
The cooperation effect in the Au–Pd/LDH for promoting photocatalytic selective oxidation of benzyl alcohol†
Zhitong
Wang
a,
Yujie
Song
a,
Junhua
Zou
a,
Liuyi
Li
 b,
Yan
Yu
b and
Ling
Wu
*a
b,
Yan
Yu
b and
Ling
Wu
*a
aState Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou, 350116, China. E-mail: wuling@fzu.edu.cn
bKey Laboratory of Eco-materials Advanced Technology, Fuzhou University, Fuzhou, 350116, China
First published on 20th November 2017
Abstract
Materials with different Au–Pd ratios bimetallic alloy nanoparticles (NPs) supported on MgAl–LDH (Au–Pd/LDH) were prepared by an impregnation–reduction method and developed as photocatalysts for the selective oxidation of benzyl alcohol to benzaldehyde under visible light irradiation. A typical sample (Au9–Pd1/LDH) showed efficient photocatalytic activity with 91.1% conversion and 99% selectivity. The conversion was greatly increased from 3.6% for Au/LDH and 1.5% for Pd/LDH, respectively. Moreover, it was four times higher than that for Au9–Pd1/TiO2. The ESR spectrum demonstrated that the main reactive species were superoxide radicals (O2˙−) generated by the transfer of hot electrons from the Au–Pd alloy NPs to the surface adsorbed molecular oxygen. The formation of hot electrons was attributed to localized surface plasmon resonance (LSPR) of the alloy NPs. Based on XPS analysis, a synergistic electron effect in the Au–Pd alloy was observed due to the transfer of electrons from Pd to Au atoms. This effect enhances the adsorption ability of Pd atoms to oxygen molecules and facilitates the formation of O2˙−. The results from CO2-TPD and in situ FT-IR analyses indicated that benzyl alcohol could be efficiently activated at the surface base sites on the LDH via surface coordination to form a metal-alkoxide intermediate. Here, the intermediate could be more easily oxidized by O2˙− to form benzaldehyde. Finally, a possible mechanism based on the cooperation effect between the alloy NPs and the surface basic sites on the LDH was proposed to illustrate the photocatalytic process.
Introduction
The shortage of fossil fuels and the resulting environmental pollution from the combustion of fossil fuel have become major obstacles to social development. The utilization of solar as a potential sustainable energy to drive chemical reactions has attracted more and more attention because sunlight is abundant, sustainable and environmentally friendly.1 Supported metal catalysts can be used as photocatalysts for many chemical transformations, such as water splitting,2 nitrobenzene reduction3 and alcohol oxidation.4 Among these, bimetallic catalysts, such as Au–Pd, exhibit effective activity compared to their single metal counterparts because Au–Pd alloy NPs have a synergistic electron effect that can improve the catalytic performance.5 The supports used for Au–Pd alloy NPs are generally some typical semiconductors, such as TiO2 (ref. 6) and BiVO4,7 or some excellent electronic conductors, like graphene.8 Under visible light irradiation, hot electrons are produced by Au–Pd alloy NPs by localized surface plasmon resonance (LSPR) effects.9 The electrons can be injected into the conduction band of a semiconductor or the surface of graphene to initiate the reaction process. The efficient separation and transfer of the electrons are conducive to achieving high efficiency in the plasmon-mediated catalysis process.As for the catalytic performances of supported metal catalysts, the nature of the support plays a crucial role, especially its surface active sites, such as the acidic or basic sites.10 The surface acidic or basic sites may activate the reactant molecules by chemisorption and the formation of surface complexes, promoting the reactant conversion. However, there are few reports on the interaction between the support itself and the reactant molecules. It is valuable to develop new types of supported metal catalysts that can achieve higher activity by common promotion of metal NPs and the surface active sites.
Layered double hydroxides (LDHs) are a large family of two-dimensional (2D) anionic clay materials with the general formula of [M2+1−xM3+x(OH)2][An−x/n·mH2O] (where M2+ and M3+ are divalent and trivalent metals, respectively, and An− is the interlayer anion), consisting of positively charged brucite-like layers and charge-balancing interlayer anions (Fig. S1†).11 LDHs have been widely used in the fields of catalysis and biology, and in magnetic and optical functional materials, due to their unique structural properties, composition and pore structure.12 In particular, as solid base catalysts, LDHs have been extensively studied in Knoevenagel condensation, aldol condensation and Claisen–Schmidt condensation.13 This is because there are large number of basic sites on LDH surfaces. Besides, as a substrate, LDH has a restricted nano-size effect for the supported metal particles on its surface. This effect can effectively disperse the metal particles and prevent them from agglomerating during the prepared process.14 Very recently, it was reported that Au/NiAl–LDH showed good catalytic performance for the selective oxidation of alcohols to aldehydes and ketones.15 It is the basic sites on the surface of the LDH that can promote the activation of alcoholic hydroxyl groups and then promote the oxidation of alcohols.
Herein, we prepared a series of bimetallic alloy Au–Pd/LDH by an impregnation–reduction method and used them as photocatalysts for the selective oxidation of benzyl alcohol (BA) to benzaldehyde (BAD). The Au–Pd/LDH show efficient catalytic performances. A 91.1% conversion for BA and 99% selectivity for BAD were obtained over the typical sample Au9–Pd1/LDH. Based on the results from XRD, XPS, HR-TEM, in situ FT-IR and CO2-TPD analyses, it was found that the enhancement of activity was due to the cooperative interaction between the Au–Pd alloy NPs and the basic sites on LDH. The active species (O2˙−) in the oxidation reaction process was detected by ESR. Finally, a possible mechanism is proposed herein to illustrate the cooperation effect in Au–Pd/LDH in the process of BA oxidation.
Experimental section
Catalysts preparation
The precursor Mg–Al LDH (LDH) crystals were prepared by the procedure reported by Sasaki, with a few modifications.16 Briefly, 0.015 mol of Mg(NO3)2·6H2O, 0.0075 mol of Al(NO3)3·9H2O and 0.0195 mol of hexamethylenetetramine were co-dissolved in 60 ml of deionized water. The mixture was transferred to an autoclave and heated at 140 °C for 24 h. The product was obtained by centrifugation, washed several times with deionized water and then dried in air.Au9–Pd1/LDH, with the molar ratio of Au/Pd of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, was prepared by an impregnation–reduction method reported by Noritaka Mizuno.17 LDH (1.0 g) was dispersed in 30 ml of an aqueous solution containing HAuCl4·4H2O (7.50 mM) and H2PdCl4 (0.83 mM). After stirring the suspension for 12 h, the solid was obtained by centrifugation and then washed with deionized water. Then, the solid was redispersed in 25 mL water. Under stirring, 35 mg of NaBH4 was added into the suspension. After 2 h, a dark-grey powder was obtained by centrifugation, which was then washed several times with deionized water, followed by vacuum drying. The same procedure was used to prepare Au1–Pd1/LDH and Au1–Pd9/LDH.
:1, was prepared by an impregnation–reduction method reported by Noritaka Mizuno.17 LDH (1.0 g) was dispersed in 30 ml of an aqueous solution containing HAuCl4·4H2O (7.50 mM) and H2PdCl4 (0.83 mM). After stirring the suspension for 12 h, the solid was obtained by centrifugation and then washed with deionized water. Then, the solid was redispersed in 25 mL water. Under stirring, 35 mg of NaBH4 was added into the suspension. After 2 h, a dark-grey powder was obtained by centrifugation, which was then washed several times with deionized water, followed by vacuum drying. The same procedure was used to prepare Au1–Pd1/LDH and Au1–Pd9/LDH.
Au/LDH was prepared as follows. First, LDH (1.0 g) was dispersed in 30 ml of an aqueous solution containing HAuCl4·4H2O (8.3 mM). Then, 0.15 mL of 30% ammonia solution was added. The remaining steps were the same as for Au9–Pd1/LDH. Finally, a purple powder was obtained.
Pd/LDH was prepared as follows. First, LDH (1.0 g) was dispersed in 30 ml of an aqueous solution containing H2PdCl4 (8.3 mM). Then, an aqueous solution of NaOH (1.0 M) was used to adjust the pH of the suspension to 10. The remaining steps were the same as for Au9–Pd1/LDH. Finally, a dark grey powder was obtained.
For comparison, Au9–Pd1/TiO2 was also prepared according to the literature procedure.18
Catalysts characterization
A Bruker D8 Advance X-ray diffractometer with Cu Kα radiation operated at 40 kV and 40 mA was used to obtain the powder X-ray diffraction (XRD) patterns of the samples. The morphologies of the samples were detected by a Hitachi New Generation SU8010 field emission scanning electron microscope (FESEM). A JEOL model JEM 2010 EX microscope at an accelerating voltage of 200 kV was used to obtain the transmission electron microscopy (TEM) images, high-resolution TEM images and element mapping. A Nicolet Nexus 670 Fourier transform infrared spectrometer was used to record the Fourier transform infrared (FT-IR) spectra of LDHs at a resolution of 4 cm−1. A UV-vis spectrophotometer (Varian Cary 500) was used to obtain the UV-vis diffuse reflectance spectra (UV-vis DRS) and the data were converted to Kubelka–Munk (KM) functions. Barium sulfate as a reference was used. A PHI Quantum 2000 XPS system with a monochromatic Al Kα source and a charge neutralizer was used for the X-ray photoelectron spectroscopy (XPS). The binding energies were determined utilizing the C1s spectrum as a reference at 284.8 eV. A Brucker A300 spectrometer was used to record the electron spin resonance (ESR) signals.TG-DSC thermostability analysis of the sample was carried out on a STA 449C thermogravimetric analyzer (Netzsch, Germany), with 20 mg of a catalyst and a background atmosphere of air. The temperature-programmed desorption (TPD) of CO2 was conducted on a Micromeritics AutoChem II. About 20 mg of the samples were loaded in a quartz reactor and heated in He for 120 min at 200 °C, followed by treating with CO2 in flow (50 mL min−1) for 60 min at 80 °C. Weakly adsorbed probe molecules were removed by flowing He for 60 min at 80 °C, and then the desorption process was carried out from 80 °C to 500 °C at 5 °C min−1 in the He flow.
A Nicolet Nexus 670 FT-IR spectrometer was used to obtain the in situ infrared spectra of BA adsorbed on the catalysts at a resolution of 4 cm−1. A total of 32 scans were performed to obtain each spectrum. First, the powder samples (MgAl–LDH mixed with KBr at a mass ratio of 50%) were pressed into a sheet and then placed on a self-supporting IR disc, and then the disc was placed into the sample holder. In order to remove surface contaminants, the disc was treated under 6 × 10−4 Torr at 200 °C for 2 h. Then, 10 μL of BA was spiked into the cell with a syringe via the septum after the disc had cooled to room temperature. After waiting for 30 min, the FT-IR spectra of the samples were collected. The physisorbed BA was removed by a further evacuation at 150 °C for 3 min under 6 × 10−4 Torr, and then, more FT-IR spectra were taken.
