
Efficient photocatalytic water reduction by a cobalt(II) tripodal iminopyridine complex†
C.-F.
Leung
 *a,
S.-C.
Cheng
b,
Y.
Yang
a,
J.
Xiang
ac,
S.-M.
Yiu
b,
C.-C.
Ko
*b and
T.-C.
Lau
b
*a,
S.-C.
Cheng
b,
Y.
Yang
a,
J.
Xiang
ac,
S.-M.
Yiu
b,
C.-C.
Ko
*b and
T.-C.
Lau
b
aDepartment of Science and Environmental Studies, The Education University of Hong Kong, 10 Lo Ping Road, Tai Po, Hong Kong, China. E-mail: cfleung@eduhk.hk
bDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China. E-mail: vinccko@cityu.edu.hk
cDepartment of Chemical and Environmental Engineering, Yangtze University, Jingzhou, China
First published on 20th November 2017
Abstract
The visible light-driven catalytic water reduction by a cobalt(II) tripodal iminopyridine complex [Co(tachpy3)](ClO4)2 (1) (tachpy3 = cis,cis-1,3,5-tris(pyridine-2-carboxaldimino)-cyclohexane) has been investigated in aqueous acetonitrile. The catalysis is found to be homogeneous and an impressive H2 TON of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440 has been achieved, which is far better than that by a structurally related complex [Co(trenpy3)](ClO4)2 (2) (trenpy3 = tris-[4-(2-pyridyl)-3-azabut-3-enyl]amine) bearing identical donor groups. A comparison between the two complexes reveals that the catalytic properties are sensitive to the change in ligand rigidity and thus the coordination geometry, as reflected by their respective reaction potentials and H2 TONs. The catalysis of 1 proceeds via a penta-coordinate species formed by the protonation of the corresponding CoI state. The results in spectroscopic and electrochemical studies are supplemented by DFT computation.
440 has been achieved, which is far better than that by a structurally related complex [Co(trenpy3)](ClO4)2 (2) (trenpy3 = tris-[4-(2-pyridyl)-3-azabut-3-enyl]amine) bearing identical donor groups. A comparison between the two complexes reveals that the catalytic properties are sensitive to the change in ligand rigidity and thus the coordination geometry, as reflected by their respective reaction potentials and H2 TONs. The catalysis of 1 proceeds via a penta-coordinate species formed by the protonation of the corresponding CoI state. The results in spectroscopic and electrochemical studies are supplemented by DFT computation.
Introduction
In the recent decade, the development of functional materials for tapping solar energy by the light-driven conversion of earth abundant materials, e.g. water and carbon dioxide, into storable solar fuels has gained increasing momentum.1–7 In particular, the catalytic generation of hydrogen by the reduction of water has been widely investigated, as H2 is an energy vector used in fuel cells. For economic reasons, the development of molecular water reduction catalysts (WRCs) based on first-row transition metals (e.g. Co, Cu, Fe and Ni) has received much attention.8–39 Co WRCs are commonly studied, due to their desirable catalytic properties. For example, Co oxime catalysts were found to function at low overpotentials.15 Recently, enhanced H2-evolution performance was achieved using a Co–NHC catalyst,49 while Co polypyridyl catalysts exhibit higher stability.17–19,25,50,51 In contrast, photo-driven H2 generation with Co catalysts is, however, usually less efficient compared to that with other metal catalysts.16,24,28,36There have been only a few studies on Co iminopyridine WRCs despite their fast catalytic rates.20–23 We report herein the photocatalytic water reduction by a cobalt(II) tripodal iminopyridine complex [Co(tachpy3)](ClO4)2 (1) (tachpy3 = cis,cis-1,3,5-tris(pyridine-2-carboxaldimino)-cyclohexane) in aqueous acetonitrile (Scheme 1).451 is found to be an active molecular WRC. To shed light on the factors affecting the reactivity, a structurally related complex [Co(trenpy3)](ClO4)2 (2) (trenpy3 = tris-[4-(2-pyridyl)-3-azabut-3-enyl]amine)46 which bears the same metal centre and donor groups is also investigated. The catalytic properties, e.g. stability and reaction potential, are compared using photo- and electrochemical methods supplemented with DFT computation. The coordination geometry and the more rigid ligand structure in 1 are suggested to contribute to its enhanced WRC reactivity.
 | ||
| Scheme 1 Structures of 1, 2 and IrPS+. | ||
Results and discussion
The H2 content in the headspace of a sealed reaction tube containing an acetonitrile solution (5 mL) of catalyst 1 (1 μM), cyclometallated Ir photosensitizer IrPS+ (0.2 mM), 0.2 M triethylamine (TEA) and water (10% v/v) was determined by GC-TCD. H2 is produced upon irradiation with visible light (λ > 420 nm), but the yield of H2 levels off after 8 h of irradiation (10 μmol). The control experiment in the absence of the catalyst generates less H2 (4 μmol).No H2 could be detected in the absence of IrPS+, H2O or TEA (Fig. 1). Both the production rate and yield of H2 decrease, when the amount of H2O is lowered from 10% to 2.5% (Fig. S2†); increasing [H2O] to 15% results in no further improvement in the H2 yield. Cyclometallated Ir photosensitizers are known to decompose via the dissociation of the diimine ligand from the unstable reduced state.47,48 In an attempt to restore the H2 production, a fresh solution of IrPS+ (0.2 mM) was added to the reaction solutions after the first 16 h of photolysis. The activity of 1 was restored and a comparable amount of H2 (12.3 μmol) was produced after another 16 h of irradiation, indicating that the decomposition of IrPS+ is the major cause for the loss of activity. H2 production was maintained and a total H2 yield of 109 μmol was attained after 5 cycles of IrPS+ addition (overall 80 h of irradiation), whereas only 27.4 μmol H2 was recorded in the control experiment without the catalyst, leading to an effective TON of 16440 (vs. catalyst). The performance of several reported Co WRCs is shown in Table 1. Co catalysts bearing pentadentate polypyridyl ligands are, so far, the most efficient among all Co WRCs reported.50–52 A H2 TON of 11000 was reported for [CoII(aPPy)(Br)]Br (aPPy = bis-2,2′′-bipyridine-6-yl(pyridine-2-yl)methanol), using a ReI photosensitizer and ascorbic acid/ascorbate as the sacrificial donor under aqueous conditions,50 while a higher TON (30000) has been achieved when a secondary sacrificial electron donor was provided.51 A H2 TON of 20000 has recently been reported also in acetonitrile for Co(TMPA)Cl2 (TMPA = 1-(6-substituted-pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methane amine).52 The H2 TON for 1 is therefore amongst the highest reported for Co WRCs without the use of a secondary sacrificial donor. A similar yield (9.2 μmol) and response to H2O concentration have been observed for 2 in the first catalytic cycle (16 h) under the same conditions (Fig. 1 and S2†). However, the yield of H2 in the subsequent cycles is similar to that of the control experiment when additional IrPS+ was provided, reflecting that the catalyst has been deactivated after the first cycle. The change in the H2 yield during the first hour of photolysis at varied concentrations of 1 has been recorded (Fig. 2). At the selected concentrations, there was no observable lag phase and H2 yield increases linearly with time, indicating that the catalytic reaction is homogeneous.40 The first order rate constant (kinitial), obtained as the initial rate of H2 production in the first hour, exhibits linear dependence on [1]. A second order rate constant k2 of 0.25 h−1 was obtained accordingly.
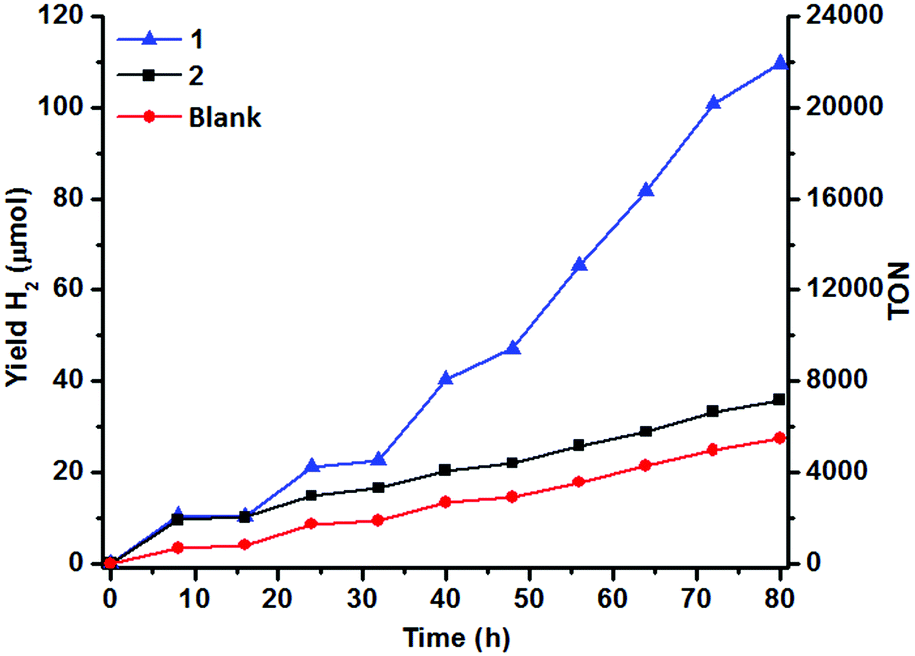 | ||
| Fig. 1 Hydrogen evolution of 1 and 2 (1 μM) in aqueous (10% v/v) acetonitrile at 25 °C. [IrPS+] = 0.2 mM and [TEA] = 0.2 M upon photoexcitation. | ||
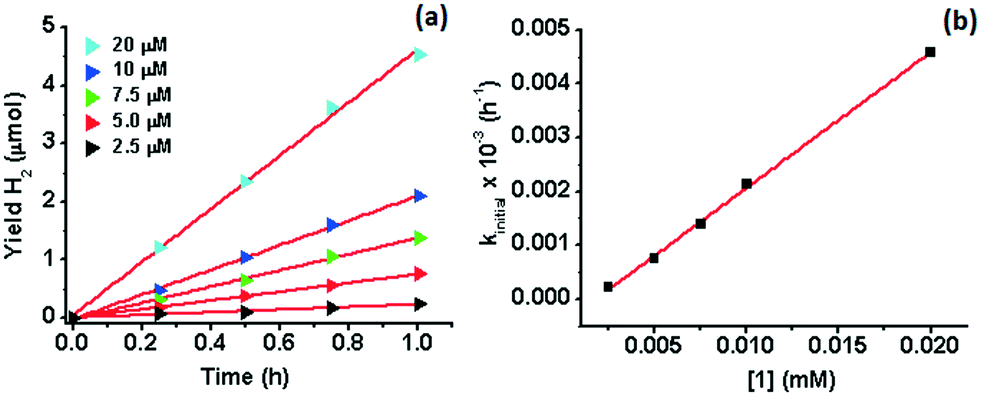 | ||
| Fig. 2 a) Hydrogen yields during the first hour of photolysis at varied concentrations of [1] in acetonitrile containing 10% water (v/v), 0.2 mM IrPS+ and 0.2 M TEA at 28 °C; b) plot of kinitial against [1]; slope = 0.2521 ± 0.0043, y-intercept = (−4.474 ± 0.47) × 10−7, R2 = 0.999. | ||
Metal-based nanoparticles generated by electro- or photochemical deposition are found to be real active species for some noble and non-noble metal WRCs.40–44 To verify whether the observed catalytic activity is caused by the colloidal particle formation rather than by 1 itself, the photolysis was performed in the presence of Hg0 (Fig. S3†). The activity of 1 does not change significantly (<20%) in the presence of Hg0 (0.5 mL). The temporal change of the particle size during the photoreactions (first 8 h) has also been monitored using dynamic light scattering (DLS) measurement (Fig. S4†). It falls in line with the above result; particle formation and significant change in particle size were not observed during the photoreactions in comparison with a fresh reaction solution, indicating that the catalysis is homogeneous. Similar results are also observed for 2.
The change of the absorption spectrum of acetonitrile solution containing IrPS+ (0.25 mM) and TEA (6.25 mM) upon irradiation with visible light (λ > 420 nm) is shown in Fig. S5.† New absorption bands are observed at 463, 497 and 532 nm after 120 s, which are consistent with those reported for electrochemically generated IrPS0.44 The quenching rate constants (kq) for IrPS+ with TEA, 1 and 2 have been determined with Stern–Volmer plots (Fig. S6†) at varied concentrations of the quenchers (kq = 1.2 × 109, 4.8 × 108 and 1.3 × 108, respectively). The measured kq for IrPS+ with TEA is comparable to the recently reported value (1 × 109).44 Thus, the photoreaction is believed to proceed predominantly by the reductive quenching of photoexcited IrPS+ to produce IrPS0, which then reduces the Co catalysts (CoII to CoI) to mediate proton reduction. The quantum yields of H2 generation over the first 2 h for 1 and 2 are similar (φ = 0.56), as determined using a chemical actinometer.
To reveal the nature of active species and redox processes involved, cyclic voltammetry (CV) of 1 and 2 was performed in acetonitrile (Fig. 3 and Table 2). In the CV of 1 (Fig. 3a), quasi-reversible and reversible couples are observed at 0.47 and −0.68 V vs. SCE, tentatively assigned to the CoIII/II and CoII/I processes. The quasi-reversible couples at −1.50 and −1.80 V are assigned to the ligand-centred L0/˙− and CoI/0 processes. The CV of 2 exhibits similar CoIII/II and CoII/I couples at 0.45 V and −0.98 V vs. SCE (Fig. 3b, red line). An irreversible L0/˙− couple (Ep,c = −1.61 V vs. SCE) is observed on further cathodic scanning (Fig. 3b, black line). In the subsequent anodic scan, it is noted that CoIII/II and CoII/I couples became less reversible. The irreversible L0/˙− process is probably associated with the deactivation of 2 during the photocatalysis. A comparison between the redox potentials of the two complexes reveals a substantial difference of E(CoII/I) by 0.30 V, while E(CoIII/II) and E(L0/˙−) are less sensitive to the change in the coordination sphere. Thus, tachpy3 is suggested to better stabilize both the coordination environment and the CoI state than trenpy3.
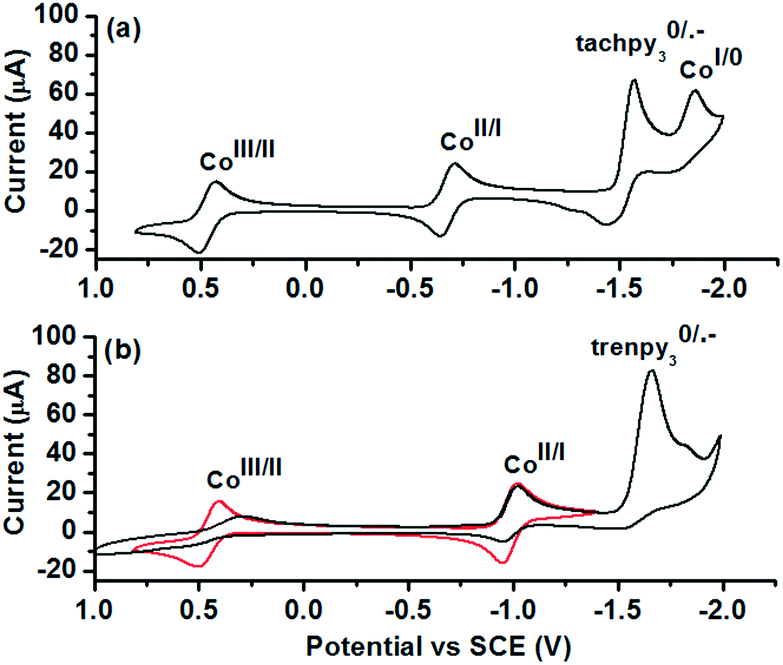 | ||
| Fig. 3 CVs of 1 mM (a) 1 and (b) 2 in 0.1 M nBu4NPF6 acetonitrile solution under an Ar atmosphere. Scan rate = 100 mV s−1. | ||
On addition of one mole equivalent of 4-bromoanilinium triflate (BAN), a new peak is observed in the CV of 1 at a slightly positive potential of the CoII/I couple (onset potential = −0.55 V). The peak current increases at higher BAN concentrations (1–6 mM), suggesting the catalytic reduction of protons via the CoI state (Fig. 4). A similar catalytic wave is also observed for 2 at a slightly positive potential of the corresponding CoII/I couple (onset potential = −0.76 V) in the presence of BAN. As [BAN] increases, the peak current of the wave increases, while the onset potential is anodically shifted. When aqueous citric acid (CA) was added instead of BAN, similar catalytic waves are also observed in the CVs at the same potentials (Fig. S7†). In the absence of the catalyst, both acids give rise to negligible reduction currents (Fig. S8†).
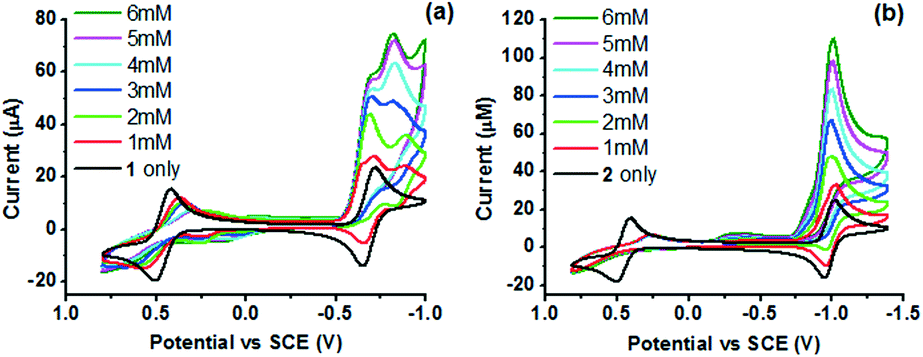 | ||
| Fig. 4 CVs of 1 mM (a) 1 and (b) 2 at varied concentrations of 4-bromoanilinium triflate ([BAN] = 1, 2, 3, 4, 5 and 6 mM) in 0.1 M nBu4NPF6 acetonitrile solution under an Ar atmosphere. Scan rate = 100 mV s−1. | ||
To characterize the intermediate involved in the protonation of the CoI state, controlled-potential electrolysis was performed (−1.0 V vs. SCE) with 0.1 M nBu4NPF6 acetonitrile solution containing 1 (1 mM). The solution was then diluted (×10) and analysed with ESI-MS (Fig. S9 and S10†). The observed single peak at m/z = 629.5 in the MS is assigned to the N2-adduct [Co(L) + H + PF6 + N2]+ (supported by the isotopic pattern) and suggests the formation of dicationic [CoI(η5-HL)(N2)]2+, which bears in the presence of solvent moisture a protonated tachpy3 ligand. Such a N2 adduct is not observed in the MS of the corresponding [CoII(L)]2+ (Fig. S10†). Under photochemical conditions, the more negative reductive IrPS0 state (−1.36 V vs. SCE)44 should in principle be sufficiently reducing to generate, in the presence of a proton source, similar five-coordinate [CoI(η5-HL)]2+. The formation of [CoI(η5-HL)]2+ upon protonation of [CoI(L)]+ is consistent with the observed new peak near the CoII/I couple and also with the DFT calculation (Fig. 6). CoIII hydride [CoIII(H)(η5-L)]2+ may then be formed via an intramolecular PCET process and further react to produce H2.15 The presence of a CoIII–H intermediate is supported by the CV of 1 (Fig. 4 and S7†). A new quasi-reversible couple is observed at −0.85 V vs. SCE, upon addition of 1 mole equivalent of the acids, and assigned to the [CoIII/II(H)(η5-L)]2+/+ process. A similar CoIII/II(H) couple was reported for a Co oxime WRC.15
X-ray crystal data obtained in this (Fig. S1, Table S1 and S2†) and previous work45 show that the CoII complexes 1 and 2 exhibit a trigonal prismatic and a distorted octahedral geometry, respectively. The energies of the possible spin states of the two complexes in acetonitrile were calculated by DFT methods.61–69 The high-spin states of 1 and 2 (with 3 unpaired d electrons) are predicted to be more stable than the low-spin states by 42.2 and 48.7 kJ mol−1, respectively. It is expected that only the high-spin state would be observed at room temperature. The measured effective magnetic moments (μeff) of 1 and 2 at room temperature are 5.11 and 4.54 BM, respectively, which are typical for a high-spin d7 configuration. The high-spin state nature of the two complexes at room temperature is also revealed in the UV-vis absorption spectra. By comparison of the absorption features, the experimental absorption spectra match with the calculated spectra of the high-spin states of the complexes, rather than the low-spin ones. The difference of the transition energies of the absorption features of the two complexes is reproduced by the calculation, but the energies are overestimated by 0.2–0.4 eV (Fig. S11†). Overestimation of absorption energy is commonly observed for other Co complexes.70,71
The optimized structures of 1 at the high-spin Co0, CoI and CoII states (Fig. 5 and S12, Tables S3 and S4†) exhibit a trigonal prismatic geometry. In contrast, an octahedral geometry is energetically favored for the low-spin CoII state. All optimized structures of 2 adopt a distorted octahedral geometry. The optimized structures for both complexes in the CoII state are therefore consistent with the experimental results from X-ray crystallography and spectrophotometry. A close look at the optimized structures of 1 and 2 gives possible explanations for the different coordination geometries. When comparing the degrees of freedom of the three imine N atoms, the tris(imino)cyclohexane group is less flexible compared to the tris(2-iminoethyl)amine moiety. Also, due to the partially filled Co dσ* orbitals, which are anti-bonding (Co–N) in nature, in the high spin state of 1 (in the oxidation state of Co0, CoI and CoII), their structures would have much longer Co–N bond lengths than the corresponding optimized structures in the low spin state. Thus, with longer Co–N in the high-spin state of 1, the C–N bonds between the imine and the cyclohexyl group will be strained if the complex adopted an (distorted) octahedral geometry. As a result, the distorted octahedral structure in the high spin state is much more unstable with respect to that in the trigonal prismatic geometry.
 | ||
| Fig. 5 Optimized structures of 1 in the a) CoII and b) CoI oxidation states. | ||
The reduction potentials of the two CoII/I redox couples were calculated using the differences of the Gibbs free energy of the optimized structures with CoII and CoI metal centers. The values of the calculated Co(II/I) reduction potentials for 1 and 2 are close to the experimental values and the large difference in reduction potentials observed in the experiment can be reproduced (Table 3). The calculated CoI/0 reduction potential for 1 is also consistent with the measured value. Given the fundamental difference in the coordination geometry for both the measured and optimized structures of the two complexes, the stabilized E(CoII/I) in 1 is attributed to its trigonal prismatic geometry.
| Calculated E°(CoII/I)/V vs. SCE | Calculated E°(CoI/0)/V vs. SCE | |
|---|---|---|
| 1 | −0.67 | −1.83 |
| 2 | −0.89 | — |
When the CoI state of 1 is optimized by the DFT method with an added proton, it is shown that the pyridine moieties of tachpy3 are more likely to be protonated. The protonation causes the pyridine moiety to dissociate from the CoI center, while the other five nitrogen-donor atoms are only slightly affected (Table S5†). Subsequently, a penta-coordinate structure [CoI(η5-HL)]2+ is obtained, in line with our experimental data (Fig. 6 and S10†). On the other hand, further reduction of the singly positively charged CoI state of 1 leads to an optimized structure which reveals the neutral doubly reduced [CoI(L˙−)]0 state and supports the assignment of ligand-centred reduction. The extra spin densities corresponding to the added electron would be mainly residing on the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)–C moiety of the two iminopyridine functions closer to the CoI centre, with a smaller amount also on the aromatic rings, (Fig. S13†), while much lower spin density would be located on the one further away.
C(H)–C moiety of the two iminopyridine functions closer to the CoI centre, with a smaller amount also on the aromatic rings, (Fig. S13†), while much lower spin density would be located on the one further away.
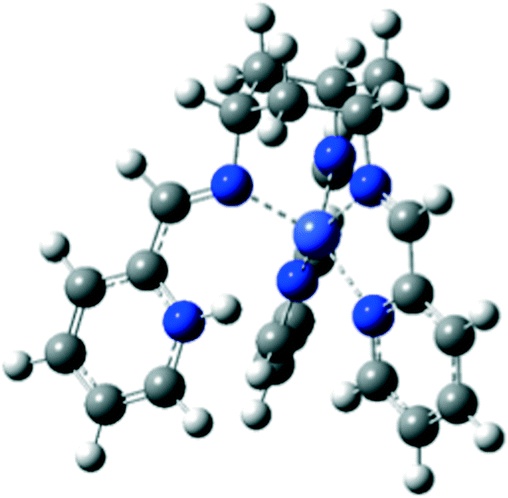 | ||
| Fig. 6 Optimized structures of 1 in the CoI state in the presence of one mole equivalent of proton. | ||
The above results demonstrate that replacing the tris(2-iminoethyl)amine moiety in 2 with tris(imino)cyclohexane in 1 has significantly enhanced the catalyst stability and lowered the thermodynamic barrier for proton reduction. 1 is found to be an active homogeneous WRC with impressive catalytic performance. The Co–N bond in the singly reduced [CoI(L)]+ state of 1 is destabilized in the presence of a proton source to give the five-coordinate [CoI(η5-HL)]2+, which may then give [CoIII(H)(η5-L)]2+ to produce H2. Further investigation on the reaction mechanism is now in progress and will be published in due course.
Experimental
Materials and methods
Compounds 1 and 2 were prepared according to literature procedures.45–47IrPS+ was synthesized using a modified literature method using 4,4′-dimethyl-2,2′-dipyridyl.55 4-Bromoanilinium triflate (BAN) was prepared by the addition of trifluoromethane sulfonic acids into a stirred solution of 4-bromoaniline in diethyl ether followed by recrystallization from acetonitrile/diethyl ether. 0.7 M aqueous citric acid (CA) was prepared using Mini-Q water. All other chemicals (Sigma Aldrich) were obtained commercially and used without further purification.Crystallography
Single crystals of 2 were obtained by recrystallization from acetonitrile/diethyl ether. Crystal data and experimental details are given in Table S1.† X-ray diffraction data were collected at 293 K on an Oxford CCD diffractometer using graphite-monochromated Cu KR radiation (λ = 1.54178 Å) in the ω-scan mode. Absorption corrections were performed by the multi-scan method. The structure was resolved by the heavy-atom Patterson method or direct methods, refined by full-matrix least-squares using SHELX-97 and expanded using Fourier techniques.56 All non-hydrogen atoms were refined anisotropically. H atoms were generated using the program SHELXL-97. The positions of H atoms were calculated on the basis of riding mode with thermal parameters equal to 1.2 times that of the associated C atoms and participated in the calculation of final R-indices. Selected bond lengths and bond angles are given in Table S2.†Electrochemical measurement
Cyclic voltammetry (CV) was carried out on a CH Instrument Electrochemical Station Model 660C, using a glassy-carbon working electrode, platinum-wire counter electrode and saturated calomel electrode (SCE), with 0.1 M nBu4NPF6 in acetonitrile (HPLC grade, Anaqua Chemical Supply) as the supporting electrolyte. In the CV experiments, BAN (1–6 mM) and aqueous citric acid (1–7 mM) were added. Anilinium was commonly used for studying electrocatalytic proton reduction in organic solutions.26 Aqueous citric acid (CA) was added instead of BAN as an aqueous proton source to mimic the photocatalytic conditions. Their low concentrations and weak acidity (pKa = 9.6 in CH3CN for BAN53 and pKa > 5 for CA; pKa of CA = 5.0 in 30% v/v H2O/CH3CN (ref. 54)) in acetonitrile should maintain the pH close to neutral and be relatively comparable to the photochemical conditions. Controlled-potential electrolysis was conducted using an ITO-plate working electrode, platinum-wire counter electrode and saturated calomel electrode (SCE). nBu4NPF6 was recrystallized in ethanol three times and dried in vacuo at 120 °C overnight before use. Ferrocene (Fc) was used as the internal reference in all measurements.Photocatalytic hydrogen generation
Photolysis was performed using a 10 mL Quickfit test tube (Pyrex) sealed with a serrated rubber septum. In general, a degassed acetonitrile solution (5 mL) containing the catalyst (1 or 2), IrPS+, TEA and H2O was irradiated using a white LED lamp (>420 nm) obtained from PRIME LED. The photocatalytic H2 generation was performed in unbuffered aqueous acetonitrile to avoid the potential influence of the protonated sacrificial donor (TEAH+) on the reductive quenching in the catalytic process.57 The content of the headspace was then analyzed by a gas chromatograph equipped with a thermoconductivity detector (GC-TCD) and a Chrompack 5A° molecular sieve column (25 m × 0.32 mm) at 45 °C. The H2 concentration was determined with reference to a calibration curve of a hydrogen standard prepared at one atmospheric pressure using a graduated syringe fitted with a three-way valve and a rubber septum.The quantum yield for hydrogen generation was determined as follows. A reaction solution (2 mL) in a 1 cm quartz cuvette sealed with a rubber septum was degassed by bubbling argon for 20 min and then irradiated at 430 nm for 2 h using a 500 W mercury–xenon lamp equipped with a Newport monochromator (Model 77200). The hydrogen content in the headspace of the cuvette was then determined by GC-TCD. The incident light intensity at 430 nm was taken from the average values measured just before and after the photolysis experiment using ferrioxalate as a chemical actinometer and was corrected for the absorbance of the actinometer solution at the same wavelength.58 The quantum yield of hydrogen generation (ΦH2) of the catalytic system is defined by eqn (1) and calculated on the basis of the total amount of hydrogen evolved after 2 h of irradiation:59,60
| ΦH2 = 2 × H2 produced (mole)/photon absorbed (Einstein) | (1) |
Luminescence quenching measurement
All luminescence quenching experiments were carried out by adding known amounts of quencher to 0.05 mM IrPS+ solution at room temperature. The solutions were then rigorously degassed on a high-vacuum line in a two-compartment cell for at least four successive freeze-pump-thaw cycles. Steady-state luminescence spectra of each sample were then recorded on a SPEX FluoroLog 3-TCSPC spectrofluorometer. The relative emission intensity or lifetime of the characteristic emission of IrPS+ in the presence of the different concentrations of the quencher was used to calculate the quenching rate constant (kq) using the Stern–Volmer equation (eqn (2)).60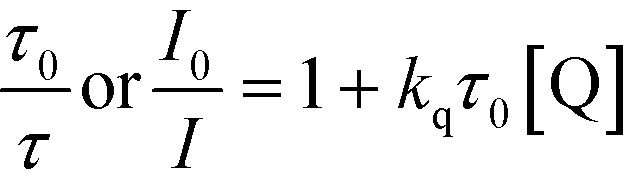 | (2) |
Dynamic light scattering
The light source for the scattering experiments was a He–Ne laser emitting vertically polarized light at a wavelength of 638 nm and operating at 4 mW. Data were collected at 20 °C with the scattering angle set to 173 degrees (non-invasive back-scatter), using an Avalanche photodiode detector connected to a correlator (Zetasizer nanoparticle analyser series, Nano ZS, ZEN 3600, Malvern Instruments).Magnetic susceptibility measurement
The molar susceptibility (XM) of 1 and 2 was determined using a Sherwood magnetic balance. The effective magnetic moment (μeff) was calculated using eqn (3), where T = temperature: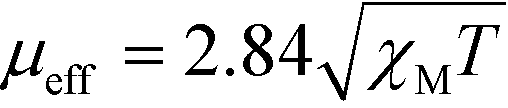 | (3) |
DFT calculations
Density functional theory (DFT) calculations on complexes 1 and 2 were carried out using the GAUSSIAN 09 program.61 The hybrid meta-GGA M06 functional and a mixed basis set of 6-31G(d) (for non-metals) and LANL2DZ effective core potential (for Co atom) were employed in the calculations.61–64 Frequency calculations were performed on the optimized structures and no imaginary frequencies were found. The solvent effect of acetonitrile was taken into account using the polarized continuum model (PCM).65 The electronic transition energies of the complexes were calculated by TD-DFT methods.66,67 The simulated UV-vis absorption spectra were generated68,69 by the combination of individual bands of Gaussian peak shapes with a full width at half maximum (FWHM) of 0.275 eV.Conclusions
Our study reveals that the Co tripodal iminopyridine complex [Co(tachpy3)](ClO4)2 (1) is an active homogeneous WRC and mediates H2 photogeneration at an impressive TON of 16440 which is amongst the highest in all Co WRCs reported in the absence of a secondary sacrificial donor. The catalysis by 1 is suggested to proceed via penta-coordinate [CoI(η5-HL)]2+ formed upon the protonation of the pyridine in [CoI(L)]+. A comparison with the structural analogue [CoII(trenpy3)](ClO4)2 (2), bearing the same donor groups, reveals that the difference in ligand rigidity and thus the coordination geometry results in significant change in catalytic properties. Complex 1 exhibits not only more anodic reaction potential but also much higher stability. Consistency between the electrochemical and computation data reveals that the coordination geometry above the metal active site, as imposed by the ligands, has significant influence on these catalytic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is financially supported by the HK Research Grants Council (project number 28300014 and 18300715) and EdUHK (04024:ECR16, 04255:SFRS8 and R3736:RG95). The Croucher Foundation is also acknowledged for the Visitorship for PRC Scholars (J. Xiang, 2016/17) in EdUHK.Notes and references
- K. Yamauchi, S. Masaoka and K. Sakai, J. Am. Chem. Soc., 2009, 131, 8404–8406 CrossRef CAS PubMed.
- H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227–2242 RSC.
- E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378–10388 CrossRef CAS PubMed.
- J. Xie, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Inorg. Chem., 2012, 51, 6376–6384 CrossRef CAS PubMed.
- A. M. Appel, D. L. DuBois and M. R. DuBois, J. Am. Chem. Soc., 2005, 127, 12717–12726 CrossRef CAS PubMed.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- H. I. Karunadasa, E. Montalvo, Y. J. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- I. Bhugun, D. Lexa and J.-M. Saveánt, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- S. Ott, M. Kritikos, B. Akermark, L. C. Sun and R. Lomoth, Angew. Chem., Int. Ed., 2004, 43, 1006–1009 CrossRef CAS PubMed.
- F. Gartner, B. Sundararaju, A. E. Surkus, A. Boddien, B. Loges, H. Junge, P. H. Dixneuf and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9962–9965 CrossRef PubMed.
- S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Angew. Chem., Int. Ed., 2010, 49, 8033–8036 CrossRef CAS PubMed.
- F. Wang, W.-J. Liang, J.-X. Jian, C.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2013, 52, 8134–8138 CrossRef CAS PubMed.
- T. Yu, Y. Zeng, J. Chen, Y.-Y. Li, G. Yang and Y. Li, Angew. Chem., Int. Ed., 2013, 52, 5631–5635 CrossRef CAS PubMed.
- M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310–8313 CrossRef CAS PubMed.
- X. L. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS PubMed.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- J. R. Long, C. J. Chang and Y. Sun, J. Am Chem. Soc., 2013, 2, 9212–9215 Search PubMed.
- A. Rodenberg, M. Orazietti, B. Probst, C. Bachmann, R. Alberto, K. K. Baldridge and P. Hamm, Inorg. Chem., 2015, 54, 646–657 CrossRef CAS PubMed.
- B. Shan, T. Baine, X. Zhao and R. H. Schmehl, Inorg. Chem., 2013, 20, 3164–3170 Search PubMed.
- B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133, 18070–18073 CrossRef CAS PubMed.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed.
- C.-F. Leung, Y.-Z. Chen, H.-Q. Yu, S.-M. Yiu, C.-C. Ko and T.-C. Lau, Int. J. Hydrogen Energy, 2011, 36, 11640–11645 CrossRef CAS.
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed.
- W. M. Singh, T. Baine, S. Kudo, S. L. Tian, X. A. N. Ma, H. Y. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed.
- P.-A. Jacques, V. Artero, J. Pécaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed.
- L. L. Efros, H. H. Thorp, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 1992, 31, 1722–1724 CrossRef CAS.
- M. P. McLaughlin, T. M. McCormick, R. Eisenberg and P. L. Holland, Chem. Commun., 2011, 47, 7989–7991 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- M. P. Stewart, M. H. Ho, S. Wiese, M. L. Lindstrom, C. E. Thogerson, S. Raugei, R. M. Bullock and M. L. Helm, J. Am. Chem. Soc., 2013, 135, 6033–6046 CrossRef CAS PubMed.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2013, 136, 356–366 CrossRef PubMed.
- W. Zhang, J. Hong, J. Zheng, Z. Huang, J. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680–20683 CrossRef CAS PubMed.
- J. Han, W. Zhang, T. Zhou, X. Wang and R. Xu, RSC Adv., 2012, 2, 8293–8296 RSC.
- H. Cui, J. Wang, M. Hu, C. Ma, H. Wen, X. Song and C. Chen, Dalton Trans., 2013, 42, 8684–8691 RSC.
- H. N. Kagalwala, E. Gottlieb, G. Li, T. Li, R. Jin and S. Bernhard, Inorg. Chem., 2013, 52, 9094–9101 CrossRef CAS PubMed.
- Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659–14669 CrossRef CAS PubMed.
- J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS PubMed.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- H. Junge, Z. Codolà, A. Kammer, N. Rockstroh, M. Karnahl, S. P. Luo, M. M. Pohl, J. Radnik, S. Gatla and S. Wohlrab, J. Mol. Catal. A: Chem., 2014, 395, 449–456 CrossRef CAS.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak and M. Robert, J. Am. Chem. Soc., 2012, 134, 6104–6107 CrossRef PubMed.
- L. A. Berben and J. C. Peters, Chem. Commun., 2010, 46, 398–400 RSC.
- S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- L. Chen, G. Chen, C. F. Leung, S. M. Yiu, C. C. Ko, E. Anxolabéhère-Mallart, M. Robert and T. C. Lau, ACS Catal., 2015, 5, 356–364 CrossRef CAS.
- R. A. D. Wentworth, P. S. Dahl, C. J. Huffman, W. O. Gillium, W. E. Streib and J. C. Huffman, Inorg. Chem., 1982, 21, 3060–3063 CrossRef CAS.
- E. Larsen, G. N. La Mer, B. E. Wagner, J. E. Parks and R. H. Holm, Inorg. Chem., 1972, 11, 2652–2667 CrossRef CAS.
- L. L. Tinker, N. D. McDaniel, P. N. Curtin, C. K. Smith, M. J. Ireland and S. Bernhard, Chem. – Eur. J., 2007, 13, 8726–8732 CrossRef CAS PubMed.
- Y.-J. Yuan, Z.-T. Yu, J.-G. Cai, C. Zheng, W. Huang and Z.-G. Zou, ChemSusChem, 2013, 6, 1357–1365 CrossRef CAS PubMed.
- K. Kawano, K. Yamauchi and K. Sakai, Chem. Commun., 2014, 50, 9872–9875 RSC.
- C. Bachmann, M. Guttentag, B. Spingler and R. Alberto, Inorg. Chem., 2013, 52, 6055–6061 CrossRef CAS PubMed.
- C. Bachmann, B. Probst, M. Guttentag and R. Alberto, Chem. Commun., 2014, 50, 6737–6739 RSC.
- J. Wang, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Catal. Sci. Technol., 2016, 6, 8482–8489 CAS.
- R. T. Edidin, J. M. Sullivan and J. R. Norton, J. Am. Chem. Soc., 1987, 109, 3945–3953 CrossRef CAS.
- J. Barbosa, J. L. Beltrán and V. Sanz-Nebot, Anal. Chim. Acta, 1994, 288, 271–278 CrossRef CAS.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- G. M. Sheldrick, SHELX-97, University of Göttingen, Göttingen, Germany, 1998 Search PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283 CrossRef CAS.
- S. L. Murov, I. Carmicheal and G. L. Hug, Handbook of photochemistry, Marcel Dekker Inc., New York, 2nd edn, 1993 Search PubMed.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Press, New York, 2006 Search PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- Creating UV/Visible Plots from the Results of Excited States Calculations, http://gaussian.com/uvvisplot/, (accessed September 2017) Search PubMed.
- Convoluting UV-Vis spectra using oscillator strengths, http://gausssum.sourceforge.net/GaussSum_UVVis_Convolution.pdf, (accessed September 2017) Search PubMed.
- G. Cioncoloni, H. M. Senn, S. Sproules, C. Wilson and M. D. Symes, Dalton Trans., 2016, 45, 15575–15585 RSC.
- M. Rudolph, T. Ziegler and J. Autschbach, Chem. Phys., 2011, 391, 92–100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1565200 contains the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cy01524k |
| This journal is © The Royal Society of Chemistry 2018 |