
Dehydrocoupling of dimethylamine–borane promoted by manganese(II) m-terphenyl complexes†
Helen R.
Sharpe
 a,
Ana M.
Geer
a,
Toby J.
Blundell
a,
Fiona R.
Hastings
a,
Michael W.
Fay
b,
Graham A.
Rance
b,
William
Lewis
a,
Alexander J.
Blake
a and
Deborah L.
Kays
*a
a,
Ana M.
Geer
a,
Toby J.
Blundell
a,
Fiona R.
Hastings
a,
Michael W.
Fay
b,
Graham A.
Rance
b,
William
Lewis
a,
Alexander J.
Blake
a and
Deborah L.
Kays
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Deborah.Kays@nottingham.ac.uk
bNanoscale and Microscale Research Centre, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 20th November 2017
Abstract
Two- and three-coordinate manganese m-terphenyl complexes are precatalysts for the dehydrogenation of dimethylamine–borane (Me2NH·BH3) affording one equivalent of molecular hydrogen and half an equivalent of [Me2N–BH2]2. Experimental studies into the nature of the catalyst indicate that small changes in the coordination environment give rise to significant differences in the reaction mechanism, occurring through a homogeneous mechanism for two-coordinate precatalysts, whilst for the three-coordinate species a heterogeneous mechanism takes place where nanoparticles are responsible for the catalysis.
Introduction
The catalytic dehydrocoupling of amine–boranes has gained considerable significance in the last decade because of their potential application as hydrogen storage materials.1 These molecules contain a high weight percentage of hydrogen, which, in conjunction with the electronegativity difference between nitrogen and boron (inducing protic N–H and hydridic B–H bonds), permits the release of hydrogen at high temperatures or mediated by a catalyst. Furthermore, amine–boranes have applications as hydrogen transfer reagents2 and precursors to BN-based ceramics and polymeric materials.3 Most of the catalysts for this transformation contain precious metals, which are expensive, scarce and toxic.4 Recently, abundant first row transition elements have been used as catalysts for dehydrogenation reactions, although many of these require photoirradiation in order to initiate the reaction. There has been a special interest in iron precatalysts;5 in particular the use of carbonyl species, [CpFe(CO)2]2 and CpFe(CO)2I (Cp = η5-C5H5), where structural variations in the precatalysts result in a homogeneous or heterogeneous reaction mechanism.5g A limited number of cobalt precatalysts have also been reported to dehydrocouple amine–boranes; these include Cp*Co(CO)I2 (Cp* = η5-C5Me5), which is active under aerobic conditions.6 Manganese catalysts are underutilised for dehydrogenation reactions. CpMn(CO)3 catalyses amine–borane dehydrocoupling (Fig. 1),7 although the reaction requires photoactivation and long reaction times. | ||
| Fig. 1 Previous examples of manganese complexes used for amine–borane dehydrogenation and m-terphenyl manganese complexes 1–3 (Xyl = 2,6-Me2C6H3, Mes = 2,4,6-Me3C6H2, Tmp = 2,4,5-Me3C6H2) used in this work. | ||
More recently, an N-heterocyclic phosphenium manganese catalyst (Fig. 1) has been shown to be active for ammonia borane dehydrogenation, with substrate activation through an unusual ligand-centred pathway, where the substrate transfers hydrogen to the phosphorus atom and one nitrogen atom of the phosphenium ligand.8 There is an increasing interest in catalysts based on manganese due to its high natural abundance and biocompatibility.9 In this context, we have recently disclosed manganese(II) m-terphenyl complexes as effective catalysts for the cyclotrimerisation of aliphatic isocyanates under mild conditions, operating through a Lewis acid mechanism.10 Herein, we report the dehydrogenation/dehydrocoupling of dimethylamine–borane (Me2NH·BH3) catalysed by two- and three-coordinate manganese(II) m-terphenyl complexes (1–3) under mild conditions (Scheme 1). Small changes in the metal coordination sphere have considerable impact on the reaction, which can operate either through a homogeneous or heterogeneous mechanism.
 | ||
| Scheme 1 Dehydrocoupling of Me2NH·BH3 using manganese(II) m-terphenyl precatalysts 1–3. | ||
Results and discussion
The two-coordinate complex (2,6-Xyl2C6H3)2Mn (1) was synthesised via the reaction of MnBr2 with one equivalent of [2,6-Xyl2C6H3Li]2 (Xyl = 2,6-Me2C6H3) in a mixture of toluene and THF with concomitant formation of the lithium halide. Crystallisation was achieved by slow cooling of saturated hexane (Fig. 2) obtaining single crystals suitable for X-ray diffraction; the Mn–C bond lengths and C–Mn–C angle for 1 [Mn(1)–C(1) = 2.089(3) Å, Mn(1)–C(23) = 2.087(3) Å and C(1)–Mn(1)–C(23) 169.57(15)°] are within the ranges of other two-coordinate manganese terphenyl complexes in literature.11 The synthesis of (2,6-Mes2C6H3)2Mn (2; Mes = 2,4,6-Me3C6H2) and (2,6-Tmp2C6H3)2Mn(THF) (3, Tmp = 2,4,5-Me3C6H2) were achieved by literature procedures.10,11b In order to compare the steric impact of the m-terphenyl ligands in these complexes, the percent buried volume (%VBur) and steric maps were calculated from the solid state structure (Fig. S1, ESI†).12 Varying the flanking group from 2,6-Xyl to 2,6-Mes has negligible impact on the %VBur of the ligands on 1 and 2 (42.0 and 42.7, respectively). However, for complex 3, the %VBur of the m-terphenyl ligand dramatically decreases to 37.5, indicating a significant reduction in the steric shielding around the metal centre. In particular, the ortho substituents on the outer aromatic ring provide greater steric coverage to the metal centre in 1 and 2. | ||
| Fig. 2 Molecular structure of (2,6-Xyl2C6H3)2Mn (1) with anisotropic displacement parameters set at 50% probability. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) for 1: Mn(1)–C(1) 2.089(3), Mn(1)–C(23) 2.087(3), C(1)–Mn(1)–C(23) 169.57(15), Ar–Ar dihedral angle 93.91(8). | ||
Initial dehydrocoupling experiments between 1–3 (5 mol%) and Me2NH·BH3 were performed in C6D6 at room temperature (rt); the appearance of dihydrogen bubbles in the NMR tube suggesting an immediate reaction. However, under these reaction conditions all three precatalysts gave poor conversion to products (entries 1–3, Table 1). Analysis of these reactions by 1H and 11B NMR spectroscopy after 16 h revealed conversions of 15–38%, where the major product Me2NH–BH2–NMe2–BH3 (4) was assigned by 1H and 11B NMR spectroscopy [δB 2.3 (t, JBH = 109 Hz, BH2), −13.1, (q, JBH = 97 Hz, BH3)]. In the 11B NMR spectrum, the BH3 moiety was concealed by the precursor Me2NH·BH3 (Fig. S10, ESI†).13 This linear diborazane (4) is a key intermediate in the formation of the cyclic dimer [Me2N–BH2]2 (5) for catalytic systems including Ti,13 Fe,5g Sc,14 Ir15 and Rh.16 Increasing the temperature to 60 °C improved activity and selectivity for all three precatalysts (entries 4–6, Table 1). Precatalysts 1, 2 and 3 completely converted Me2NH·BH3 into 5 in ca. 2 h, 4 h and 12 h, respectively. The higher proportion of 5 to the linear species 4 in the reactions performed at 60 °C compared with those at room temperature is in agreement with the latter being an intermediate to the cyclic dimer, as previously described by Manners et al.5g Precatalysts 1 and 3 display high conversions when lowering the catalyst loading to 2 mol%, whilst for 2 the conversion drops to 75% and is accompanied by a larger amount of 4 (entries 7–9, Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | T (°C) | t (h) | Conversionb (%) | Product ratioc,d4/5/6e |
a Reaction conditions: 10 mg of catalyst, 0.6 mL of C6D6.
b Determined by 1H NMR and 11B NMR spectroscopy.
c Ratio by 1H NMR and 11B NMR.
d Small amounts of HB(NMe2)2 (<3%) were also detected by 11B NMR.
e Me2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2 (6).
f Samples were heated at 60 °C in an oil bath, after 16 h the progress was monitored by NMR. BH2 (6).
f Samples were heated at 60 °C in an oil bath, after 16 h the progress was monitored by NMR.
|
|||||
| 1 | 1 (5) | rt | 16f | 15 | 75/24/0 |
| 2 | 2 (5) | rt | 16f | 33 | 84/10/3 |
| 3 | 3 (5) | rt | 16f | 38 | 69/28/1 |
| 4 | 1 (5) | 60 | 1.7 | 94 | 0/97/2 |
| 5 | 2 (5) | 60 | 3.8 | 96 | 1/95/1 |
| 6 | 3 (5) | 60 | 12 | 96 | 2/94/2 |
| 7 | 1 (2) | 60 | 16f | 94 | 1/98/1 |
| 8 | 2 (2) | 60 | 16f | 75 | 4/95/1 |
| 9 | 3 (2) | 60 | 16f | 99 | 0/99/0 |

To gain further insight into the dehydrocoupling of Me2NH·BH3 using precatalysts 1–3 the progress of the reactions was monitored by 1H NMR and 11B NMR spectroscopy in C6D6 at 60 °C (Fig. 3 and ESI†) and by measuring the amount of H2 formed with a gas burette (Fig. 4); no induction period was observed when undertaking these experiments. Small quantities (ca. 2%) of the diaminoborane HB(NMe2)2 and the monomeric aminoborane Me2NBH2 (6) were observed in these reactions. Significantly, monitoring the reaction of Me2NH·BH3 with precatalyst 3 (5 mol%, C6D6, 60 °C) by NMR provided mechanistic insight. Free THF is observed by 1H NMR upon addition of Me2NH·BH3, indicating that the first step of the reaction is likely amine–borane coordination to the metal centre displacing labile THF. Therefore, differences in the reaction mechanisms are likely due to the changes in the m-terphenyl ligands. Although ammonia has been shown to cleave a M–C bond in (2,6-Dipp2C6H3)2Mn (Dipp = 2,6-iPr2C6H3),17 protolytic cleavage of a metal-bound m-terphenyl ligand as part of the catalytic cycle has been discounted due to the lack of evidence of the formation of protonated ligand during the catalysis via1H NMR. It is conceivable that the Mn(II) centre will behave as a Lewis acid in the homogeneous reaction, similar to that seen for other low-coordinate transition metal aryl precatalysts,10,18 with initial coordination of Me2NH·BH3 to the metal centre though interaction of the BH3 unit, similar to that for manganese half-sandwich complexes,7 and catalysis occurring via a mechanism reminiscent of that for Manners' Fe(II)-catalysed system.5g
 | ||
| Fig. 3 (a) Conversion (%) vs. time (h) for the dehydrogenation of Me2NH·BH3 using 1 (5 mol%) at 60 °C. (b) Conversion (%) vs. time (h) for the dehydrogenation of Me2NH·BH3 using 3 (5 mol%) at 60 °C. In both cases 6 and HB(NMe2)2 were observed in the 11B NMR spectra in small quantities (<2%). | ||
 | ||
| Fig. 4 H2 equivalents (molH2 per molMe2NH·BH3) vs. time (h) for the dehydrogenation of Me2NH·BH3 using 2 (7 mol%) in toluene at 60 °C. | ||
Recycling studies were carried out with catalyst 2 (7 mol%, toluene, 60 °C), by addition of three consecutive loadings of Me2NH·BH3 after the catalysis (Fig. 4). Comparable conversions are obtained on the second run, but the third run shows a significant decrease in the catalytic activity.19 Although no colour change is observed the decrease in catalyst activity indicates decomposition of the air and moisture sensitive manganese complexes; other likely contributions are dilution effects as successive equivalents of Me2NH·BH3 substrate are added (solution in 1 mL of toluene), along with build-up of by-products.
It is important to note that whilst no colour change is observed upon the addition of Me2NH·BH3 to solutions of precatalysts 1 or 2, the reaction mixture instantly turns from a colourless solution to a dark suspension when Me2NH·BH3 is added to 3. This observation provides a strong indication of the formation of manganese nanoparticles (NPs), a heterogeneous catalyst for the dehydrocoupling reaction involving precatalyst 3. The absence of an induction period could indicate that the NPs are formed immediately upon addition of the amine–borane; no induction period was similarly reported by Morris et al. in the dehydrogenation of ammonia–borane by iron NPs.20 In order to assess whether the catalysts are homogeneous or heterogeneous the mercury drop test was performed for 1–3 with 5 mol% catalyst loading at 60 °C (entries 1–3, Table 2).21 The reactions with precatalysts 1 and 2 in the presence of mercury reached similar conversions to those without after 2 and 4 hours, respectively, indicative of a homogeneous catalyst. Moreover, the presence of an excess of cumene, which can act as a radical trap,18,22 has no effect on the reaction rate and conversion when using precatalysts 1 and 2 indicating that these reactions are not radical-mediated. However, when precatalyst 3 is used in the presence of mercury the conversion from Me2NH·BH3 to products is considerably lower (Table 2), suggesting that the active catalyst is heterogeneous.20,23 Amine–boranes have been used as effective reducing agents in the chemical synthesis of NPs;24 for instance Me2NH·BH3 can be used as a reductant to form Sn, Ni, Pd, Pt and Fe NPs.20,25 This result is, however, unexpected for manganese(II) due to its large negative reduction potential which has made the synthesis of Mn NPs via chemical reduction challenging.26
| Entry | Cat. (mol%) | Time | Conversionb (%) | Product ratioc,d4/5/6 |
|---|---|---|---|---|
| a Reaction conditions: 10 mg cat., 60 °C, 16 h, 0.6 mL of C6D6. b Determined by 1H NMR and 11B NMR spectroscopy. c Ratio by 1H NMR and 11B NMR. d Small amounts of HB(NMe2)2 (<3%) were also detected by 11B NMR. | ||||
| 1 | 1 (5%) | 2 h | 92 | 0/98/2 |
| 2 | 2 (5%) | 4 h | 90 | 40/60/1 |
| 3 | 3 (5%) | 16 h | 29 | 14/83/1 |
To probe the structure and chemical composition of the heterogeneous catalyst, samples of the reaction mixture were analysed using transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM) and energy dispersive X-ray (EDX) spectroscopy. In an attempt to minimise the potential effect of exposure of the sample to the atmosphere, a few drops of a solution of the catalytically active species, obtained after heating 3 (5 mol%) and Me2NH·BH3 in C6D6 (0.6 mL) at 60 °C for 16 h and diluting with ca. 2 mL benzene, was deposited onto a copper-mounted carbon film under an inert atmosphere. STEM (Fig. 5a) and TEM (Fig. S2, ESI†) images indicate the formation of small ∼10 nm Mn NPs, a consequence of the rapid decomposition of the manganese precatalysts 3 into nanoparticles, which readily assemble into large sub-micron aggregates, consistent with the absence of an effective mechanism for the stabilisation of discrete NPs. Furthermore, EDX spectroscopic analysis (Fig. 5b and d) confirmed the presence of both manganese (75 at%) and oxygen (22 at%), with the apparent excess of manganese indicative of the formation of manganese NPs passivated by a thin layer of a manganese oxide, an artefact of the unavoidable exposure of the sample to atmospheric oxygen during its introduction to the microscope. Complementary analysis of the post-reaction mixture by dynamic light scattering (DLS) indicated that large particles (ca. 800–900 nm in hydrodynamic diameter) were indeed present in solution (Fig. 5c), supporting the microscopy observations. The apparent offset in determined aggregate size is expected based on fundamental differences in how the absolute value of NP size is determined, with DLS typically overestimating NP size owing to the greater amount of light scattered by larger particles within the distribution.
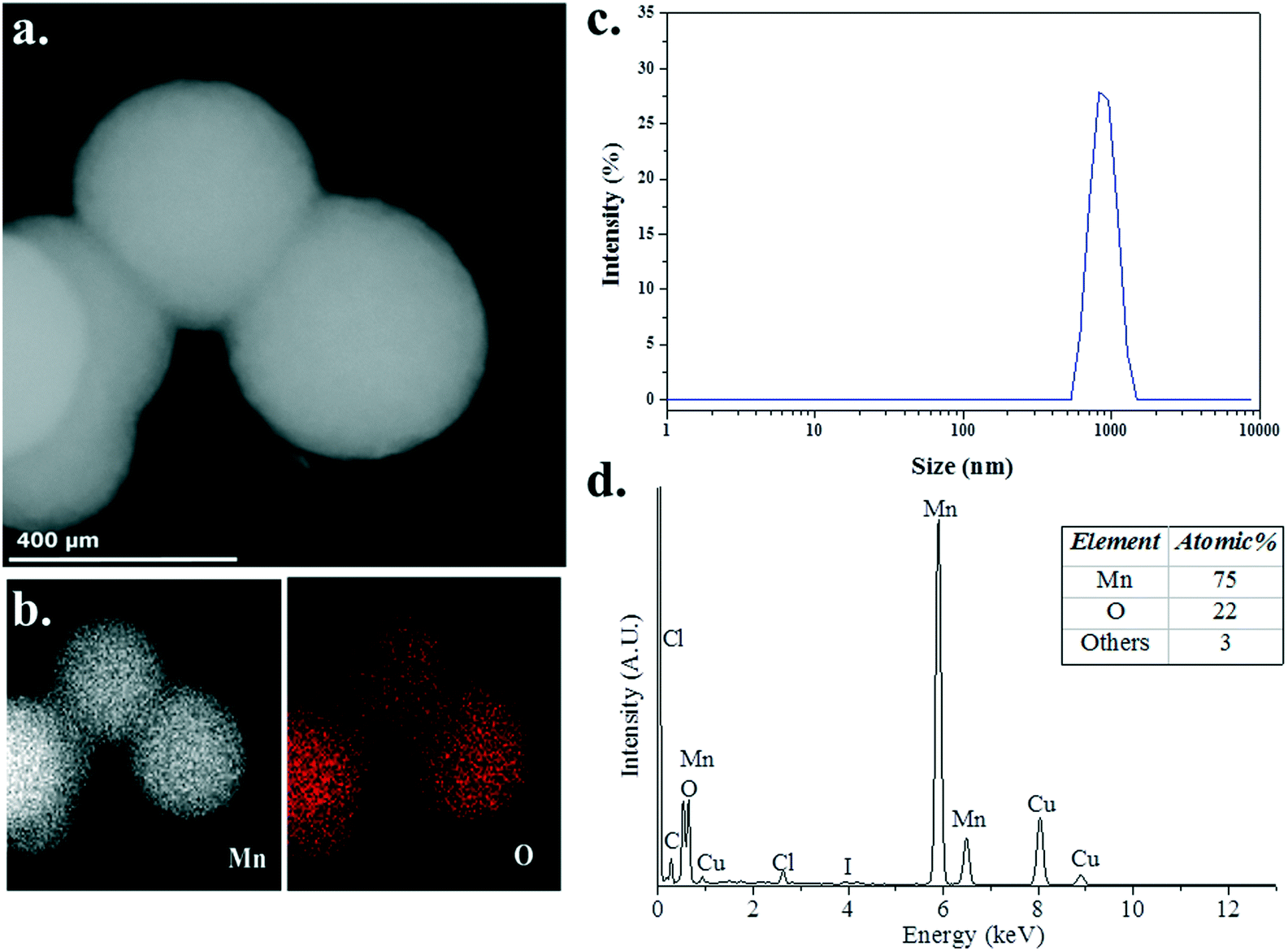 | ||
| Fig. 5 (a) Dark field STEM micrograph of the heterogeneous catalyst. (b) EDX spectroscopic mapping confirming the presence of Mn (white) and O (red) in the NPs. (c) Particle size distribution of hydrodynamic diameter as determined by DLS. (d) Energy dispersive X-ray (EDX) spectrum of manganese NPs on TEM grid with table of atomic% of elements (inset). Copper and carbon peaks originate from the sample support (mesh and film respectively). | ||
Conclusions
Our results indicate that two distinct mechanisms operate when using 1–3 as precatalysts for the dehydrocoupling of Me2NH·BH3. In the case of 1 and 2 experimental evidence indicates that a homogenous catalyst is the active species, whilst for 3 poisoning experiments together with electron microscopy and light scattering measurements indicate that the true catalyst is heterogeneous and most likely formed from the rapid decomposition of the manganese complex into NPs. However, an important question remains: why do two mechanisms exist (homogeneous vs. heterogeneous) for precatalysts 1–3? A similar observation has been observed for iron carbonyl complexes in the dehydrocoupling of Me2NH·BH3 under photoirradiation, where CpFe(CO)2I operates through a homogeneous mechanism and [CpFe(CO)2]2 through a heterogeneous mechanism.5g The authors postulate that the low formal oxidation state of Fe(I) could aid the reduction to Fe NPs in the presence of Me2NH·BH3. However, in 1–3 the manganese centres are in the +II oxidation state, and therefore the difference in reaction mechanisms is more likely due to the steric differences of the ligands. The bulky m-terphenyls in 1 and 2 can make the manganese centre resistant to reduction, whilst, the lower steric demands of the Tmp substituent could render the metal centre in 3 more susceptible to reduction, as reflected in the %VBur of the m-terphenyl ligand.27In summary, two- and three-coordinate manganese m-terphenyl complexes are precatalysts for the dehydrocoupling of dimethylamine–borane. Small changes in the ligand used to support the manganese centre produce drastic changes in the reaction mechanism for dehydrocoupling catalysis using m-terphenyl precatalysts, indicating that this is an important consideration in the use of these complexes for catalysis.
Experimental
General methods
All compounds prepared herein are air and moisture sensitive; therefore, all reactions and manipulations were performed by using standard Schlenk line and glovebox equipment under an atmosphere of purified argon or nitrogen. Iso-hexane (contains <5% n-hexane) and n-pentane were dried by passing through a column of activated 4 Å molecular sieves. THF and toluene were freshly distilled over sodium benzophenone ketyl (THF) or molten potassium (toluene) under nitrogen. All solvents were degassed in vacuo and stored over a potassium mirror (iso-hexane, n-pentane, toluene) or activated 4 Å molecular sieves (THF) prior to use. Benzene-d6 was dried over potassium and degassed with three freeze/pump/thaw cycles prior to use. NMR spectroscopy were performed on Bruker AV400, AV(III)400, AV(III)400HD or AV(III)600 spectrometers. Chemical shifts are quoted in ppm relative to neat TMS (1H), and 0.04 M BF3·OEt2 (11B). UV/visible spectroscopy was obtained in a 10 mm quartz cell modified with a Young's tap using a Lambda 750 spectrophotometer. Magnetic moments were calculated using Evans' method.28 [2,6-Xyl2C6H3Li]2,29 (2,6-Tmp2C6H3)2Mn(THF)10 and (2,6-Mes2C6H3)2Mn11b were prepared following the procedures described in the literature. Elemental microanalysis was performed by Mr. Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK. Me2NH·BH3 is commercially available and was transferred directly into the glovebox and stored at −30 °C. Transmission electron microscopy (TEM) measurements were performed at the Nanoscale and Microscale Research Centre, University of Nottingham using a JEOL 2100F transmission electron microscope (field emission electron gun source, information limit 0.19 nm). Scanning transmission electron microscopy (STEM) images were acquired using the JEOL digital STEM system. Energy dispersive X-ray (EDX) spectra were recorded using an Oxford Instruments 30 mm2 Si(Li) detector or an Oxford Instruments X-Max 80 SDD running on an INCA microanalysis system. TEM/STEM/EDX samples were prepared by casting several drops of the reaction mixture onto copper-mesh holey-carbon films and dried under an inert atmosphere. Dynamic light scattering (DLS) measurements were carried out at the Nanoscale and Microscale Research Centre, University of Nottingham. Samples were prepared as a suspension in dry degassed benzene in a sealed quartz cuvette and intensity particle size distributions acquired using a Malvern Zetasizer Nano-ZS at 25 °C. %VBur was calculated by employing SambVca and using the metal as the centre of the sphere; default settings were sphere radius 3.5 Å, mesh spacing 0.10 Å, hydrogen atoms included.12Safety warning: Dehydrocoupling of Me2NH·BH3 with catalysts 1–3 evolves H2(g), for the closed systems between 6 and 10 mL of H2.
Synthesis of (2,6-Xyl2C6H3)2Mn (1)
Toluene (30 mL) and THF (3 mL) were added to a mixture of [2,6-Xyl2C6H3Li]2 (500 mg, 0.860 mmol) and MnBr2 (183 mg 0.860 mmol) and stirred overnight at room temperature. The solvent was removed in vacuo and the resulting pale off white solid was extracted into toluene (20 mL). Filtration and storage at −30 °C for 24 hours resulted in pale pink crystals suitable for X-ray diffraction studies (144 mg, 27%). 1H NMR (C6D6, 400 MHz, 25 °C): δH/ppm −6.26 (s, br, Δν1/2 = 1020 Hz), 0.40 (s), 0.97 (s), 10.34 (s, br, Δν1/2 = 1200 Hz), 15.55 (s, br, Δν1/2 = 5600 Hz), 26.35 (s, br, Δν1/2 = 2832 Hz). μeff (Evans, C6D6, 25 °C) = 5.32 μB. UV/vis (THF) λmax/nm (ε/dm3 mol−1 cm−1): 369 (141), 356 (207), 322 (429). MS (ESI) m/z: calcd for C44H42Mn [M + H]+ 626.2742, found: 626.2745 (err [ppm] = 0.50). Elemental analysis C44H42Mn (625.758): calcd. C 84.46 H 6.77; found C 84.31, H 6.54%.Standard procedure for the catalytic dehydrocoupling of Me2NH·BH3
In a Young's NMR tube 5 or 2 mol% of precatalyst (1–3) and Me2NH·BH3 were dissolved in C6D6 (0.6 mL). The reactions were either carried out at room temperature or heated to 60 °C in a sealed NMR tube (closed system). Conversion was quantified by integration of 1H and 11B NMR spectra. See Table S1 (ESI†) for quantities of reagents used.Standard procedure for the catalytic dehydrocoupling of Me2NH·BH3 monitored by H2 evolution
7 mol% of precatalyst 2 (57.0 mg, 0.0835 mmol) was dissolved in toluene (3.5 mL) under argon in a Schlenk flask connected to a gas burette filled with paraffin oil. The reaction was heated to 60 °C and, after the vapour pressure of the solvent was reached, 1 mL of a toluene solution of Me2NH·BH3 (70.1 mg,1.19 mmol) was syringed into the reaction vessel via a silicon septum. The mixture was stirred (400 rpm) during the run. Upon injection, the reaction was monitored measuring the volume increase in the gas burette, zeroing the data to the point where the volume of oil began to decrease. To test the recyclability of the system, a second and third loading of Me2NH·BH3 (70.1 mg, 1.19 mmol) dissolved in toluene (1 mL) were syringed into the reaction via a silicon septum when the catalysis was finished and monitored by H2 evolution. It must be noted that upon addition of successive Me2NH·BH3 loadings the volume increases, from 3.5 mL (first run) to 4.5 mL (second run) and 5.5 mL (third run), leading to a decrease in the concentration of the catalyst and therefore can account for some of the decrease in catalyst activity.Standard procedure for in situ NMR reaction monitoring for the catalytic dehydrocoupling of Me2HN·BH3
In a Young's NMR tube 5 mol% of precatalyst (1–3) was dissolved in C6D6 (0.6 mL) and cooled to −30 °C after which Me2NH·BH3 in C6D6 (0.2 mL) was added and the sample cooled to −78 °C to prevent the reaction from initiating. The sample was transferred to the NMR spectrometer and monitored directly at 60 °C. Conversion was quantified by integration of 1H and 11B NMR spectra. See Table S1† for quantities of reagents used.Mercury drop test
In a Young's NMR tube 5 mol% of precatalyst (1–3) and Me2NH·BH3 were dissolved in C6D6 (0.6 mL), hydrogen bubbles were immediately observed. After 5 min, Hg (ca. 1 drop) was added to the reaction and it was heated at 60 °C. The reaction was monitored by 1H and 11B NMR at the times indicated in Table 2. See Table S1† for quantities of reagents used.Radical trap test
In a Young's NMR tube 5 mol% of precatalyst 1 or 2 was dissolved in C6D6 (0.4 mL) with cumene (1: 11 μL, 0.0800 mmol; 2: 10 μL, 0.0735 mmol) and cooled to −30 °C after which Me2NH·BH3 in C6D6 (0.2 mL) was added and the sample cooled to −78 °C to prevent the reaction from initiating. The sample was transferred to the NMR spectrometer and monitored directly at 60 °C. Conversion was quantified by integration of 1H and 11B NMR spectra. See Table S1† for quantities of reagents used.Transmission electron microscopy (TEM)
The sample for TEM analysis was prepared under a nitrogen atmosphere in a glove box. The reaction solution of 3 (5 mol% catalyst loading) and Me2NH·BH3 in C6D6 (0.6 mL), after heating at 60 °C for 16 h, was diluted with ca. 2 mL of benzene. An aliquot of this solution was spotted onto a lacey carbon coated copper grid and the solvent allowed to evaporate. The grid was then transferred to the TEM holder under an inert atmosphere after which the air tight cover was placed on the holder. The TEM holder was quickly put inside the TEM microscope, resulting in the exposure of the sample in air for ca. 5 seconds until high vacuum was achieved.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of the University of Nottingham, the EPSRC and the Leverhulme Trust. We also thank Dr. Huw Williams and Mr. Rhys Lodge, University of Nottingham for helpful discussions and Mr. Stephen Boyer (London Metropolitan University) for elemental analyses.Notes and references
- (a) S. Bhunya, T. Malakar, G. Ganguly and A. Paul, ACS Catal., 2016, 6, 7907 CrossRef CAS; (b) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817 CrossRef CAS PubMed; (c) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079 CrossRef CAS PubMed; (d) A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023 CrossRef CAS PubMed.
- (a) E. M. Leitao, N. E. Stubbs, A. P. Robertson, H. Helten, R. J. Cox, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2012, 134, 16805 CrossRef CAS PubMed; (b) Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 5493 CrossRef CAS.
- (a) J. C. Koepke, J. D. Wood, Y. Chen, S. W. Schmucker, X. Liu, N. N. Chang, L. Nienhaus, J. W. Do, E. A. Carrion, J. Hewaparakrama, A. Rangarajan, I. Datye, R. Mehta, R. T. Haasch, M. Gruebele, G. S. Girolami, E. Pop and J. W. Lyding, Chem. Mater., 2016, 28, 4169 CrossRef CAS; (b) H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078 CrossRef CAS PubMed; (c) S. Frueh, R. Kellett, C. Mallery, T. Molter, W. S. Willis, C. King'ondu and S. L. Suib, Inorg. Chem., 2011, 50, 783 CrossRef CAS PubMed.
- (a) M. A. Esteruelas, P. Nolis, M. Olivan, E. Oñate, A. Vallribera and A. Velez, Inorg. Chem., 2016, 55, 7176 CrossRef CAS PubMed; (b) A. Kumar, N. A. Beattie, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2016, 55, 6651 CrossRef CAS PubMed; (c) M. A. Esteruelas, A. M. López, M. Mora and E. Oñate, ACS Catal., 2015, 5, 187 CrossRef CAS; (d) M. Rosello-Merino, J. Lopez-Serrano and S. Conejero, J. Am. Chem. Soc., 2013, 135, 10910 CrossRef CAS PubMed; (e) L. J. Sewell, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2012, 134, 3598 CrossRef CAS PubMed.
- (a) F. Anke, D. Han, M. Klahn, A. Spannenberg and T. Beweries, Dalton Trans., 2017, 46, 6843 RSC; (b) N. T. Coles, M. F. Mahon and R. L. Webster, Organometallics, 2017, 36, 2262 CrossRef CAS; (c) A. Glüer, M. Förster, V. R. Celinski, J. Schmedt auf der Günne, M. C. Holthausen and S. Schneider, ACS Catal., 2015, 5, 7214 CrossRef; (d) C. Lichtenberg, L. Viciu, M. Adelhardt, J. Sutter, K. Meyer, B. de Bruin and H. Grützmacher, Angew. Chem., Int. Ed., 2015, 54, 5766 CrossRef CAS PubMed; (e) A. Schäfer, T. Jurca, J. Turner, J. R. Vance, K. Lee, V. A. Du, M. F. Haddow, G. R. Whittell and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 4836 CrossRef PubMed; (f) P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153 CrossRef CAS PubMed; (g) J. R. Vance, A. Schäfer, A. P. Robertson, K. Lee, J. Turner, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2014, 136, 3048 CrossRef CAS PubMed; (h) R. T. Baker, J. C. Gordon, C. W. Hamilton, N. J. Henson, P. H. Lin, S. Maguire, M. Murugesu, B. L. Scott and N. C. Smythe, J. Am. Chem. Soc., 2012, 134, 5598 CrossRef CAS PubMed.
- (a) S. Todisco, L. Luconi, G. Giambastiani, A. Rossin, M. Peruzzini, I. E. Golub, O. A. Filippov, N. V. Belkova and E. S. Shubina, Inorg. Chem., 2017, 56, 4296 CrossRef CAS PubMed; (b) J. K. Pagano, J. P. Stelmach and R. Waterman, Dalton Trans., 2015, 44, 12074 RSC; (c) T. P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310 CrossRef CAS PubMed.
- (a) S. Muhammad, S. Moncho, E. N. Brothers and A. A. Bengali, Chem. Commun., 2014, 50, 5874 RSC; (b) T. Kakizawa, Y. Kawano, K. Naganeyama and M. Shimoi, Chem. Lett., 2011, 40, 171 CrossRef CAS.
- M. Gediga, C. M. Feil, S. H. Schlindwein, J. Bender, M. Nieger and D. Gudat, Chem. – Eur. J., 2017, 23, 11560 CrossRef CAS PubMed.
- (a) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS; (b) D. A. Valyaev, G. Lavigne and N. Lugan, Coord. Chem. Rev., 2016, 308, 191 CrossRef CAS; (c) J. Emsley, Nature's Building Blocks: An A–Z. Guide to the Elements – Manganese, Oxford University Press, Oxford, 2001 Search PubMed.
- H. R. Sharpe, A. M. Geer, H. E. L. Williams, T. J. Blundell, W. Lewis, A. J. Blake and D. L. Kays, Chem. Commun., 2017, 53, 937 RSC.
- (a) C. Ni, J. C. Fettinger, G. J. Long and P. P. Power, Dalton Trans., 2010, 39, 10664 RSC; (b) D. L. Kays and A. R. Cowley, Chem. Commun., 2007, 1053 RSC.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286 CrossRef CAS.
- M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831 CrossRef CAS PubMed.
- M. S. Hill, G. Kociok-Kohn and T. P. Robinson, Chem. Commun., 2010, 46, 7587 RSC.
- H. C. Johnson, A. P. Robertson, A. B. Chaplin, L. J. Sewell, A. L. Thompson, M. F. Haddow, I. Manners and A. S. Weller, J. Am. Chem. Soc., 2011, 133, 11076 CrossRef CAS PubMed.
- (a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS PubMed; (b) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962 RSC.
- C. Ni, H. Lei and P. P. Power, Organometallics, 2010, 29, 1988 CrossRef CAS.
- H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake and D. L. Kays, Angew. Chem., Int. Ed., 2017, 56, 4845 CrossRef CAS PubMed.
- Rate constants could not be determined due to solubility problems at catalyst loadings higher than 7 mol%.
- J. F. Sonnenberg and R. H. Morris, ACS Catal., 2013, 3, 1092 CrossRef CAS.
- (a) R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed; (b) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS.
- K. J. Gallagher and R. L. Webster, Chem. Commun., 2014, 50, 12109 RSC.
- M. Zahmakiran and S. Ozkar, Inorg. Chem., 2009, 48, 8955 CrossRef CAS PubMed.
- S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, ChemSusChem, 2011, 4, 317 CrossRef PubMed.
- O. Lidor-Shalev and D. Zitoun, RSC Adv., 2014, 4, 63603 RSC.
- J. F. Bondi, K. D. Oyler, X. Ke, P. Schiffer and R. E. Schaak, J. Am. Chem. Soc., 2009, 131, 9144 CrossRef CAS PubMed.
- R. C. Smith, T. Ren and J. D. Protasiewicz, Eur. J. Inorg. Chem., 2002, 2779 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003 RSC.
- S. Hino, M. M. Olmstead, J. C. Fettinger and P. P. Power, J. Organomet. Chem., 2005, 690, 1638 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected spectra, %VBur calculations, NMR reaction monitoring, crystallographic data and CIF files. CCDC 1548834. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cy02086d |
| This journal is © The Royal Society of Chemistry 2018 |