Hydrogen peroxide was detected by a reported DPD/POD method.19 This was based on the horseradish peroxidase (POD)-catalyzed oxidation by H2O2 of N,N-diethyl-p-phenylenediamine (DPD). The oxidation of DPD is assumed to consist of the oxidation by H2O2 of POD to a higher valent state, which in turn oxidizes two molecules of DPD to the radical cation, DPD˙+. The radical cation DPD˙+ is then stabilized by resonance and forms a fairly stable colour, with one absorption maxima at 510 nm and one at 550 nm.
The photocatalytic selective oxidation of BA was carried out in a 15 ml quartz tube. Here, 0.1 mmol BA, 24 mg of catalyst and 1.5 mL benzotrifluoride (BTF) were well mixed in the quartz tube. The used BTF was saturated with pure molecular oxygen. The reactor was irradiated by a 300 W Xe lamp (Beijing Perfectlight Co. Ltd., PLS-SXE300c) with a 420 nm cut-off filter. After 5 h of light irradiation, the mixture was centrifuged to completely remove the catalyst particles. The remaining solution was analyzed with an Agilent gas chromatograph (GC-7820). The conversion of BA and the selectivity for BAD were defined as the follows:
| Conversion (%) = [(C0 − CBA)/C0] × 100 |
| Selectivity (%) = [CBAD/(C0 − CBA)] × 100 |
Results and discussions
XRD analysis
As revealed in Fig. 1, the XRD pattern of the LDH shows the characteristic peaks for the (003), (006), (222), (225) and (228) lattice planes at 2θ = 11.7°, 23.5°, 34.8°, 39.5° and 47.0°, respectively. This indicates that the LDH with a pure phase and high crystallinity was successfully prepared (PDF card No. 51-1525). The basal spacing value (d003) of LDH was 0.75 nm, indicating that the interlayer anions were CO32−. After the loading with noble metals, the typical diffraction peaks of the cubic structure Au at about 38.2° (111) and 44.3° (200) can be observed in Fig. 1b. Moreover, the typical diffraction peaks of the cubic structure Pd at about 40.1° (111) and 46.7° (200) cannot be seen clearly in Fig. 1c, which may be attributed to the peaks coincidence with LDH. In Fig. 1d, a weak peak can be observed at around 38.7° lying between the corresponding (111) peak positions of Au/LDH and Pd/LDH, suggesting the formation of Au–Pd alloy NPs.20–23 For all the samples, the diffraction peaks of LDH show no obvious changes, indicating that the structure of the LDH was stable.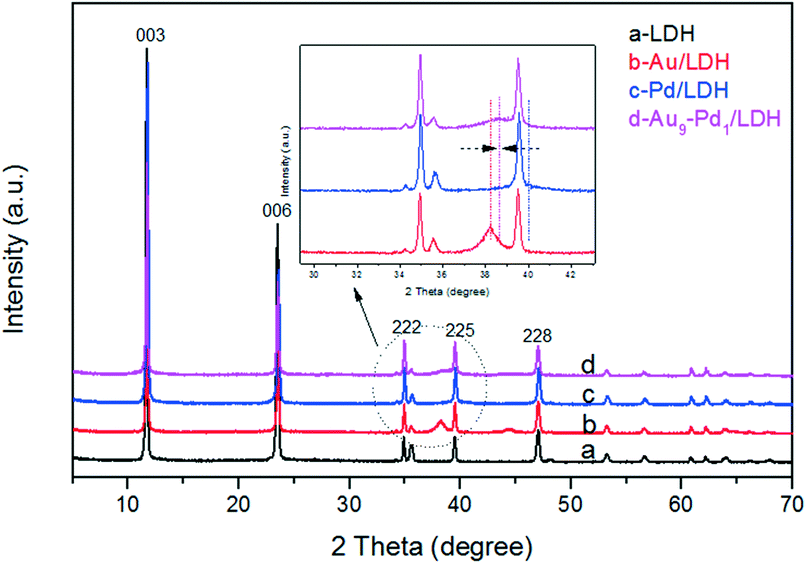 | ||
| Fig. 1 The samples XRD patterns: LDH (a), Au/LDH (b), Pd/LDH (c) and Au9–Pd1/LDH. | ||
SEM and TEM analyses
Fig. S2a–d† show the SEM images of LDH, Au/LDH, Pd/LDH and Au9–Pd1/LDH, respectively. It can be observed that the prepared LDH has a hexagonal plate-like morphology, with an average lateral size ranging from 4–6 um. The LDH maintains its original structure after loading with the metal, which is in line with the XRD results. Moreover, the metal particles cannot be observed on the surface of the LDH due to the uniform distribution and small particle sizes of the metal particles.The TEM analysis of the prepared Au9–Pd1/LDH is shown in Fig. 2. It can be observed that the prepared bimetallic Au–Pd alloy NPs are highly dispersed on the surface of the LDH, with average sizes of 4.9 nm (Fig. 2b). Fig. 2c presents the HR-TEM image of the Au9–Pd1/LDH. The measured lattice spacing of Au–Pd alloy NPs was 0.231 nm, which was just between the values of Au(111) (0.235 nm) and Pd(111) (0.225 nm) and consistent with the XRD results and the values reported in the reference. This could serve as strong evidence for the formation of Au–Pd alloy NPs.4 In addition, the elemental mapping analysis (Fig. 2d) showed that the distribution of Au and Pd in the same region almost overlap with each other, indicating that there were no specific core–shell or other heterostructures for the as-prepared Au–Pd alloyed NPs.
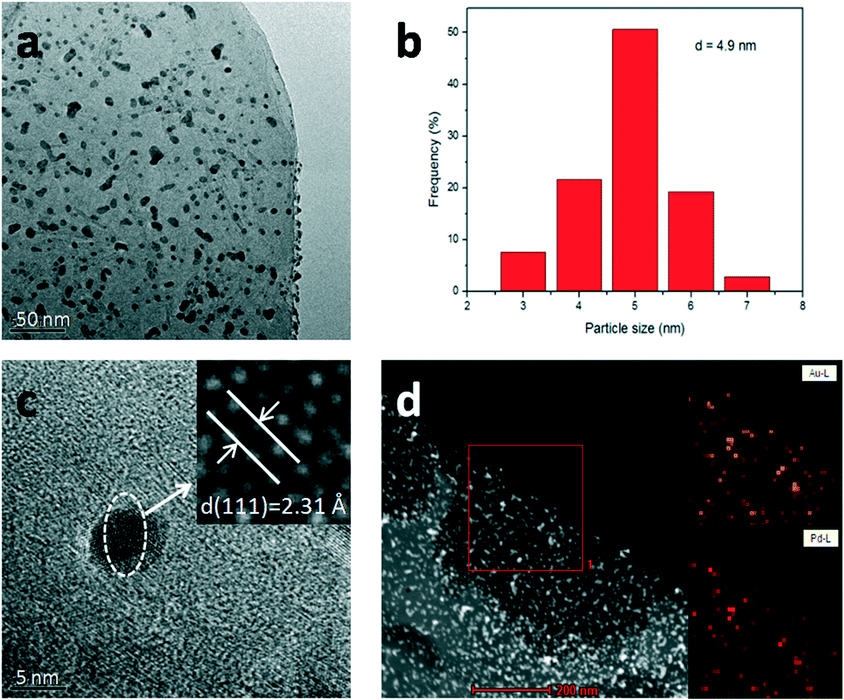 | ||
| Fig. 2 TEM analyses of Au9–Pd1/LDH. TEM image (a), size distribution (b), HR-TEM image (c) and element mapping (d). | ||
FT-IR and TG analyses
The FT-IR spectra of the LDH, Au/LDH, Pd/LDH and Au9–Pd1/LDH are shown in Fig. S3a.† We can see that the LDH shows a strong broad absorption band between 3500 and 3100 cm−1 due to the stretching vibration of hydroxyl groups and interlayer water molecules.24 The strong peak at 1357 cm−1 and a weak one at 778 cm−1 can be assigned to the symmetric stretching vibrations and out-of-plane deformation of the interlayer carbonate ions. The other peaks below 800 cm−1 are the vibrations of metal–O bonds and metal–OH bonds in the LDH lattice.25 The Au/LDH, Pd/LDH and Au9–Pd1/LDH show similar spectra with LDH. The thermostability of LDH was analyzed by TG. The result is shown in Fig. S3b.† When the temperature was below 200 °C, the structure of LDH does not change. When the temperature was increased to 250–450 °C, LDH loses interlayer water, accompanied by CO2 generation. When heated to 450–500 °C, CO32− was completely converted to CO2, resulting in the formation of bimetallic composite oxide (LDO).Optical absorption property
The optical absorption properties of the prepared samples were investigated by DRS (Fig. 3), where it was obvious that the LDH exhibited a weak light absorption above 400 nm. This can prove that the LDH itself does not contribute to the visible light absorption. After the loading with metal NPs on the LDH, the samples showed greatly enhanced light absorption. In particular, Au/LDH showed obvious light absorption at about 520 nm, due to the LSPR effect of Au NPs. As can be seen in Fig. 3b, the peak at a wavelength of 330 nm is the characteristic absorption peak of Pd NPs. For Au–Pd/LDH, the characteristic peak of Au at about 520 nm is broad and weak due to the fusion of Au atoms with Pd atoms, and accompanies a colour change of the sample from purple to grey.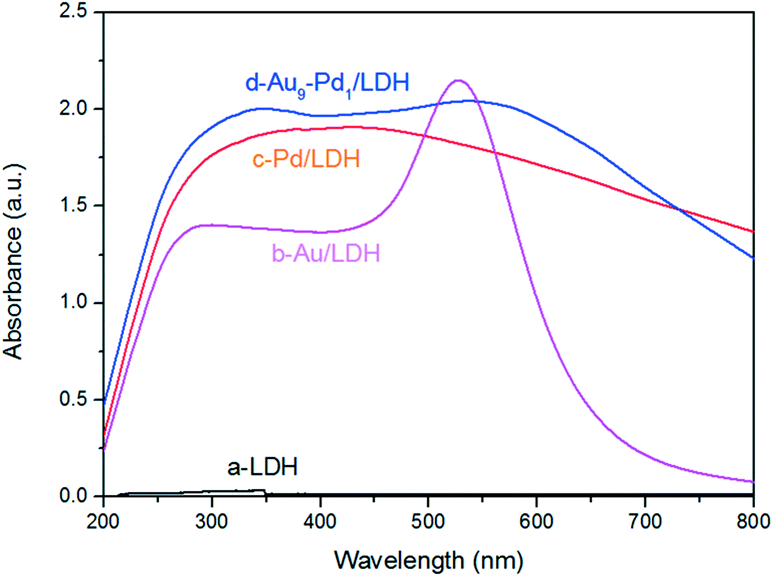 | ||
| Fig. 3 DRS spectra of LDH (a), Au/LDH (b), Pd/LDH (c) and Au9–Pd1/LDH (d). | ||
Performance of Au–Pd/LDH in the photocatalytic selective oxidation of BA
The photocatalytic activities of the as-prepared samples were evaluated by the photocatalytic oxidation of BA at room temperature under visible light irradiation (λ ≥ 420 nm). As shown in Fig. 4, the catalytic performances of the samples were greatly promoted under the light irradiation, indicating that light does play an important role in the catalytic reaction. The conversions for Au/LDH and Pd/LDH were only 3.6% and 1.5%, respectively. All the bimetallic Au–Pd/LDHs exhibited enhanced activity compared to their single metal. Among these, with the increase of Pd content, the catalytic activity also decreases. When Au/Pd was 1:9, the conversion under the same conditions was only 5.2%. Au9–Pd1/LDH exhibited the highest conversion (91.1%) and the highest selectivity (99%). In order to further elucidate why Au9–Pd1/LDH shows the best catalytic activity, FT-IR spectra of CO adsorption on the catalysts were recorded to reveal the bimetallic distribution on the catalysts surface. In Fig. 5, two new CO adsorbed peaks can be observed at 1954 cm−1 and 2062 cm−1 for both samples, corresponding to the linear and bridge adsorption on highly dispersed Pd atoms and aggregated Pd atoms, respectively.10 However, the intensity of the bridge adsorption peak for Au9–Pd1/LDH is significantly reduced. This indicates that the Pd atoms in Au9–Pd1/LDH exist mainly in the form of highly dispersed Pd atoms. It is reported that the highly dispersed Pd atoms by Au atoms are the active sites that promote the catalytic activity and selectivity effectively.26,27 Therefore, Au9–Pd1/LDH showed the best activity among all the catalysts. This is also consistent with the results shown in Fig. 4.
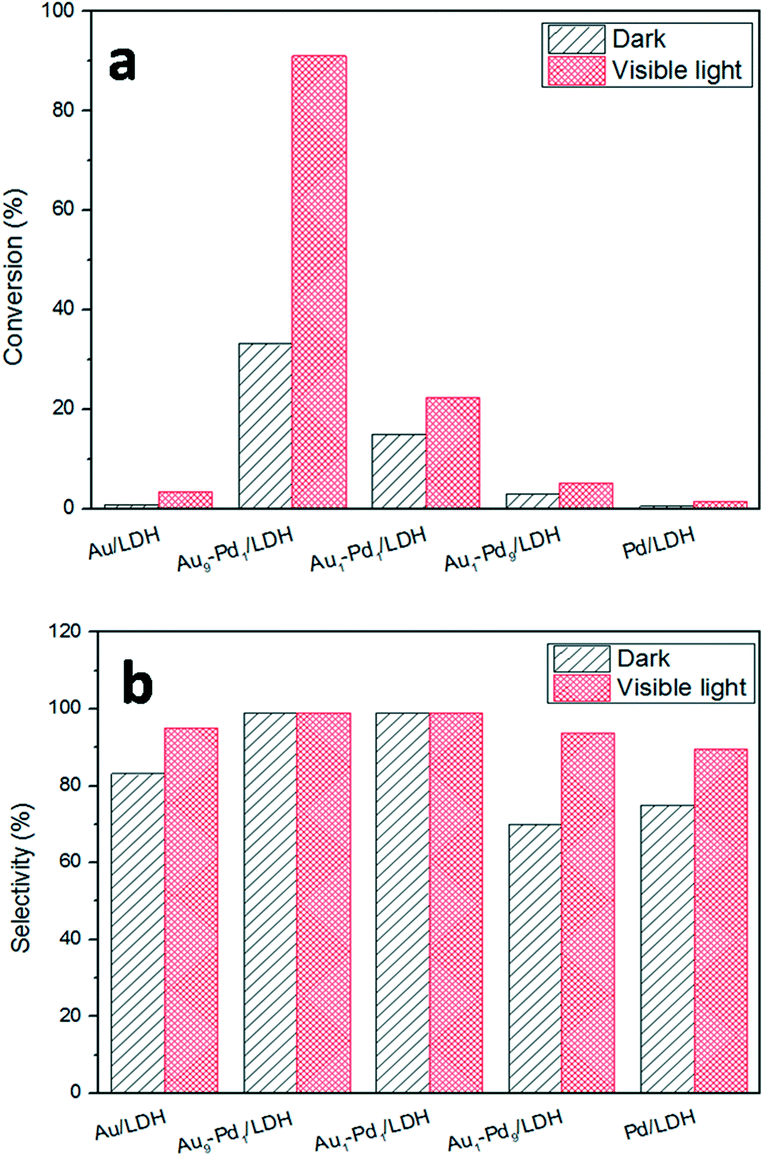 | ||
| Fig. 4 The photocatalytic conversions (a) and selectivity (b) over different Au:Pd molar ratios catalysts. | ||
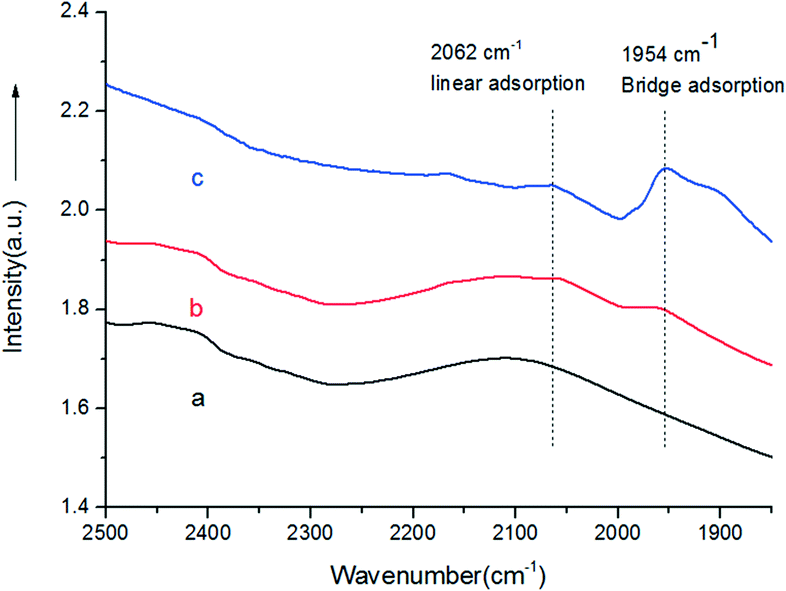 | ||
| Fig. 5 FT-IR spectra of Au9–Pd1/LDH before (a) and after (b) adsorbing CO, and CO adsorbed on the Pd/LDH (c). | ||
Electron properties of the Au–Pd alloy NPs
The surface element composition and the corresponding valence state can often be investigated by XPS. In order to confirm whether there is a synergistic electron effect between Au and Pd, XPS measurements were carried out. Fig. 6a shows the Au 4f spectra of Au/LDH and Au9–Pd1/LDH. The peaks of Au 4f7/2 show a negative shift from 83.88 eV for Au/LDH to 83.69 eV for Au9–Pd1/LDH, suggesting that the electron cloud density of Au species was increased in Au9–Pd1/LDH. In contrast, as can be seen in Fig. 6b, the Pd 3d5/2 peaks show a positive shift from 335.36 eV for Pd/LDH to 335.91 eV for Au9–Pd1/LDH, indicating that the electron cloud density of Pd species in Au9–Pd1/LDH is decreased. These results reveal that the fusion of Au with Pd leads to the charge transfer from Pd to Au. Considering that the electro-negativity of single Au is larger than that of single Pd, the charge transfer is reasonable.7,28 This synergistic electron effect causes Pd atoms to be positively charged and Au atoms to be negatively charged. The positively charged Pd atoms lead to a more intense interaction between Pd atoms and nucleophilic reactant molecules, such as oxygen molecules, which promotes the catalytic activity.10,29,30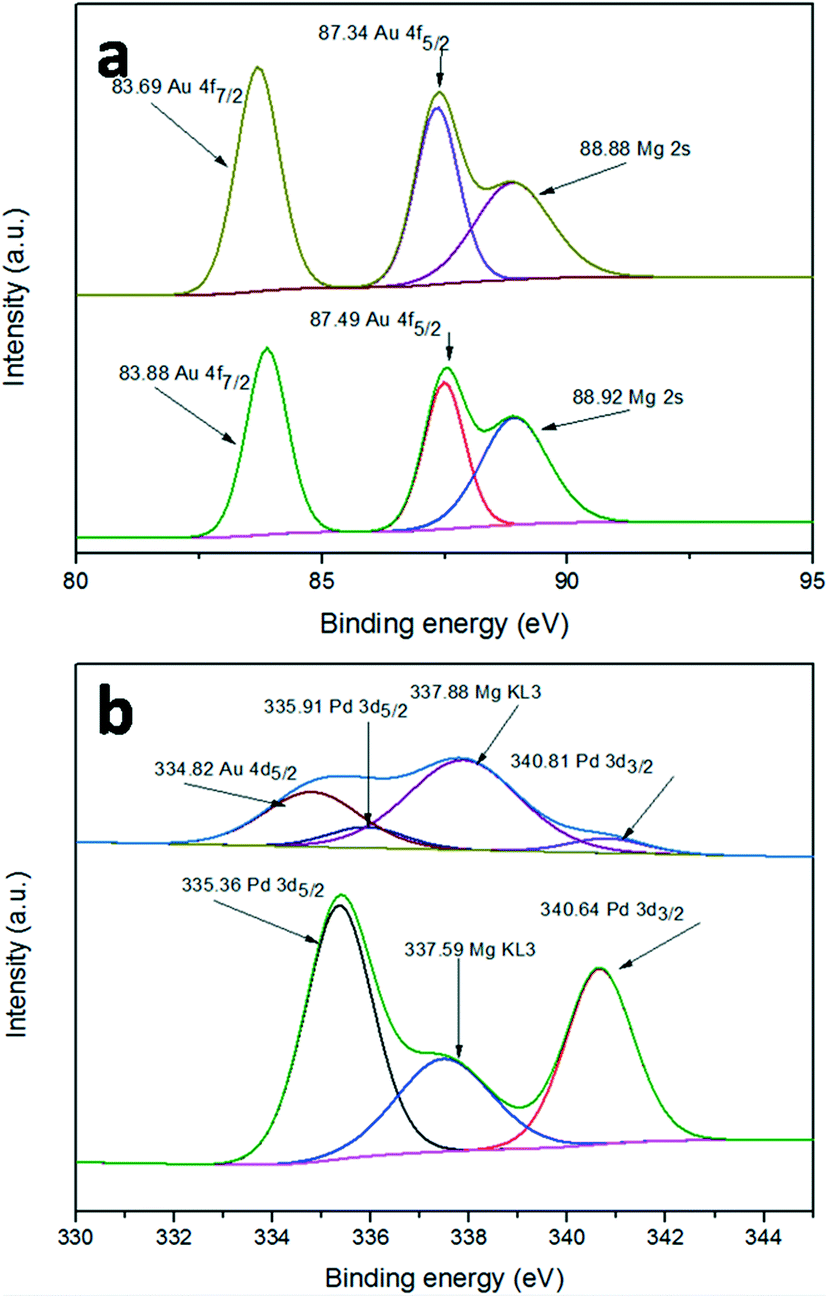 | ||
| Fig. 6 XPS spectra of (a) Au 4f for Au/LDH (down) and Au9–Pd1/LDH (up), (b) Pd 3d for Pd/LDH (down) and Au9–Pd1/LDH (up). | ||
Support effect of the Au–Pd catalysts
As we all know, the surface basic sites of a catalyst can greatly affect the catalytic activity. For the purpose of comparison, TiO2 was chosen as a support to load Au–Pd NPs for the oxidation of BA. The results are shown in Fig. 7. It is obvious that the catalytic activity of Au9–Pd1/LDH (91.1%) exhibits a four times higher activity than that of Au9–Pd1/TiO2 (22.1%), revealing that the basic sites on the support have a marked promotion effect on the reaction process. To gain more insights into the differences between the supports, the basicity of the supports was determined, and the results are shown in Fig. S4.† The CO2-TPD profile of the LDH support could be deconvoluted into three main desorption peaks, reaching a maximum desorption rate at about 297 °C, 345 °C and 395 °C, which can be attributed to the weak (α-OH groups), moderate (β-Mg–O pairs) and strong basic sites (γ-O2− anions), respectively.31 The moderate basic sites show the strongest signal of CO2 desorption, indicating that the metal sites as the main basic sites have the high basicity. These metal sites may act as efficient active sites for the absorption of reagent molecules. For comparison, the CO2-TPD curve of TiO2 indicated that little CO2 can be adsorbed on its surface, due to the absence of basic sites.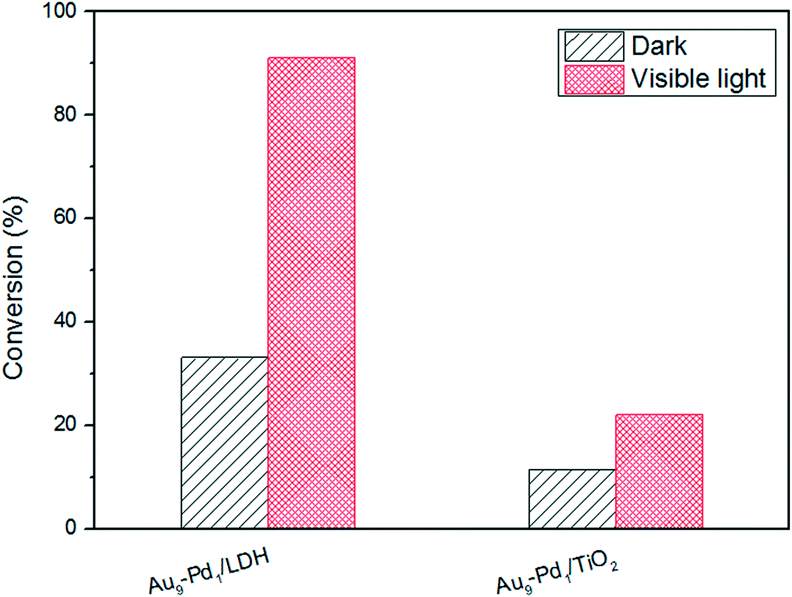 | ||
| Fig. 7 The photocatalytic conversions for BA over different supports. | ||
In order to explore the role of the surface basic sites on the surface of LDH, in situ FT-IR spectra were carried out for the BA absorption experiments to reveal the interactions between the support and reagent molecules. According to the TG analysis, the structure of LDH does not change when the temperature is below 200 °C. So a desorption pretreatment temperature of LDH pf 200 °C was used in the in situ FT-IR experiments. The experimental results are shown in Fig. 8. The in situ FT-IR spectra of BA/LDH (Fig. 8(1)) indicated that BA could be chemically adsorbed on the surface of LDH by surface coordination to form surface complexes. It is worth noting that the characteristic C–O band of BA adsorbed on the LDH surface (1040 cm−1) showed a negative shift compared with free BA (1080 cm−1). However, the other characteristic bands of adsorbed BA showed no change. These results indicated that the surface metal-alkoxide intermediate was formed by a –C–O–Mg– coordinate bond between BA and LDH.32 However, the results of the in situ FT-IR spectra of BA/TiO2 (Fig. 8(2)) revealed that BA may not be adsorbed on the surface of TiO2 due to the absence of basic sites. Considering the abundant basic sites of LDHs and the results of the in situ FT-IR analysis, we suggest that these surface basic sites can act as the active sites for the chemisorption and activation of reagent molecules to form a metal-alkoxide intermediate, facilitating the reaction progress. It is because there are few basic sites on the surface of Au9–Pd1/TiO2 that the BA molecules may not be activated by the basic sites to form a metal-alkoxide intermediate, resulting in a decrease in the photocatalytic activity compared with that of Au9–Pd1/LDH.
 | ||
| Fig. 8 In situ FT-IR spectra of LDH (1) and TiO2 (2) discs before and after adsorbing BA. Conditions: (a) after degassing at 200 °C for 2 h. (b) Adsorption for 30 min at RT (physisorption + chemisorption). (c) Further evacuation of excess probe molecules at 150 °C for 1 min under 6 × 10−4 Torr (chemisorption). | ||
Combined with the above results, we can see that there was a cooperation effect between the Au–Pd alloy and the basic sites on LDH. When there was only single metal, either Au or Pd, loaded on LDH, Au/LDH and Pd/LDH showed very weak activity. In comparison with Au9–Pd1/LDH, Au9–Pd1/TiO2 showed a dramatic decrease in the catalytic activity. The Au–Pd alloy NPs and LDH were thus indispensable in the efficient photocatalytic transformation of BA.
The role of oxygen in the progress of photocatalysis
For the purpose of gaining an in-depth understanding of the photocatalytic process, we studied the role of oxygen in the reactions (Fig. 9). The conversion of BA over Au9–Pd1/LDH in an air atmosphere decreased from 91.1% to 28.1%. Meanwhile, the conversions for all the other samples showed huge decreases under the same conditions.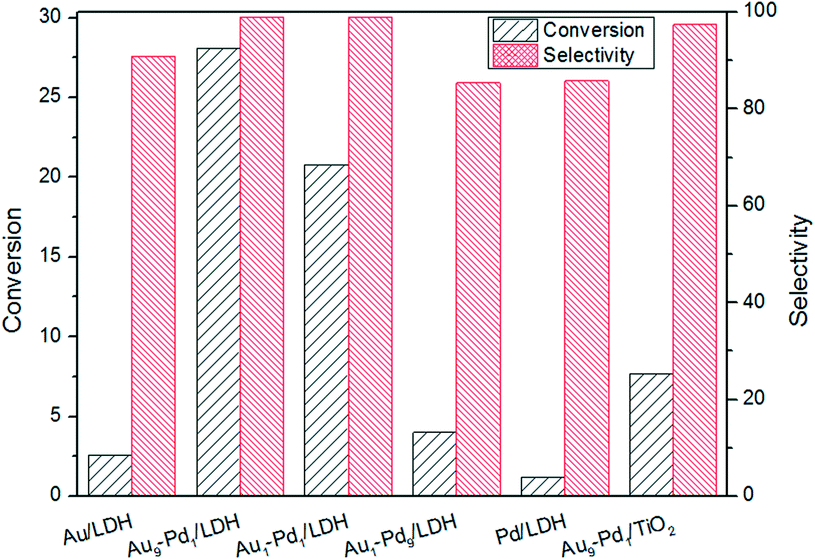 | ||
| Fig. 9 The photocatalytic conversions and selectivity over different catalysts at the air atmosphere. | ||
This confirms that the oxygen molecules play an important role in the BA oxidation. We conducted ESR measurements for the system. The ESR spectra of the irradiated reaction system containing Au9–Pd1/LDH and BA in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a trapping agent showed typical signals of the DMPO–O2˙− adduct, thus confirming the formation of O2˙− radicals during the photocatalytic reaction (Fig. 10). Moreover, no O2˙− signal was detected in the dark. This gives further confirmation that the photocatalytic oxidation of BA truly follows the photogenerated O2˙− radical-mediated oxygenation pathway. It is possible that the O2˙− radical in this system can be generated via the transfer of photoinduced hot electrons from the Au–Pd alloy to surface oxygen molecules under visible light irradiation. The synergistic electron effect in Au–Pd alloy NPs can efficiently facilitate the formation of O2˙− due to the enhanced adsorption ability of Pd atoms to oxygen molecules.
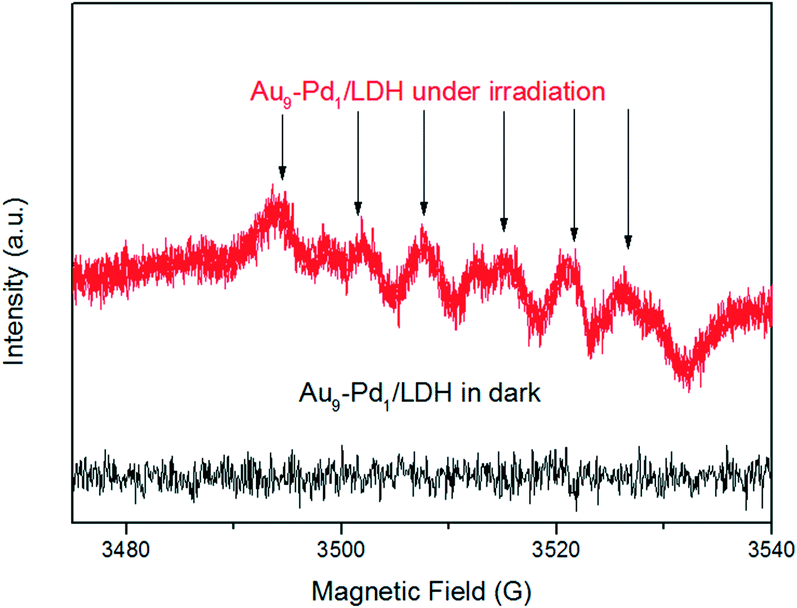 | ||
| Fig. 10 ESR spectra of radical adducts trapped by DMPO (DMPO–O2˙−) in the Au9–Pd1/LDH dispersion solution. | ||
Catalysts stability
Stability texts were also carried out to investigate the repeatability of the catalysts. The results are very interesting. When the catalyst was used for the first time to catalyze the reaction, it exhibited remarkable activity, but when we used the catalyst recovered by centrifugation for the second time in the reaction, the activity was greatly reduced, with almost only 5% conversion. Then, we dispersed the catalyst in a NaOH solution (2 M) and kept stirring for 2 h, whereby the “reactivation catalyst” showed a 44% conversion. Moreover, the “reactivation catalyst” stirred for 4 h had the same activity as the first used catalyst. This interesting phenomenon serves as strong evidence to prove that the surface basic sites on the LDH surface have an excellent promoting effect on the reaction process. Through the “reactivation”, the basic sites consumed in the reaction can be regenerated.Possible oxidation mechanisms
According to the above experimental results and related reports,14 a possible photocatalytic mechanism based on the cooperation between the Au–Pd alloy NPs and the basic sites on the LDH surface was proposed. First, the BA can be chemisorbed on the basic sites (Mg–OH) on the surface of LDH, and then form a metal-alkoxide intermediate at the interface through surface coordination. Under visible light irradiation, the Au atoms in alloy NPs will adsorb the light to excite the electrons due to the LSPR effect, resulting in Au atoms with hot electrons at the alloy surface.33 Then, the hot electrons transfer from Au atoms to oxygen molecules adsorbed on neighbouring Pd sites to produce O2˙− radicals. The superoxide radicals attack the intermediates to form BAD (Scheme 1).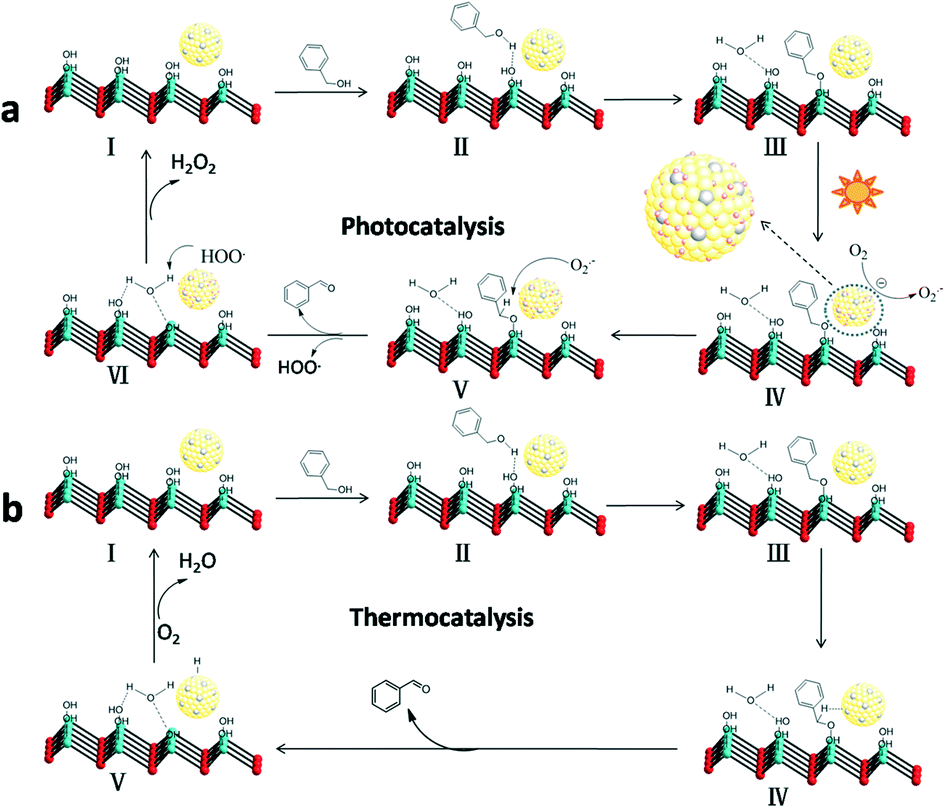 | ||
| Scheme 1 Possible mechanism for the selective oxidation of BA to BAD over the as-prepared Au–Pd/LDH. Photocatalysis (a), thermocatalysis (b) (light blue spheres: Mg/Al atoms; red spheres: oxygen atoms; yellow spheres: Au–Pd alloy NPs). | ||
Ultimately, the active sites are recovered with the formal liberation of H2O2.34 The formation of hydrogen peroxide in the reaction process was detected by a reported DPD/POD method (Fig. S5†).19 Meanwhile, we noticed that Au9–Pd1/LDH also had a higher activity (33.2%) in the dark conditions and room temperature. There may be a thermocatalytic effect in the process of oxidation. In the thermocatalytic mechanism, BA also attacks the basic sites. The basic sites promote the abstraction of protons from BA molecules to form the metal-alkoxide intermediate. Then, the intermediate undergoes β-hydride elimination with coordinate Au–Pd alloy active centres to afford the BAD.35 Finally, the metal hydride is oxidized by O2 to recover the active sites. It should be noted that the oxidation process from BA to BAD is photothermal synergistic catalysis. In order to further explore what kind of mechanism is dominant in the present catalytic reaction, the thermocatalytic oxidation of BA to BAD over the Au9–Pd1/LDH was also carried out under different temperatures, without light irradiation. The results are shown in Fig. S6.† The thermal catalytic conversion of Au9–Pd1/LDH at 80 °C (86.7%) was still lower than that of photocatalytic conversion under light and room temperature (91.1%), indicating that photocatalysis did have a dominating effect in the present catalytic process.
Conclusions
We demonstrated that the cooperation effect in Au–Pd/LDH can efficiently promote photocatalytic activity for BA oxidation. The synergistic electron effect in the Au–Pd alloy is caused by the transfer of electrons from Pd to Au atoms, which enhances the adsorption ability of Pd atoms to adsorbed oxygen molecules and thereby facilitates the formation of O2˙−. Meanwhile, the basic sites on the surface of the LDH can efficiently active the BA molecules to form a metal-alkoxide intermediate by surface coordination. The intermediate can be more easily oxidized by O2˙− to form BAD, promoting the catalytic reaction. The Au–Pd alloy NPs and the basic sites on the LDH are indispensable in the efficient photocatalytic transformation of BA to BAD. We believe that the present work will provide intensive insights for research into novel metal catalysts for the photocatalytic selective oxidation of BA.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51672048), the major science and technology projects of Fujian Province (2015YZ0001-1).Notes and references
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- M. Hmadeh, V. Hoepfner, E. Larios, K. Liao, J. Jia, M. Joseyacaman and G. A. Ozin, ChemSusChem, 2014, 7, 2104 CrossRef CAS PubMed.
- A. Tanaka, Y. Nishino, S. Sakaguchi, T. Yoshikawa, K. Imamura, K. Hashimoto and H. Kominami, Chem. Commun., 2013, 49, 2551–2553 RSC.
- T. Jiang, C. Jia, L. Zhang, S. He, Y. Sang, H. Li, Y. Li, X. Xu and H. Liu, Nanoscale, 2015, 7, 209–217 RSC.
- S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793–5801 CrossRef CAS PubMed.
- S. Ren, R. Tiruvalam, H. Qian, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopezsanchez, R. Bechstein, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6284 CrossRef PubMed.
- J. Zhang, Y. Lu, L. Ge, C. Han, Y. Li, Y. Gao, S. Li and H. Xu, Appl. Catal., B, 2017, 204, 385–393 CrossRef CAS.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, J. Phys. Chem. C, 2014, 118, 5299–5308 CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- J. Feng, C. Ma, P. J. Miedziak, J. K. Edwards, G. L. Brett, D. Li, Y. Du, D. J. Morgan and G. J. Hutchings, Dalton Trans., 2013, 42, 14498–14508 RSC.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- D. G. Evans and X. Duan, Chem. Commun., 2006, 485–496 RSC.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Catal., 2004, 221, 474–482 CrossRef CAS.
- W. Fang, J. Chen, Q. Zhang, W. Deng and Y. Wang, Chemistry, 2011, 17, 1247–1256 CrossRef CAS PubMed.
- J. Wang, X. Lang, B. Zhaorigetu, M. Jia, J. Wang, X. Guo and J. Zhao, ChemCatChem, 2014, 6, 1737–1747 CrossRef CAS.
- L. Li, R. Ma, Y. Ebina, A. Nobuo Iyi and T. Sasaki, Chem. Mater., 2005, 17, 4386–4391 CrossRef CAS.
- X. Jin, K. Taniguchi, K. Yamaguchi and N. Mizuno, Chem. Sci., 2016, 7, 5371–5383 RSC.
- K. Taniguchi, X. Jin, K. Yamaguchi and N. Mizuno, Chem. Commun., 2015, 51, 14969–14972 RSC.
- H. Bader, V. Sturzenegger and J. Hoigné, Water Res., 1988, 22, 1109–1115 CrossRef CAS.
- L. Zhang, J. Zhang, Q. Kuang, S. Xie, Z. Jiang, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2011, 133, 17114–17117 CrossRef CAS PubMed.
- Y. W. Lee, M. Kim, Y. Kim, S. W. Kang, J. H. Lee and S. W. Han, J. Phys. Chem. C, 2010, 114, 7689–7693 CAS.
- Y. W. Lee, N. H. Kim, Y. L. Kang, K. Kwon, A. Minjung Kim and W. H. Sang, J. Phys. Chem. C, 2008, 112, 6717–6722 CAS.
- Y.-H. Chen, Y.-H. Tseng and C.-S. Yeh, J. Mater. Chem., 2002, 12, 1419–1422 RSC.
- Q. Wang, X. Zhang, C. J. Wang, J. Zhu, Z. Guo and D. O'Hare, J. Mater. Chem., 2012, 22, 19113 RSC.
- S. Kumar, M. A. Isaacs, R. Trofimovaite, L. Durndell, C. M. A. Parlett, R. E. Douthwaite, B. Coulson, M. C. R. Cockett, K. Wilson and A. F. Lee, Appl. Catal., B, 2017, 209, 394–404 CrossRef CAS.
- M. Chen, D. Kumar, C. W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS PubMed.
- D. Wang, A. Villa, F. Porta, L. Prati and D. S. Su, J. Phys. Chem. C, 2008, 112, 8617 CAS.
- Q. Chen, S. Tanaka, T. Fujita, L. Chen, T. Minato, Y. Ishikawa, M. Chen, N. Asao, Y. Yamamoto and T. Jin, Chem. Commun., 2014, 50, 3344–3346 RSC.
- W. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22–27 CrossRef CAS.
- Q. Xiao, S. Sarina, E. Jaatinen, J. Jia, D. P. Arnold, H. Liu and H. Zhu, Green Chem., 2014, 16, 4272–4285 RSC.
- J. I. Di Cosimo, C. R. Apesteguı, M. J. L. Ginés and E. Iglesia, J. Catal., 2000, 190, 261–275 CrossRef CAS.
- S. Liang, L. Wen, S. Lin, J. Bi, P. Feng, X. Fu and L. Wu, Angew. Chem., Int. Ed., 2014, 53, 2951–2955 CrossRef CAS PubMed.
- Q. Xiao, S. Sarina, A. Bo, J. Jia, H. Liu, D. P. Arnold, Y. Huang, H. Wu and H. Zhu, ACS Catal., 2014, 4, 1725–1734 CrossRef CAS.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- S. Nishimura, A. Takagaki and K. Ebitanj, Green Chem., 2013, 15, 2026 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy02006f |
| This journal is © The Royal Society of Chemistry 2018 |