
QSAR modeling of cumulative environmental end-points for the prioritization of hazardous chemicals
Paola
Gramatica
 *,
Ester
Papa
and
Alessandro
Sangion
*,
Ester
Papa
and
Alessandro
Sangion
QSAR Research Unit on Environmental Chemistry and Ecotoxicology, Department of Theoretical and Applied Sciences (DiSTA), University of Insubria, Varese, Italy. E-mail: paola.gramatica@uninsubria.it; Web: http://www.qsar.it
First published on 1st December 2017
Abstract
The hazard of chemicals in the environment is inherently related to the molecular structure and derives simultaneously from various chemical properties/activities/reactivities. Models based on Quantitative Structure Activity Relationships (QSARs) are useful to screen, rank and prioritize chemicals that may have an adverse impact on humans and the environment. This paper reviews a selection of QSAR models (based on theoretical molecular descriptors) developed for cumulative multivariate endpoints, which were derived by mathematical combination of multiple effects and properties. The cumulative end-points provide an integrated holistic point of view to address environmentally relevant properties of chemicals.
Environmental significanceThe majority of existing compounds has not been sufficiently well characterized for environmental behaviour and potential human or ecological toxicity. Screening methods are needed to prioritize the most hazardous chemicals. In this way, it is possible to reduce costs, time and number of sacrificed animals. Here we present our approach, based on the combination of Principal Component Analysis (PCA) and Quantitative Structure Activity Relationship (QSAR) models, applied to study environmental persistence, aquatic toxicity and PBT behaviour of different classes of organic chemicals. Such an approach may be particularly useful for the identification of new emerging pollutants and also for planning a priori the synthesis of safer alternatives to undesired chemicals, according to the benign structural design approach of Green Chemistry. |
Introduction
Thousands of chemicals that are used in the world may have potential harmful effects to humans and ecosystems. However, due to experimental costs, scarcity of resources and slow chemical assessment procedures, information on physico-chemical properties, reactivity and biological activities of a large part of existing and commercialized compounds is limited.1–3 Hence the majority of existing compounds, including the High Production Volume (HPV) chemicals, has not been sufficiently well characterized for their environmental behaviour and potential to cause human or ecologic toxicity.4 Most recent approaches to chemical hazard and risk assessment, such as those proposed in the European legislation REACH (Registration Evaluation Authorization and restriction of Chemicals),5 as well as US-TSCA6,7 (https://www.epa.gov/tsca-screening-tools), call for the reduction of such extensive data gaps by using testing and non-testing strategies. However, these approaches must be efficiently applied to contain experimental costs, time and number of sacrificed animals, since it would be impossible to measure properties and activities for all chemicals to which humans and ecosystems are possibly exposed, as well as to test all the required endpoints. In particular, the adoption of adequate screening strategies, also based on methods alternative to animal testing, has been recognized worldwide as a main need to prioritize the most hazardous compounds.7–12 Moreover, these approaches may be particularly challenging for the identification of new emerging pollutants.Hazard is an inherent property of chemicals and is directly related to the molecular structure.13,14 Approaches based on Quantitative Structure Activity Relationships (QSARs) are suitable to identify the potential intrinsic hazard of chemicals in a preliminary hazard-based priority setting phase for rapid screening of large data sets and also before synthesis (i.e. a priori).15,16 QSARs can be applied to detect, as early as possible, potential dangerous properties of not yet tested compounds and possibly to prevent undesirable effects. In this way, it is possible to focus financial resources on the most hazardous chemicals for further evaluation in more comprehensive assessment phases that involve environmentally relevant properties, exposure and toxicity evaluation.14,17
Moreover QSAR methodologies can be applied for planning a priori the synthesis of safer alternatives to undesired chemicals according to the benign by structural design approach (i.e. principle 4 of Green Chemistry).18,19
The validation of QSAR models for their predictive performances on external chemicals, and the quantification of the Applicability Domain (AD), are particularly relevant when QSAR models are applied for prioritization purposes in the screening of large data sets.20–27 QSAR modelling is not a “Push a button and find a correlation”28 procedure, but is a complex process that involves different steps, with limits that should be clear to the users, and that should be characterized case by case. The application of any model to any chemical can lead to erroneous predictions and misleading conclusions. The reliability of predictions from validated QSAR models is guaranteed only for chemicals that belong to the model AD, which is determined by the structures and the responses of the compounds in the training set. Interpolated predictions are more reliable than extrapolations.
Plethora of QSAR models for many different single endpoints of environmental relevance are available in literature, in addition to models included in various public and proprietary tools (for instance: EPISUITE,29 VEGA,30 T.E.S.T.,31 QSARToolbox,32 TOPKAT,33 VirtualToxLab,34–36 DTU QSAR Database,37 QsarDB,38 QSARINS-Chem,39,40etc.).
However, in some cases, multivariate endpoints generated by the combination of multiple effects and properties may provide an integrated view of a studied phenomenon, and be more informative than the endpoints taken singularly. This is the case, for instance, of the environmental persistence, where it would be useful to have a unique endpoint able to give indication of the overall environmental persistency, in addition to the single half-lives in each environmental compartment. Another example is represented by an index able to indicate the toxicity of a compound considering the aquatic trophic chain as a complex system (i.e. by combining effects measured for multiple species) instead of the single toxicities estimated in each organism.
The derivation and the modelling of cumulative endpoints have been the distinctive approach and the main focus of the research pursued by our research group (i.e. the QSAR Research Unit of the University of Insubria) in the last 20 years. Here we review a selection of QSAR models based on theoretical molecular descriptors developed following this unique approach, which provides a holistic (i.e. based on the integration of multiple endpoints) view to study environmental properties of chemicals.
Cumulative indexes: PCA rankings as macro endpoints for QSAR models
The behaviour of chemicals in the environment and their impact on humans and wildlife strongly depend on properties inherent in the molecular structure such as physico-chemical properties, chemical reactivity and biological activity. Most of these properties are in some way related to each other and their cumulative effect contributes to the environmental fate and biological effects of chemicals. For instance, if we think about persistence, we cannot forget to mention the lifetime (half-life) of a chemical in different environmental compartments (i.e. air, water, soil and sediment). The final overall persistence in the environment depends cumulatively on the persistence of chemicals in each single medium.41Therefore, it is crucial to find an effective way to understand, rationalize, and interpret correlations and covariance among the individual properties characterizing a phenomenon, in order to address it from a holistic point of view. In this complex context, multivariate statistical analysis methods are fundamental to extract the meaningful information from data. In particular, exploratory data analysis such as Principal Component Analysis (PCA)42 can combine the available information generating ranking or grouping of the studied chemicals according to various properties, reactivities, or activities, analysed together.
PCA consists of a linear combination of the original variables describing the studied system. These combinations explain, to the highest possible degree, the principal variation in the original data, therefore the enclosed information. The first principal component (PC1) accounts for the maximum amount of possible variance in the studied variables, while subsequent PCs account for successively smaller quantities of the original variance.43
In particular, the PC1 scores, i.e. the coordinates of objects along the first principal component, rank compounds along the direction of maximum variance by linear combination of the information encoded into the variables used to feed the PCA. The linear combination is a new macro-variable, which describes a complex aspect of a studied system or phenomenon and depends on the covariances among the original variables and on their weights along each component. The ranking of the chemicals along PC1, i.e. their relative position, is from now on referred to as cumulative index, which can be modelled as a new macro-endpoint by the QSAR approach. These models allow to predict behaviours of chemicals which depend on the variation of the cumulative index (for instance the overall persistence in the environment), and not of each single variable (e.g. persistence in different environmental media). Therefore, this approach based on PCA is useful to screen and rank compounds according to cumulative indexes. The additional advantage is that, in the absence of data for the single variables, the QSAR models possibly derived for the cumulative indexes can be used for the direct prediction of the macro-endpoints from the chemical structure, or to screen macro properties of chemicals before their synthesis.
PCA has been widely applied by our research group to derive cumulative indexes in many environmental contexts (e.g. tropospheric degradability of Volatile Organic Compounds (VOCs) by the main oxidants OH and nitrate radicals and ozone together: Atmospheric Persistence Index (ATPIN),44,45 environmental partitioning tendency and leaching of pesticides46,47). Over 40 models developed for single and cumulative endpoints have been implemented, so far, for application in our software QSARINS, and in particular in the module QSARINS-Chem, where models are listed with the respective QSAR Model Reporting Format (QMRF).39,40
Here we present some examples of indexes (i.e. the macro-variables generated by PCA), which were derived to describe the potential overall environmental persistence (Global Half-Life Index (GHLI)), the PBT behaviour (PBT index) of heterogeneous organic chemicals of possible environmental concern and the aquatic ecotoxicity (Aquatic Toxicity Index (ATI)) of Pharmaceuticals and Personal Care Products (PPCPs).
Screening of Persistent Organic Pollutants (POPs) by Global Half-Life Index (GHLI)
Environmental persistence is very often recognized as an undesired property; it increases when degradation by physical, chemical and biological processes is slow and the molecule can persist unaltered in the environment for a long time. Recalcitrant chemicals can accumulate in the environment and into organisms and then they can exert their chronic adverse effects.48Degradation half-lives in various compartments are regarded to as key parameters to assess persistence and environmental fate; in general, a substance is defined as persistent if any of its half-lives exceeds specific criteria in four environmental media.41 Chemical degradation half-life was defined by Mackay et al.49 as a quasi-intensive property, which is independent of quantity and is a characteristic of a molecule within a defined environmental medium.
However, experimental degradation half lives in all the environmental media are not always available because they are not easily measurable. When data of persistence are available in different media multivariate analysis can help to analyse and combine this information to have a general overview of the overall environmental persistence of chemicals. In a study of 2007 we aimed to extract and model only the intensive aspect (the characteristics of the molecule) of the chemical half-life, catching the structural features of a chemical that are related to its intrinsic ability or tendency to persist in the environment, independently of its partitioning properties and environmental conditions.
Available half-life data for degradation in air, water, sediment, and soil,50 for a set of 250 organic chemicals of heterogeneous classes, were combined by PCA (the cumulative variance explained by PC1 and PC2 was 94%). To cover as much as possible of the environmental persistence range the dataset contained data for non-persistent, easy degradable chemicals as well as for contaminants already recognized as POPs. The PC1 score i.e. the ranking of the studied organic pollutants along PC1 according to their relative overall half-life, was named Global Half-Life Index (GHLI).51 The biplot (i.e. plot of the PCA scores and loadings) of the first and second components is reported in Fig. 1, where the chemicals (points) are distributed according to their overall environmental persistence, represented by the linear combination of their half-lives in the four selected media. The loadings (i.e. segments with origin in zero) show the importance of each variable in PC1–2. The half-lives in different media (the segments) are oriented in the same direction along PC1, which captures a very significant part of the total information (E.V. 78%). Therefore, the ranking of chemicals along PC1, i.e. the GHLI, can be interpreted as a new macro-variable, which gives information on the tendency of chemicals to persist in the four environmental media. PC1 discriminates chemicals with regard to persistence: chemicals with high half-life values in all the media are located to the right of the PCA plot (Fig. 1), in the zone of global higher persistence (very persistent chemicals anywhere); chemicals with a lower global half-life fall to the left of the graph, not being persistent in any medium. PC2, although less informative (E.V. 16%), is also interesting: it separates the compounds more persistent in air (upper parts in Fig. 1, region 1), i.e., those that could have higher Long-Range Transfer (LRT) potential, from chemicals more persistent in water, soil, and sediment (region 3 in Fig. 1).
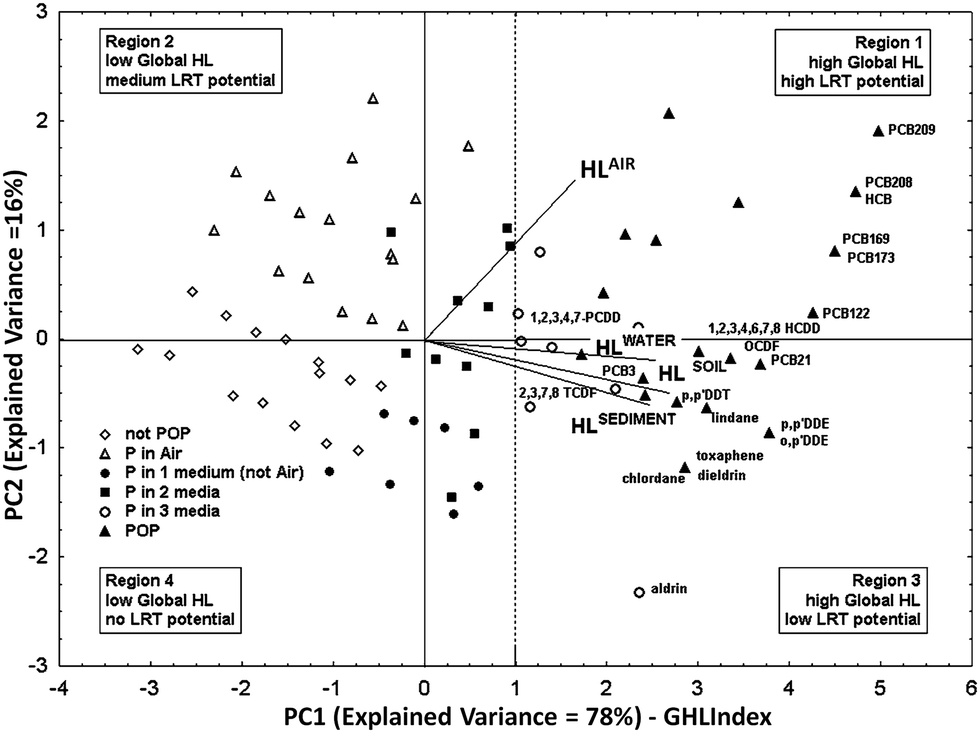 | ||
| Fig. 1 Principal component analysis on half-life data for 250 organic compounds in various compartments (air, water, sediment, and soil) (PC1–PC2: explained variance = 94%) (permission from Gramatica and Papa, EST, 2007 (ref. 51)). | ||
A deeper analysis of the distribution of the studied chemicals confirmed experimental evidences. In fact most of the compounds recognized as POPs by the Stockholm Convention52 are located to the right of the PC1–2 plot, among the very persistent chemicals in all the compartments (full triangles in Fig. 1). Highly chlorinated PCBs and hexachlorobenzene are among the most persistent compounds in this reference scenario; they are grouped in region 1 owing to their global high persistence, especially in air. The less chlorinated PCBs (PCB 3 and PCB 21), p,p′-DDT, p,p′-DDE and o,p′-DDE, highly chlorinated dioxins and some dioxin-like compounds, as well as pesticides toxaphene, lindane, chlordane, dieldrin, and aldrin fall in region 3. These compounds are highly persistent chemicals in all compartments but in the air.
This global index GHLI was then taken as response variable and modelled using the QSAR approach based on DRAGON53 theoretical molecular descriptors.51 The original set of available data was firstly randomly split into training and prediction sets to develop a model verified for its ability to predict external chemicals, not participating to model development28 (i.e. 50% of the compounds (125 compounds) were used for OLS model development, while the other 50% was put into the prediction set to validate the developed QSPR model). The original GHLI model was highly stable and externally predictive as verified by various statistical parameters (R2 and QExt2 > 0.8).
The GHLI model has been recently redeveloped by using descriptors calculated by the freely available online PaDEL-Descriptor software54 and is now implemented in the module QSARINS-Chem40 of the software QSARINS39 (http://www.qsar.it). This model has the same external predictivity of the original one51 (Q2 external functions (QFn2) = 0.83–0.84; Concordance Correlation Coefficient (CCC) = 0.90;55,56 RMSEext = 0.71; MAEext = 0.56).
Statistics of the full GHLI model, applicable in QSARINS, are here reported i.e. number of training molecules (n), determination coefficients (R2), Qleave one out2 (QLOO2), RMSE and MAE, in eqn (1).
| GHL index = −0.57 + 0.01 × MW − 0.15 × MaxHBa + 0.75 × minsCl − 0.05 × nBondsS2 − 0.43nHBDonLipinski, n = 250; R2 = 0.85; QLOO2 = 0.84; RMSE = 0.69; MAE = 0.55 | (1) |
All are bi-dimensional descriptors independent of chemical conformation and easily calculable starting only from the topological graph. The descriptors take account of the different structural properties involved in defining environmental persistence tendency, such as chemical size (MW, as more complex chemicals are generally expected to be more persistent than simpler) and various electronic features (MaxHBa, minsCl, nBondsS2, nHBDonLipisnki more related to a compound's ability to form electrostatic and dipole–dipole interactions in the surrounding media determining lower persistency).
The application of the GHLI model allows for fast preliminary identification and prioritization of not yet known POPs, just from the knowledge of their molecular structure. The proposed multivariate approach, rigorously verified for external prediction ability on chemicals not participating in model development, is particularly useful not only to screen and to make an early prioritization of environmental persistence for pollutants already on the market, but also for planning the synthesis of new compounds, which could represent safer alternatives and replacement solutions for recognized POPs. No method other than QSAR is applicable to detect a priori the potential persistence of not yet synthesized compounds. In addition, this preliminary screening can be refined in a further step to obtain a more realistic evaluation of the potential persistence of chemicals in a specific medium using partition coefficients.
Prioritization of PBTs (Persistent Bioaccumulative and Toxic Chemicals) by Insubria PBT index
Chemicals that are at the same time Persistent, Bioaccumulative, and Toxic (PBT) are considered Substances of Very High Concern (SVHC), which require authorization for use and plan for safer alternatives by REACH,5 due to the potential risk they pose to humans and ecosystems.In a pro-active approach to chemical assessment, more chemicals should be screened and evaluated for the hazard posed by their potential PBT behaviour. Unfortunately, little is known about undesired properties of many existing substances. In the last years, several screening works1,57–71 showed that many, among commercial chemicals, might be PBTs.
This highlights the importance of approaches able to identify potential PBTs, among the existing chemicals. Moreover the so called “benign by design” approach described in the Green Chemistry principles18,19 should be applied for planning the synthesis of safe chemicals and safer alternatives to dangerous compounds.
Currently, the identification of potential PBT (or POP) candidates relies mainly on determining if specific properties of a chemical exceed threshold values for each property related to the PBT behaviour (commonly, half-life in various compartments for P, BCF for B, and a number of toxicity evidences for T).72,73 The most widely used tool for PBT assessment is the US-EPA PBT Profiler (2006), which is easily run as web application.74 The PBT Profiler screens chemicals on the basis of individual P, B, and T properties, calculated by QSAR models and compared to cut off values for each of the three endpoints.
As an alternative approach to the US-EPA PBT Profiler for PBT prioritization, we developed and proposed a new tool for the screening of chemicals for their potential cumulative PBT behaviour,58 as an inherent property of a compound. The PBT behaviour of 180 chemicals was studied by PCA using as input variables the GHLI commented above,51 which represented the chemical persistence (P), the bioconcentration factor (BCF)75 as an indicator for bioaccumulation (B), and the aquatic acute toxicity (T) to Pimephales promelas76 (Fig. 2).
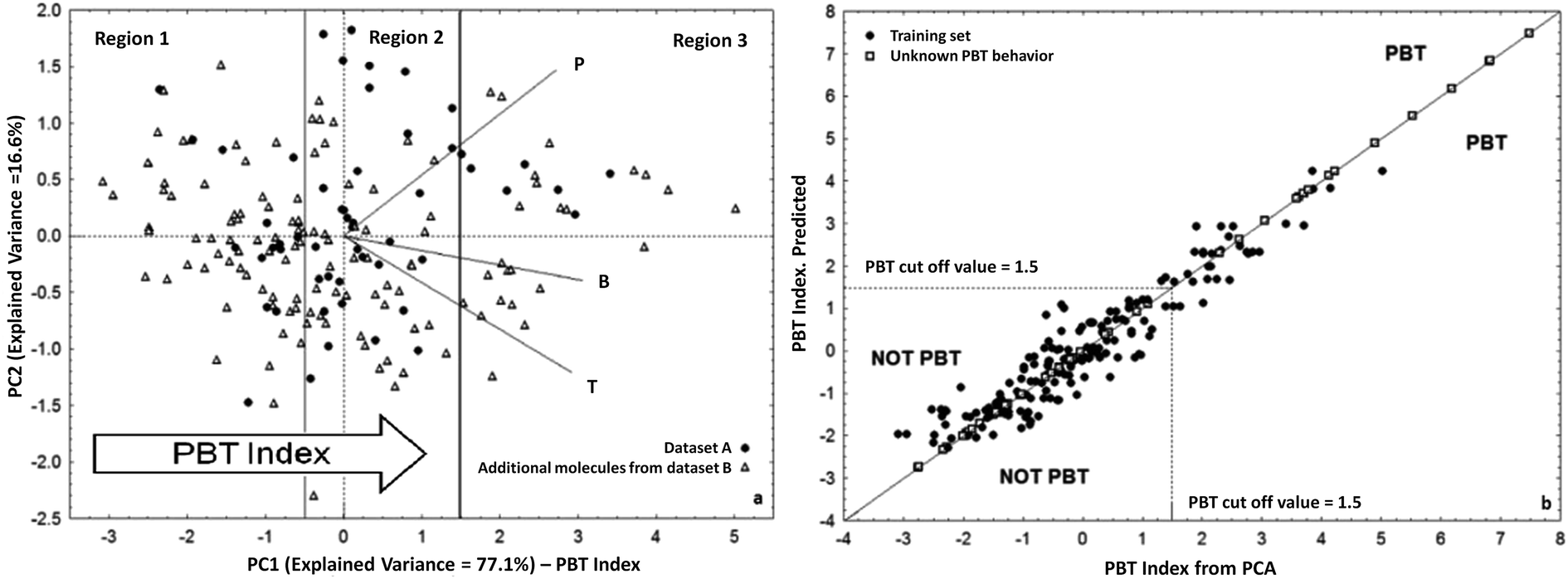 | ||
| Fig. 2 (a) PCA of persistence, bioaccumulation, and toxicity for 180 heterogeneous chemicals and definition of the PBT index. Segments of different lengths are the loadings i.e. weight of the original variables in PC1–2 space; (b) scatter plot of the QSAR model of the PBT index. (Reproduced by permission of The Royal Society of Chemistry from Papa and Gramatica, Green Chem. 2010 (ref. 58)). | ||
According to the orientation of the loadings in PCA (Fig. 2a), the PC1 score, which explains more than 77% of the total variance, is an index of the potential PBT behaviour, i.e. PBT compounds are ranked on the right side of the plot. The cut-off value for PBTs was fixed at 1.5. This value was arbitrarily chosen after comparison with the thresholds of the criteria applied for PBTs and very P very B (vPvB), also by US-PBT Profiler.74
The PBT index was then modelled by Multiple Linear Regression based on the Ordinary Least Squares (MLR-OLS) using the QSAR approach and four simple DRAGON molecular descriptors, with verified external predictivity (QFn2 >0.8) (scatter plot in Fig. 2b).
The PBT index model has been recently implemented in the QSARINS software using freely calculable PaDEL Descriptors54 for a wider applicability. The external predictivity has been verified by various statistical parameters: QFn2 = 0.88–0.90; CCCext = 0.94; RMSEext = 0.5; MAEext = 0.4.
The equation of the full PBT index, applicable in QSARINS, is reported as follows:
| PBT index = −1.46 + 0.64 × nX + 0.22 × nBondsM − 0.39 × nHBDonLipinski − 0.06 × MAXDP2, n = 180; R2 = 0.89; QLOO2 = 0.88; RMSE = 0.51; MAE = 0.4 | (2) |
The descriptors, selected for the best model by the Genetic Algorithm (GA) procedure, are (in descending order of importance): nX (number of halogen atoms), nBondsM (number of multiple bonds), nHDonLipinski (number of donor atoms for H bonds), and MAXDP2 (maximal electrotopological positive variation). All of these descriptors are mono- or bi-dimensional and independent of chemical conformation, thus easily calculable from the topological graph (2D sketch) or from the SMILES string.
These descriptors take into account different chemical properties. The most important descriptors, nX and nBondsM, which encode for substitution with halogens and unsaturation, are known to increase the PBT behaviour of chemicals. On the contrary, MAXDP2 and nHDonLipinski are inversely related to the PBT index. These last two descriptors are related to a compound's ability to form electrostatic and dipole–dipole interactions, as well as hydrogen bonds in the surrounding media.
Recently the PBT index model was applied for the screening of large data sets such as hundreds of heterogeneous chemicals (part 1 of the series “Early PBT assessment and prioritization in Gramatica et al. 2015 (ref. 61)), Personal Care Product (PCP) ingredients (part 2 in Cassani and Gramatica 2015 (ref. 69)), Flame Retardants (FRs) (part 3 in Gramatica et al. 2016 (ref. 70)), and pharmaceuticals (part 4 in Sangion and Gramatica 2016 (ref. 71)).
In all these screening studies, we have compared results generated by the Insubria PBT index model with those obtained by the US-EPA PBT Profiler. The two approaches are very different because the PBT index gives combined information on the PBT behaviour, while the PBT Profiler predicts P, B and T properties separately. The good agreement among conceptually different approaches (>70%) corroborates the results, so that predictions by consensus should be considered as more reliable than those obtained by a single model. Predictions in agreement by two methods were flagged as reliable, and used for PBT prioritization.
It is interesting to note that in the screening of FRs (Fig. 3) some supposed “safer alternatives” to the banned FRs, which are already in commerce, were detected as intrinsically hazardous for their PBT properties (i.e. they are “regrettable substitutions”). This suggests that, following the principle no. 4 of Green Chemistry, reliable predictive QSAR models should more often applied before synthesis and from the very beginning of the product development process. In this way it would be possible to avoid the continuous placing on the market, and consequently in the environment, of compounds that will be identified as PBTs, only several years after their use.
 | ||
| Fig. 3 (a) Graph of the agreement between Insubria PBT index and US-EPA PBT Profiler for the flame retardants study (in the graph are labelled banned flame retardants, halogen flame retardants (HFRs), and halogen-free flame retardants (HFFRs)) (Permission from Gramatica et al., J. Hazard. Mater. 2016 (ref. 70)), (b) Insubria graph for the AD of Flame Retardant model. | ||
We have verified that the Insubria PBT index model is, in the majority of the screenings, more conservative in screening PBTs. Moreover, the analysis of the disagreements among different approaches, based on experimental evidences, supported our results in most of the cases.
Some main interesting results obtained by applying the PBT index model are summarized below:
(1) More than 300 out of 2780 chemicals of potential environmental concern,61 included in the QSARINS-Chem module of the software QSARINS,39,40 were predicted as PBTs by consensus with the US EPA PBT Profiler (agreement >82%).
(2) Eight (with interpolated reliable predictions) out of 534 PCPs, were prioritized as PBTs by consensus of two methods: they are mainly UV filters as benzo-triazoles.69
(3) In the screening of 128 FRs (Fig. 3), of which some are banned and some other are already on the market as substitutes, 30 FRs, which are supposed “safer alternatives,” are predicted as PBTs by both modelling tools in agreement.70
(4) Thirty-five out of 1267 pharmaceutical ingredients of various therapeutic categories were included in a priority list of potential PBTs, while 83% of the screened pharmaceuticals were predicted as non-PBTs by consensus.71
In the application of the PBT index model for screening sets of new chemicals it is possible to verify if a particular compound is into the applicability domain of the model by the Insubria graph, as that reported in Fig. 3b.
The Insubria graph is a modification of the so called Williams plot (plot of hat diagonal values from leverage matrix vs. standardized residuals) which can be used to screen the domain for chemicals without experimental data.
Aquatic Toxicity Index (ATI) of Pharmaceuticals and Personal Care Products (PPCPs)
In the last decades the increased demand for food and health provision and in general the improvements in life style conditions has led to an enhancement in the use of Pharmaceuticals and Personal Care Products (PPCPs).77,78 The irrational use of these products is well documented,78,79 while overuse of medicine is assessed to push up health care costs and have negative consequences for societies and individuals.79 Moreover, PPCPs are detected in the environment with increasing level measure in more than 71 countries all over the world and representing a threat for wildlife and ecosystems.80–89Environmental risk assessment of pharmaceutical products is regulated by EMEA90 while PCPs undergo the REACH regulation; anyway, both risk assessment procedures require a high amount of experimental data.
Despite more than 5000 PPCPs ingredients are in the market only few of them have been tested for their environmental toxicity in the aquatic environment.91
In our recent studies, an approach based on the combination of toxicities in different trophic levels has been proposed for the prioritization of PPCPs for their potential simultaneous toxicity on various aquatic organisms.92,93 Acute toxicity in key species representing the main aquatic trophic level in a simplified aquatic ecosystem (i.e. Pseudokirchneriella subcapitata for primary producer level, Daphnia magna for the crustacean level and Pimephales promelas or Oncorhynchus mykiss for fish level) were selected as target species for the toxicity endpoints. Experimental data were collected from literature and public databases94–97 and underwent through a curation process. Data were filtered for measured effect, experimental procedure and time of exposure, (i.e. growth rate inhibition at 96 h in algae, immobilization at 48 h in daphnia and mortality at 96 h in fish) in order to obtain homogeneous datasets based on a well-defined endpoints.98 It is important to note that after this filtering and curation phase only small or medium datasets were retained (between 20 and 125 chemicals for each end-point) highlighting the lack of ecotoxicity data for PPCPs. New externally validated QSAR models, specific to predict acute toxicity for PCPs92 and pharmaceuticals93 were developed according to the OECD principles for the validation of QSARs using the QSARINS software.39 These OLS models of each endpoint, based on theoretical molecular descriptors calculated by the freely available online PaDEL-Descriptor software and selected by a GA, were statistically robust and externally predictive (CCCext![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 55,56 range: 85–95% and QFn225 range: 70–90%).
55,56 range: 85–95% and QFn225 range: 70–90%).
QSAR models must be used carefully since, as it has been mentioned in the introduction, the application of any model to any chemicals can lead to misleading results. Madden et al.99 have highlighted this problem for the ECOSAR100 models when applied to pharmaceuticals. In fact, many ECOSAR models were developed using, as training sets, small sets of industrial chemicals mainly of simple chemical structure with only a single functional group, while pharmaceuticals are complex chemicals often with a plurality of functional groups. Sangion and Gramatica93 have verified that the RMSE of QSARs developed on training sets of pharmaceuticals is about 1log unit lower than the RMSE calculated for ECOSAR on the same set of compounds (i.e. 0.5 for our models vs. 1.3 for ECOSAR models).
As the second step, the proposed specific models were applied to predict each single acute aquatic toxicities for about 500 PCPs and about 1200 pharmaceuticals without experimental data, verifying the high statistical quality and also the wide structural applicability domain of each model by the Insubria graphs.28 The applicability to new PPCPs was very high: 95–98% of the screened PCPs and 76–96% of the pharmaceuticals were into the AD with reliable, not extrapolated, predictions. This is not surprising since the new models were developed on training sets containing a wide variety of molecular structure reflecting the high complexity of pharmaceuticals.
Example of the Insubria graphs to verify the AD of two models to the large data sets are reported in Fig. 4.
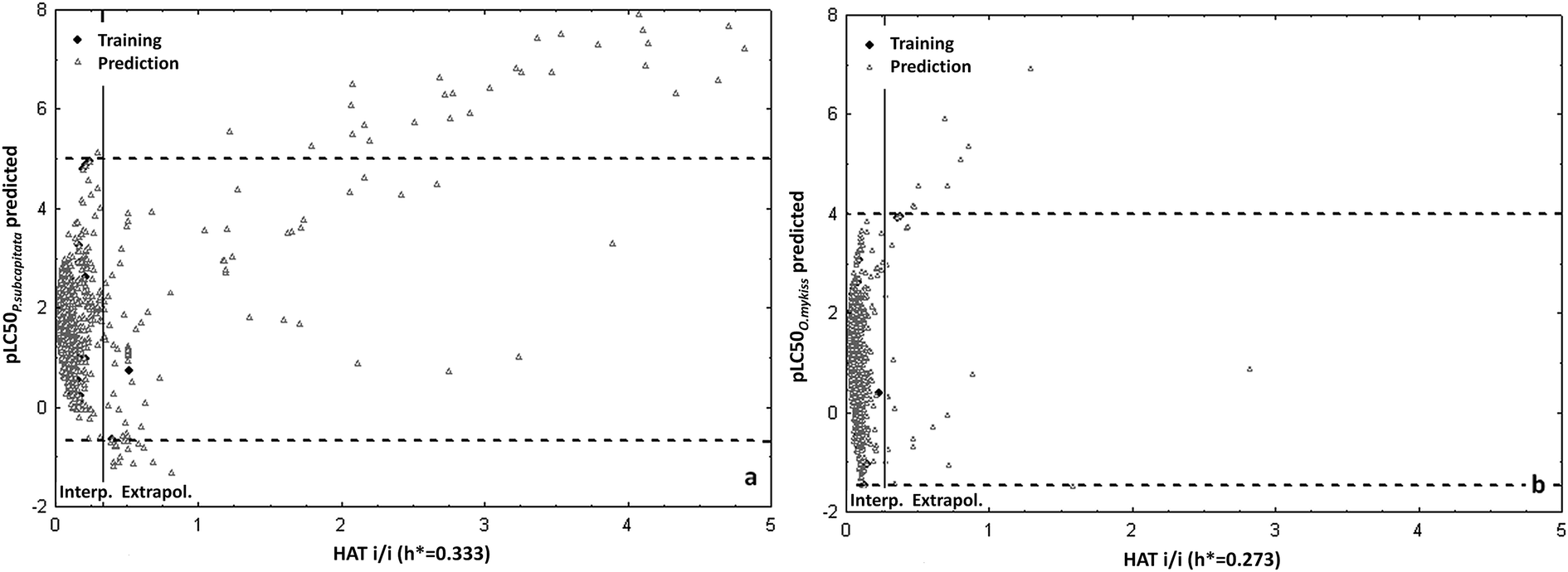 | ||
| Fig. 4 (a) Insubria graphs to verify the applicability domain to chemicals without experimental data of the P.subcapitata model for pharmaceuticals; (b) Insubria graphs to verify the applicability domain to chemicals without experimental data of the O.mykiss model for pharmaceuticals. The training set domain is the box into the vertical (for structure) and horizontal (for response) lines: the chemicals out of these spaces are extrapolations of the models, while those into the boxes are interpolations, thus more reliable. Permission from Sangion and Gramatica, Environ. Int. 2016.93 | ||
After filling the data gaps with the QSAR models for each aquatic species, the predictions for the different endpoints have been combined by PCA and a trend of cumulative acute aquatic toxicity was highlighted on the first principal component. This trend, named Aquatic Toxicity Index (ATI), allows to rank the compounds according to their overall toxicity in all three trophic levels. In the biplot of Fig. 5a, the most dangerous PCPs for all the studied aquatic organisms are located in the right zone, because all the original variables (i.e. the endpoints) are oriented in that direction.
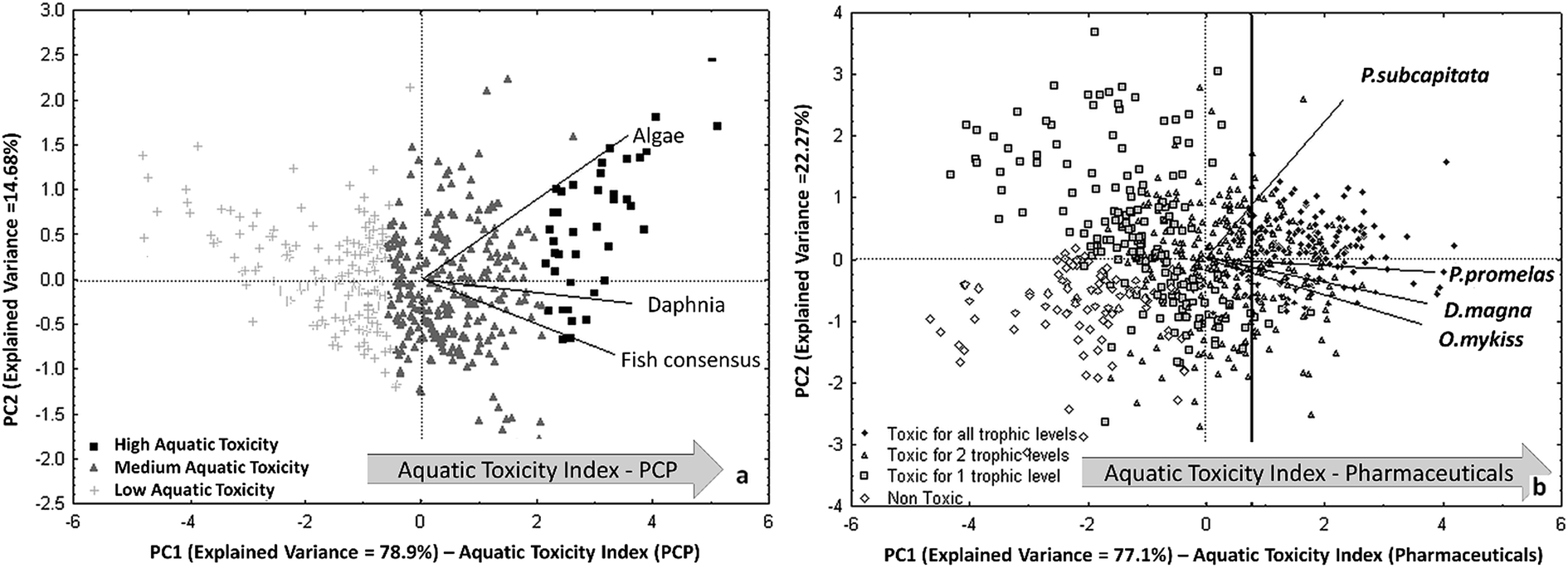 | ||
| Fig. 5 (a) PCA of experimental and predicted aquatic toxicity data for 484 PCPs (reproduced by permission of The Royal Society of Chemistry from Gramatica et al., Green Chem. 2016 (ref. 92)); (b) PCA biplot of the four studied ecotoxicity data for 706 pharmaceuticals. Identification of the Aquatic Toxicity Index (ATI) for both scenarios (permission from Sangion and Gramatica, Environ. Int. 2016 (ref. 93)). | ||
A priority list of 40 most hazardous PCPs has been then proposed: it includes mainly UV-filters (in particular benzo-triazoles), phthalates and fragrances. The priority list of pharmaceuticals is larger and included, with a precautionary approach, 143 chemicals of various therapeutic classes with high toxicity for all the trophic levels, resulting in a ATI value >0.74 (vertical line in Fig. 5b).
It is interesting to note that in both PCA, shown in Fig. 5a and b, the toxicity on algae is relevant along the PC2. The second component can be interpreted as a macro-variable able to differentiate compounds more toxic on algae from those more toxic to organisms in higher trophic levels.
This ATI for both data sets was modelled by non-commercial PaDEL Descriptor structural descriptors to obtain validated and predictive models of eqn (3) and (4), respectively.
| ATIPCP = −14.00 + 0.34 × XlogP + 17.97 × Mp + 0.02 × TIC1, n = 484; R2 = 0.93; QLOO2 = 0.93; RMSE = 0.39; MAE = 0.31; QFn2 = 0.91–0.94; CCCext = 0.96–0.97; RMSEext = 0.38–0.43; MAEext = 0.31–0.33 | (3) |
| ATIpharmaceuticals = −1.87 + 0.62 × CrippenlogP + 0.07 × SaaCH − 0.05 × SHBint2, n = 706; R2 = 0.81; QLOO2 = 0.81; RMSE = 0.68; MAE = 0.54; QFn2 = 0.79–0.82; CCCext = 0.89–0.90; RMSEext = 0.67–72; MAEext = 0.55–0.58 | (4) |
Moreover, the GA selected, in both equations, logP-related parameters (i.e. XlogP and CrippenlogP) as the most relevant descriptors in modeling and predicting the overall aquatic toxicity: this confirmed the well-known relevance of lipophilicity to model toxicity.97 Other selected descriptors with positive coefficients are related to molecular dimension (TIC1), mean atomic polarizabilities scaled on carbon atom (Mp) and aromaticity (SaaCH), while SHBint2 with negative sign is related to the possibility to form hydrogen bonds and has a reducing influence on the toxicity.
These models can be used to interpret toxicity in the whole aquatic ecosystem independently of the considered species. Moreover they can rank and prioritize existing and new PPCPs for their overall hazard for the aquatic environment.
Conclusions
We have presented here some examples of how explorative analysis by PCA combined to QSAR modelling can be useful to screen, rank and predict complex behaviour of chemicals in the environment, such as the environmental persistence in multiple media, the potential PBT behaviour of heterogeneous chemical classes and the aquatic toxicity at different trophic levels for PPCPs.QSAR models generated for the cumulative indexes, which are derived from the combination of multiple endpoints, link these new macro-endpoints to the potential hazard inherent in the chemical structure and can be proposed for preliminary priority setting purposes both for compounds without experimental data and for new chemicals (i.e. before synthesis). This approach is summarized in Fig. 6.
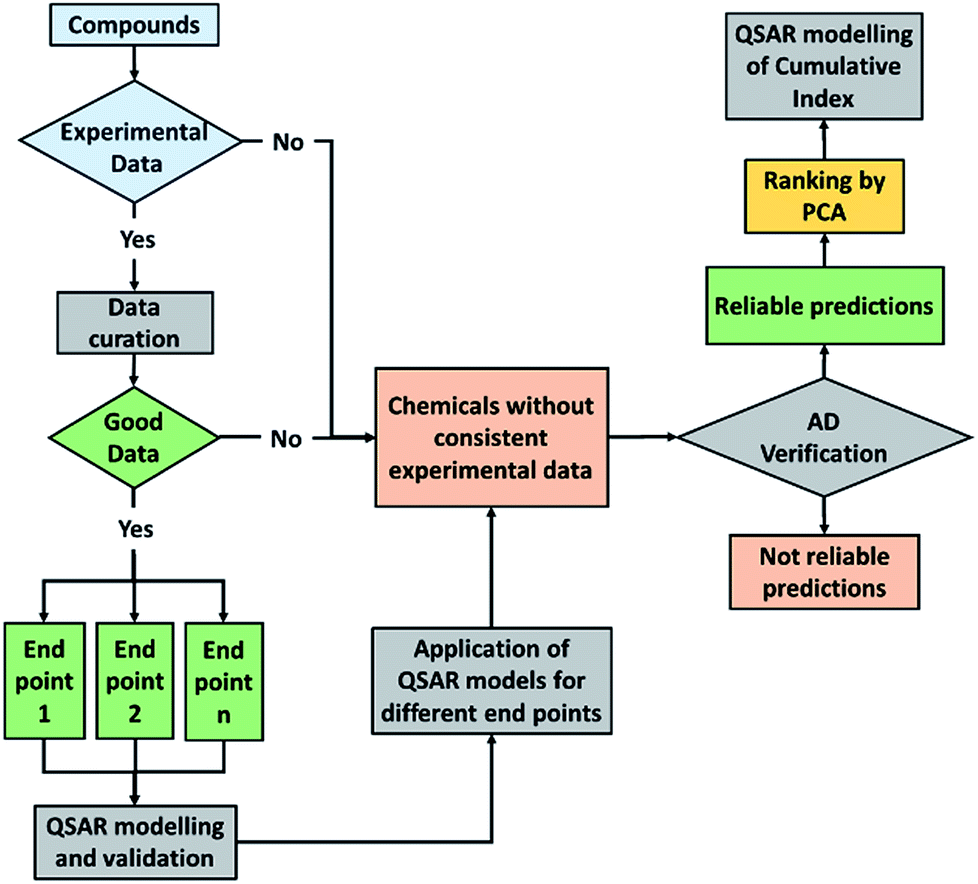 | ||
| Fig. 6 Conceptual scheme of the QSAR modelling of PCA end-points. | ||
This prioritization provides useful information to reduce the number of tested chemicals, thus reducing time, costs and animal tests. In addition this approach is helpful to avoid the synthesis, the commercialization and the release in the environment of harmful compounds, which would be recognized as dangerous only after evidence of human health concerns was manifested. This is the basis of the “benign by design” approach of Green Chemistry.
The case study of Flame Retardants, addressed in the paragraph dedicated to PBT screening, and in particular of the compounds that were introduced in the market as safer alternatives to the banned PBDE is an emblematic example. Flame retardant alternatives that are identified as potential PBTs on the basis of their molecular structure (i.e. by QSAR) may be experimentally highlighted as PBTs and regulated in few years. Therefore, we want to stress again that QSAR models can predict undesired potential behaviour before the synthesis of chemicals.
In conclusion, environmental and industrial scientists and regulators should be more confident in screening approaches based on the molecular structure of compounds such as QSAR, which is powerful to identify and prioritize hazardous chemicals before and after synthesis. The availability and easy applicability of these models for cumulative end points of environmental relevance in software such as QSARINS-Chem is an additional benefit to the researchers' and regulators' community.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. C. Muir and P. H. Howard, Environ. Sci. Technol., 2006, 40, 7157–7166 CrossRef CAS PubMed.
- J. A. Arnot, T. N. Brown, F. Wania, K. Breivik and M. S. McLachlan, Environ. Health Perspect., 2012, 120, 1565–1570 CrossRef PubMed.
- P. P. Egeghy, R. Judson, S. Gangwal, S. Mosher, D. Smith, J. Vail and E. A. Cohen Hubal, Sci. Total Environ., 2012, 414, 159–166 CrossRef CAS PubMed.
- R. Judson, A. Richard, D. J. Dix, K. Houck, M. Martin, R. Kavlock, V. Dellarco, T. Henry, T. Holderman, P. Sayre, S. Tan, T. Carpenter and E. Smith, Environ. Health Perspect., 2009, 117, 685–695 CrossRef CAS PubMed.
- EC Regulation, Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH). Regulation (EC) No. 1907/2006 of the European Parliament and of the Council, 2006 Search PubMed.
- L. Shi, W. Tong, H. Fang, Q. Xie, H. Hong, R. Perkins, J. Wu, M. Tu, R. M. Blair, W. S. Branham, C. Waller, J. Walker and D. M. Sheehan, SAR QSAR Environ. Res., 2002, 13, 69–88 CrossRef CAS PubMed.
- US EPA, Procedures for Prioritization of Chemicals for Risk Evaluation Under the Toxic Substances Control Act, 82 FR 4825, 2016 Search PubMed.
- G. Schaafsma, E. D. Kroese, E. L. J. P. Tielemans, J. J. M. Van de Sandt and C. J. Van Leeuwen, Regul. Toxicol. Pharmacol., 2009, 53, 70–80 CrossRef CAS PubMed.
- R. S. Judson, K. A. Houck, R. J. Kavlock, T. B. Knudsen, M. T. Martin, H. M. Mortensen, D. M. Reif, D. M. Rotroff, I. Shah, A. M. Richard and D. J. Dix, Environ. Health Perspect., 2010, 118, 485–492 CrossRef CAS PubMed.
- T. Höfer, I. Gerner, U. Gundert-Remy, M. Liebsch, A. Schulte, H. Spielmann, R. Vogel and K. Wettig, Arch. Toxicol., 2004, 78, 549–564 CrossRef PubMed.
- F. Pederson, J. de Bruijn, S. Munn and K. Van Leeuwen, Assessment of additional testing needs under REACH Effects of (Q)SARS, risk based testing and voluntary industry initiatives, European Commision, 2003 Search PubMed.
- K. van der Jagt, S. Munn, J. Torslov and J. de Bruijn, Alternative approaches can reduce the use of test animals under REACH, Eurpoean Commission, 2004 Search PubMed.
- National Research Council, Division on Earth and Life Studies, Board on Environmental Studies and Toxicology, Board on Chemical Sciences and Technology and Committee on the Design and Evaluation of Safer Chemical Substitutions: A. Framework to Inform Government and Industry Decisions, A Framework to Guide Selection of Chemical Alternatives, National Academies Press, 2014 Search PubMed.
- D. Mackay, J. Hubbarde and E. Webster, QSAR Comb. Sci., 2003, 22, 106–112 CAS.
- J. M. McKim, S. P. Bradbury and G. J. Niemi, Environ. Health Perspect., 1987, 71, 171–186 CrossRef CAS PubMed.
- E. J. Matthews, N. L. Kruhlak, D. R. Benz, J. Ivanov, G. Klopman and J. F. Contrera, Regul. Toxicol. Pharmacol., 2007, 47, 136–155 CrossRef CAS PubMed.
- G. M. Klecka, D. C. Muir, P. Dohmen, S. J. Eisenreich, F. A. Gobas, K. C. Jones, D. Mackay, J. V. Tarazona and D. van Wijk, Integr. Environ. Assess. Manage., 2009, 5, 535–538 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- J. B. Zimmerman and P. T. Anastas, Science, 2015, 347, 1198–1199 CrossRef CAS PubMed.
- A. Tropsha, Mol. Inf., 2010, 29, 476–488 CrossRef CAS PubMed.
- S. Weaver and M. P. Gleeson, J. Mol. Graphics Modell., 2008, 26, 1315–1326 CrossRef CAS PubMed.
- T. I. Netzeva, A. G. Saliner and A. P. Worth, Environ. Toxicol. Chem., 2006, 25, 1223–1230 CrossRef CAS PubMed.
- D. Horvath, G. Marcou and A. Varnek, J. Chem. Inf. Model., 2009, 49, 1762–1776 CrossRef PubMed.
- P. Gramatica, Mol. Inf., 2014, 33, 311–314 CrossRef CAS PubMed.
- P. Gramatica and A. Sangion, J. Chem. Inf. Model., 2016, 56, 1127–1131 CrossRef CAS PubMed.
- T. I. Netzeva, A. P. Worth, T. Aldenberg, R. Benigni, M. T. D. Cronin, P. Gramatica, J. S. Jaworska, S. Kahn, G. Klopman, C. A. Marchant, G. Myatt, N. Nikolova-Jeliazkova, G. Y. Patlewicz, R. Perkins, D. W. Roberts, T. W. Schultz, D. T. Stanton, J. J. M. van de Sandt, W. D. Tong, G. Veith and C. H. Yang, ATLA, Altern. Lab. Anim., 2005, 33, 155–173 CAS.
- P. Gramatica, QSAR Comb. Sci., 2007, 26, 694–701 CAS.
- P. Gramatica, S. Cassani, P. P. Roy, S. Kovarich, C. W. Yap and E. Papa, Mol. Inf., 2012, 31, 817–835 CrossRef CAS PubMed.
- US EPA, Estimation Programs Interface Suite™ for Microsoft® Windows, United States Environmental Protection Agency, Washington, DC, USA, 2012 Search PubMed.
- E. Benfenati, A. Manganaro and G. Gini, VEGA-QSAR: AI inside a platform for predictive toxicology, 2013 Search PubMed.
- US EPA, User's Guide for T.E.S.T. (version 4.2) (Toxicity Estimation Software Tool) A Program to Estimate Toxicity from Molecular Structure, 2016 Search PubMed.
- OECD, QSAR Toolbox, 2017 Search PubMed.
- BIOVIA Corporate Americas, TOPKAT (TOxicity Prediction by Komputer Assisted Technology), BIOVIA Corporate Americas Search PubMed.
- A. Vedani and M. Smiesko, ATLA, Altern. Lab. Anim., 2009, 37, 477–496 CAS.
- A. Vedani, M. Dobler, Z. Hu and M. Smieško, Toxicol. Lett., 2015, 232, 519–532 CrossRef CAS PubMed.
- A. Vedani, M. Dobler and M. Smieško, Toxicol. Appl. Pharmacol., 2012, 261, 142–153 CrossRef CAS PubMed.
- Division of Diet, Disease Prevention and Toxicology, National Food Institute, Technical University of Denmark, Danish (Q)SAR Database, http://qsar.food.dtu.dk Search PubMed.
- V. Ruusmann, S. Sild and U. Maran, J. Cheminf., 2015, 7, 32 CAS.
- P. Gramatica, N. Chirico, E. Papa, S. Cassani and S. Kovarich, J. Comput. Chem., 2013, 34, 2121–2132 CrossRef CAS.
- P. Gramatica, S. Cassani and N. Chirico, J. Comput. Chem., 2014, 35, 1036–1044 CrossRef CAS PubMed.
- D. Mackay, D. M. Hughes, M. L. Romano and M. Bonnell, Integr. Environ. Assess. Manage., 2014, 10, 588–594 CrossRef PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
- I. T. Jolliffe, Principal Component Analysis, Springer-Verlag, New York, 2002 Search PubMed.
- P. Gramatica, P. Pilutti and E. Papa, Atmos. Environ., 2004, 38, 6167–6175 CrossRef CAS.
- P. Gramatica, P. Pilutti and E. Papa, SAR QSAR Environ. Res., 2002, 13, 743–753 CrossRef CAS PubMed.
- P. Gramatica and A. Di Guardo, Chemosphere, 2002, 47, 947–956 CrossRef CAS PubMed.
- P. Gramatica, E. Papa and B. Battaini, Int. J. Environ. Anal. Chem., 2004, 84, 65–74 CrossRef CAS.
- R. Boethling, K. Fenner, P. Howard, G. Klecka, T. Madsen, J. R. Snape and M. J. Whelan, Integr. Environ. Assess. Manage., 2009, 5, 539–556 CAS.
- D. Mackay, L. S. McCarty and M. MacLeod, Environ. Toxicol. Chem., 2001, 20, 1491–1498 CrossRef CAS PubMed.
- D. Mackay, W.-Y. Shiu, K.-C. Ma and S. C. Lee, Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals, 2nd edn, CRC Press, 2006 Search PubMed.
- P. Gramatica and E. Papa, Environ. Sci. Technol., 2007, 41, 2833–2839 CrossRef CAS PubMed.
- UNEP, Stockholm Convention on persistent organic pollutants (POPs), 2014 Search PubMed.
- Talete, DRAGON for Windows (Software for Molecular Descriptor Calculations), Talete srl, 2007 Search PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- N. Chirico and P. Gramatica, J. Chem. Inf. Model., 2011, 51, 2320–2335 CrossRef CAS PubMed.
- N. Chirico and P. Gramatica, J. Chem. Inf. Model., 2012, 52, 2044–2058 CrossRef CAS PubMed.
- P. H. Howard and D. C. G. Muir, Environ. Sci. Technol., 2010, 44, 2277–2285 CrossRef CAS PubMed.
- E. Papa and P. Gramatica, Green Chem., 2010, 12, 836–843 RSC.
- T. Öberg and M. S. Iqbal, Chemosphere, 2012, 87, 975–981 CrossRef PubMed.
- S. Strempel, M. Scheringer, C. A. Ng and K. Hungerbühler, Environ. Sci. Technol., 2012, 46, 5680–5687 CrossRef CAS PubMed.
- P. Gramatica, S. Cassani and A. Sangion, Environ. Int., 2015, 77, 25–34 CrossRef CAS PubMed.
- H. P. H. Arp, T. N. Brown, U. Berger and S. E. Hale, Environ. Sci.: Processes Impacts, 2017, 19, 939–955 CAS.
- M. Zachary and G. M. Greenway, SAR QSAR Environ. Res., 2009, 20, 145–157 CrossRef CAS PubMed.
- F. Pizzo, A. Lombardo, A. Manganaro, C. I. Cappelli, M. I. Petoumenou, F. Albanese, A. Roncaglioni, M. Brandt and E. Benfenati, Environ. Res., 2016, 151, 478–492 CrossRef CAS PubMed.
- M. Nendza, S. Gabbert, R. Kuehne, A. Lombardo, A. Roncaglioni, E. Benfenati, R. Benigni, C. Bossa, S. Strempel, M. Scheringer, A. Fernandez, R. Rallo, F. Giralt, S. Dimitrov, O. Mekenyan, F. Bringezu and G. Schueuermann, Regul. Toxicol. Pharmacol., 2013, 66, 301–314 CrossRef PubMed.
- E. Rorije, E. Verbruggen, A. Hollander, T. Traas and M. Janssen, Identifying potential POP and PBT substances: Development of a new Persistence/Bioaccumulation-score, RIVM, 2011 Search PubMed.
- G. Stieger, M. Scheringer, C. A. Ng and K. Hungerbuehler, Chemosphere, 2014, 116, 118–123 CrossRef CAS PubMed.
- S. Ortiz de García, G. P. Pinto, P. A. García-Encina and R. I. Mata, J. Environ. Manage., 2013, 129, 384–397 CrossRef PubMed.
- S. Cassani and P. Gramatica, Sustainable Chem. Pharm., 2015, 1, 19–27 CrossRef CAS.
- P. Gramatica, S. Cassani and A. Sangion, J. Hazard. Mater., 2016, 306, 237–246 CrossRef CAS PubMed.
- A. Sangion and P. Gramatica, Environ. Res., 2016, 147, 297–306 CrossRef CAS PubMed.
- M. Matthies, K. Solomon, M. Vighi, A. Gilman and J. V. Tarazona, Environ. Sci.: Processes Impacts, 2016, 18, 1114–1128 CAS.
- C. Rauert, A. Friesen, G. Hermann, U. Jöhncke, A. Kehrer, M. Neumann, I. Prutz, J. Schönfeld, A. Wiemann, K. Willhaus, J. Wöltjen and S. Duquesne, Environ. Sci. Eur., 2014, 26, 9 CrossRef.
- US EPA, PBT profiler;Persistent, Bioaccumulative, and Toxic Profiles Estimated for Organic Chemicals On-line, http://www.pbtprofiler.net/, accessed, September 8, 2015 Search PubMed.
- P. Gramatica and E. Papa, QSAR Comb. Sci., 2005, 24, 953–960 CAS.
- E. Papa, F. Villa and P. Gramatica, J. Chem. Inf. Model., 2005, 45, 1256–1266 CrossRef CAS PubMed.
- United Nations, World Population Prospects The 2006 Revision, Department of Economic and Social Affairs, 2007 Search PubMed.
- WHO, The World Medicines Situation 2011, 2011 Search PubMed.
- J. Busfield, Soc. Sci. Med., 2015, 131, 199–206 CrossRef PubMed.
- IWW, Pharmaceuticals in the environment: occurrence, effects, and options for action, Research project funded by the German Federal Environment Agency (UBA) within the Environmental Research Plan No. 371265408, 2014 Search PubMed.
- K. Kümmerer, Pharmaceuticals in the Environment: Sources, Fate, Effects and Risks, Springer Science & Business Media, 2013 Search PubMed.
- F. A. Weber, T. aus der Beek, A. Bergmann, A. Carius, G. Grüttner, S. Hickmann, I. Ebert, A. Hein, A. Küster, J. Rose, J. Koch-Jugl and H.-C. Stolzenberg, Pharmaceuticals in the environment – the global perspective, 2014 Search PubMed.
- E. Zuccato, S. Castiglioni, R. Bagnati, M. Melis and R. Fanelli, J. Hazard. Mater., 2010, 179, 1042–1048 CrossRef CAS PubMed.
- S. R. Hughes, P. Kay and L. E. Brown, Environ. Sci. Technol., 2013, 47, 661–677 CrossRef CAS PubMed.
- A. Barra Caracciolo, E. Topp and P. Grenni, J. Pharm. Biomed. Anal., 2015, 106, 25–36 CrossRef CAS PubMed.
- K. Fent, A. A. Weston and D. Caminada, Aquat. Toxicol., 2006, 76, 122–159 CrossRef CAS PubMed.
- P. P. Fong and A. T. Ford, Aquat. Toxicol., 2014, 151, 4–13 CrossRef CAS PubMed.
- J. L. Oaks, M. Gilbert, M. Z. Virani, R. T. Watson, C. U. Meteyer, B. A. Rideout, H. L. Shivaprasad, S. Ahmed, M. J. I. Chaudhry, M. Arshad, S. Mahmood, A. Ali and A. A. Khan, Nature, 2004, 427, 630–633 CrossRef CAS.
- J. P. Sumpter, A. C. Johnson, R. J. Williams, A. Kortenkamp and M. Scholze, Environ. Sci. Technol., 2006, 40, 5478–5489 CrossRef CAS PubMed.
- EMEA, Guideline on The Environmental Risk Assessment of Medicinal Products for Human USE Doc. Ref. EMEA/CHMP/SWP/4447/00, 2006 Search PubMed.
- K. E. Arnold, A. R. Brown, G. T. Ankley and J. P. Sumpter, Philos. Trans. R. Soc., B, 2014, 369, 20130569 CrossRef PubMed.
- P. Gramatica, S. Cassani and A. Sangion, Green Chem., 2016, 18, 4393–4406 RSC.
- A. Sangion and P. Gramatica, Environ. Int., 2016, 95, 131–143 CrossRef CAS PubMed.
- US EPA, ECOTOX User Guide: ECOTOXicology Database System, version 4.0. Available: http:/www.epa.gov/ecotox/, 2015 Search PubMed.
- M. Cassotti, D. Ballabio, V. Consonni, A. Mauri, I. V. Tetko and R. Todeschini, ATLA, Altern. Lab. Anim., 2014, 31–41 CAS.
- NOAA, NCCOS Pharmaceuticals in the environment, http://products.coastalscience.noaa.gov/peiar/search.aspx Search PubMed.
- H. Sanderson and M. Thomsen, Toxicol. Lett., 2009, 187, 84–93 CrossRef CAS PubMed.
- OECD, Principles for the validation, for regulatory purposes, of (Quantitative) Structure-Activity Relationship Models, 2004 Search PubMed.
- J. C. Madden, S. J. Enoch, M. Hewitt and M. T. D. Cronin, Toxicol. Lett., 2009, 185, 85–101 CrossRef CAS PubMed.
- US EPA, The ECOSAR (ECOlogical Structure Activity Relationship) Class Program, version 1.11, 2012 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |