12. Several efficient strategies that utilize synthetic and biosynthetic approaches have been utilized to develop small molecule rapamycin analogs or for synthesizing hybrid compounds containing a partial rapamycin structure to improve pharmacokinetic properties. Herein, we report selected case studies related to the synthesis of rapamycin-derived compounds and hybrid molecules to explore their biological properties.
Dr. Shiva Krishna Reddy Guduru took his doctoral studies under the guidance of Professor Prabhat Arya at the Dr. Reddy's Institute of Life Sciences (DRILS), an associate institute of the University of Hyderabad. His PhD thesis work was focused on the development of novel methods to build tetrahydroquinoline-derived, natural product inspired macrocyclic architectures and rapamycin-derived hybrid natural products. Currently he is working as a postdoctoral research associate with Dr. Damian Young in Baylor College of Medicine; his research mainly focuses on Fragment Based Drug Discovery (FBDD). He has a strong passion for undertaking research projects at the interface of modern organic synthesis, medicinal chemistry and chemical biology. |
Professor Prabhat Arya moved back to India in 2009 to establish a program at the interface of stereoselective organic synthesis and cell signaling following a long academic career at the NRC Canada. His research is focused on developing synthesis methods to allow building a chemical toolbox with compounds that are more close to bioactive natural products and can be classified as natural product-inspired and hybrid natural products. His ultimate interest is in addressing challenging biological questions that are related to protein–protein interactions and pathways with an extensive use of small molecules that are emerging from his group. Prior to move back to India (which he does not regret so far), he was also an Adjunct Professor at the Department of Biochemistry, McGill University, OHRI Ottawa, the Department of Chemistry, University of Ottawa, and the Department of Biochemistry, University of Ottawa. |
Introduction
Rapamycin (also known as sirolimus), a secondary metabolite produced by the soil bacterium Streptomyces hygroscopicus, was first isolated in Easter Island (Rapa Nui) in the 1970s.1,2 Rapamycin is a 31-membered polyketide macrolactone that embeds the following features: (a) a cyclohexyl moiety derived from the shikimic acid pathway, (b) a 6-membered hemiacetal, (c) a pipecolinyl ring and (d) a substituted triene moiety (Fig. 1). The biological activity of rapamycin was originally discovered in a screening for novel antifungal agents and was observed to be mainly active against Candida albicans. Later, it was also discovered to be an antibiotic with significant immunosuppressant properties. Since this discovery, rapamycin has been used successfully for preventing rejection in organ transplantation. Rapamycin is currently approved for the treatment of cardiovascular diseases by the US Food and Drug Administration.3 | ||
| Fig. 1 Structure of rapamycin and its analogs. | ||
mTOR structure and biological function
A key feature of rapamycin is its inhibition of the mammalian target of rapamycin (mTOR) protein, which is a 289 kDa intracellular serine/threonine protein kinase. mTOR belongs to the phosphoinositide-3-kinase (PI3K)-related kinase family that plays a central role in various cellular processes including cell growth and proliferation, protein synthesis and autophagy.4 mTOR contains a central regulatory catalytic core of at least two functionally distinct multi-protein complexes commonly known as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). These two can be distinguished from each other based on their unique compositions and substrates (see Fig. 2). In 2013, Yang and co-workers solved the crystal structure at 3.2 Å of a truncated ∼1500-amino acid mTOR–mammalian lethal with SEC13 protein 8 (mLST8) complex which contained the FAT, FRB, kinase and FATC domains, as well as the structures of this complex bound to an ATP transition state analogue. The structure shows that the kinase domain adopts a canonical protein kinase conformation and provides a model for the inhibition of mTORC1 by rapamycin–FKBP12.5 More recently, cryo-EM structures of mTORC1 at different resolutions (5.9 Å, 4.4 Å) and the TOR–Lst8 complex at 6 Å, crystallized from the thermotolerant yeast Kluyveromyces marxianus, were published. When combined with biochemical analyses, these structures provide an accurate topological interpretation of human mTOR which improves understanding of the assembly and function of mTORC1.6–8 Initially, mTORC1 and mTORC2 were identified on the basis of their differential sensitivity to the inhibitory effects of rapamycin, with mTORC1 identified as the rapamycin-sensitive and mTORC2 as the rapamycin-insensitive complex.9,10 However, treatment with high doses of rapamycin can also indirectly inhibit mTORC2 activity in a cell specific manner.11 | ||
| Fig. 2 (a) mTOR domain structure: mTOR consists of tandem HEAT repeats, a central FAT domain, a FRB domain, a catalytic kinase domain and a FATC domain. Rapamycin binds with its intracellular receptor, FKBP12, and the resulting complex then binds with the FRB domain of mTOR. The binding of rapamycin–FKBP12 to the FRB domain disrupts the association of mTOR with the mTORC1 specific component, Raptor, and thus decouples mTORC1 from its substrates, thereby blocking mTORC1 signaling. (b) mTORC1 and mTORC2 composition and regulation: mTORC1 contains mTOR, Raptor, PRAS40, mLST8 (also known as GβL) and Deptor which regulate cell growth through S6K and 4E-BP1 effectors. mLST8 attaches to the mTOR kinase domain in both complexes, where it seems to be essential for their assembly. Deptor acts as an inhibitor of both mTORC1 and mTORC2 complexes. Other protein partners differ between these two complexes. mTORC2 contains Protor, Sin1 and Rictor which regulate the prosurvival kinase Akt/PKB and PDK1. | ||
mTOR, in particular, mTORC1 plays a key role as a nutrient and energy sensor, both at the cellular and whole-body levels.12,13 The result of mTORC1 signalling in metabolically important organs such as liver, adipose tissue, muscle and hypothalamus coordinates whole-body metabolism during the food intake intervals and starvation. In a healthy physiological state, glucose homeostasis is regulated by glucose by its conversion into glycogen when glucose levels are high and, reversely, glycogen conversion into glucose when blood glucose levels are low. These responses are strongly controlled by insulin and its downstream signalling to mTORC1 and mTORC2. Excess glucose can also be stored as triglycerides in the white adipose tissue (WAT). mTORC1 promotes adiposity14 and lipid accumulation by up-regulating transcription factors such as peroxisome proliferator-activated receptor-γ (PPARγ) and sterol regulatory element-binding protein 1 (SREBP1) and SREBP2.15 Chronic overfeeding results in continuously high levels of mTOR signaling, which disturbs whole-body metabolic regulation and eventually leads to obesity (excessive lipid accumulation) and type 2 diabetes (loss of insulin responsiveness). Abnormalities in metabolic homeostasis and growth control lead to diabetes and cancer, respectively, and, it is not surprising that mTOR plays a role in their aetiology. However, an additional aspect in the TOR story is its role in lifespan extension. Long-term suppression of mTOR signaling (which is stimulated by nutrients) mimics dietary restriction, a well-documented means of lifespan prolongation.16,17 Adult mice treated with rapamycin have shown increased lifespan with better health indicators compared to an untreated control group.18 Though TOR inhibition has been shown to increase the lifespan of yeast, flies and worms, proving similar lifespan extension in vertebrates opens opportunities for developing mTOR inhibitors. Major clinical benefits from mTOR inhibition are expected to be seen in cancer treatment, management of diabetes and associated complications,19 lifespan extension and amelioration of age-related disorders.20
Biological activity of rapamycin
Rapamycin binds to FK506 binding proteins (FKBPs), which belong to a specific family of immunophilins known as cytosolic binding proteins. The protein within this family which is most important for mediation of the immunosuppressive effects of rapamycin is FKBP12, a 12 kDa protein which functions as cis/trans peptidylpropyl isomerase.21 Rapamycin binds to FKBP12 and inhibits its isomeric activity. However, the rapamycin–FKBP12 complex alone is insufficient to mediate an immunosuppressive effect. Rapamycin becomes biologically active only when the FKBP12–rapamycin complex binds with its intracellular target, FRB domain, a small (11 kDa) hydrophobic binding domain present on the 289 kDa protein FRAP. FKBP12–rapamycin binding to the FRB domain leads to protein–protein interaction (PPI) stabilization of the complex which blocks various signal transduction pathways, in turn inhibiting cell cycle progression from the G1 to the S phase in various cell types allowing rapamycin to exert its biological actions.22,23In 1996, Choi and co-workers solved the X-ray crystallographic structure of the ternary complex FKBP12–rapamycin–FRB at 2.7 Å resolution (Fig. 3). Later, in 1999, Liang and co-workers crystallized the ternary complexes of rapamycin derivatives and their structures were solved at 1.85 and 2.2 Å resolutions.24 The FKBP12 protein consists of an α, β sheet made-up of five antiparallel β strands, and a short α helix. Rapamycin binds to the hydrophobic pocket generated between an α helix and a β sheet. The FRB domain of FRAP is composed of a bundle of four α helices with rapamycin binding to the hydrophobic pocket formed by helices α1 and α4. It is evident that rapamycin interacts with both (i.e. FKBP12 and FRB) receptor proteins. Moreover, experimental studies have shown that the FKBP12 protein is unable to bind with FRB in the absence of rapamycin.25,26 To obtain deeper insight into the interaction network responsible for the binding of rapamycin with FKBP12 and FRB, Sironi and co-workers recently reported molecular dynamics (MD) simulations and free energy calculations of this ternary system.27 In particular, they calculated the binding free energy between the components of the FKBP12–rapamycin–FRB ternary system, and, also performed computational alanine scanning (CAS) to evaluate the contribution of each amino acid at the protein–protein and protein–rapamycin interface to the overall binding energy. This free energy study has given clear understanding of protein–protein interaction stabilization with rapamycin at an atomic level.
 | ||
| Fig. 3 The structure of a ternary complex of FKBP12–rapamycin–FRB: (A) surface model and (B) ribbon model; red color – FKBP12, green color (small molecule) – rapamycin and purple color – FRB (PDB code: 1NSG). Molecular model images were generated using the Maestro v9.6 software package (Schrodinger LLC). | ||
Rapamycin is also known to suppress interleukin-2 (IL-2) induced T cell proliferation by downregulating the expression of genes necessary for key processes required for cell cycle progression. This results in immunosuppression, which is desirable to prevent allograft rejection in patients post-transplantation.28,29 In general, rapamycin is tolerated well and does not exhibit renal toxicity which is associated with other immunosuppressants such as cyclosporine or FK506.30 In 1999, rapamycin was approved for prevention of graft rejection in kidney transplant recipients by the US FDA. Another clinical application of rapamycin is vascular smooth muscle growth inhibition, and, in 2002, it was approved as an anti-restenosis agent following balloon angioplasty in coronary arterial stents.31 Recently, there is increasing interest in developing rapamycin as an anticancer drug supported by increased understanding of the mTOR function and the preponderance of tumour suppressors or oncoproteins [phosphoinositide-dependent protein kinase 1 (PDK1), PI3K, PTEN, AKT, TSC1 and TSC2] in the mTOR signaling network. Multiple studies have shown that rapamycin can also provide therapeutic benefit in experimental models of several age-linked neurodegenerative diseases,32 such as Alzheimer's which is common,33 Parkinson's34 and also Huntington's.35,36 Other studies have demonstrated that rapamycin is able to extend the lifespan of several species including mice.18,37 Because of the multiplicity of mTOR downstream signalling pathways, diverse molecular mechanisms have been proposed to explain rapamycin's effects in these studies.
Due to the important role that rapamycin plays in modulating mTOR signalling events and its structural complexity, several synthetic efforts have been dedicated to obtaining simpler structural analogs by synthetic as well as semi- and bio-synthetic approaches. The ultimate goal of these efforts is the discovery of novel small molecules with improved pharmacokinetic properties. Another objective of these synthetic endeavours is the development of strong intellectual property positions compared to those of the parent molecule. Herein, we report selected case studies on the synthesis of rapamycin analogs and hybrid molecules to explore their biological activities (total synthesis and biological evaluation of rapamycin and its analogs by various research groups) that have not been covered in earlier review articles.3,38
Rapamycin total synthesis efforts
Five total syntheses of rapamycin have been published to date from the research groups of Nicolaou,39 Schreiber,40 Danishefsky,41 Smith42 and, more recently Ley.43 Nicolaou and co-workers were the first to successfully achieve rapamycin synthesis. The retrosynthetic analysis of their approach is shown in Scheme 1.44 The disconnection of the triene system in rapamycin (F1) [Stille palladium-catalyzed coupling] was suggested to provide bis(vinyl iodide) 1.1 and distannylethene 1.2 as the two potential precursors. Further, the disconnection of an amide group and the ester moiety in 1.1 and the opening of a lactol ring reveal 1.3, 1.4 and 1.5 as advanced key intermediates. The most complex of the latter three fragments is compound 1.5 which could be put together using an Evans aldol reaction (for C34–C35 bond formation) and chromium–nickel coupling (for C28–C29 bond formation). After functional group manipulations, this path afforded compounds 1.6–1.8 as potential building blocks. Thus, a strategy was devised that led to the construction of intermediates 1.6–1.8, with final elaboration to complete the synthesis of rapamycin. | ||
| Scheme 1 Nicolaou's approach to rapamycin synthesis. | ||
In 1993, Schreiber and co-workers also achieved the total synthesis of rapamycin (F1) and C32-(S)-dihydrorapamycin (2.1).40 As shown in their retrosynthesis (Scheme 2), key reactions involved were the Mukaiyama macrocyclization, aldol reaction, Wittig olefination and Evans–Tischenko fragment coupling reaction. The first disconnection of rapamycin at the C10 carbon and oxygen bond (Mukaiyama macrocyclization) provided the key intermediate 2.2 which was obtained through cyclisation of intermediate 2.3 by aldol reaction. This acyclic compound 2.3 was obtained by Wittig reaction of two key intermediates 2.4 and 2.5.45 Fragment 2.4 was obtained from samarium mediated Evans–Tischenko fragment coupling reaction.
 | ||
| Scheme 2 Schreiber's approach to rapamycin and C32-(S)-dihydrorapamycin synthesis. | ||
In the same year, Danishefsky and co-workers succeeded in the total synthesis of rapamycin (F1) via a titanium-mediated aldol macrocyclization reaction.41 As shown in Scheme 3, key disconnections at the ester and C27–C28 bond (macroaldolization mediated by TiCl3(OiPr) in rapamycin (F1)) were expected to provide intermediates 3.1 and 3.2. Intermediate 3.1 was further disconnected (condensation) to give fragment 3.3, which could be readily obtained from L-pipecolic acid and aldehyde 3.4.
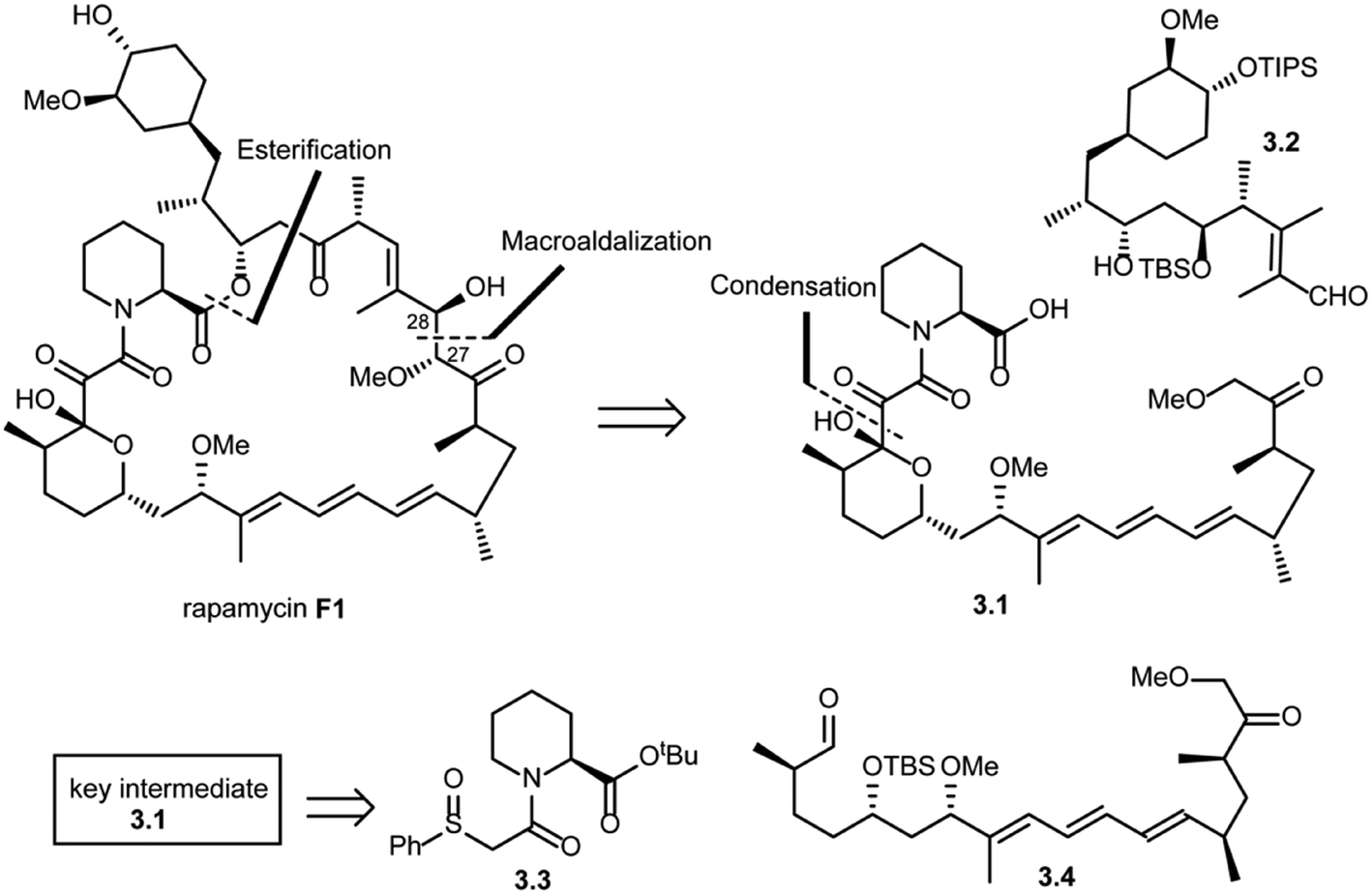 | ||
| Scheme 3 Danishefsky's approach to the total synthesis of rapamycin. | ||
Amos B. Smith's research group also reported the total synthesis of rapamycin (F1) and 27-demethoxyrapamycin (4.1) in 1995.42,46,47 As shown in Scheme 4, disconnection of the triene system (Pd(0) catalyzed Stille coupling) and ester group (intermolecular acylation at C34) in rapamycin F1 was suggested to provide compound 4.2 (vinyl iodide fragment) and 4.3 (vinyl tributyl stannane fragment). Further disconnection of compound 4.2 provided compounds 4.4 (N-acetyl pipecolic acid) and 4.5 as key intermediates (protocol reported by Golec et al.48). Disconnection of compound 4.3 at the C33–C32 bond and C27–C26 revealed compounds 4.6, 4.7 and 4.8.
 | ||
| Scheme 4 Smith's approach to the total synthesis of rapamycin. | ||
The most recent synthesis of rapamycin was reported by Ley and co-workers. Their synthesis is not as linear as the previous syntheses and their retrosynthetic analysis is shown in Scheme 5.49 The formation of the rapamycin macrocycle was successfully achieved by employing a transannular catechol-templated Dieckmann-like reaction. Further, the disconnection at the central olefin (C20–C21) of the triene moiety through Pd0-catalyzed Stille coupling afforded the simplified C10–C20 lactone 5.1 and C21–C42 vinyl stannane 5.2. For the latter stannane moiety (5.2), Ley's team envisioned the sequential carbanionic coupling of 5.3, 5.4, and 5.6, whose synthesis was planned to highlight the chemistry that was developed by them in the past.
 | ||
| Scheme 5 Ley's approach to rapamycin synthesis. | ||
Synthesis and biological evaluation of rapamycin analogs
During the biosynthesis of rapamycin, the amino acid L-pipecolate is incorporated into the macorcyclic architecture. In 2004, Ritacco and co-workers investigated the use of precursor-directed biosynthesis to create new rapamycin analogs by substituting unusual L-pipecolate derivatives in place of the normal amino acid.50 It is postulated in their study that the L-pipecolate analog (±)-nipecotic acid inhibits the biosynthesis of L-pipecolate, thereby limiting the supply of this molecule for rapamycin biosynthesis. They used (±)-nipecotic acid for the precursor-directed biosynthetic studies to reduce L-pipecolate availability, and to enhance the incorporation of the pipecolate analogs into the rapamycin architecture. By using this method, they produced two new sulfur-containing rapamycin analogs, 20-thiarapamycin (6.1) (yield: ∼100 mg per litre) and 15-deoxo-19-sulfoxylrapamycin (6.2) (yield: ∼10 mg per litre) (Scheme 6). These two analogs were tested in a FKBP12 binding assay, i.e. expressed as the percent inhibition of 3H-labelled FK506–FKBP12 (control) complex formed. Rapamycin was examined as a control for the binding affinity. In this assay, 20-thiarapamycin (6.1) had a 50% inhibitory concentration (IC50) of 53.6 nM, while rapamycin had an IC50 of 1.6 nM, whereas 15-deoxo-19-sulfoxylrapamycin (6.2) had an IC50 of 800 nM (rapamycin IC50 4.9 nM). | ||
| Scheme 6 Rapamycin structure, 20-thiarapamycin and 15-deoxo-19-sulfoxylrapamycin. | ||
In 2005, Sheridan and co-workers from Biotica Technology Ltd. reported the mutasynthesis of rapamycin analogs through the manipulation of the gene governing starter unit biosynthesis.51 Rapamycin, FK506, and F520 are biogenetically related natural products which are synthesized by a mixed polyketide synthase (PKS)/nonribosomal peptide synthetase (NRPS) system. The dihydroxycyclohexane moiety of rapamycin originates from the incorporation of a 4,5-dihydroxycyclohex-1-enecarboxylic acid (DHCHC) starter unit derived from shikimic acid. The immediate product of the rapamycin PKS is pre-rapamycin 7.2, which is then modified by a series of two cytochrome P450 monooxygenases (RapJ and RapN) and three O-methyltransferases (RapI, RapM and RapQ). When the region of DNA including the genes thought to encode these enzymes was excised (rapKIJMNOQL) from the rapamycin biosynthetic gene cluster, no production of rapamycin was observed from the resulting strain, Streptomyces hygroscopicus MG2-10. The mutant strain MG2-10 was then independently complemented with full-length copies of each of the genes which had been removed. Surprisingly, the production of pre-rapamycin 7.2 was only observed when rapK was reintroduced and expressed in the strain. The researchers first carried-out a series of experiments in an attempt to explain the lack of production of the rapamycin macrocycle by S. hygroscopicus MG2-10. These included feeding experiments in which exogenous pseudostarter carboxylic acid 7.1 was added to the fermentation medium to verify that the supply of this component was not a limiting factor. The addition of pseudostarter carboxylic acid 7.1 to the fermentation medium led to the efficient production of pre-rapamycin 7.2. Furthermore, the addition of other carboxylic acids in place of pseudostarter carboxylic acid 7.1, as reported for the wild-type organism, was found to lead to the specific production of pre-rapamycin analogs in which these non-natural starter acids were incorporated, in most cases, after prior hydroxylation. Cyclohexanecarboxylic acid 7.3, cyclohex-1-enecarboxylic acid 7.5, and cycloheptanecarboxylic acid 7.7 were all fed separately to S. hygroscopicus MG2-10, which led to the production of analogs 7.4, 7.6, and 7.8 respectively (Scheme 7). Compounds 7.6 (22 mg) and 7.8 (77 mg) were isolated from the fermentation broth by preparative chromatography and their structures were confirmed using high-resolution FT-ICR-MSn. Compound 7.4 (100 mg) was isolated and its structure was confirmed by a combination of high-resolution FT-ICR-MSn and multidimensional NMR spectroscopy experiments. These analogs offer great potential as chemical genetic probes of the molecular pathways involved in rapamycin functioning, and as new agents with enhanced therapeutic properties.
 | ||
| Scheme 7 (a) Re-establishment of pre-rapamycin 7.2 production in S. hygroscopicus MG2-10 by feeding the pseudostarter unit 4.1. (b and c) Exclusive production of 7.4–7.8 as analogs of pre-rapamycin by feeding cyclohexanecarboxylic acid, cyclohex-1-enecarboxylic acid, and cycloheptanecarboxylic acid to S. hygroscopicus MG2-10. | ||
From the same company, Lanceron and co-workers reported an expeditious route to obtain fluorinated rapamycin analogs by utilizing a mutasynthesis approach. They synthesized 6 flouorohydrins (fluorinated starter units) which were fed and then incorporated S. hygroscopicus MG-210 at various incorporation levels (Scheme 8).52,53 Biological evaluation of these fluorinated rapamycin analogs led to the discovery of the importance of the hydrogen bond of the hydroxyl group on carbon 40 with FKBP12. This was performed by measuring the binding of these analogs to FKBP12 and FRAP in the ternary complex and its impact on the biological activity of rapamycin.
 | ||
| Scheme 8 The corresponding pre-rapamycin analogs obtained by a mutasynthesis approach. | ||
Holt and co-workers synthesized over 100 rapamycin analogs by using a selective acid-catalyzed nucleophilic substitution reaction at the C-16 methoxy group of rapamycin.54,55 This unique transformation allowed the selective manipulation of the rapamycin effector domain which further affects FKBP12 binding, and, the biological activity. Rapamycin C-16-modified analogs of both R and S configurations were shown to have high affinities for FKBP12, yet, these analogs displayed a wide range of potencies in splenocyte and yeast proliferation assays (see Table 1).56,57 In the ternary complex, the C-16 methoxy group of rapamycin was found to be in very close proximity to FRAP, at the interface of FRAP and FKBP12.
|
|
||||
|---|---|---|---|---|
| R | Compound | FKBP Ki (nM) | T cell IC50 (nM) | Yeast IC12 (nM) |

|
1 | 0.6 | 1 | 7 |

|
4 | 1 | 30 | 20 |

|
5a | 3.5 | 10 | 10 |

|
6a | 1 | 4 | 4 |

|
6b | 1 | 4 | 4 |

|
7a | 4.5 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
170 |

|
7b | 3.7 | 2500 | 220 |

|
8a | 9 | 1000 | 225 |

|
8b | 38 | >1000 | 400 |

|
9 | 7 | >1000 | >1000 |

|
10a | 3 | 20 | 12.5 |

|
10b | 6 | 20 | 3 |
|
11a | 10 | 11 | 28 |

|
11b | 1.5 | 70 | 28 |

|
12 | 1 | 2 | 8 |

|
13 | 29 | 300 | 210 |

|
14a | 5 | 6 | 6 |

|
15a | 7 | 50 | 20 |

|
16a | 1 | 45 | 6 |

Nicolaou and co-workers designed and synthesized rapamycin-based high affinity binding FKBP12 ligands and named them rapamycin–peptides (Rap-P) by using peptide tether chemistry.58 The designed small molecules contained three peptide cassettes, D-homoPhe-Gly-Sar 9.2. This approach allowed the synthesis of a 21-membered macrocyclic ring 9.5 starting from 9.2. Similarly, starting from D-homoPhe-(Gly)39.3 and D-homoPhe-(Gly)49.4 led to 24- and 27-membered rings 9.6 and 9.7, respectively. The retrosynthesis of rapamycin–peptides is shown in Scheme 9. These hybrid molecules were evaluated where they exhibited powerful binding properties, but, unlike rapamycin, showed no activity in IL-6 dependent B-cell proliferation, and, in contrast to FK506, also showed no activity in the IL-2 reporter assay. The modular nature of these designed molecules should make the generation of a series of compounds with the effector domains for targeting either calcineurin or FRAP (TOR/RAFT1) or both possible, as potential chemical probes and immunosuppressive agents.
 | ||
| Scheme 9 Molecular design and retrosynthetic analysis of FKBP12 ligands RAP-P (9.5–9.7). | ||
Chemically induced dimerization (CID) is a biological process in which a dimerizer (chemical or biological entity) brings two proteins in close proximity.59,60 An application of CID is the induction of protein translocation by bringing the protein of interest (POI) to a specific cellular location by addition of the dimerizer in order to stimulate or inhibit signal transduction induced by the protein of interest (Fig. 4).61 Rapamycin also serves as an inducer of the dimerization of FKBP12 and FRB which activates enzyme activity inside an intact cell.62 CID is a powerful technique to investigate various biological processes.63–65 Through the careful choice of POIs and targets, various biological systems were studied, for example, lipids as targets to study KCNQ ion channels,66 Ca2+ influx,67 clathrin-coated endocytic pits,68 receptor mediated endocytosis,69 PTEN tumor suppressors70 and Rab5a-positive endosomes.70 Small GTPases as targets were used to study cell-surface receptor CD25,71 axonal growth cones,72 neutrophil migration,73 Ras signalling by organelle targeting motifs74 and other biological systems like ZAP70 tyrosine kinase,75 Akt kinase in apoptosis,76 β-arrestin-2 and vasopressin receptors,77 GTPase activity,78 and plant hormone gibberellin A3 (GA3) based CID.79,80 Schreiber and co-workers developed a method for inducible gene expression and translocation by using non-toxic derivatives of rapamycin.81 However, rapamycin is an inhibitor of cell proliferation which mediates its effects by binding to FRAP. To limit FRAP binding, the non-toxic derivatives of rapamycin were designed with bulky substituents at the C16-position and synthesized by using the method previously reported by Luengo and co-workers (Scheme 10). The isosteric isopropoxy and methallyl groups, substituents with the non-natural C16-configuration, abolished both binding to FRAP and inhibition of T cell proliferation. The binding proteins for these derivatives were identified from the libraries of cDNAs encoding mutants of the FRB domain of FRAP by using a mammalian three-hybrid transcription assay. The targeting of mutations was guided by the structure of the FKBP12–rapamycin–FRB ternary complex. Three compensatory mutations within the FRB domain, along one face of an α-helix in a highly rapamycin-binding pocket, together restore binding of the rapamycin derivatives. Using this mutant FRB domain, the library of non-toxic rapamycin derivatives was screened, which led to the identification of a non-toxic rapamycin derivative (3R) that induced targeted gene expression in Jurkat T cells with an EC50 below 10 nM.81 Another derivative (9R) was used to bring a cytosolic protein to the plasma membrane, mimicking a process involved in many signalling pathways. Recent advances like photoactivatable derivatives of rapamycin provide attractive tools for basic science research.82–84 However rapamycin, rapalogs or even caged rapamycin derivatives serving as great tools for CID, due to rapamycin's high affinity to its binding partners have some limitations such as the lack of switching off the signal, toxicity, and cost of the dimerizer and non-productive interactions often occur because of the fusion of the dimerizer with the abundant endogenous FKBP.
 | ||
| Fig. 4 Schematic representation of CID experiment in living cells: one protein component (here FRB) anchoring on a plasma membrane at the desired site of the protein of action takes place. The other protein component (FKBP) is attached to the protein of interest (POI). In the absence of a dimerizer (rapamycin), the POI–FKBP complex freely diffuses into the cytoplasm. Upon addition of a dimerizer (rapamycin) to the cells, it passes through the plasma membrane and first binds to FKBP forming the FKBP–rapamycin complex. Then, the FKBP–rapamycin complex moves towards the plasma membrane along with the POI and binds to FRB to form the FKBP–rapamycin–FRB complex. POI translocation to the desired functional site (in this case the plasma membrane) induces or inhibits cellular function. | ||
 | ||
| Scheme 10 C16-substituted derivatives of rapamycin. | ||
The long half-life of rapamycin (62 to 82 hours in humans) has further complicated its use, increasing the probability of associated adverse side effects. In this context, Abbott Laboratories initiated a program that was aimed at identifying rapamycin analogs with shorter in vivo half-lives and improved side effect profiles. Their efforts were focused mainly on the introduction of novel stable functionalities at C40 that would allow them to identify analogs with attractive physicochemical attributes. This study led to the design of zotarolimus F1.2, which has a tetrazole substituent at C40 (Fig. 1). The Abbott team reported in vitro antiproliferative activities and in vivo immunosuppressive activities of zotarolimus, which has the potential for an improved safety profile by virtue of its shorter in vivo half-life. Zotarolimus was shown to be mechanistically similar to rapamycin in terms of its high binding affinity to FKBP12 and showed comparable potency for inhibiting the in vitro proliferation of both human and rat T cells.85 Rat pharmacokinetic studies with intravenous dosing demonstrate terminal elimination half-lives of 9.4 hours for zotarolimus compared to the 14.0 hours for rapamycin. When these two drugs were orally administered, half-lives of 7.9 hours for zotarolimus and 33.4 hours for rapamycin were seen. Consistent with its shorter half-life, zotarolimus shows a statistically significant 4-fold reduction in potency for systemic immunosuppression in 3 rat disease models. In addition to rat models, also pharmacokinetic studies were performed in cynomolgus monkey to validate the half-life difference between zotarolimus and rapamycin. The in vitro inhibitory effect on human coronary artery smooth muscle cell proliferation by zotarolimus was comparable to that of rapamycin. Drug-eluting stents for the local delivery of zotarolimus to the vessel wall of coronary arteries are in clinical development.86–88 The pharmacological profile of zotarolimus suggests that it may be more useful for preventing restenosis with reduced potential for causing systemic immunosuppression or other side effects.
The other rapamycin analogs with improved pharmacokinetic properties compared to rapamycin are everolimus (F1.3), temsirolimus (F1.4) and ridaforolimus (F1.5), which are synthesized by derivatization of rapamycin at its C-40 position (Fig. 1). Everolimus (Afinitor; Novartis) is a hydroxyethyl ether derivative developed for oral administration to inhibit mTOR, the therapeutic target for metastatic renal cell carcinoma.89,90 Everolimus was recently reported to increase progression-free survival in patients with advanced pancreatic neuroendocrine tumours.91 Temsirolimus (Torisel; Wyeth) is a water-soluble ester derivative for oral or intravenous formulations. In 2007, temsirolimus was approved for the treatment of renal cell carcinoma by the US FDA and the European Medicines Agency.92 Patients with renal cell carcinoma who had poor prognosis were found to have longer overall survival and progression-free survival when treated with temsirolimus compared to controls treated with interferon alfa.93 Temsirolimus was also seen to improve outcome in patients who failed to show improvement with everolimus therapy. Temsirolimus was also efficacious in the treatment of refractory mantle cell lymphoma.94 Ridaforolimus (Merck/Ariad), also known as deforolimus, was designed by substitution of the secondary alcohol moiety at C-40 with a polar phosphate group. Ridaforolimus was designed to improve aqueous solubility and oral delivery and was effective in advanced sarcomas.95,96 Ridaforolimus administration reduced the risk of progression or death by 28% in patients with advanced soft tissue and bone sarcomas potentiating prior cytotoxic chemotherapy.97
Wu and co-workers developed a parallel synthesis method to generate a 200-membered library of bifunctional cyclic peptides as FK506 and rapamycin analogs, which were referred to as “rapalogs”. Each rapalog consists of two domains: a common FKBP-binding moiety and a variable effector domain. The peptides were chosen because a diverse structural library could be generated from a small set of natural and unnatural amino acid building blocks.98 The synthesis of cyclic peptides 11a–n was initiated with the preparation of key building blocks 11a–c from commercially available N-Fmoc-L-pipecolinic acid, 11.1. The carboxyl group was protected as an allyl ester by treatment with allyl bromide under basic conditions. Removal of the Fmoc group gave a free amine moiety, which was then acylated with dihydro-4,4-dimethyl-2,3-furandione to obtain the corresponding alcohols. Coupling of the alcohol group with three different N-Fmoc amino acids followed by de-protection of the allyl group with Pd(PPh3)4 afforded the building block 11a–c in good yields. Fifteen cyclic peptides (11a–n) were then synthesized in parallel on a Rink amide resin. The building block 11.2, N-Fmoc-Glu-α-allyl ester, was then coupled to the amino group of Rink resin using a coupling reagent, O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU). After deprotection of the Fmoc group, the N-terminal amine was further acylated with 10 different N-Fmoc amino acids (R1). Further addition of building blocks 11a–c and L-Ala–L-Ala were carried out using standard peptide chemistry that yielded 11.3. Prior to cyclization of peptides, the C-terminal allyl group was de-protected by treatment with a catalytic amount of Pd(PPh3)4 in the presence of N-methylaniline, and the N-terminal Fmoc group was then deprotected using piperidine. The cyclization of peptides was successfully achieved by using (benzotriazole-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBop) as the coupling reagent. Finally, treatment with 50% trifluoroacetic acid (TFA) in dichloromethane released the peptides (11a–n) from the resin with the de-protected amino acid side chains. The resulting crude peptides were then quickly passed through the silica gel column to remove salts and then used directly in various activity assays (Scheme 11). These rapalogs (11a–n) were tested for binding to FKBP12 by a fluorescence polarization competition assay. The results showed that FKBP12 binds to most of the rapalogs with a high affinity (KI values within the nanomolar to low micromolar range), making a large repertoire of composite surfaces for potential recognition of macromolecular targets like proteins.99,100
 | ||
| Scheme 11 Solid-phase synthesis of rapalogs (11a–n). | ||
More recently, Arya and co-workers have synthesized rapamycin fragment-derived macrocycles 12.1.101 As shown in the retrosynthesis (Scheme 12), Arya's team developed a stereoselective approach to the synthesis of rapamycin-derived pyran fragment 12.2. The key reactions in their approach included the Paterson aldol reaction, stereoselective β-hydroxy carbonyl reduction and an intramolecular regio- and stereoselective oxy-Michael addition.
 | ||
| Scheme 12 Arya's approach to rapamycin fragment-derived macrocycles 12.1. | ||
As shown in Scheme 13, the Paterson aldol reaction of aldehyde 12.6 and a keto fragment 12.7 using (−)-DIPCl provided β-hydroxy carbonyl compound 13.1 as a single diastereomer. This was then subjected to a stereoselective carbonyl reduction with Et2BOMe and NaBH4 resulting in the 1,3-diol. The diol was protected with 2,2-dimethoxypropane followed by removal of the silyl group using TBAF giving alcohol 13.2. This alcohol 13.2 was oxidized and subjected to the Horner–Wittig reaction to obtain α,β-unsaturated ethyl ester 13.3. Upon treatment under mildly acidic conditions, it gave the free 1,3-diol. Treatment with potassium tert-butoxide led to a regio- and stereoselective intramolecular oxy-Michael addition, giving 13.4 as a single diastereomer. With this key fragment in hand, the next step in the plan was to explore the chemical space around the pyran scaffold by obtaining various macrocycles. The ethyl ester of 13.4 was hydrolyzed to obtain a free carboxylic acid which was then coupled with various pipecolic amino acid building blocks of secondary amine 12.3 with EDC·HCl and HOBT to obtain 13.5 in good yields. Compound 13.5 was treated with various acid chlorides (R2COCl) giving key starting materials to explore the ring closing metathesis approach. Finally, the bis-allyl intermediates were treated with Grubbs' second generation catalyst giving rapamycin fragment-derived macrocycles 12.1(a–d) in good yields.101 Molecular docking studies were performed with these macrocycles to predict key binding interactions with human immunophilin FKBP-12 and the FKBP12–rapamycin associated protein complexed with human immunophilin. The docking studies showed good binding interactions of rapamycin fragment-derived macrocycles with both targets. Biological studies with these rapamycin fragment-derived macrocycles 12.1(a–d) are in progress and are expected to be reported in future publications.
 | ||
| Scheme 13 Synthesis of rapamycin fragment-derived macrocycles 12.1(a–d). | ||
mTOR inhibitors other than rapamycin
In addition to rapamycin and rapalogs, several small molecules have also demonstrated potential to inhibit the mTOR pathway in some cancer cells.3,102,103 This new generation of small molecules are ATP-competitive inhibitors of mTOR kinase such as PP244, INK126, INK128, XL388 Torin 1, Torin 2, AZD8055, AZD2014, WYE-354, WYE-600, WYE-687, WYE-125132, Ku-0063794, OSI-027, CC-115 and CC-223 (structures are not shown).104–119 These small molecules inhibit mTOR kinase activity by competing with ATP for binding to the kinase domain in mTOR. In addition, dual mTOR/PI3K inhibitors such as NVPBEZ235, BGT226, XL765, GDC0980, SF1126, PKI587, PF04691502, GDC-0980, PKI-402, and GSK2126458 also effectively inhibit mTORC1 and mTORC2 (structures are not shown).120–125 When compared to rapamycin and rapalogs, these small molecules show lower specificity and are likely to elicit off-target effects on related kinases with a correspondingly higher toxicity profile. A recent study showed that anthramycin, a secondary metabolite of Streptomyces, suppressed mTOR signaling and was effective at inhibiting HCC cell proliferation with mTOR activation.126 Eriocalyxin B, a natural ent-kaurane diterpenoid, exhibits anti-tumor activity through the suppression of the Akt/mTOR/p70S6K signaling pathway in breast cancer.Concluding remarks
Inhibitors of the mTOR pathway have found significant use in the clinic and are being actively developed by several research groups. Rapamycin is a potent inhibitor of the mTOR pathway and is currently used as an immunosuppressant. However, pharmacological limitations such as poor solubility and long half-lives have spurred ongoing efforts to develop rapamycin analogs with improved properties. The complex chemical structure of rapamycin piqued the interest of several research groups who have, over the years, presented several elegant synthetic methodologies to synthesize rapamycin and provided pathways for analog development. Evaluation of rapamycin and its analogs has enhanced our understanding of the mTOR pathway, while simultaneously providing a larger arsenal of small molecules as potential immunosuppressive, anti-cancer and neuroprotective agents for age-related neurodegenerative diseases. In addition to rapamycin, other small molecules also serving as mTOR inhibitors give more opportunity to understand the mTOR pathway at a molecular level. Despite the progress reported by these studies, there remains a need to synthesize rapamycin-based compounds that have improved pharmacokinetic properties and small molecules other than rapamycin that have improved specificity towards the mTOR complex or more potent dual PI3–mTOR inhibitors which enhance our current understanding of the downstream regulation of the mTOR pathway. In the future, these novel small molecules may also serve as potential clinical therapeutics and may increase the human life span.Abbreviations
| mTOR | Mammalian target of rapamycin |
| FKBPs | FK506 binding proteins |
| WAT | White adipose tissue |
| PPARγ | Peroxisome proliferator-activated receptor-γ |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| SREBP2 | Sterol regulatory element-binding protein 2 |
| PPI | Protein–protein interaction |
| FRB domain | FKBP12–rapamycin binding domain |
| FRAP | FKBP12–rapamycin associated protein |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This review article was made possible with grant support from the CSIR funding agency to SKRG and DST (SR/S1/OC-30/2010) and DBT (102/IFD/SAN/PR 2862/2010-11) to PA. We thank Girdhar Singh Deora for helping us obtain molecular model images from the protein database and Sudhir Raghavan for proof reading.References
- S. N. Sehgal, H. Baker and C. Vezina, J. Antibiot., 1975, 28, 727–732 CrossRef CAS PubMed.
- C. Vezina, A. Kudelski and S. N. Sehgal, J. Antibiot., 1975, 28, 721–726 CrossRef CAS PubMed.
- D. Benjamin, M. Colombi, C. Moroni and M. N. Hall, Nat. Rev. Drug Discovery, 2011, 10, 868–880 CrossRef CAS PubMed.
- T. Schmelzle and M. N. Hall, Cell, 2000, 103, 253–262 CrossRef CAS PubMed.
- H. Yang, D. G. Rudge, J. D. Koos, B. Vaidialingam, H. J. Yang and N. P. Pavletich, Nature, 2013, 497, 217–223 CrossRef CAS PubMed.
- C. H. S. Aylett, E. Sauer, S. Imseng, D. Boehringer, M. N. Hall, N. Ban and T. Maier, Science, 2016, 351, 48–52 CrossRef CAS PubMed.
- D. Baretić, A. Berndt, Y. Ohashi, C. M. Johnson and R. L. Williams, Nat. Commun., 2016, 7, 11016 CrossRef PubMed.
- H. Yang, J. Wang, M. Liu, X. Chen, M. Huang, D. Tan, M.-Q. Dong, C. C. L. Wong, J. Wang, Y. Xu and H.-W. Wang, Protein Cell, 2016, 7, 878–887 CrossRef CAS PubMed.
- D. D. Sarbassov, S. M. Ali, D.-H. Kim, D. A. Guertin, R. R. Latek, H. Erdjument-Bromage, P. Tempst and D. M. Sabatini, Curr. Biol., 2004, 14, 1296–1302 CrossRef CAS PubMed.
- E. Jacinto, R. Loewith, A. Schmidt, S. Lin, M. A. Rüegg, A. Hall and M. N. Hall, Nat. Cell Biol., 2004, 6, 1122–1128 CrossRef CAS PubMed.
- D. D. Sarbassov, S. M. Ali, S. Sengupta, J.-H. Sheen, P. P. Hsu, A. F. Bagley, A. L. Markhard and D. M. Sabatini, Mol. Cell, 2006, 22, 159–168 CrossRef CAS PubMed.
- R. C. Russell, C. Fang and K.-L. Guan, Development, 2011, 138, 3343–3356 CrossRef CAS PubMed.
- D. M. Sabatini, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11818–11825 CrossRef CAS PubMed.
- P. Polak, N. Cybulski, J. N. Feige, J. Auwerx, M. A. Rüegg and M. N. Hall, Cell Metab., 2008, 8, 399–410 CrossRef CAS PubMed.
- K. Düvel, J. L. Yecies, S. Menon, P. Raman, A. I. Lipovsky, A. L. Souza, E. Triantafellow, Q. Ma, R. Gorski and S. Cleaver, Mol. Cell, 2010, 39, 171–183 CrossRef PubMed.
- M. Kaeberlein, R. W. Powers, K. K. Steffen, E. A. Westman, D. Hu, N. Dang, E. O. Kerr, K. T. Kirkland, S. Fields and B. K. Kennedy, Science, 2005, 310, 1193–1196 CrossRef CAS PubMed.
- P. Kapahi, B. M. Zid, T. Harper, D. Koslover, V. Sapin and S. Benzer, Curr. Biol., 2004, 14, 885–890 CrossRef CAS PubMed.
- D. E. Harrison, R. Strong, Z. D. Sharp, J. F. Nelson, C. M. Astle, K. Flurkey, N. L. Nadon, J. E. Wilkinson, K. Frenkel and C. S. Carter, Nature, 2009, 460, 392–395 CAS.
- K. Sataranatarajan, M. M. Mariappan, M. J. Lee, D. Feliers, G. G. Choudhury, J. L. Barnes and B. S. Kasinath, Am. J. Pathol., 2007, 171, 1733–1742 CrossRef CAS PubMed.
- K. Cao, J. J. Graziotto, C. D. Blair, J. R. Mazzulli, M. R. Erdos, D. Krainc and F. S. Collins, Sci. Transl. Med., 2011, 3, 89ra58 CAS.
- J. J. Siekierka, S. H. Y. Hung, M. Poe, C. S. Lin and N. H. Sigal, Nature, 1989, 341, 755–757 CrossRef CAS PubMed.
- J. Chen, X. F. Zheng, E. J. Brown and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4947–4951 CrossRef CAS.
- S. N. Sehgal, Clin. Biochem., 1998, 31, 335–340 CrossRef CAS PubMed.
- J. Liang, J. Choi and J. Clardy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 736–744 CrossRef CAS.
- L. A. Banaszynski, C. W. Liu and T. J. Wandless, J. Am. Chem. Soc., 2005, 127, 4715–4721 CrossRef CAS PubMed.
- B. E. Bierer, P. S. Mattila, R. F. Standaert, L. A. Herzenberg, S. J. Burakoff, G. Crabtree and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 9231–9235 CrossRef CAS.
- S. Chaurasia, S. Pieraccini, R. De Gonda, S. Conti and M. Sironi, Chem. Phys. Lett., 2013, 587, 68–74 CrossRef CAS.
- G. Woerly, N. Brooks and B. Ryffel, Clin. Exp. Immunol., 1996, 103, 322–327 CAS.
- J. Gonzalez, T. Harris, G. Childs and M. B. Prystowsky, Blood Cells, Mol., Dis., 2001, 27, 572–585 CrossRef CAS PubMed.
- T. F. Andoh, E. A. Burdmann, N. Fransechini, D. C. Houghton and W. M. Bennett, Kidney Int., 1996, 50, 1110–1117 CrossRef CAS PubMed.
- C. A. Thompson, Am. J. Health-Syst. Pharm., 2003, 60, 1210–1212 Search PubMed.
- J. Bové, M. Martínez-Vicente and M. Vila, Nat. Rev. Neurosci., 2011, 12, 437–452 CrossRef PubMed.
- A. Caccamo, M. A. Maldonado, S. Majumder, D. X. Medina, W. Holbein, A. Magrí and S. Oddo, J. Biol. Chem., 2011, 286, 8924–8932 CrossRef CAS PubMed.
- C. Malagelada, Z. H. Jin, V. Jackson-Lewis, S. Przedborski and L. A. Greene, J. Neurosci., 2010, 30, 1166–1175 CrossRef CAS PubMed.
- S. Sarkar, G. Krishna, S. Imarisio, S. Saiki, C. J. O'Kane and D. C. Rubinsztein, Hum. Mol. Genet., 2008, 17, 170–178 CrossRef CAS PubMed.
- A. S. Tsvetkov, J. Miller, M. Arrasate, J. S. Wong, M. A. Pleiss and S. Finkbeiner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16982–16987 CrossRef CAS PubMed.
- V. N. Anisimov, M. A. Zabezhinski, I. G. Popovich, T. S. Piskunova, A. V. Semenchenko, M. L. Tyndyk, M. N. Yurova, M. P. Antoch and M. V. Blagosklonny, Am. J. Pathol., 2010, 176, 2092–2097 CrossRef CAS PubMed.
- E. I. Graziani, Nat. Prod. Rep., 2009, 26, 602–609 RSC.
- K. Nicolaou, T. Chakraborty, A. Piscopio, N. Minowa and P. Bertinato, J. Am. Chem. Soc., 1993, 115, 4419–4420 CrossRef CAS.
- D. Romo, S. D. Meyer, D. D. Johnson and S. L. Schreiber, J. Am. Chem. Soc., 1993, 115, 7906–7907 CrossRef CAS.
- C. M. Hayward, D. Yohannes and S. J. Danishefsky, J. Am. Chem. Soc., 1993, 115, 9345–9346 CrossRef CAS.
- A. B. Smith III, S. M. Condon, J. A. McCauley, J. L. Leazer Jr, J. W. Leahy and R. E. Maleczka Jr, J. Am. Chem. Soc., 1995, 117, 5407–5408 CrossRef.
- M. L. Maddess, M. N. Tackett, H. Watanabe, P. E. Brennan, C. D. Spilling, J. S. Scott, D. P. Osborn and S. V. Ley, Angew. Chem., 2007, 119, 597–603 CrossRef.
- K. Nicolaou, A. D. Piscopio, P. Bertinato, T. K. Chakraborty, N. Minowa and K. Koide, Chem. – Eur. J., 1995, 1, 318–333 CrossRef CAS.
- D. Romo, D. D. Johnson, L. Plamondon, T. Miwa and S. L. Schreiber, J. Org. Chem., 1992, 57, 5060–5063 CrossRef CAS.
- A. B. Smith, S. M. Condon, J. A. McCauley, J. L. Leazer, J. W. Leahy and R. E. Maleczka, J. Am. Chem. Soc., 1997, 119, 947–961 CrossRef CAS.
- A. B. Smith, S. M. Condon, J. A. McCauley, J. L. Leazer, J. W. Leahy and R. E. Maleczka, J. Am. Chem. Soc., 1997, 119, 962–973 CrossRef CAS.
- M. J. Batchelor, R. J. Gillespie, J. M. Golec and C. J. Hedgecock, Tetrahedron Lett., 1993, 34, 167–170 CrossRef CAS.
- S. V. Ley, M. N. Tackett, M. L. Maddess, J. C. Anderson, P. E. Brennan, M. W. Cappi, J. P. Heer, C. Helgen, M. Kori and C. Kouklovsky, Chem. – Eur. J., 2009, 15, 2874–2914 CrossRef CAS PubMed.
- F. V. Ritacco, E. I. Graziani, M. Y. Summers, T. M. Zabriskie, K. Yu, V. S. Bernan, G. T. Carter and M. Greenstein, Appl. Environ. Microbiol., 2005, 71, 1971–1976 CrossRef CAS PubMed.
- M. A. Gregory, H. Petkovic, R. E. Lill, S. J. Moss, B. Wilkinson, S. Gaisser, P. F. Leadlay and R. M. Sheridan, Angew. Chem., 2005, 117, 4835–4838 CrossRef.
- R. J. Goss, S. Lanceron, A. Deb Roy, S. Sprague, M. Nur-e-Alam, D. L. Hughes, B. Wilkinson and S. J. Moss, ChemBioChem, 2010, 11, 698–702 CrossRef CAS PubMed.
- R. J. Goss, S. E. Lanceron, N. J. Wise and S. J. Moss, Org. Biomol. Chem., 2006, 4, 4071–4073 CAS.
- J. I. Luengo, A. Konialian-Beck, L. W. Rozamus and D. A. Holt, J. Org. Chem., 1994, 59, 6512–6513 CrossRef CAS.
- J. I. Luengo, D. S. Yamashita, D. Dunnington, A. K. Beck, L. W. Rozamus, H.-K. Yen, M. J. Bossard, M. A. Levy, A. Hand and T. Newman-Tarr, Chem. Biol., 1995, 2, 471–481 CrossRef CAS PubMed.
- D. A. Holt, J. I. Luengo, D. S. Yamashita, H. J. Oh, A. L. Konialian, H. K. Yen, L. W. Rozamus, M. Brandt and M. J. Bossard, J. Am. Chem. Soc., 1993, 115, 9925–9938 CrossRef CAS.
- B. Bierer, P. Somers, T. Wandless, S. Burakoff and S. Schreiber, Science, 1990, 250, 556–559 CrossRef CAS PubMed.
- T. Chakraborty, H. Weber and K. Nicolaou, Chem. Biol., 1995, 2, 157–161 CrossRef CAS PubMed.
- J. Choi, J. Chen, S. L. Schreiber and J. Clardy, Science, 1996, 273, 239–242 CAS.
- D. Spencer, T. Wandless, S. Schreiber and G. Crabtree, Science, 1993, 262, 1019–1024 CAS.
- P. J. Belshaw, S. N. Ho, G. R. Crabtree and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4604–4607 CrossRef CAS.
- M. Putyrski and C. Schultz, FEBS Lett., 2012, 586, 2097–2105 CrossRef CAS PubMed.
- A. Fegan, B. White, J. C. T. Carlson and C. R. Wagner, Chem. Rev., 2010, 110, 3315–3336 CrossRef CAS PubMed.
- S. Voß, L. Klewer and Y.-W. Wu, Curr. Opin. Chem. Biol., 2015, 28, 194–201 CrossRef PubMed.
- R. DeRose, T. Miyamoto and T. Inoue, Pflügers Arch., 2013, 465, 409–417 CrossRef CAS PubMed.
- B.-C. Suh, T. Inoue, T. Meyer and B. Hille, Science, 2006, 314, 1454–1457 CrossRef CAS PubMed.
- P. Varnai, B. Thyagarajan, T. Rohacs and T. Balla, J. Cell Biol., 2006, 175, 377–382 CrossRef CAS PubMed.
- R. Zoncu, R. M. Perera, R. Sebastian, F. Nakatsu, H. Chen, T. Balla, G. Ayala, D. Toomre and P. V. De Camilli, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3793–3798 CrossRef CAS PubMed.
- N. Abe, T. Inoue, T. Galvez, L. Klein and T. Meyer, J. Cell Sci., 2008, 121, 1488–1494 CrossRef CAS PubMed.
- M. Rahdar, T. Inoue, T. Meyer, J. Zhang, F. Vazquez and P. N. Devreotes, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 480–485 CrossRef CAS PubMed.
- F. Castellano, P. Montcourrier and P. Chavrier, J. Cell Sci., 2000, 113, 2955–2961 CAS.
- M. Fivaz, S. Bandara, T. Inoue and T. Meyer, Curr. Biol., 2008, 18, 44–50 CrossRef CAS PubMed.
- T. Inoue and T. Meyer, PLoS One, 2008, 3, e3068 Search PubMed.
- T. Komatsu, I. Kukelyansky, J. M. McCaffery, T. Ueno, L. C. Varela and T. Inoue, Nat. Methods, 2010, 7, 206–208 CrossRef CAS PubMed.
- I. A. Graef, L. J. Holsinger, S. Diver, S. L. Schreiber and G. R. Crabtree, EMBO J., 1997, 16, 5618–5628 CrossRef CAS PubMed.
- B. Li, S. A. Desai, R. A. MacCorkle-Chosnek, L. Fan and D. M. Spencer, Gene Ther., 2002, 9, 233 CrossRef CAS PubMed.
- S. Terrillon and M. Bouvier, EMBO J., 2004, 23, 3950–3961 CrossRef CAS PubMed.
- S. C. Phua, C. Pohlmeyer and T. Inoue, ACS Chem. Biol., 2012, 7, 1950–1955 CrossRef CAS PubMed.
- T. Miyamoto, R. DeRose, A. Suarez, T. Ueno, M. Chen, T.-p. Sun, M. J. Wolfgang, C. Mukherjee, D. J. Meyers and T. Inoue, Nat. Chem. Biol., 2012, 8, 465 CrossRef CAS PubMed.
- M. Skwarczynska, M. Molzan and C. Ottmann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E377–E386 CrossRef CAS PubMed.
- S. D. Liberles, S. T. Diver, D. J. Austin and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7825–7830 CrossRef CAS.
- A. V. Karginov, Y. Zou, D. Shirvanyants, P. Kota, N. V. Dokholyan, D. D. Young, K. M. Hahn and A. Deiters, J. Am. Chem. Soc., 2011, 133, 420–423 CrossRef CAS PubMed.
- N. Umeda, T. Ueno, C. Pohlmeyer, T. Nagano and T. Inoue, J. Am. Chem. Soc., 2011, 133, 12–14 CrossRef CAS PubMed.
- K. A. Brown, Y. Zou, D. Shirvanyants, J. Zhang, S. Samanta, P. K. Mantravadi, N. V. Dokholyan and A. Deiters, Chem. Commun., 2015, 51, 5702–5705 RSC.
- Y.-W. Chen, M. L. Smith, M. Sheets, S. Ballaron, J. M. Trevillyan, S. E. Burke, T. Rosenberg, C. Henry, R. Wagner and J. Bauch, J. Cardiovasc. Pharmacol., 2007, 49, 228–235 CrossRef CAS PubMed.
- P. W. Serruys, S. Silber, S. Garg, R. J. van Geuns, G. Richardt, P. E. Buszman, H. Kelbæk, A. J. van Boven, S. H. Hofma and A. Linke, N. Engl. J. Med., 2010, 363, 136–146 CrossRef CAS PubMed.
- M. B. Leon, L. Mauri, J. J. Popma, D. E. Cutlip, E. Nikolsky, C. O'Shaughnessy, P. A. Overlie, B. T. McLaurin, S. L. Solomon and J. S. Douglas, J. Am. Coll. Cardiol., 2010, 55, 543–554 CrossRef CAS PubMed.
- A. C. Yeung, M. B. Leon, A. Jain, T. R. Tolleson, D. J. Spriggs, B. T. Mc Laurin, J. J. Popma, P. J. Fitzgerald, D. E. Cutlip and J. M. Massaro, J. Am. Coll. Cardiol., 2011, 57, 1778–1783 CrossRef PubMed.
- R. J. Motzer, B. Escudier, S. Oudard, T. E. Hutson, C. Porta, S. Bracarda, V. Grünwald, J. A. Thompson, R. A. Figlin and N. Hollaender, Lancet, 2008, 372, 449–456 CrossRef CAS.
- R. J. Motzer, B. Escudier, S. Oudard, T. E. Hutson, C. Porta, S. Bracarda, V. Grünwald, J. A. Thompson, R. A. Figlin and N. Hollaender, Cancer, 2010, 116, 4256–4265 CrossRef CAS PubMed.
- J. C. Yao, M. H. Shah, T. Ito, C. L. Bohas, E. M. Wolin, E. Van Cutsem, T. J. Hobday, T. Okusaka, J. Capdevila and E. G. de Vries, N. Engl. J. Med., 2011, 364, 514–523 CrossRef CAS PubMed.
- B. I. Rini, Clin. Cancer Res., 2008, 14, 1286–1290 CrossRef CAS PubMed.
- G. Hudes, M. Carducci, P. Tomczak, J. Dutcher, R. Figlin, A. Kapoor, E. Staroslawska, J. Sosman, D. McDermott and I. Bodrogi, N. Engl. J. Med., 2007, 356, 2271–2281 CrossRef CAS PubMed.
- G. Hess, R. Herbrecht, J. Romaguera, G. Verhoef, M. Crump, C. Gisselbrecht, A. Laurell, F. Offner, A. Strahs and A. Berkenblit, J. Clin. Oncol., 2009, 27, 3822–3829 CrossRef CAS PubMed.
- S. P. Chawla, A. P. Staddon, L. H. Baker, S. M. Schuetze, A. W. Tolcher, G. Z. D'Amato, J.-Y. Blay, M. M. Mita, K. K. Sankhala and L. Berk, J. Clin. Oncol., 2012, 30, 78–84 CrossRef CAS PubMed.
- M. Mita, K. Sankhala, I. Abdel-Karim, A. Mita and F. Giles, Expert Opin. Invest. Drugs, 2008, 17, 1947–1954 CrossRef CAS PubMed.
- G. D. Demetri, S. P. Chawla, I. Ray-Coquard, A. Le Cesne, A. P. Staddon, M. M. Milhem, N. Penel, R. F. Riedel, B. Bui-Nguyen and L. D. Cranmer, J. Clin. Oncol., 2013, 31, 2485–2492 CrossRef CAS PubMed.
- X. Wu, L. Wang, Y. Han, N. Regan, P.-K. Li, M. A. Villalona, X. Hu, R. Briesewitz and D. Pei, ACS Comb. Sci., 2011, C13, 486–495 CrossRef PubMed.
- G. M. Dubowchik, J. L. Ditta, J. J. Herbst, S. Bollini and A. Vinitsky, Bioorg. Med. Chem., 2000, 10, 559–562 CrossRef CAS.
- P. D. Braun and T. J. Wandless, Biochemistry, 2004, 43, 5406–5413 CrossRef CAS PubMed.
- S. K. R. Guduru, R. Jimmidi, G. S. Deora and P. Arya, Org. Lett., 2015, 17, 480–483 CrossRef CAS PubMed.
- S. V. Bhagwat and A. P. Crew, Curr. Opin. Investig. Drugs, 2010, 11, 638–645 CAS.
- J. Liu, H.-Q. Li, F.-X. Zhou, J.-W. Yu, L. Sun and Z.-H. Han, Tumor Biol., 2017, 39, 1–15 Search PubMed.
- D. J. Richard, J. C. Verheijen and A. Zask, Curr. Opin. Drug Discovery Dev., 2010, 13, 428–440 CAS.
- C. García-Echeverría, Bioorg. Med. Chem. Lett., 2010, 20, 4308–4312 CrossRef PubMed.
- S.-Y. Sun, Cancer Lett., 2013, 340, 1–8 CrossRef CAS PubMed.
- D. S. Mortensen, S. M. Perrin-Ninkovic, G. Shevlin, J. Zhao, G. Packard, S. Bahmanyar, M. Correa, J. Elsner, R. Harris and B. G. Lee, J. Med. Chem., 2015, 58, 5323–5333 CrossRef CAS PubMed.
- D. S. Mortensen, S. M. Perrin-Ninkovic, G. Shevlin, J. Elsner, J. Zhao, B. Whitefield, L. Tehrani, J. Sapienza, J. R. Riggs and J. S. Parnes, J. Med. Chem., 2015, 58, 5599–5608 CrossRef CAS PubMed.
- Z. Zeng, Y. X. Shi, T. Tsao, Y. Qiu, S. M. Kornblau, K. A. Baggerly, W. Liu, K. Jessen, Y. Liu and H. Kantarjian, Blood, 2012, 120, 2679–2689 CrossRef CAS PubMed.
- B. Blaser, L. Waselle, A. Dormond-Meuwly, M. Dufour, D. Roulin, N. Demartines and O. Dormond, BMC Cancer, 2012, 12, 86 CrossRef CAS PubMed.
- M. R. Janes, C. Vu, S. Mallya, M. Shieh, J. J. Limon, L.-S. Li, K. A. Jessen, M. B. Martin, P. Ren and M. B. Lilly, Leukemia, 2013, 27, 586–594 CrossRef CAS PubMed.
- L. Willems, N. Chapuis, A. Puissant, T. Maciel, A. Green, N. Jacque, C. Vignon, S. Park, S. Guichard and O. Herault, Leukemia, 2012, 26, 1195–1202 CrossRef CAS PubMed.
- P. J. Houghton, R. Gorlick, E. A. Kolb, R. Lock, H. Carol, C. L. Morton, S. T. Keir, C. P. Reynolds, M. H. Kang and D. Phelps, Pediatr. Blood Cancer, 2012, 58, 191–199 Search PubMed.
- S. V. Bhagwat, P. C. Gokhale, A. P. Crew, A. Cooke, Y. Yao, C. Mantis, J. Kahler, J. Workman, M. Bittner and L. Dudkin, Mol. Cancer Ther., 2011, 10, 1394–1406 CrossRef CAS PubMed.
- J. M. García-Martínez, J. Moran, R. G. Clarke, A. Gray, S. C. Cosulich, C. M. Chresta and D. R. Alessi, Biochem. J., 2009, 421, 29–42 CrossRef PubMed.
- D. S. Mortensen, J. Sapienza, B. G. Lee, S. M. Perrin-Ninkovic, R. Harris, G. Shevlin, J. S. Parnes, B. Whitefield, M. Hickman and G. Khambatta, Bioorg. Med. Chem. Lett., 2013, 23, 1588–1591 CrossRef CAS PubMed.
- K. G. Pike, K. Malagu, M. G. Hummersone, K. A. Menear, H. M. Duggan, S. Gomez, N. M. Martin, L. Ruston, S. L. Pass and M. Pass, Bioorg. Med. Chem. Lett., 2013, 23, 1212–1216 CrossRef CAS PubMed.
- D. Fantus, H. Dai, Y. Ono, A. Watson, S. Yokota, K. Mohib, O. Yoshida, M. A. Ross, S. C. Watkins, B. Ramaswami, A. Valusjkikh, D. M. Rothstein and A. W. Thomson, Transplantation, 2017, 101, 2830–2840 CrossRef PubMed.
- H. Liao, Y. Huang, B. Guo, B. Liang, X. Liu, H. Ou, C. Jiang, X. Li and D. Yang, Am. J. Cancer Res., 2015, 5, 125–139 CAS.
- P. Baumann, S. Mandl-Weber, F. Oduncu and R. Schmidmaier, Exp. Cell Res., 2009, 315, 485–497 CrossRef CAS PubMed.
- M. C. Manara, G. Nicoletti, D. Zambelli, S. Ventura, C. Guerzoni, L. Landuzzi, P.-L. Lollini, S.-M. Maira, C. García-Echeverría and M. Mercuri, Clin. Cancer Res., 2010, 16, 530–540 CrossRef CAS PubMed.
- J. R. Garlich, P. De, N. Dey, J. D. Su, X. Peng, A. Miller, R. Murali, Y. Lu, G. B. Mills and V. Kundra, Cancer Res., 2008, 68, 206–215 CrossRef CAS PubMed.
- A. Molckovsky and L. L. Siu, J. Hematol. Oncol., 2008, 1, 20 CrossRef PubMed.
- R. Marone, D. Erhart, A. C. Mertz, T. Bohnacker, C. Schnell, V. Cmiljanovic, F. Stauffer, C. Garcia-Echeverria, B. Giese and S.-M. Maira, Mol. Cancer Res., 2009, 7, 601–613 CrossRef CAS PubMed.
- A. Zask, J. C. Verheijen and D. J. Richard, Expert Opin. Ther. Pat., 2011, 21, 1109–1127 CrossRef CAS PubMed.
- T. Hayashi, T. Yamashita, H. Okada, N. Oishi, H. Sunagozaka, K. Nio, Y. Hara, Y. Asahina, M. Yoshida, T. Hashiba, T. Suda, T. Shirasaki, Y. Igarashi, K. Miyanouchi, M. Honda and S. Kaneko, Anticancer Res., 2017, 37, 3397–3403 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |