
A β-carboline derivative-based nickel(II) complex as a potential antitumor agent: synthesis, characterization, and cytotoxicity†‡
Jing-Mei
Yang§
a,
Yan-Hong
Zhu§
a,
Sheng
Chen
a,
Xing
Lu
a,
Yi-Ming
Wu
a,
Feng-E
Ma
a,
Liang-Ping
Li
a,
Yang
Yang
a,
Zhen-Hao
Shi
a,
Kun-Yuan
Huang
a,
Xue
Hong
a,
Ping
Jiang
*b and
Yan
Peng
 *a
*a
aState Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmacy, Guangxi Normal University, No. 15 Yucai Road, Guilin 541004, China. E-mail: pengyan630@aliyun.com; Fax: +86 773 2120958; Tel: +86 773 2120958
bShanghai Mental Health Center, Shanghai Institute of Mental Health, Shanghai Jiao Tong University School of Medicine, 600 Wan Ping Nan Road, Shanghai 200030, P.R. China. E-mail: jiangping413@126.com; Fax: +86 21 64387986; Tel: +86 21 64387250
First published on 24th November 2017
Abstract
A novel nickel(II) complex of 6-methoxy-1-pyridine-β-carboline (4a) was synthesized and characterized. The cytotoxicities of the complex towards six cancer cell lines, including MGC-803, Hep G2, T24, OS-RC-2, NCI-H460, and SK-OV-3, and human normal liver cell line HL-7702 were investigated. The IC50 values for MGC-803, Hep G2, T24, OS-RC-2, NCI-H460 and SK-OV-3 were generally in the micromolar range (3.77–15.10 μM), lower than those of ligand 4 and cisplatin. Furthermore, 4a (6 μM) significantly induced cell cycle arrest at the S phase, and caused the down-regulation of p-AKT, cyclin E, cyclin A and CDK2 and the up-regulation of p27. Various experiments showed that 4a induced apoptosis, activated caspase-3, increased the levels of reactive oxygen species (ROS) and enhanced the intracellular [Ca2+]c levels in MGC-803. In addition, the expression of intrinsic apoptotic proteins, including cytochrome c and apaf-1, increased. Further intrinsic apoptosis was triggered via executive molecular caspase-9 and caspase-3. In short, 4a exerted its cytotoxic activity primarily through inducing cell cycle arrest at the S phase and intrinsic apoptosis.
1. Introduction
Cisplatin, oxaliplatin and carboplatin are widely used as anticancer agents in clinical oncology.1 Cisplatin mainly crosslinks the pyrimidine and purine bases in DNA to trigger DNA damage responses,2 and the anti-tumor effect of cisplatin primarily results from the apoptosis induced by DNA damage.3 However, platinum-based anticancer chemotherapy is exposed to severe adverse effects.4 For instance, systemic toxicities of cisplatin like nephrotoxicity, neurotoxicity, ototoxicity, and emetogenesis inflict serious injuries to patients during treatment, which severely restrict its efficacy.5,6 Moreover, the intrinsic and acquired resistance hinders the efficacy of cisplatin in numerous cancers.7–9 Therefore, much effort has been devoted to developing novel antitumor drugs, which would be more efficient, less toxic, and more specific.As we know, nickel plays a key role in the biological system. In 1975, the enzyme urease containing nickel was discovered.10 Since then, many nickel-containing or nickel-dependent enzymes have also been discovered.10,11 A number of nickel(II) complexes were reported to possess notable anti-HIV,12 anticonvulsant,13 antibacterial,14,15 antifungal,16,17 anti-inflammatory,18 anti-leishmania,19 and antioxidant20–22 activities, as well as antiproliferative activity towards diverse cell lines.23–25
Harmine, a natural β-carboline, is widely distributed in fungi,26 plants,27–29 and mammalian and marine invertebrates.30,31 The biological and pharmacological activities of β-carbolines include antimicrobial,32 antithrombotic,33 parasiticidal,34 anti-HIV,35 anti-Alzheimer,36 and antitumor activities.37 The antitumor activity of β-carbolines is mediated through various mechanisms, such as intercalation into DNA,38 inhibition of topoisomerases I and II39,40 and kinase Eg5,41 blocking cell mitosis via inhibition of cyclin-dependent kinases (CDKs),42,43 and targeting specific cancer signaling pathways.
The antitumor activity of β-carboline complexes has caught the interest of medicinal chemists. The effect of an iridium(III) β-carboline complex on autophagy was studied44 and a ruthenium(II) β-carboline complex was found to induce p53-mediated apoptosis.45,46 Here, a novel nickel and β-carboline complex 4a was synthesized and characterized. Its cytotoxicity towards tumor cells and the underlying mechanisms were studied.
2. Results
2.1. Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol/DMSO from 6-methoxy-1-pyridine-β-carboline (4) and Ni(NO3)2·6H2O.
:1 methanol/DMSO from 6-methoxy-1-pyridine-β-carboline (4) and Ni(NO3)2·6H2O.
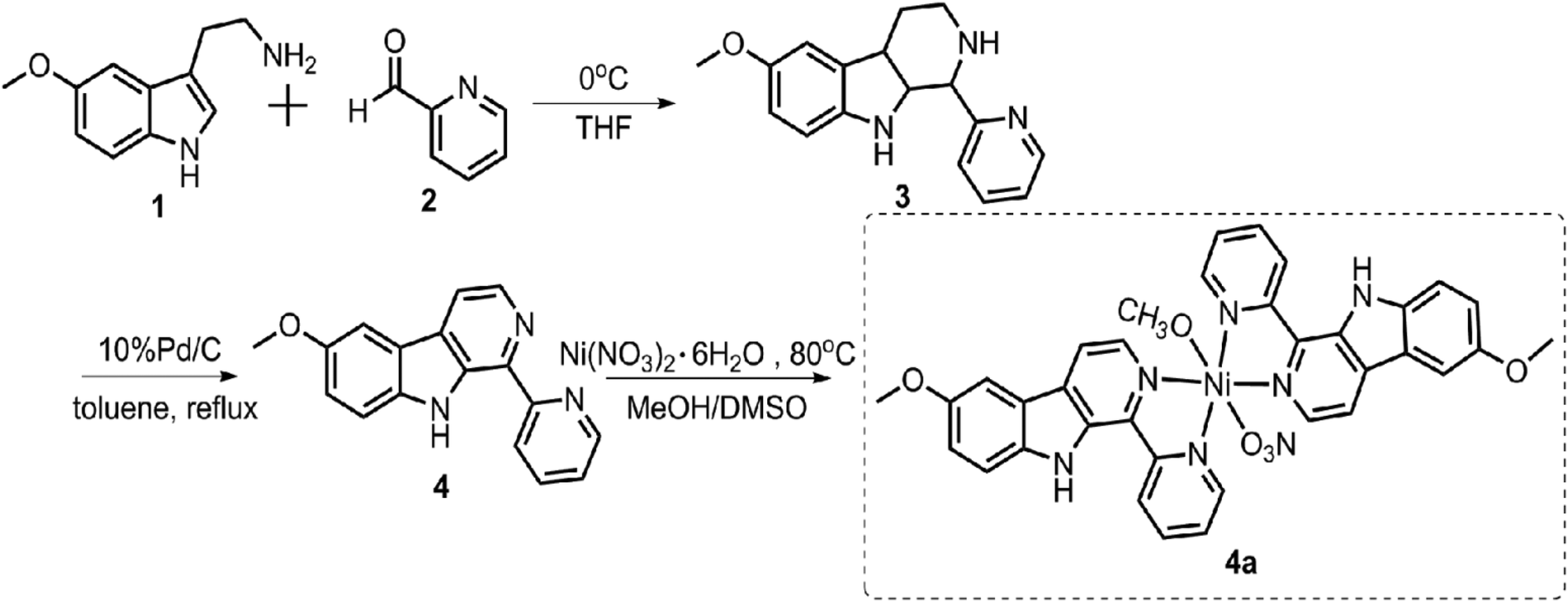 | ||
| Scheme 1 Synthesis route to 4a. | ||
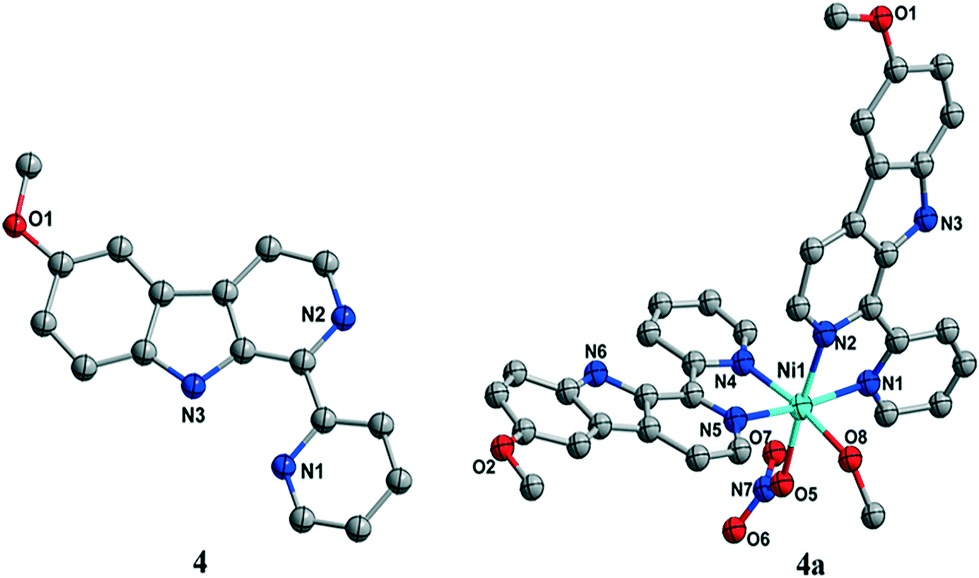 | ||
| Fig. 1 X-ray crystal structures of ligand 4 and complex 4a. | ||
2.2. In vitro cytotoxicity test by MTT assay
The inhibition rates and IC50 values of 4a against seven cell lines (MGC-803, Hep G2, T24, OS-RC-2, NCI-H460, SK-OV-3 and HL-7702) were determined by the MTT method, and they were compared with those of 4 and cisplatin. The IC50 (MGC-803, Hep G2, T24, OS-RC-2, NCI-H460 and SK-OV-3) values (Fig. 2 and Table S5†) of 4a were lower than those of 4 and cisplatin. Specifically, the IC50 value of 4a for MGC-803 was 3.77 ± 0.06 μM, which is 4.6 times lower than that of cisplatin (17.57 ± 0.03 μM).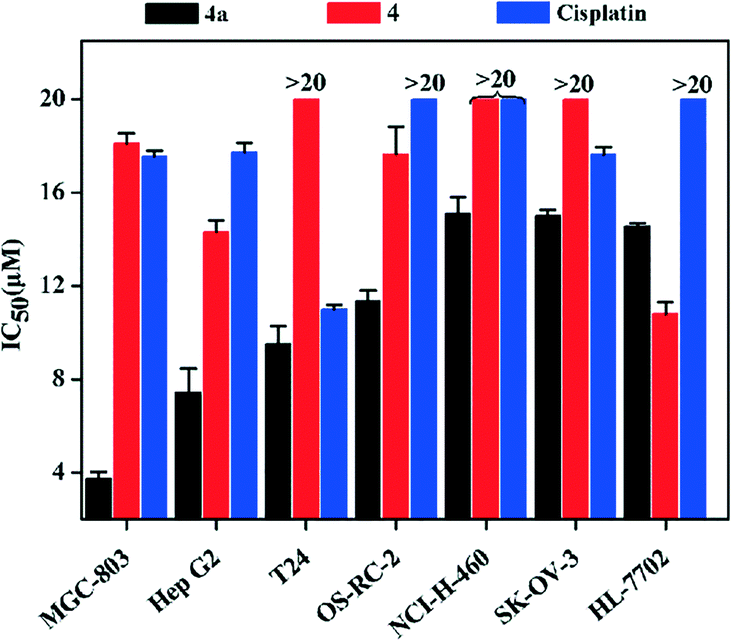 | ||
| Fig. 2 IC50 (μM) values of 4a, 4 and cisplatin for seven human cell lines. | ||
2.3. Cell cycle assay
To further study the mechanism underlying the inhibitory activity of 4a on MGC-803 cells, we investigated its effect on cell cycle progression. Hence, we monitored the cell cycle phase distribution of MGC-803 cells treated with 2.0 μM, 4.0 μM, and 6.0 μM concentrations of 4a for 48 h. The cell cycle distribution was examined by measuring the DNA content of nuclei labeled with propidium iodide (PI) using flow cytometry. The cell cycle analysis in Fig. 3 shows that after treating the MGC-803 cells with 4a for 48 h, the proportion of cells in the S phase increased to 33.34% and 53.74% for the 4.0 μM and 6.0 μM groups, respectively. At the same time, the proportions of cells in the G2 phase in the treatment groups were lower than that in the control. This result indicated that the MGC-803 cells were blocked in the S phase after treatment with 4a for 48 h.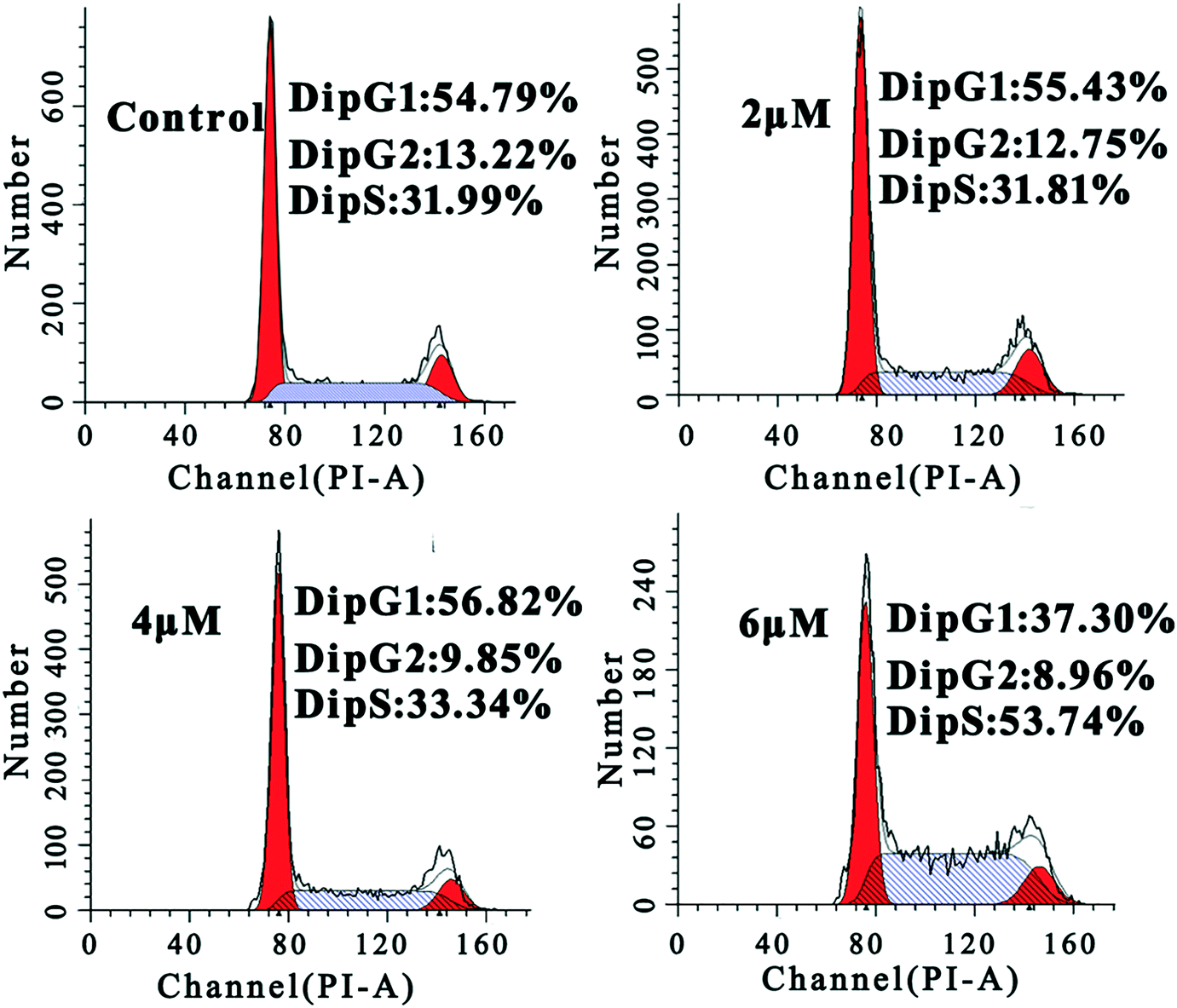 | ||
| Fig. 3 FACS analysis of the cell cycle of MGC-803 cells by PI staining after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. | ||
2.4. Study on the mechanism of S phase arrest
To investigate the mechanism of cell cycle arrest induced by 4a, we examined the changes of the cyclin E1–CDK2 complex and cyclin A2–CDK2 complex in the cell cycle regulation system.48Fig. 4 indicates that 4a down-regulated cyclin E1, cyclin A2 and CDK2 in MGC-803, resulting in cell cycle S phase arrest. Suppression of Akt phosphorylation resulted in a distinct increase of p27 expression. The inhibitory protein p27 acts on two subtypes of cyclin and cycle-dependent kinase complexes to inhibit CDK2 activity.49 P27 was negatively regulated by oncogene p-AKT and blocked the cell cycle in the S phase through the Akt–p27–CDK2 pathway.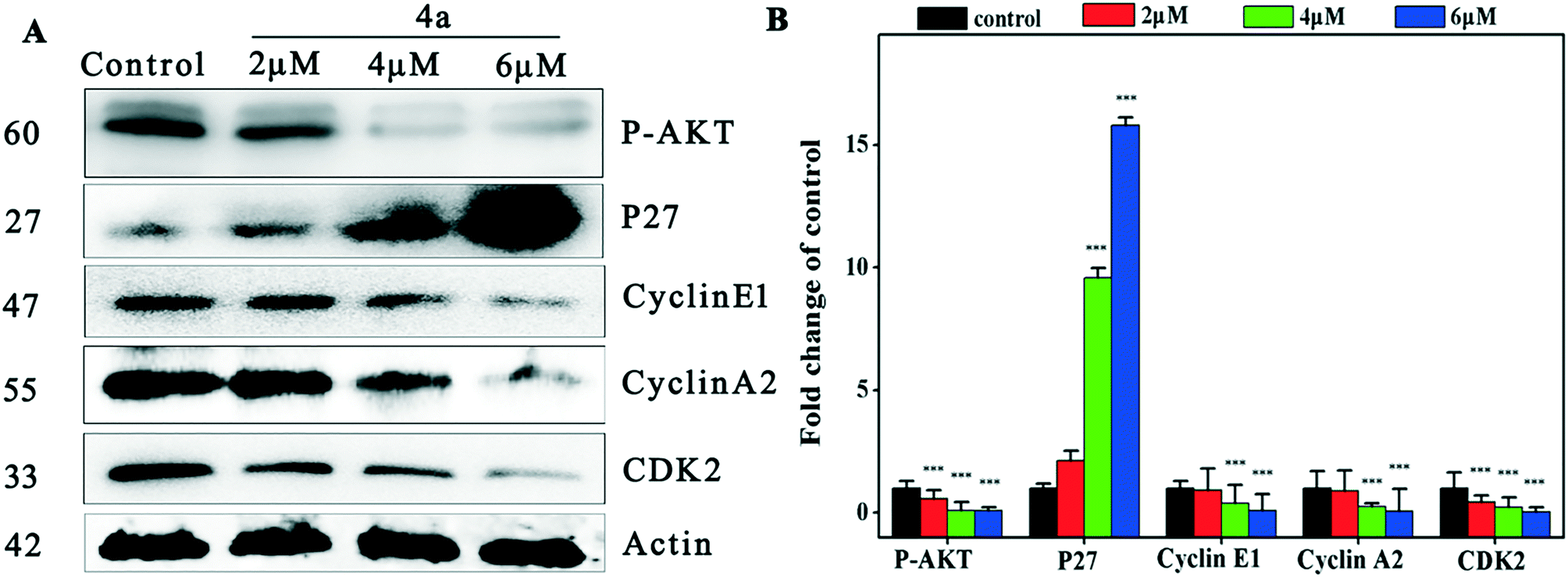 | ||
| Fig. 4 Detection of S phase-related proteins of MGC-803 cells by western blot after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. (A) Western blot was used to determine the expression of p-AKT, p27, cyclin E1, cyclin A2, CDK2 and actin in MGC-803 cells treated with 4a (2 μM, 4 μM, and 6 μM) for 48 h. (B) Densitometric analysis of p-AKT, p27, cyclin E1, cyclin A2 and CDK2 proteins normalized with actin. The relative expression of each protein is represented by the density of the protein band/density of the actin band. The mean SD was from three independent measurements. ***P < 0.001, compared to the control. | ||
2.5. Cell apoptosis assay
The interaction of MGC-803 cells with 4a was further investigated by PE-Annexin V/7-AAD staining to assess apoptosis induction (Fig. 5). Because phosphatidylserine (PS) exposure usually precedes the loss of cell membrane integrity in apoptosis, PE-Annexin V can be combined with PS. FITC-labeled Annexin-V was used as a probe to detect the occurrence of apoptosis by flow cytometry. PE-Annexin V (+)/7-AAD (−) (Q4) staining is considered as an indicator of early apoptosis. PE-Annexin V (+)/7-AAD (+) (Q2) staining indicated necrosis and the later period of apoptosis. The apoptosis ratios (sum of ratios in Q2 and Q4) caused by 4a at different concentrations were found to be 12.7%, 17.43%, and 62.0% for the control, 4.0 μM, and 6.0 μM groups, respectively. The results in Fig. 5 suggested that apoptosis was induced after the MGC-803 cells were treated with 4a.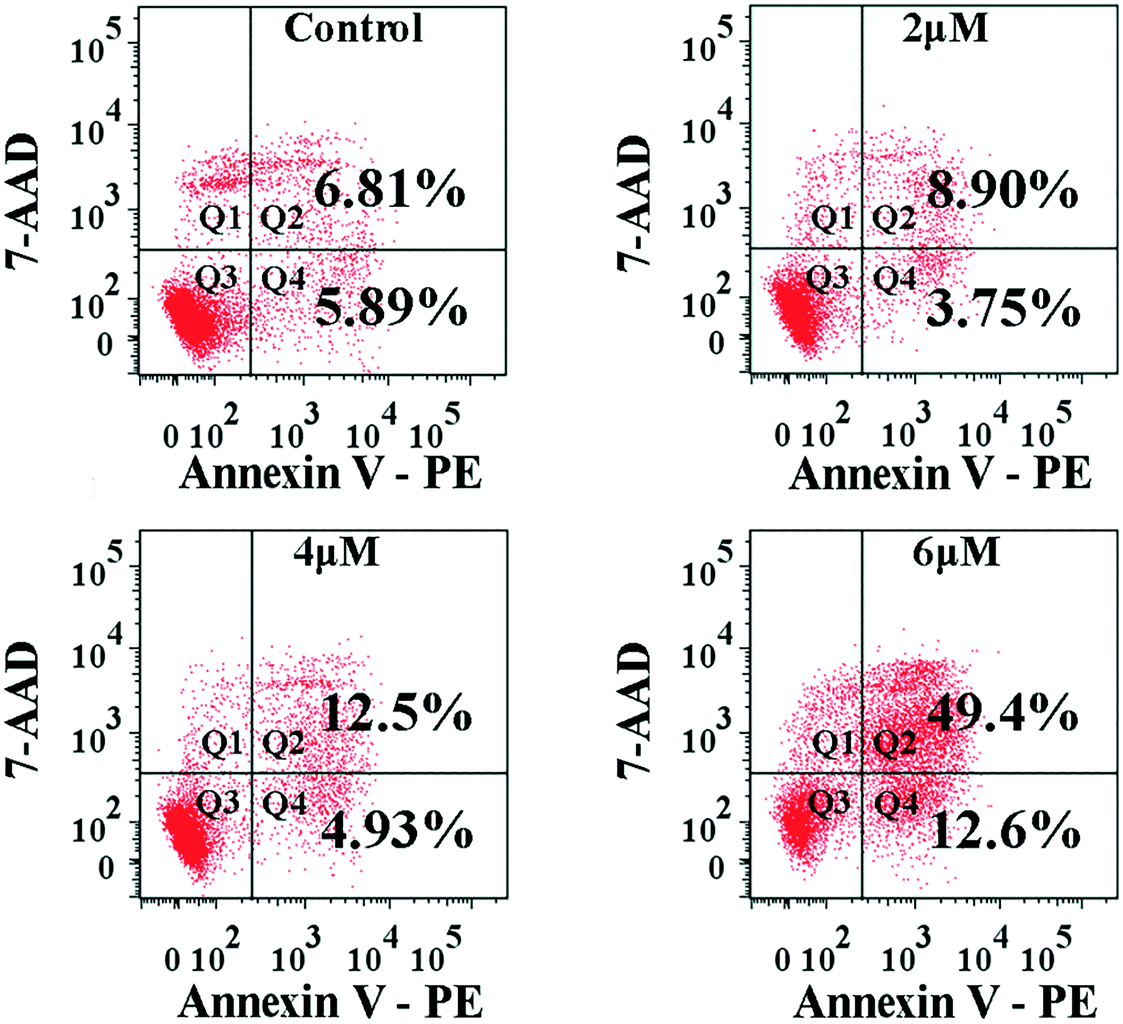 | ||
| Fig. 5 FACS analysis of the apoptosis of MGC-803 cells by Annexin V-PE/7-AAD staining after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. | ||
2.6. Assessment of caspase-3 activation in apoptosis induction
The intrinsic pathway of apoptosis is regulated by the caspase family members. It is of obvious interest to determine which of the caspase family members actively participated in the signaling after the cells were treated with 4a. Hence, flow cytometry was adopted to examine the abundance of candidate caspase members. The experiment took the advantage of FITC-DEVD-FMK (for caspase-3) probes. Fig. 6 shows that the proportion of active caspase-3 cells was 3.91%, 7.83%, 16.46%, and 57.75% for the control, 2.0 μM, 4.0 μM, and 6.0 μM groups, respectively. The results indicated that 4a induced cell apoptosis by triggering caspase-3 activation in the MGC-803 cells.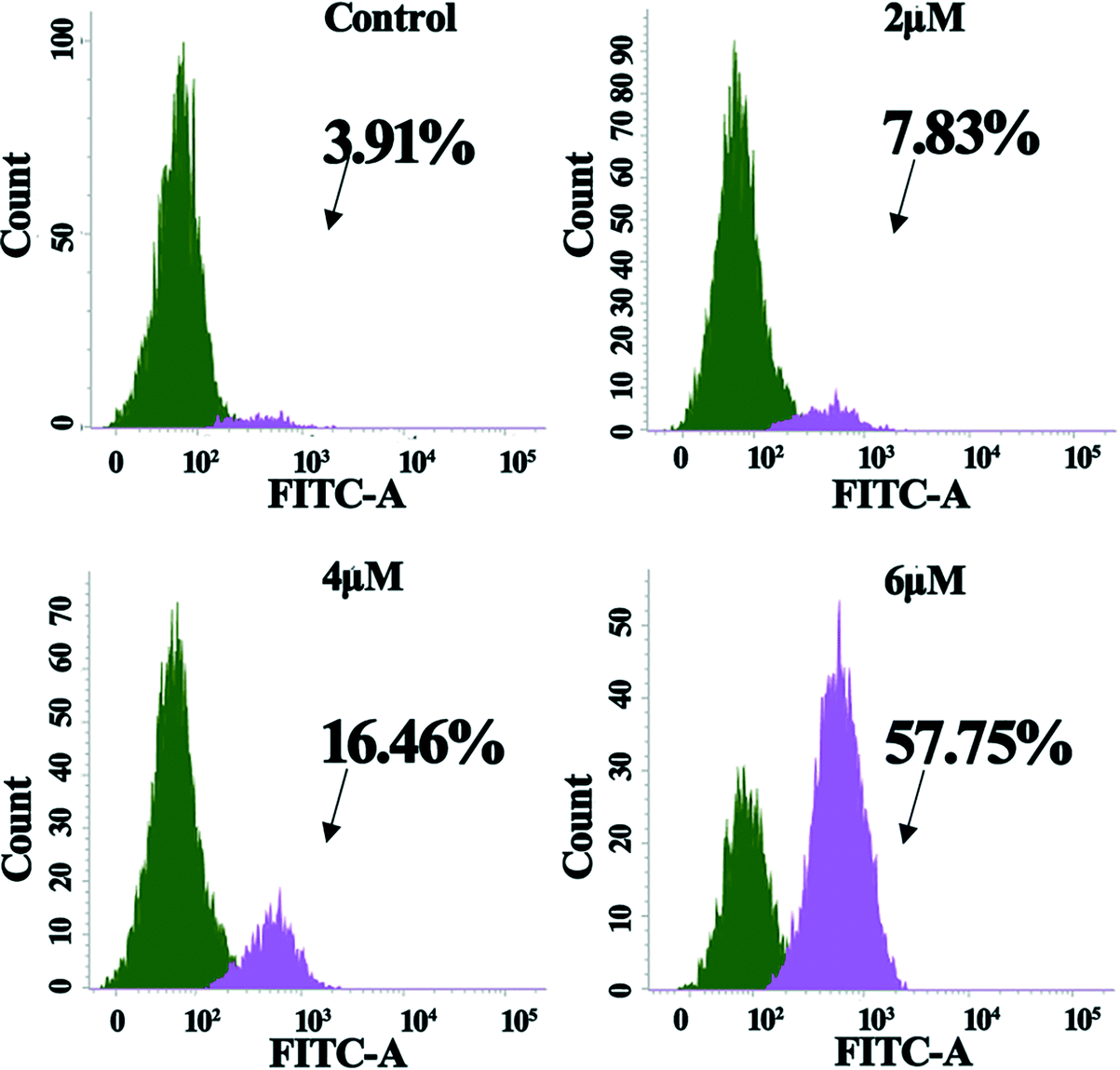 | ||
| Fig. 6 Activation of caspase-3 in MGC-803 cells after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. | ||
2.7. Reactive oxygen species (ROS) generation in cells treated with 4a
Under normal circumstances, cells maintain balanced reactive oxygen species (ROS) generation and cellular antioxidant system activity. When the balance is broken, collective generation of ROS leads to oxidative stress. Oxidative stress plays a vital role in influencing tumor cell behavior. Research found that ROS not only regulate subcellular functions but also induce cell death via the apoptotic pathway.50 Moreover, accumulating evidence suggests that oxidative stress leads to mitochondrial apoptosis via regulating the activity of the Bcl-2 family of proteins.51,52 Flow cytometry with DCFH-DA staining was carried out to examine ROS generation in MGC-803 cells after they were treated with 4a. The DCFH-DA probe could be converted to the fluorescent DCF when bound with ROS. Fig. 7 shows that compared with the control, the level of ROS generation in the MGC-803 cells increased significantly after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. Therefore, the cell apoptosis induced by 4a was closely related to the ROS-mediated caspase-initiated apoptosis pathway.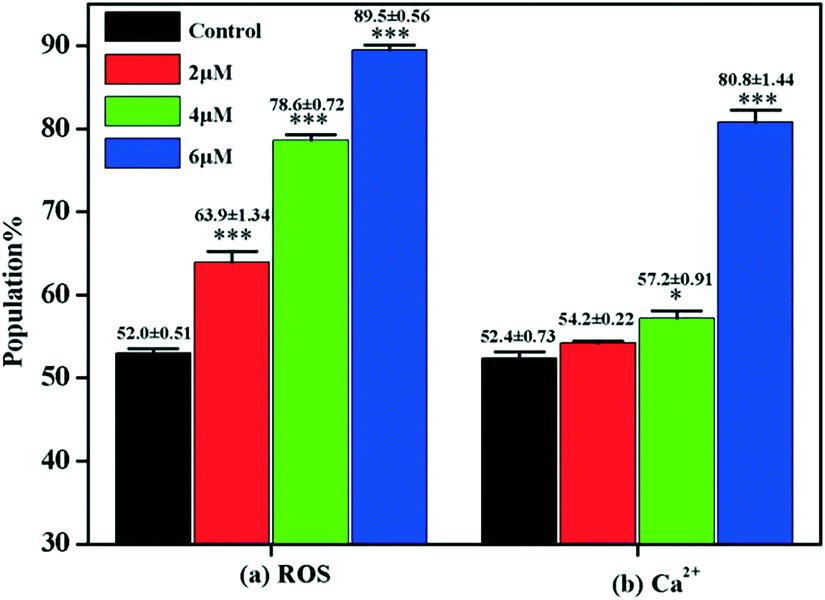 | ||
| Fig. 7 (a) Increased generation of reactive oxygen species in the MGC-803 cells after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. (b) Intracellular levels of [Ca2+]c in the MGC-803 cells after treatment with 4a (2.0 μM, 4.0 μM, and 6.0 μM) for 48 h. *P < 0.05, ***P < 0.001, compared to the control. | ||
2.8. Ca2+ release in cell apoptosis
