
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysiological modes of action across species and toxicants: the key to predictive ecotoxicology†
Roman
Ashauer
 *ab and
Tjalling
Jager
c
*ab and
Tjalling
Jager
c
aEnvironment Department, University of York, Heslington, York YO10 5NG, UK. E-mail: roman.ashauer@york.ac.uk
bToxicodynamics Ltd, York YO10 4PE, UK
cDEBtox Research, De Bilt, The Netherlands
First published on 1st November 2017
Abstract
As ecotoxicologists we strive for a better understanding of how chemicals affect our environment. Humanity needs tools to identify those combinations of man-made chemicals and organisms most likely to cause problems. In other words: which of the millions of species are at risk from pollution? And which of the tens of thousands of chemicals contribute most to the risk? We identified our poor knowledge on physiological modes of action (how a chemical affects the energy allocation in an organism), and how they vary across species and toxicants, as a major knowledge gap. We also find that the key to predictive ecotoxicology is the systematic, rigorous characterization of physiological modes of action because that will enable more powerful in vitro to in vivo toxicity extrapolation and in silico ecotoxicology. In the near future, we expect a step change in our ability to study physiological modes of action by improved, and partially automated, experimental methods. Once we have populated the matrix of species and toxicants with sufficient physiological mode of action data we can look for patterns, and from those patterns infer general rules, theory and models.
Environmental significanceHumanity designs and produces ever more chemicals and urgently needs to identify those that pose the greatest risk to the millions of species living on earth. Predictive ecotoxicology would enable risk assessors to project the impact of untested chemicals on environmental organisms, yet we are currently not very proficient at this. We outline a research strategy that will deliver more effective theory and models for environmental risk assessment of chemicals. This strategy rests on mechanistic toxicokinetic-toxicodynamic modelling, complements efforts to develop quantitative adverse outcome pathways (qAOPs), and will also enable the design of more efficient quantitative structure-activity relationships (QSARs). |
Introduction
The science of ecotoxicology is challenged by two large numbers: thousands of man-made chemicals are released into the environment and millions of biological species are exposed.1 There are two central questions that we need to answer: which of the many thousands of chemicals pose the greatest risk to the environment? And which of the millions of species in the environment are most at risk? This is the central conundrum of ecotoxicology and we think that the answers cannot be found with today's approaches. Currently, there are two main lines of reasoning, each focusing on different aspects of the central conundrum.The first approach relies on high-throughput bioassays of cellular or molecular markers and promises to upscale the bioassay results to organisms and beyond.2,3 Those efforts rally around the idea of quantitative adverse outcome pathways (AOP, see also glossary in Table 1).4–6 In our view, this approach will help to solve many important problems, but continues to struggle with the central conundrum because of a poor link to the whole organism (life history traits), and because it does not have easy scalability in the species dimension. We use the term scalability as it is used in engineering: the ability of a process to accommodate a growing amount of work. Implementation of a method consumes most of the costs and effort, applying it thousands or millions of times adds little extra costs. We can see that high-throughput testing will likely go a long way to screening large numbers of chemicals, i.e. we have scalability in the many chemicals dimension, and quantitative AOPs will aid interpretation and extrapolation, but the results will initially tell us something only about a limited number of biological species. To extrapolate to other, untested, species requires new knowledge that is not available yet. Simply put, we think these approaches, as they currently are put together, do not have scalability in the biological species dimension. Extrapolating quantitative AOPs across species requires the assumption that molecular pathways and functions are conserved across biological species. Moreover, it requires that they are conserved not only qualitatively, but also that the quantitative response is the same across biological species. For most species we do not have this information, and for those where we know something already, we can see that a notable fraction of receptors and target sites is not conserved between species.7,8 However, because a large fraction of receptors and pathways is conserved across species, the promise is that building blocks of quantitative AOPs can be reused – eventually making their development quick and cheap. However we haven't reached that point yet. The resources required to build quantitative AOPs for a new species are substantial, and more importantly, they are larger than simply doing apical toxicity tests with a range of model toxicants in the new species. To put it more bluntly: currently the most efficient way of establishing if a previously untested species is vulnerable to chemical pollutants might be to simply perform traditional toxicity testing of life-history traits with a dozen or so carefully selected model toxicants – and not by embarking on a large research programme with the aim to build quantitative AOPs around that species. When will that situation change? The answer hinges on how much quantitative AOPs differ amongst species – something that we do not know very much about.
| Term | Abbreviation | Meaning |
|---|---|---|
| Toxicokinetics | TK | What an organism does to a chemical, including uptake, biotransformation, distribution and elimination. Process that links the external concentration to a change in the concentration at the target site, often studied over time |
| Toxicodynamics | TD | What a chemical does to an organism. Process that links the concentration at the target site to toxicity. Encompasses all kinds of effects (e.g. on growth, reproduction, behavior, survival, etc.) & often studied over time |
| Adverse outcome pathway | AOP | A way to organise and structure toxicological knowledge. Diagrams with boxes and arrows connecting cellular, physiological and individual level variables. Sometimes viewed as a framework for (eco)toxicity |
| Dynamic energy budget model with toxicity module | DEBtox | A famility of models following from DEB theory. Used to simulate how organisms acquire and use energy to live, grow and reproduce, and how chemicals change those energy flows |
| Physiological mode of action | pMoA | A distinct way in which a chemical interferes with the energy fluxes in an organism, and thereby affects life-history traits. Different pMoAs are defined within DEBtox |
The second approach to tackle the conundrum of large numbers views ecotoxicology as chemical stress ecology.9,10 A large body of literature, including some high level reviews, calls for more ecological relevance and realism in the environmental risk assessment of chemicals.11 This school of thought recognizes that, in the environment, chemical stress is just one of many stress factors that influence an organism, a population, community or ecosystem.12 Ecological impacts of toxicants are conditional on environmental variables as well as ecological factors. Also, there is clear evidence that the environment is degraded in many locations,13 and the question arises how much of that environmental degradation and biodiversity loss can be attributed to man-made, synthetic chemicals. Ecology provides tools to study stress at different biological scales (organisms, populations, communities, ecosystems) and there are concepts of what makes a biological species vulnerable to toxicants.14 However these approaches are all limited by a lack of quantitative models for multiple stressors, in particular how organisms respond to chemical stress in realistic environmental situations, which will inevitably include multiple stress factors. As individual organisms are the building blocks of populations, communities and ecosystems, and as they are well defined systems subject to the laws of mass and energy conservation as well as evolution, we have good reasons to start building theory and models for the effects of toxicants on the organism level.15 If we had a quantitative model to predict effects of toxicant exposure on organisms' life history traits under multiple stress, then ecology provides us with the theory and tools to extrapolate those effects to populations,16 communities17 and ecosystems.18
We take a closer look at the current limitations of ecotoxicological theory and models, and we will illuminate the interface of the first and second approach: effects of toxicants at the level of the whole organism's life history (Fig. 1). We will explain how progress on both challenges, the development of quantitative adverse outcome pathways and ecologically-relevant multiple-stress assessments, is limited by the same issue: a poor understanding of how the physiological mode of action19 (pMoA) varies across toxicants and biological species.
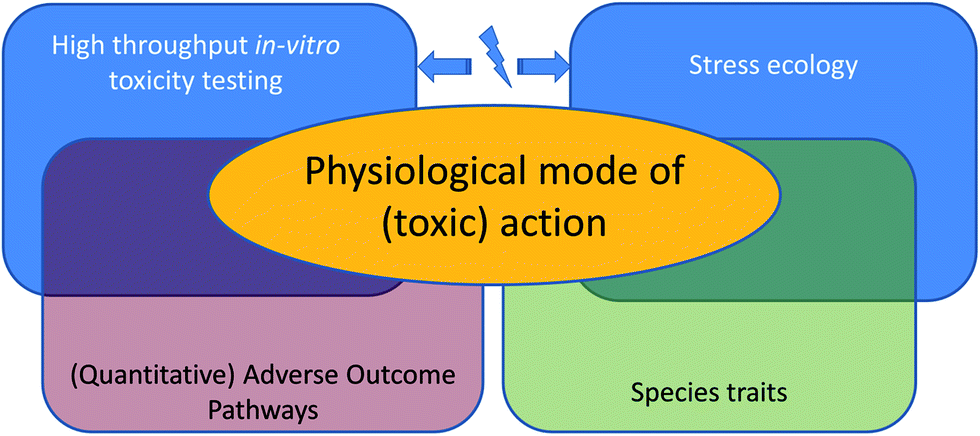 | ||
| Fig. 1 Two contrasting approaches to the challenges in ecotoxicology drift further and further apart and differ in their respective aims (see text for details). We propose to study physiological modes of toxic action because that will enable us to solve some of the challenges ahead and will mechanistically connect both approaches. | ||
How can we quantitatively link toxicity across levels of biological organization?
Upscaling from cellular toxicity to organism-level effects – missing theory
There is a gap in our understanding and theoretical frameworks. What we have are tools to study toxicity at the cellular and molecular level,20,21 but no general theory or generic models to link these effects to changes in the life-history traits over ontogeny (as would be required to move to the population level, and higher). Despite repeated attempts to derive toxic effects on life-history traits of organisms based on cellular toxicity22–24 we still lack a systematic way to do this and we lack a testable, quantitative theory of ecotoxicology that helps bridge the cellular to whole organism divide. This is a huge knowledge gap in ecotoxicology.15 We need to develop the missing theory that explains macroscopic (organism) toxicity as a consequence of microscopic (cell) toxicity, just as statistical mechanics (theory) explain the macroscopic gas law in physical chemistry based on molecular interactions (Fig. 2).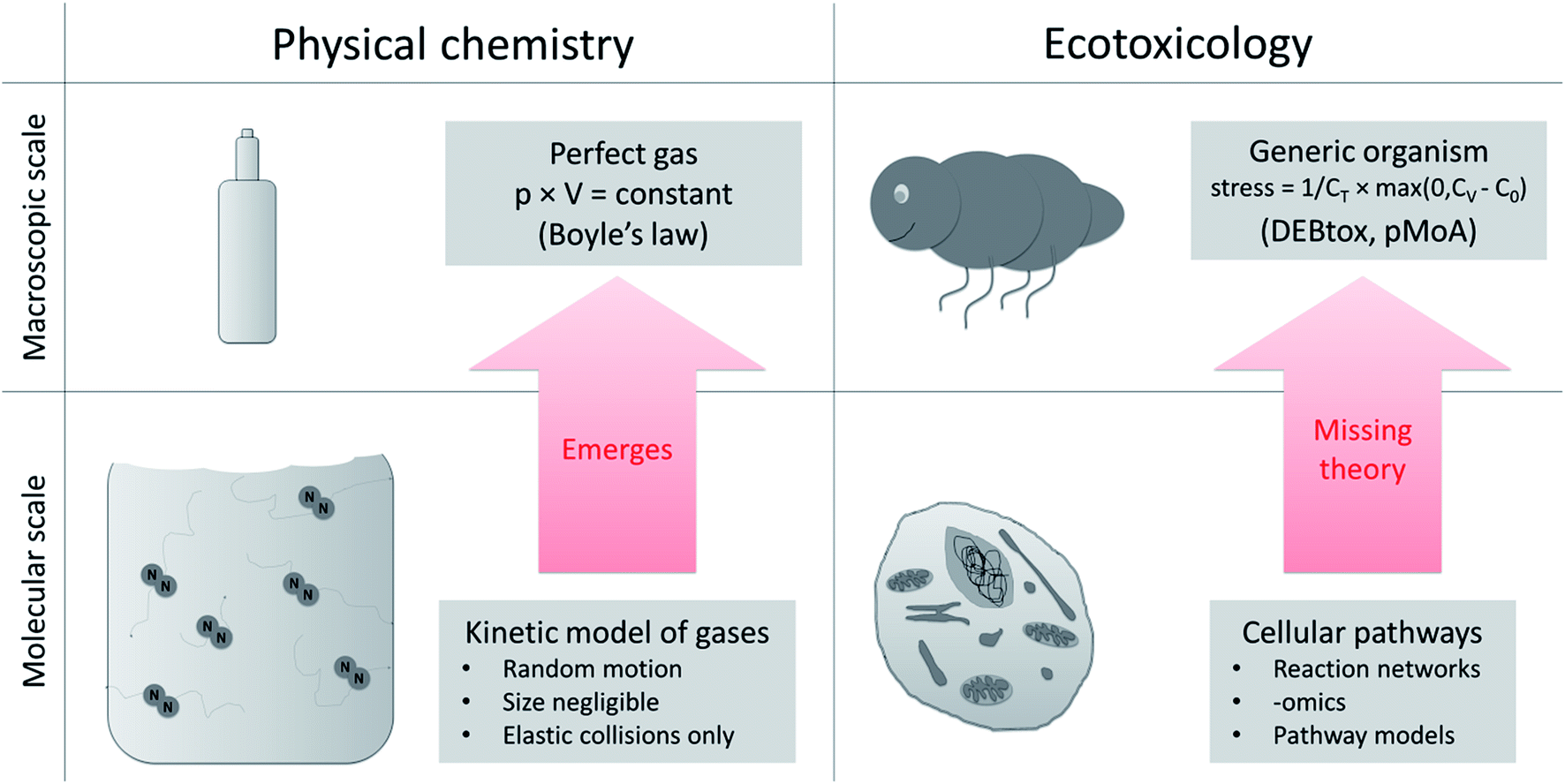 | ||
| Fig. 2 Upscaling from molecular to macroscopic scale in physical chemistry and ecotoxicology. The equations that describe a macroscopic system (perfect gas, e.g. Boyle's law) derive from the equations of the molecular scale model (kinetic model of gases). In ecotoxicology we have models at the cellular and the organism level, but the connecting theory is missing. At the macroscopic scale DEBtox and its pMoAs provide a starting point (stress: degree of stress on DEB model parameter, CT: internal tolerance concentration, CV: internal concentration of toxicant, C0: threshold38). | ||
The development of AOPs promises to fill parts of this gap, but there are very few quantitative AOPs to date4,25–28 and those that have been developed do not provide a general method for scaling cellular toxicity up to the level of the organism's life history. As a first step (necessary but not sufficient), we argue here that significant advances can only be made by exchanging the traditional organism level dose-response models with biologically based dose-response models, namely those based on energy-budget considerations. There are many shortcomings of the traditional descriptive dose-response models and the associated summary statistics like LD50, ECx or NOEC values,29,30 but it is these crude metrics that in vitro and in silico methods aim to predict. Is it really the best way forward to build quantitative models around AOPs or cellular bioassays with the aim to predict adverse outcomes by proxy of LD50, ECx or NOEC values? We can do better by using energy-budget models instead, and predicting the parameters of those models. Why is this better? Simply put, organisms require resources to grow, develop and reproduce; it is these traits that we ultimately require to link AOPs to ecological theory, and upscale to the population level and higher. In particular for sub-lethal responses, like changes in growth and reproduction, it is the acquisition and use of resources (or in general: energy) that links the different life-history traits and determines how they develop over ontogeny. Changes in energy-demanding traits, such as growth and reproduction, as a consequence of toxicant or environmental stress, logically imply changes in the energy budget. Hence we can model organism's life history using dynamic energy budget (DEB) theory,31 and we can describe toxic effects on life-history traits of organisms (growth, reproduction) as changes in their energy allocation.32,33 Energy-budget models can identify where energy allocation has changed due to a stressor – the physiological mode of action - and by how much. This type of physiological information offers far more opportunities to link to the microscopic level, as well as to the population level, than descriptive summary statistics like the ECx values.15 We propose that predicting the energy-budget parameters from sub-organismal bioassays is likely far more robust and accurate than predicting ECx values because those model parameters have a clear biological interpretation.
Toxicodynamic model parameters reflect biochemistry – key to upscaling?
When analyzing toxicity data for a set of compounds or species, one can separate the information relating to toxicokinetics from that relating to toxicodynamics.34 Accounting for variation in toxicity due to toxicokinetics is necessary if we want to learn about toxicodynamics. Accounting for toxicokinetics can be achieved by approximating the internal dose based on physical-chemical properties, by measuring body burdens or cell internal concentrations, or by using calibrated toxicokinetics models. The key in all these approaches is that the driving variable for the toxicodynamic model is a proxy for the concentration at the target site. A recent study in the freshwater arthropod Gammarus pulex, and across 14 toxicants, identified patterns in toxicodynamic model parameters for the endpoint survival34 (Fig. 3). The toxicodynamic model represents the organism level, and it was an important finding that parameter values of the TD model clustered according to the chemical class of the toxicants. The toxicants covered five chemical classes (organophosphates, carbamates, baseline toxicants, uncouplers, reactive toxicants), each representing a distinct molecular initiating event and toxicity pathway at the cellular level. Finding those distinct cellular toxicity pathways reflected in organism level apical endpoint data, even if it is just for the endpoint survival, demonstrates that toxicokinetic-toxicodynamic modelling could provide the key to quantitatively link cellular toxicity pathways to organism level apical endpoints (e.g. changes in life-history traits). It means that using TK-TD models to characterize toxicity at the organismal level could improve in vitro to in vivo toxicity extrapolation and quantitative adverse outcome pathways.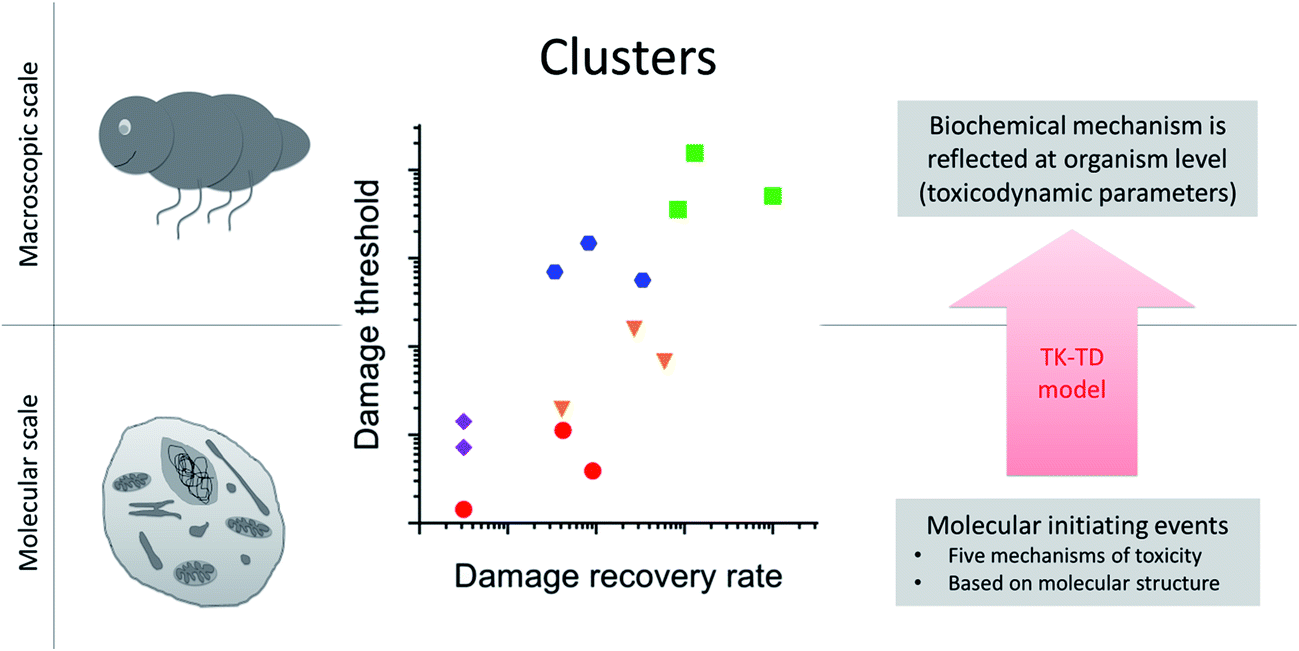 | ||
| Fig. 3 Upscaling from molecular to macroscopic scale with toxicokinetic-toxicodynamic (TK-TD) models at the organism level. Toxicodynamic model parameters cluster according to the biochemical mechanism of toxicity (adapted from Ashauer et al. 201634). In this example toxicokinetics and toxicodynamics were accounted for separately using a TK-TD model at the organism level. The toxicants were from five chemical classes (organophosphates, carbamates, baseline toxicants, uncouplers, reactive toxicants), each representing a distinct molecular initiating event and toxicity pathway at the cellular level (different symbols in plot). Finding those distinct cellular toxicity pathways reflected in organism level apical endpoint data (as clusters in the plot), even if it is just for the endpoint survival, demonstrates that biochemistry is reflected at the organism level in the values of TD model parameters. | ||
TKTD model for organism level effects on growth and reproduction
The endpoint measured in the example above was survival, and the toxicokinetic-toxicodynamic modelling used the General Unified Threshold model of Survival (GUTS).35 However, survival (i.e. mortality), although important for population dynamics, is unlikely to be the most sensitive endpoint, and in practice it is often more relevant to understand toxicant effects on sub-lethal endpoints, such as the life-history traits growth and reproduction. Hence we need a mechanism-based toxicodynamic model for growth and reproduction, and, as already explained, this requires us to focus on the energy budget. A family of TKTD models originating from DEB theory31,36 has been developed over the last decades,37 and is currently the most advanced framework for such thinking, modelling and data analysis. In these models, effects of toxicants are modelled as changes in energy allocation. The way in which a toxicant alters the dynamic energy budget is termed the physiological mode of action (pMoA).19,38 The five that have been commonly used in ecotoxicology are a decrease in assimilation, an increase in the costs for maintenance, growth or reproduction, and a direct hazard to the embryo.38Physiological mode of action in different combinations of species and toxicant
Fitting an energy-budget model to measured data on growth and reproductive output under chemical stress yields the pMoA – a descriptor that allows comparing very different biological species and toxicants. How does the pMoA vary across species and toxicants? Are there patterns? We have collected a crude overview from previously published studies in Fig. 4. The first observation is that the matrix of species vs. toxicants is very sparsely populated; clearly, there is a need for more (and more structured) experimental effort and perhaps some coordination amongst researchers. We need to populate the pMoA matrix, make it quantitative, and we need to systematically expand the species and toxicants covered. Understanding how pMoA varies across species and toxicants will not only provide the framework for better predictive ecotoxicology (e.g. read-across, QSARs) but also provides evidence to test one of the key assumptions (implicitly) underlying the adverse outcome pathway concept: the same molecular initiating event leads to the same pMoA, and thus to the same patterns of effects on life history (or ‘adverse outcome’).4 Interestingly, this important assumption, or rather hypothesis, is a matter of debate within the AOP literature with some arguing that perturbation of a single molecular initiating event can lead to different adverse outcomes.39 If AOPs are explicitly defined as being chemical agnostic, meaning that it is the molecular initiating event that triggers the adverse outcome pathway, then different chemicals that trigger the same molecular initiating event must lead to the same adverse outcome.4 This would suggest that chemicals of the same class – triggering the same molecular initiating event – should result in the same pMoA. Is this hypothesis supported by the evidence? The matrix in Fig. 4 is too sparsely populated to properly answer this question, but the little evidence therein does not provide strong support for this hypothesis. For example, the baseline toxicants should lead to the same pMoA in a given biological species. What we see in Fig. 4 is that the baseline toxicants lead to different pMoAs in D. magna, including effects on maintenance for pyridine, and direct effects on reproduction for fluoranthene, pyrene and 3,4-dichloroaniline. Firstly, it should be noted that it is very hard to distinguish between the pMoAs reproduction costs (R) and hazard to embryo (H) in practice; both only affect reproduction and do so with a different shape of the relationship between internal concentration and the reproduction rate. This means that the pMoA for 3,4-dichloroaniline might be the same as for fluoranthene and pyrene, which would only leave pyridine as the odd one out amongst the baseline toxicants in D. magna. When considering the baseline toxicants in C. elegans we find that two of them exhibit the same pMoA (G + R) whereas atrazine (M) is the outlier. But perhaps atrazine does not act via baseline toxicity in C. elegans whole life cycle tests? If this were the case, there is more hope for general patterns of pMoAs emerging.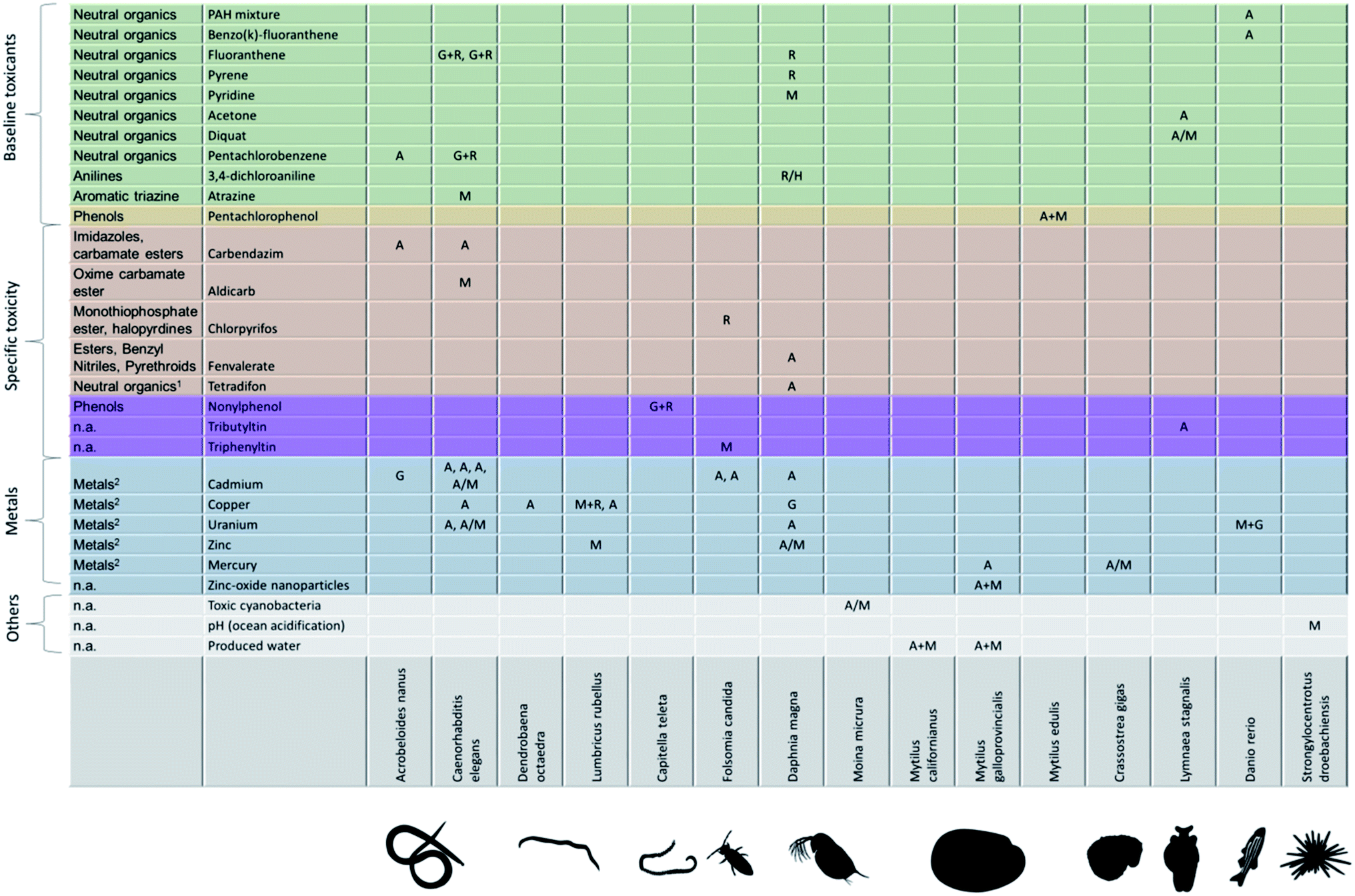 | ||
| Fig. 4 Currently known physiological modes of action across toxicants and biological species. Physiological mode of action: M = maintenance, A = assimilation, G = growth costs, R = reproduction costs, H = hazard to embryo (list of studies in ESI†). First column: ECOSAR class (Ecological Structure Activity Relationships (ECOSAR) Predictive Model v1.11, US EPA); (1): insecticide, inhibits oxidative phosphorylation; (2): metals classified by us; n.a.: not applicable. Physiological modes of action are extracted from the scientific literature: A. nanus,19C. elegans,23,48–54D. octaedra,55L. rubellus,56–58C. teleta,59F. candida,60–62D. magna,32,57,63–67M. micrura,68M. californianus,69M. galloprovincialis,57,69,70M. edilus,57C. gigas,57L. stagnalis,71–73D. rerio,57,74S. droebachiensis.75 | ||
In Fig. 4 we list the chemical class from ECOSAR (Ecological Structure Activity Relationships Predictive Model v1.11, US EPA) in the first column. This classification is based on the molecular structure and existing toxicity data. It is important to realize that the mode of toxic action classification of chemicals itself is subject to variation depending on which method is used.40
We advocate populating the pMoA matrix until we can see patterns. From those emerging patterns we can then derive new classification schemes for chemicals, which will then correspond to the chemicals' physiological mode of action. Development of new quantitative structure activity relationships will follow. In addition to populating the pMoA matrix, we also need to map AOPs onto pMoAs. In other words: for each AOP we should carry out experiments with toxicants triggering the same molecular initiating event and calibrate an energy-budget model to test how widely the assumption of AOPs being chemical agnostic holds.
AOPs and pMoAs are complementary, in our view. AOPs are detailed at the molecular/cellular level, but sketchy about the ‘adverse outcome’, i.e., the effects on the life-history traits of the organism. The pMoAs (and the associated energy-budget model) provide a direct link to growth, development and reproduction, over the entire life cycle of the organism, and thereby a direct connection to higher levels of biological organization. However, the pMoAs are extremely sketchy at the sub-individual level as they consider rather abstract, lumped, energy fluxes such as assimilation and maintenance. We propose to populate the species & chemicals matrix to test the hypothesis that similarly acting chemicals will result in the same pMoA. The outcome of this exercise is totally open. It is also conceivable that one molecular initiating event will influence several energetic fluxes, and the chemical will thus have a pMoA that is made up of multiple energy fluxes. The AOP framework has not yet resulted in quantitative models that are general enough to test the above hypothesis.
Towards better in vitro to in vivo toxicity extrapolation
We suggest a research program with the aim to establish quantitative models that predict ecologically-relevant, sub-lethal, organism-level endpoints (i.e., life-history traits) based on input from in vitro assays. In order to anchor such an in vitro to in vivo toxicity extrapolation (IVIVE), we first need to select a suitable mathematical TD model for the organism. We think that ‘DEBtox’ models are currently the most suitable candidates. Here, we define DEBtox broadly as a TKTD model that includes some form of DEB model to link a toxicokinetic model to life-history traits over ontogeny.37 DEBtox models offer very simple rules to model toxicant effects, so that only one or two model parameters are affected by the toxicant (the pMoA). We need to find in vitro bioassays that are predictive for the pMoA as well as for the actual value of the model parameters governing the toxic effect (Fig. 5). It helps that the DEBtox model parameters that we want to predict with bioassays are time independent and have a biological interpretation, which can guide bioassay selection. The research programme leading to improved IVIVE comprises calibrating DEBtox for a large number of toxicants, and then establishing bioassays that show a strong correlation with the values of the model parameters (or the stress factors on the affected energetic process) across the toxicants. Once that quantitative link is established, the bioassays can be used to predict DEBtox model parameters, which in turn can be used for predicting toxicity in various prioritization, hazard or risk assessment schemes.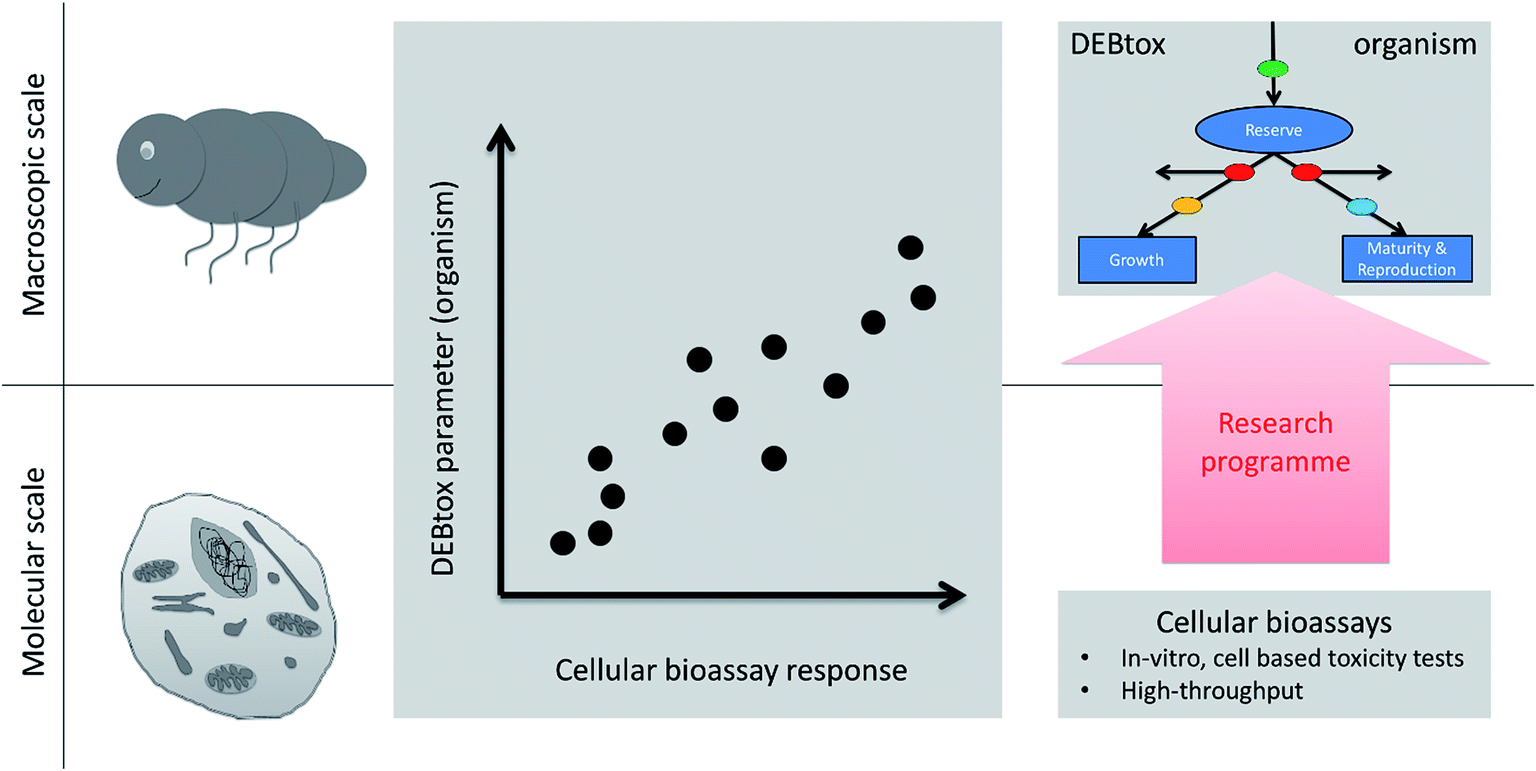 | ||
| Fig. 5 Upscaling from molecular to macroscopic scale with DEBtox. As DEBtox is an organism model, its parameters have a biological interpretation and we can develop bioassays that predict changes in those DEBtox parameters. That combination is a framework for high-throughput ecotoxicity testing which would account for reduced growth or reproduction due to toxic chemicals changing energy budgets. There must be a link between cellular toxicity and changes in energy budgets – the trick is to find it. Hence we propose a research programme to that end. | ||
We view the proposed research programme into pMoAs as complementary to the development of AOPs and AOP-based quantitative models. We can simply view the pMoA in DEBtox terminology as the ‘adverse outcome’ in AOP terminology. What is missing in the AOP concept, is the explicit use of a dynamic biological model to describe the organism and the physiological context within which an adverse outcome manifests itself. DEBtox models can fill this gap. Note however, that different AOPs can map onto the same pMoA. For example we can imagine many pathways by which a toxicant can affect the assimilation process, or each of the other pMoAs.
The first key challenge for IVIVE is that for most combinations of toxicant and biological species we do not know the pMoA yet (Fig. 4). The second is that it can be difficult to identify the pMoA from, typically noisy, experimental data. The third challenge is that DEBtox parameterization requires rather extensive animal testing with growth and reproductive output measured over time for a good part of the life cycle, and with sufficiently strong chemical effects.
At the modelling side, there are several practical issues that need to be addressed. Firstly, DEBtox is not a single model but rather a family of closely-related models.37 It is highly unlikely that different DEBtox models will identify a different pMoA, but the quantitative comparison of TD parameter values is best served by selecting a single model for all analyses. Further aspects that require more research are the identification of the relevant dose metric at the target site (e.g., the membrane concentration for baseline toxicants) and the quantitative link between the level of target occupation and the associated energy flux in the DEBtox model. Currently, most applications have relied on the linear-with-threshold relationships as presented by Kooijman and Bedaux.41 However, there are no strong theoretical reasons to dismiss other possible relationships. Further complications arise because we cannot observe the energy fluxes, and thus the pMoAs, themselves. They are derived from observations on growth and reproduction over time, and linked to the underlying fluxes with auxiliary assumptions. This may hamper the identification of patterns in pMoAs across species and chemicals and constitutes one more reason why we need high-throughput testing with whole organisms (see next section below).
Many animals require some modification of the DEB model to fit their life cycles; biological reality is often more complex than can be captured by the simplicity of generic models. This requires more parameters to be fitted, and thus more extensive testing efforts. However, this only needs to be done once in detail for each species to build the DEB model. After that, the DEB part of the TD model remains the same, and only the toxicological part needs to be calibrated for each toxicant. In general, partial life-cycle testing suffices to identify the pMoA and to fit the toxicological parameters, following growth and reproduction over a substantial part of the life cycle (starting with juveniles, and continuing until a number of reproduction events has been observed). An often-overlooked aspect in such tests is that toxicants may affect the investment per offspring. These differences are not only extremely relevant to identify and quantify the pMoA in the energy-budget context, it is also essential for an accurate prediction of population-level effects.16 From a practical perspective, it will be important to start with species that have very little variation between individuals (e.g., clones), and that do not require a sexual partner (e.g., parthenogenetic species). To prove the concept, and to allow high-throughput testing, we would suggest starting with a small animal species that has a fast life cycle, such as some daphnids, rotifers, nematodes or even protozoans. Once such a proof-of-concept is firmly established, and patterns in pMoA's and model parameters identified, subsequent testing with other animal species can likely be more focused.
Looking forward
Ecotoxicology and environmental risk assessment of chemicals will benefit from the shift towards using the biologically meaningful models at the individual level that we advocate. Dose-response modelling, and the use of simple summary statistics (LC50s, ECx values, NOECS, etc.), carries too many limitations.29 Replacing traditional dose-response models with data analysis using DEBtox or other biologically-meaningful models will have a positive impact on several aspects of ecotoxicology (Table 2). The development of efficient quantitative structure-activity relationships, in vivo to in vitro toxicity extrapolation, and high-throughput testing will all benefit from using biologically meaningful model parameters as descriptors of toxicity instead of traditional summary statistics. However, new types of databases, populated with biologically meaningful model parameters for different combinations of species and compound, are needed to support this paradigm shift.| Which aspect of ecotoxicology? | Traditional workflow | Proposed new workflow |
|---|---|---|
| Toxicity test design and data analysis | Fitting of sigmoidal dose-response curve and extraction of summary statistic (e.g. EC50, ECx or NOEC values) for one observation time point (usually at the end of the test) and each endpoint separately. Loss of information | Fitting of toxicokinetic-toxicodynamic models (e.g. DEBtox) to time-series of multiple, physiologically related endpoints (e.g. growth & reproduction) observed throughout the toxicity test. Extraction of biologically meaningful model parameter values |
| Entries in ecotoxicology databases | Summary statistics such as EC50, ECx, NOEC, or similar values | Values of TKTD model parameters and raw data |
| Use in chemical risk assessment | Comparison with environmental concentrations | Simulate effects using environmental concentration time series as input |
| Development of new quantitative structure activity relationships (QSARs) | Benchmarking against summary statistics (EC50, ECx or NOEC values, etc.) limits progress due to lost information and lumping of toxicokinetics (related to physical-chemical properties) with toxicodynamics (related to toxicophore and toxic mechanism) | Aiming to predict TKTD model parameters is more likely to succeed because they have biological meaning and capture more information. Toxicokinetic and toxicodynamic parameters can be predicted separately |
| Development of in vitro to in vivo toxicity extrapolation (IVIVE) and high throughput testing (HTT) methods | Benchmarking against summary statistics (EC50, ECx or NOEC values, etc.) limits progress due to lost information and lumping of toxicokinetics (related to physical-chemical properties) with toxicodynamics (related to toxicophore and toxic mechanism) | Aiming to predict TKTD model parameters is more likely to succeed because they have biological meaning and capture more information. Toxicokinetics and toxicodynamics can be separated. In vitro assays can be developed to predict toxicodynamic parameters |
There is a large gap in our theory and modelling capability when it comes to linking across scales of biological organization, but there must be a link between what happens at the cellular level and what happens on the energy budget, the tricky part is to find it. To fill this gap, the bottleneck might, surprisingly, not be a lack of detailed molecular understanding, but rather the effort required to generate suitable data on whole organism life history traits under chemical stress. Technological innovation is needed to achieve the large number of toxicity tests that we envision. There are already methods, often based on image analysis, to speed up ecotoxicity testing.42–45 As far as we know none of those have led to the high-throughput organism level data generation capability that we need, but it might be only one step away (e.g. C. elegans growth assay46).
Generally, we need to make greater efforts to lay open, discuss and research the validity of assumptions of models in ecotoxicology. Models are simply tools to deduce quantitative conclusions from a set of assumptions and data, nothing more. It is good to remember that most ecotoxicity models fall into the category of ‘phenomenological’ models,47 although DEB models at least consider some fundamental physics, such as the inclusion of the factor ‘time’ and adherence to the laws for energy and mass conservation. But perhaps it would be misleading to require that models in biology must be based on fundamental physics. Or, to use Jeremy Gunawardena's words:47 “Keep it simple. Including all the biochemical details may reassure biologists but it is a poor way to model.” It is more important that models are fit for purpose and that means finding the right level of abstraction for the question asked. In ecotoxicology, the individual organism response is what connects the research programs aiming at high-throughput testing and those efforts aimed at increasing ecological realism and relevance, including multiple stressors. Hence it appears that a useful level of abstraction for ecotoxicology models and theory is the individual organism and its energy budget – and that means we need to study physiological modes of action.
Conflicts of Interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by CEFIC-LRI (LRI-ECO39-YORK: Review, ring-test and guidance for TKTD modelling) and the European Union under the 7th Framework Programme (project acronym TOXKINBIO, contract #PCIG14-GA-2013-630706). Animal silhouettes in Fig. 4 originate from http://phylopic.org/ under creative commons 3.0 licenses (CC-BY 3.0, CC-BY-SA 3.0, CC BY-NC-SA 3.0) and public domain licenses mark 1.0 and CC0 1.0.References
- A. J. Hendriks, Environ. Sci. Technol., 2013, 47, 3546–3547 CrossRef CAS PubMed.
- G. Ankley, R. S. Bennet, R. J. Erickson, D. J. Hoff, M. W. Hornung, R. D. Johnson, D. R. Mount, J. W. Nichols, C. L. Russom, P. K. Schmieder, J. A. Serrano, J. E. Tietge and D. L. Villeneuve, Environ. Toxicol. Chem., 2010, 29, 730–741 CrossRef CAS PubMed.
- R. Huang, M. Xia, S. Sakamuru, J. Zhao, S. A. Shahane, M. Attene-Ramos, T. Zhao, C. P. Austin and A. Simeonov, Nat. Commun., 2016, 7, 10425 CrossRef CAS PubMed.
- K. J. Groh, R. N. Carvalho, J. K. Chipman, N. D. Denslow, M. Halder, C. A. Murphy, D. Roelofs, A. Rolaki, K. Schirmer and K. H. Watanabe, Chemosphere, 2015, 120, 764–777 CrossRef CAS PubMed.
- V. E. Forbes and N. Galic, Environ. Int., 2016, 91, 215–219 CrossRef CAS PubMed.
- Q. Zhang, S. Bhattacharya, M. E. Andersen and R. B. Conolly, J. Toxicol. Environ. Health, Part B, 2010, 13, 253–276 CAS.
- L. Gunnarsson, A. Jauhiainen, E. Kristianssn, O. Nerman and D. G. J. Larsson, Environ. Sci. Technol., 2008, 42, 5807–5813 CrossRef CAS PubMed.
- C. A. LaLone, D. L. Villeneuve, L. D. Burgoon, C. L. Russom, H. W. Helgen, J. P. Berninger, J. E. Tietge, M. N. Severson, J. E. Cavallin and G. T. Ankley, Aquat. Toxicol., 2013, 144–145, 141–154 CrossRef CAS PubMed.
- P. J. Van den Brink, Environ. Sci. Technol., 2008, 42, 8999–9004 CrossRef CAS PubMed.
- R. Relyea and J. Hoverman, Ecol. Lett., 2006, 9, 1157–1171 CrossRef PubMed.
- SCENIHR (Scientific Committee on Emerging and Newly Identified Health Risks), SCHER (Scientific Committee on Health and Environmental Risks) and SCCS (Scientific Committee on Consumer Safety), Addressing the New Challenges for Risk Assessment, European Commission, Brussels, 2012 Search PubMed.
- R. B. Schäfer, B. Kühn, E. Malaj, A. König and R. Gergs, Freshwater Biol., 2016, 61, 2116–2128 CrossRef.
- S. Stehle and R. Schulz, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(18), 5750–5755 CrossRef CAS PubMed.
- M. N. Rubach, R. Ashauer, D. B. Buchwalter, H. J. de Lange, M. Hamer, T. G. Preuss, K. Töpke and S. J. Maund, Integr. Environ. Assess. Manage., 2011, 7, 172–186 CrossRef PubMed.
- T. Jager, J. Toxicol. Environ. Health, Part A, 2016, 79, 572–584 CrossRef CAS PubMed.
- B. Martin, T. Jager, R. M. Nisbet, T. G. Preuss and V. Grimm, Ecol. Appl., 2014, 24, 1972–1983 Search PubMed.
- J. R. Rohr, J. L. Kerby and A. Sih, Trends Ecol. Evol., 2006, 21, 606–613 CrossRef PubMed.
- F. De Laender, J. R. Rohr, R. Ashauer, D. J. Baird, U. Berger, N. Eisenhauer, V. Grimm, U. Hommen, L. Maltby, C. J. Meliàn, F. Pomati, I. Roessink, V. Radchuk and P. J. Van den Brink, Trends Ecol. Evol., 2016, 31, 905–915 CrossRef PubMed.
- O. Alda Álvarez, T. Jager, E. M. Redondo and J. E. Kammenga, Environ. Toxicol. Chem., 2006, 25, 3230–3237 CrossRef.
- S. Bhattacharya, Q. Zhang, P. L. Carmichael, K. Boekelheide and M. E. Andersen, PLoS One, 2011, 6, e20887 CAS.
- B. I. Escher and J. L. M. Hermens, Environ. Sci. Technol., 2002, 36, 4201–4217 CrossRef CAS PubMed.
- L. H. Heckmann, R. M. Sibly, R. Connon, H. L. Hooper, T. H. Hutchinson, S. J. Maund, C. J. Hill, A. Bouetard and A. Callaghan, Genome Biol., 2008, 9, 14 CrossRef PubMed.
- S. Swain, J. F. Wren, S. R. Stürzenbaum, P. Kille, A. J. Morgan, T. Jager, M. J. Jonker, P. K. Hankard, C. Svendsen, J. Owen, B. A. Hedley, M. Blaxter and D. J. Spurgeon, BMC Syst. Biol., 2010, 4, 32 Search PubMed.
- A. Hines, F. J. Staff, J. Widdows, R. M. Compton, F. Falciani and M. R. Viant, Toxicol. Sci., 2010, 115, 369–378 CrossRef CAS PubMed.
- J. Stadnicka-Michalak, K. Schirmer and R. Ashauer, Sci. Adv., 2015, 1, 1–8 Search PubMed.
- C. Wittwehr, H. Aladjov, G. Ankley, H. J. Byrne, J. de Knecht, E. Heinzle, G. Klambauer, B. Landesmann, M. Luijten, C. MacKay, G. Maxwell, M. E. Meek, A. Paini, E. Perkins, T. Sobanski, D. Villeneuve, K. M. Waters and M. Whelan, Toxicol. Sci., 2016, 155, 326–336 CrossRef PubMed.
- K. H. Watanabe, Z. Li, K. J. Kroll, D. L. Villeneuve, N. Garcia-Reyero, E. F. Orlando, M. S. Sepulveda, T. W. Collette, D. R. Ekman, G. T. Ankley and N. D. Denslow, Toxicol. Sci., 2009, 109, 180–192 CrossRef CAS PubMed.
- Z. Li, K. Kroll, K. Jensen, D. Villeneuve, G. Ankley, J. Brian, M. Sepulveda, E. Orlando, J. Lazorchak, M. Kostich, B. Armstrong, N. Denslow and K. Watanabe, BMC Syst. Biol., 2011, 5, 63 CrossRef PubMed.
- T. Jager, Environ. Sci. Technol., 2011, 45, 8180–8181 CrossRef CAS PubMed.
- R. Laskowski, Oikos, 1995, 73, 140–144 CrossRef.
- T. Sousa, T. Domingos, J. C. Poggiale and S. A. L. M. Kooijman, Philos. Trans. R. Soc., B, 2010, 365, 3413–3428 CrossRef PubMed.
- T. Jager, E. H. W. Heugens and S. A. L. M. Kooijman, Ecotoxicology, 2006, 15, 305–314 CrossRef CAS PubMed.
- T. Jager, Making sense of chemical stress, Application of Dynamic Energy Budget theory in ecotoxicology and stress ecology, Leanpub, Victoria, Canada, 2015, https://leanpub.com/debtox_book Search PubMed.
- R. Ashauer, I. O'Connor, A. Hintermeister and B. I. Escher, Environ. Sci. Technol., 2015, 49, 10136–10146 CrossRef CAS PubMed.
- T. Jager, C. Albert, T. G. Preuss and R. Ashauer, Environ. Sci. Technol., 2011, 45, 2529–2540 CrossRef CAS PubMed.
- R. M. Nisbet, E. B. Muller, K. Lika and S. A. L. M. Kooijman, J. Anim. Ecol., 2000, 69, 913–926 CrossRef.
- T. Jager, A. Barsi, N. T. Hamda, B. T. Martin, E. I. Zimmer and V. Ducrot, Ecol. Model., 2014, 280, 140–147 CrossRef.
- T. Jager and E. I. Zimmer, Ecol. Model., 2012, 225, 74–81 CrossRef CAS.
- D. L. Villeneuve, D. Crump, N. Garcia-Reyero, M. Hecker, T. H. Hutchinson, C. A. LaLone, B. Landesmann, T. Lettieri, S. Munn, M. Nepelska, M. A. Ottinger, L. Vergauwen and M. Whelan, Toxicol. Sci., 2014, 142, 312–320 CrossRef CAS PubMed.
- A. Kienzler, M. G. Barron, S. E. Belanger, A. Beasley and M. R. Embry, Environ. Sci. Technol., 2017, 51, 10203–10211 CrossRef CAS PubMed.
- S. A. L. M. Kooijman and J. J. M. Bedaux, Water Res., 1996, 30, 1711–1723 CrossRef CAS.
- S. K. Jung, X. Qu, B. Aleman-Meza, T. Wang, C. Riepe, Z. Liu, Q. Li and W. Zhong, Environ. Sci. Technol., 2015, 49, 2477–2485 CrossRef CAS PubMed.
- O. Bánszegi, A. Kosztolányi, G. Bakonyi, B. Szabó and M. Dombos, PLoS One, 2014, 9(6), e98230 Search PubMed.
- A. Agatz, M. Hammers-Wirtz, A. Gergs, T. Mayer and T. Preuss, Ecotoxicology, 2015, 24, 1385–1394 CrossRef CAS PubMed.
- F. Mallard, V. Le Bourlot and T. Tully, PLoS One, 2013, 8(5), e64387 CAS.
- W. A. Boyd, M. V. Smith, C. A. Co, J. R. Pirone, J. R. Rice, K. R. Shockley and J. H. Freedman, Environ. Health Perspect., 2016, 124, 586–593 Search PubMed.
- J. Gunawardena, BMC Biol., 2014, 12(29) DOI:10.1186/1741-7007-12-29.
- J. F. Wren, P. Kille, D. J. Spurgeon, S. Swain, S. R. Sturzenbaum and T. Jager, Ecotoxicology, 2011, 20, 397–408 CrossRef CAS PubMed.
- O. Alda ÁLvarez, T. Jager, S. A. L. M. Kooijman and J. E. Kammenga, Funct. Ecol., 2005, 19, 656–664 CrossRef.
- T. Jager, E. M. Gudmundsdóttir and N. Cedergreen, Environ. Sci. Technol., 2014, 48, 7026–7033 CrossRef CAS PubMed.
- A. Margerit, E. Gomez and R. Gilbin, Chemosphere, 2016, 146, 405–412 CrossRef CAS PubMed.
- O. Alda Álvarez, T. Jager, B. N. Colao and J. E. Kammenga, Environ. Sci. Technol., 2006, 40, 2478–2484 CrossRef.
- N. Cedergreen, N. J. Nørhave, C. Svendsen and D. J. Spurgeon, PLoS One, 2016, 11, e0140277 Search PubMed.
- B. Goussen, R. Beaudouin, M. Dutilleul, A. Buisset-Goussen, J.-M. Bonzom and A. R. R. Péry, Chemosphere, 2015, 120, 507–514 CrossRef CAS PubMed.
- T. Jager and C. Klok, Philos. Trans. R. Soc., B, 2010, 365, 3531–3540 CrossRef PubMed.
- C. Klok and A. M. de Roos, Ecotoxicol. Environ. Saf., 1996, 33, 118–127 CrossRef CAS PubMed.
- E. B. Muller, R. M. Nisbet and H. A. Berkley, Ecotoxicology, 2010, 19, 48–60 CrossRef CAS PubMed.
- C. Klok, Environ. Sci. Technol., 2008, 42, 8803–8808 CrossRef CAS PubMed.
- T. Jager and H. Selck, J. Sea Res., 2011, 66, 456–462 CrossRef CAS.
- T. Jager, T. Crommentuijn, C. A. M. Van Gestel and S. A. L. M. Kooijman, Environ. Sci. Technol., 2004, 38, 2894–2900 CrossRef CAS PubMed.
- N. T. Hamda, PhD thesis, Vrije Universiteit Amsterdam, 2013.
- T. Jager, T. Crommentuijn, C. A. M. van Gestel and S. Kooijman, Environ. Pollut., 2007, 145, 452–458 CrossRef CAS PubMed.
- B. T. Martin, T. Jager, R. M. Nisbet, T. G. Preuss, M. Hammers-Wirtz and V. Grimm, Ecotoxicology, 2013, 22, 574–583 CrossRef CAS PubMed.
- E. Billoir, M. L. Delignette-Muller, A. R. R. Péry and S. Charles, Environ. Sci. Technol., 2008, 42, 8978–8984 CrossRef CAS PubMed.
- B. J. Pieters, T. Jager, M. H. Kraak and W. Admiraal, Ecotoxicology, 2006, 15, 601–608 CrossRef CAS PubMed.
- T. Jager, T. Vandenbrouck, J. Baas, W. M. De Coen and S. A. L. M. Kooijman, Ecotoxicology, 2010, 19, 351–361 CrossRef CAS PubMed.
- S. Massarin, R. Beaudouin, F. Zeman, M. Floriani, R. Gilbin, F. Alonzo and A. R. R. Pery, Environ. Sci. Technol., 2011, 45, 4151–4158 CrossRef CAS PubMed.
- E. Billoir, A. da Silva Ferrão-Filho, M. Laure Delignette-Muller and S. Charles, J. Theor. Biol., 2009, 258, 380–388 CrossRef PubMed.
- E. B. Muller, C. W. Osenberg, R. J. Schmitt, S. J. Holbrook and R. M. Nisbet, Ecotoxicology, 2010, 19, 38–47 CrossRef CAS PubMed.
- E. B. Muller, S. K. Hanna, H. S. Lenihan, R. J. Miller and R. M. Nisbet, J. Sea Res., 2014, 94, 29–36 CrossRef.
- A. Barsi, T. Jager, M. Collinet, L. Lagadic and V. Ducrot, Environ. Toxicol. Chem., 2014, 33, 1466–1475 CrossRef CAS PubMed.
- E. Zimmer, PhD thesis, Vrije Universiteit Amsterdam, 2013.
- A. Barsi, PhD thesis, Vrije Universiteit Amsterdam, 2015.
- S. Augustine, B. Gagnaire, C. Adam-Guillermin and S. A. L. M. Kooijman, Aquat. Toxicol., 2012, 118, 9–26 CrossRef PubMed.
- T. Jager, E. Ravagnan and S. Dupont, J. Exp. Mar. Biol. Ecol., 2016, 474, 11–17 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The data used in Fig. 4 is available as supporting information (MS EXCEL file), including links to the original studies. See DOI: 10.1039/c7em00328e |
| This journal is © The Royal Society of Chemistry 2018 |