It is known that a close relationship exists between ROS generation and Ca2+ release at the intracellular level.53 Increased intracellular [Ca2+]c is regarded as one of the major messenger molecules in apoptosis induction.54 The intracellular [Ca2+]c of the MGC-803 cells treated with 4a was evaluated by flow cytometry using the fluorescent probe Fluo-3/AM. Fig. 7 shows that after treatment with 4a (6.0 μM), the [Ca2+]c in the MGC-803 cells markedly increased by 28.4% compared with the control. This observation, combined with the previous results, clearly shows that ROS generation is a precursor for enhancing intracellular [Ca2+]c levels. The findings here suggest that the apoptotic pathway induced by 4a occurred through ROS generation, as indicated by the enhanced intracellular [Ca2+]c levels.2.9. Effect of 4a on the apoptotic pathway
The mitochondrial pathway is one of the most important apoptosis pathways. Mitochondrial membrane permeability is modulated by the Bcl-2 family of proteins.55 Bax is a pro-apoptotic protein that localizes principally in the cytoplasm and redistributes to mitochondria in response to apoptotic stimuli, where it induces cytochrome c release. Bax is a component of the mitochondrial pore, which allows some small substances (ions and molecules) to pass through the mitochondrial membrane into the cytoplasm.56 Caspase activation also correlates with the Bax activity. Bcl-2 is an anti-apoptotic protein that localizes with apaf-1 on the mitochondrial membrane57and protects against collapse of the mitochondrial membrane, which would lead to release of mitochondrial substances such as cytochrome c and apoptotic protease activating factor-1 (apaf-1).58 As shown by the western blot results in Fig. 8, treatment of MGC-803 cells with 4a for 48 h led to the down-regulation of Bcl-2 and up-regulation of Bax, cytochrome c and apaf-1. These results suggested that 4a (2 μM, 4 μM, and 6 μM) induced apoptosis by regulating the levels of Bax, Bcl-2, cytochrome c and apaf-1.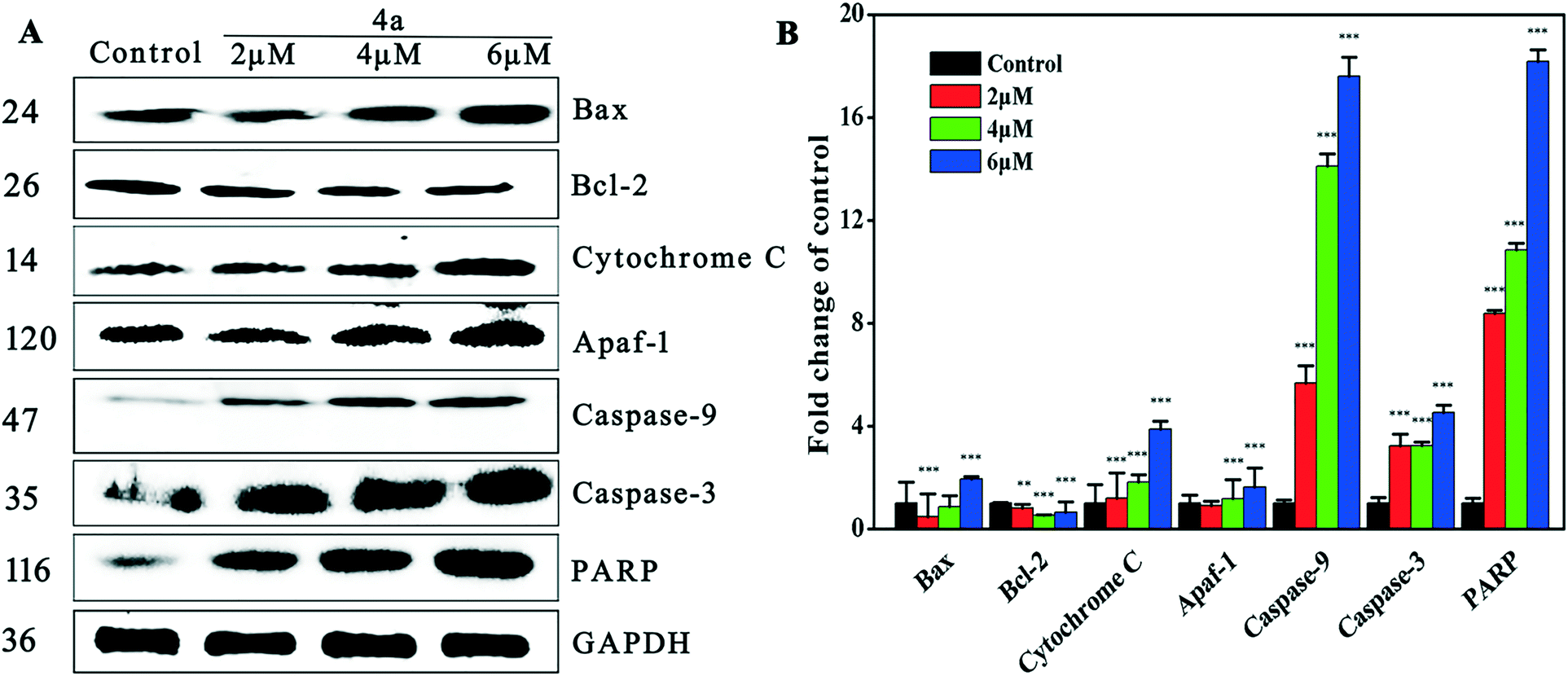 | ||
| Fig. 8 (A) Western blot was used to determine the expression of Bax, Bcl-2, cytochrome c and apaf-1 in MGC-803 cells treated with 4a (2 μM, 4 μM, and 6 μM) for 48 h. (B) Densitometric analysis of Bax, Bcl-2, cytochrome c and apaf-1 proteins normalized with GAPDH. The relative expression of each protein is represented by the density of the protein band/density of the GAPDH band. The mean SD was from three independent measurements. **P < 0.01, ***P < 0.001, compared to the control. | ||
Apoptosis signals may activate caspase and the caspase cascade. Cytochrome c binds to apaf-1, which may trigger the activation of caspase-3/9 (caspase-3 as shown in Fig. 6 and 8), leading to apoptosis.59–61 It is well known that the activation of caspase-3 during apoptosis can cause the cleavage of PARP, a major indicator of the mitochondrial pathway. Our results showed that 4a dose-dependently increased the expression of caspase-3/9 and PARP, which supports the theory that 4a induced mitochondrial apoptosis which led to caspase cascade activation in the MGC-803 cells.
3. Discussion and conclusion
In this study, a new nickel(II) complex of 6-methoxy-1-pyridine-β-carboline (4a) was synthesized and fully characterized. It was present as a deformed octahedral mono-nuclear nickel(II) complex both in the crystal state and in the aqueous solution state, suggesting the high stability of the complex to maintain the coordinated state of 6-methoxy-1-pyridine-β-carboline (4) with nickel(II). The MGC-803 cell line was most sensitive to 4a. In particular, 4a showed higher selectivity against MGC-803 cells than ligand 4 and cisplatin. 4a promoted cell apoptosis in MGC-803 cancer cells. The tightly regulated apoptosis extrinsic pathway and the mitochondrial-dependent intrinsic pathway are important in maintaining cellular homeostasis.59 High concentrations of ROS can cause damage in DNA, proteins and lipids, which results in cell death. Moreover, growing evidence suggests that oxidative stress causes mitochondrial dysfunction.62 We found that treatment of MGC-803 with 4a led to enhanced apoptosis which can be caused by reactive oxygen species (ROS)-mediated mitochondrial dysfunction. Additionally, treatment with 4a resulted in enhanced PARP cleavage and caspase activation.50Furthermore, 4a could induce cell cycle arrest at the S phase through the Akt–p27–CDK2 pathway, suggesting its different antitumor mechanism compared with cisplatin. 4a caused the down-regulation of cyclin E1, cyclin A2, CDK2, and p-AKT and the up-regulation of p27.
The relationship between the Akt–p27–CDK2 pathway and mitochondrial apoptosis can be explained by AKT. Bax is regulated by phosphorylation of Ser184 in an Akt-dependent manner and that phosphorylation inhibits Bax effects on the mitochondria by maintaining the protein in the cytoplasm, heterodimerized with anti-apoptotic Bcl-2 family members.63
Previous reports indicate that the apoptosis mechanism involved Akt inactivation, which directly correlated with Bax translocation to mitochondria, cytochrome c release to the cytosol, and the associated activation of caspase-9 and caspase-3, culminating in prominent apoptosis.63,64
We also observed that 4a decreased the expression of p-AKT and markedly suppressed serine/threonine kinase AKT/PKB (AKT) phosphorylation and kinase activity in cultured MGC-803 cells. Because p-AKT negatively regulates the expression of p27 and Bax, treatment with 4a increased the expression of p27 and Bax.
Taken together, the results demonstrated that 4a could induce apoptosis and render cell cycle arrest at the S phase in cancer cells. It should be further evaluated as a potential chemotherapeutic agent for human cancers.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This study was supported by the Natural Science Foundation of Guangxi Province (2014GXNSFAA118165 and 2015GXNSFAA139010), the State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (Guangxi Normal University) (CMEMR2015-A09, CMEMR2015-B12,ACMEMR2015-B10, and CMEMR2016-A09), the Guilin Scientific Research and Technology Development 1020 Project (20160210), the Guangxi Normal University Open Project (CMEMR2015-B12), and the Shanghai Natural Science Foundation (15ZR1435300).Notes and references
- B. Rosenberg, F. Charles and P. Kettring, Cancer, 1985, 55, 2303–2316 CrossRef CAS PubMed.
- P. U. Maheswari, D. S. M. Van, S. Smulders, S. Barends, G. P. van Wezel, C. Massera, S. Roy, D. H. Den, P. Gamez and J. Reedijk, Inorg. Chem., 2008, 47, 3719–3727 CrossRef CAS PubMed.
- A. R. Banerjee, J. A. Jaeger and D. H. Turner, Biochemistry, 1993, 32, 153–163 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202 RSC.
- M. W. English, R. Skinner, A. D. J. Pearson, L. Price, R. Wyllie and A. W. Craft, Br. J. Cancer, 1999, 81, 336–341 CrossRef CAS PubMed.
- R. Skinner, A. D. Pearson, M. W. English, L. Price, R. A. Wyllie, M. G. Coulthard and A. W. Craft, Br. J. Cancer, 1998, 77, 1677–1682 CrossRef CAS PubMed.
- E. Wong and C. M. Giandomenico, ChemInform, 1999, 30, 2451–2466 Search PubMed.
- R. D. Baird and S. B. Kaye, Eur. J. Cancer, 2003, 39, 2450–2461 CrossRef CAS PubMed.
- N. E. Dixon, C. Gazzola, R. L. Blakeley and B. Zerner, J. Am. Chem. Soc., 1975, 97, 4131–4133 CrossRef CAS PubMed.
- F. Meyer and H. Kozlowski, Compr. Coord. Chem. II, 2003, 247–554 Search PubMed.
- P. Zimmermann and C. Limberg, J. Am. Chem. Soc., 2017, 139, 4233–4242 CrossRef CAS PubMed.
- C. W. Hsu, C. F. Kuo, S. M. Chuang and M. H. Hou, BioMetals, 2013, 26, 1–12 CrossRef CAS PubMed.
- G. Morgant, N. Bouhmaida, L. Balde, N. E. Ghermani and J. D'Angelo, Polyhedron, 2006, 25, 2229–2235 CrossRef CAS.
- K. Skyrianou, E. Efthimiadou, V. Psycharis, A. Terzis, D. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2009, 103, 1617–1625 CrossRef CAS PubMed.
- R. Kurtaran, L. T. Yıldırım, A. D. Azaz, H. Namli and O. Atakol, J. Inorg. Biochem., 2005, 99, 1937–1944 CrossRef CAS PubMed.
- B. Xu, P. Shi, Q. Guan, X. Shi and G. Zhao, J. Coord. Chem., 2013, 66, 2605–2614 CrossRef CAS.
- K. Alomar, A. Landreau, M. Allain, G. Bouet and G. Larcher, J. Inorg. Biochem., 2013, 126, 76–83 CrossRef CAS PubMed.
- H. B. Shawish, Y. W. Wan, L. W. Yi, W. L. Sheng, C. Y. Looi, P. Hassandarvish, A. Y. L. Phan, W. F. Wong, H. Wang and I. C. Paterson, PLoS One, 2014, 9, e100933 Search PubMed.
- I. Ramírezmacías, C. R. Maldonado, C. Marín, F. Olmo, R. Gutiérrezsánchez, M. J. Rosales, M. Quirós, J. M. Salas and M. Sánchezmoreno, J. Inorg. Biochem., 2012, 112, 1–9 CrossRef PubMed.
- K. C. Skyrianou, F. Perdih, A. N. Papadopoulos, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 1273–1285 CrossRef CAS PubMed.
- P. Sathyadevi, P. Krishnamoorthy, E. Jayanthi, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Inorg. Chim. Acta, 2012, 384, 83–96 CrossRef CAS.
- Y. Li, Z. Y. Yang and J. C. Wu, Eur. J. Med. Chem., 2010, 45, 5692–5701 CrossRef CAS PubMed.
- A. Buschini, S. Pinelli, C. Pellacani, F. Giordani, M. B. Ferrari, F. Bisceglie, M. Giannetto, G. Pelosi and P. Tarasconi, J. Inorg. Biochem., 2009, 103, 666–677 CrossRef CAS PubMed.
- F. Bisceglie, S. Pinelli, R. Alinovi, M. Goldoni, A. Mutti, A. Camerini, L. Piola, P. Tarasconi and G. Pelosi, J. Inorg. Biochem., 2014, 140, 111–125 CrossRef CAS PubMed.
- S. Betanzos-Lara, C. Gómez-Ruiz, L. R. Barrón-Sosa, I. Gracia-Mora, M. Flores-Álamo and N. Barba-Behrens, J. Inorg. Biochem., 2012, 114, 82–93 CrossRef CAS PubMed.
- A. Teichert, J. Schmidt, A. Porzel, N. Arnold and L. Wessjohann, J. Nat. Prod., 2007, 70, 1529–1531 CrossRef CAS PubMed.
- Y. Chen, P. Kuo, H. Chan, I. Kuo, F. Lin, C. Su, M. Yang, D. Li and T. Wu, J. Nat. Prod., 2010, 73, 1993–1998 CrossRef CAS PubMed.
- D. T. Youssef, J. Nat. Prod., 2001, 64, 839–841 CrossRef CAS.
- S. L. Wong, H. S. Chang, G. J. Wang, M. Y. Chiang, H. Y. Huang, C. H. Chen, S. C. Tsai, C. H. Lin and I. S. Chen, J. Nat. Prod., 2011, 74, 2489–2496 CrossRef CAS PubMed.
- P. S. Kearns and J. A. Rideout, J. Nat. Prod., 2008, 71, 1280–1282 CrossRef CAS PubMed.
- D. Inman, W. M. Bray, N. C. Gassner, R. S. Lokey, K. Tenney, Y. Y. Shen, K. Tendyke, T. Suh and P. Crews, J. Nat. Prod., 2010, 73, 255–257 CrossRef PubMed.
- D. J. Mckenna, G. H. Towers and F. Abbott, J. Ethnopharmacol., 1984, 12, 179–211 CrossRef CAS PubMed.
- N. Sunderplassmann, V. Sarli, M. Gartner, M. Utz, J. Seiler, S. Huemmer, T. U. Mayer, T. Surrey and A. Giannis, Bioorg. Med. Chem., 2005, 13, 6094–6111 CrossRef CAS PubMed.
- T. Takeuchi, S. Oishi, T. Watanabe, H. Ohno, J. Sawada, K. Matsuno, A. Asai, N. Asada, K. Kitaura and N. Fujii, J. Med. Chem., 2011, 54, 4839–4846 CrossRef CAS PubMed.
- Y. Song, J. Wang, S. F. Teng, D. Kesuma, Y. Deng, J. Duan, J. H. Wang, R. Z. Qi and M. M. Sim, ChemInform, 2002, 12, 1129–1132 CAS.
- A. C. Castro, L. C. Dang, F. Soucy, L. Grenier, H. Mazdiyasni, M. Hottelet, L. Parent, C. Pien, V. Palombella and J. Adams, Bioorg. Med. Chem. Lett., 2003, 13, 2419–2422 CrossRef CAS PubMed.
- Y. Chen, M. Y. Qin, L. Wang, H. Chao, L. N. Ji and A. L. Xu, Biochimie, 2013, 95, 2050–2059 CrossRef CAS PubMed.
- Z. Taira, S. Kanzawas, C. Dohara, S. Ishida, M. Matsumoto and Y. Sakiya, Jpn. J. Toxicol. Environ. Health, 1997, 43, 83–91 CrossRef CAS.
- R. Cao, W. Peng, H. Chen, Y. Ma, X. Liu, X. Hou, H. Guan and A. Xu, Biochem. Biophys. Res. Commun., 2005, 338, 1557–1563 CrossRef CAS PubMed.
- Y. Song, J. Wang, S. F. Teng, D. Kesuma, Y. Deng, J. Duan, J. H. Wang, R. Z. Qi and M. M. Sim, Bioorg. Med. Chem. Lett., 2002, 12, 1129–1132 CrossRef CAS PubMed.
- P. A. Barsanti, W. Wang, Z. Ni, D. Duhl, N. Brammeier and E. Martin, Bioorg. Med. Chem. Lett., 2010, 20, 157–160 CrossRef CAS PubMed.
- Y. Song, J. Wang, S. F. Teng, D. Kesuma, Y. Deng, J. Duan, J. H. Wang, R. Z. Qi and M. M. Sim, Bioorg. Med. Chem. Lett., 2002, 12, 1129–1132 CrossRef CAS PubMed.
- Y. Song, D. Kesuma, J. Wang, Y. Deng, J. Duan, J. H. Wang and R. Z. Qi, Biochem. Biophys. Res. Commun., 2004, 317, 128–132 CrossRef CAS PubMed.
- L. He, S. Y. Liao, C. P. Tan, Y. Y. Lu, C. X. Xu, L. N. Ji and Z. W. Mao, Chem. Commun., 2014, 50, 5611–5614 RSC.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- J. S. Lan, S. S. Xie, S. Y. Li, L. F. Pan, X. B. Wang and L. Y. Kong, Bioorg. Med. Chem., 2014, 22, 6089–6104 CrossRef CAS PubMed.
- E. Yang, J. Zhang, Y. B. Chen, Y. Kang, Y. Y. Qin, Z. Li, J. K. Cheng, R. F. Hu, Y. H. Wen and Y. G. Yao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, 390–391 Search PubMed.
- D. O. Morgan, Annu. Rev. Cell Dev. Biol., 1997, 13, 261–291 CrossRef CAS PubMed.
- N. Horie, T. Mori and H. Asada, et al. Implication of CDK inhibitors p21 and p27 in the differentiation of HL-60 cells, Biol. Pharm. Bull., 2004, 7, 992–997 Search PubMed.
- F. Salehi, H. Behboudi and G. Kavoosi, RSC Adv., 2017, 68, 43141–43150 RSC.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- H. U. Simon, A. Hajyehia and F. Levischaffer, Apoptosis, 2000, 5, 415–418 CrossRef CAS PubMed.
- G. I. Giles, Curr. Pharm. Des., 2006, 12, 4427–4443 CrossRef CAS PubMed.
- V. Pachauri, A. Mehta, D. Mishra and S. J. Flora, Neurotoxicology, 2013, 35, 137–145 CrossRef CAS PubMed.
- J. C. Sharpe, D. Arnoult and R. J. Youle, Biochim. Biophys. Acta, 2004, 1644, 107 CrossRef CAS PubMed.
- E. Er, L. Oliver, P. F. Cartron, P. Juin, S. Manon and F. M. Vallette, Biochim. Biophys. Acta, 2006, 1757, 1301 CrossRef CAS PubMed.
- Y. Yun, M. Zong and W. Xu, Chem.-Biol. Interact., 2017, 262, 38–45 CrossRef PubMed.
- D. R. Green and J. C. Reed, Science, 1998, 281, 1309 CrossRef CAS PubMed.
- H. Zou, W. J. Henzel, X. Liu, A. Lutschg and X. Wang, Cell, 1997, 90, 405–413 CrossRef CAS PubMed.
- N. Inohara, T. Koseki, P. L. Del, Y. Hu, C. Yee, S. Chen, R. Carrio, J. Merino, D. Liu and J. Ni, J. Biol. Chem., 1999, 274, 14560–14567 CrossRef CAS PubMed.
- M. Zhou, Y. Li, Q. Hu, X. C. Bai, W. Huang, C. Yan, S. H. Scheres and Y. Shi, Genes Dev., 2015, 29, 2349–2361 CrossRef CAS PubMed.
- A. K. Mukherjee, A. J. Saviola, P. D. Burns and S. P. Mackessy, Apoptosis, 2015, 20, 1358–1372 CrossRef CAS PubMed.
- Z. Zhang, G. H. Lai and A. E. Sirica, Hepatology, 2004, 39, 1028 CrossRef CAS PubMed.
- F. Tsuruta, N. Masuyama and Y. Gotoh, J. Biol. Chem., 2002, 277, 14040–14047 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Experimental section. CCDC 1525103 for complex 4a contains the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7md00428a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |