
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coiled coil protein origami: from modular design principles towards biotechnological applications
Fabio
Lapenta
a,
Jana
Aupič
a,
Žiga
Strmšek
a and
Roman
Jerala
 *ab
*ab
aDepartment of Synthetic Biology and Immunology, National Institute of Chemistry, Ljubljana, Slovenia. E-mail: roman.jerala@ki.si
bEN-FIST Centre of Excellence, Ljubljana, Slovenia
First published on 5th February 2018
Abstract
The design of new protein folds represents a grand challenge for synthetic, chemical and structural biology. Due to the good understanding of the principles governing its pairing specificity, coiled coil (CC) peptide secondary structure elements can be exploited for the construction of modular protein assemblies acting as a proxy for the straightforward complementarity of DNA modules. The prerequisite for the successful translation of the modular assembly strategy pioneered by DNA nanotechnology to protein design is the availability of orthogonal building modules: a collection of peptides that assemble into CCs only with their predetermined partners. Modular CC-based protein structures can self-assemble from multiple polypeptide chains whose pairing is determined by the interaction pattern of the constituent building blocks. Orthogonal CC sets can however also be used for the design of more complex coiled coil protein origami (CCPO) structures. CCPOs are based on multiple CC modules concatenated into a single polypeptide chain that folds into a polyhedral protein cage as the peptide segments assemble into CC dimers. The CCPO strategy has hitherto led to successful de novo design of protein cages in the shape of a tetrahedron, square pyramid and triangular prism. Recent advances in the design of CC modules and design principles have enabled the construction of CCPOs that self-assemble in vivo without any apparent toxicity to human cells or animals, opening the path towards therapeutic applications. The CCPO platform therefore has potential for diverse applications in biomedicine and biotechnology, from drug delivery to molecular cages.
 Fabio Lapenta | Fabio Lapenta received his MSc in Biotechnology from the University of Bologna, Italy, in 2014 and he is currently enrolled in the Biomedicine doctoral programme at the University of Ljubljana, Slovenia. He joined Prof. Jerala's group in 2015 with an Early Stage Researcher PhD fellowship from the Marie Skłodowska-Curie ITN TOLLerant project. His research is focused on biochemistry, structural biology and protein design. |
 Jana Aupič | Jana Aupič obtained her MSc in Chemistry from the University of Ljubljana, Slovenia. As a PhD student, she joined the Department of Synthetic Biology and Immunology at the National Institute of Chemistry, Slovenia, in September 2015, under the supervision of Prof. Jerala. Her research is focused on designing novel coiled coil pairs and developing computational tools for designing and modelling CCPO structures. |
 Žiga Strmšek | Žiga Strmšek obtained his MSc in Industrial Pharmacy and BSc in Biotechnology from the University of Ljubljana, Slovenia. Since July 2015 he has been working as a PhD student at the Department of Synthetic Biology and Immunology at the National Institute of Chemistry, Slovenia. The focus of his research is presentation of protein domains on CCPO structures in order to investigate potential biotechnological applications. |
 Roman Jerala | Roman Jerala is head of the Department of Synthetic Biology and Immunology at the National Institute of Chemistry in Ljubljana, Slovenia, and professor at the University of Ljubljana. Within synthetic biology he is investigating designed modular bionanostructures, particularly coiled-coil based protein origami, mammalian cell synthetic biology and medical applications of synthetic biology and within immunology he focuses on the molecular mechanism of signaling in innate immunity and on cancer immunotherapy. |
1 Introduction
Proteins are able to fold into a large variety of three-dimensional structures underlying different functions with the number of natural folds estimated in the order of thousands.1,2 Protein tertiary and quaternary structure is determined by a large number of weak, cooperative long- and short-range interactions. The folding of polypeptide chains is largely dominated by the hydrophobic effect. Most natural proteins comprise a dense packing of non-polar residues in their hydrophobic core and adopt a specific arrangement of secondary structure elements, while the precise geometry of side chains is defined by electrostatic and van der Waals interactions.3 Amino acid sequence variations enable generation of an almost countless number of proteins. However, only a tiny fraction of possible sequences code for defined protein structures. This large sequence and fold space clearly could not have been sampled by evolution.4Prediction of proteins’ tertiary structures based on their sequence is still challenging in the absence of homologs with known tertiary structure.5 Nonetheless, recent advances in computational protein design have enabled the creation of novel protein structures with high accuracy, even without relying on the sequence homology of any template.6,7
Biomimetic design of nanoscale molecular scaffolds and design of functional molecular machines represent motivation for exploring the space of protein folds. Proteins, due to their intrinsic biocompatibility and structural plasticity, represent an appealing material for both biotechnological and therapeutic applications. Even though proteins can be easily manufactured, the complexity of their folding landscapes hinders the prospect of designing new functional protein assemblies. In contrast, DNA nanotechnology based on the straightforward base-pairing complementarity of polynucleotide chains, while offering a much lower chemical versatility, enables design of complex programmable structures with high predictability and reliability.
1.1 Modular nanotechnology and modular origami
The characteristic structural flexibility possessed by nucleic acids has been successfully repurposed to construct complex high-order three-dimensional structures.8–10 In nature, RNA can fold into defined compact structures such as e.g. aptamers or combine with polypeptide chains in the ribosome, which is one of the largest and most complicated molecular machines.11 Due to the much higher chemical variability of amino acid side chains in comparison to nucleic acids, proteins have been selected by evolution as the principal structural and functional material, while polynucleotides have been designated for the conservation and transcription of genetic information due to their straightforward base-pairing complementarity and stability. These two properties, in combination with the possibility to synthesize synthetic polynucleotides of any desired length or sequence, underlined the invention of DNA nanotechnology. Researchers in this field constructed high-order molecular shapes mainly via design of multiple DNA chains that assemble in a highly predictable manner and form defined two- and three-dimensional structures reaching up to micrometer scale.12–14 Moreover, DNA nanotechnology was able to introduce dynamic rearrangement in complex structures to design molecular machines such as molecular walkers or information processing molecular devices.15,16 DNA nanotechnology typically involves either short multiple chains’ self-assembly or a single long chain structured via addition of multiple shorter chains (DNA origami) that are slowly annealed in vitro by temperature ramping or by slow dialysis.17,18 Although it has been recently demonstrated that the design of the folding pathway of DNA nanostructures is also able to encode rapid folding of single chain knotted structures,19 the multichain assembly strategy avoids the problem of kinetic traps due to the formation of knots. However, single chain strategies have important advantages over multichain self-assembly due to independence from concentration, which in turn facilitates technological or in vivo production.The combination of the versatility of polypeptides with the robustness of the DNA nanostructure design strategy could pave the way to construct new complex protein folds. This has been achieved by concatenation of multiple coiled coil (CC) orthogonal building modules, that mimic the pairwise complementary of nucleic acids, for the construction of polyhedral protein cages.20,21 As in the case of DNA origami, the designed 3D structure is defined by long-range interactions between complementary modules that direct the final self-assembly, whereas the DNA duplex modules are replaced by dimeric CC building modules. In the first demonstration, a tetrahedral cage was designed using a set of 12 orthogonal CC units that upon slow refolding assumed a regular shape, conferring to the tetrahedral cage fold the presence of a peculiar internal cavity.20 A recent publication extended the approach to polyhedra formed from 16 and 18 units, folding into a square pyramid and a triangular prism, respectively. Additionally, the successful expression in mammalian cells and in mice showed that coiled coil protein origami (CCPO) structures are stable and do not elicit adverse reactions in vivo.21
The purpose of this review is to illustrate the field of modular protein design relying on the orthogonally interacting CC as the basic structural unit. First, the special properties and designability of the CC motif are discussed, followed by a review of reported designed orthogonal CC sets. Then we describe the successful designs of protein nanostructures using orthogonal CC sets with an emphasis on CCPO structures and potential applications.
2 Modular coiled coil units
2.1 The coiled coil motif
Coiled coils represent a highly suitable building block for building modular protein structures due to relatively well-understood rules governing their folding and specificity. Coiled coils are one of the most widespread protein structure elements in nature, estimated to be present in as much as 10% of the eukaryotic proteome,22 where they perform both structural and functional roles, acting as protein–protein interaction domains and DNA-binding domains.23 Coiled coils are described by the interaction between two or more alpha helices that in a canonical form assume a twisted left-handed supercoiled structure with a seven residue periodicity (7/2) and a pitch angle of 20° (Fig. 1).24 Those structural parameters, initially proposed by Francis Crick, impose a regular, tight side chain packing interface termed as knobs into holes, which is permitted only by a distortion of the number of residues per turn from 3.6 in normal helices to 3.5 in CCs.25–27 The seven-amino-acids periodicity that confers structural regularity to the CC motif is typically called the heptad repeat, where each residue is commonly represented as a letter in the string abcdefg (Fig. 1). The helices that compose CCs are usually highly amphipathic and exhibit a strong affinity conferred by both hydrophobic and electrostatic interactions. In canonical dimeric CCs, hydrophobic residues occupy positions a and d, and polar residues occupy positions e and g. The former are important in establishing the tight knobs into holes packing while the latter determine the formation of salt bridges between the two helices (Fig. 1).28,29 Such regularity, in addition to specificity of binding, made CCs a malleable and versatile tool in the hands of protein engineers and they have been used in multiple ways in the last few decades.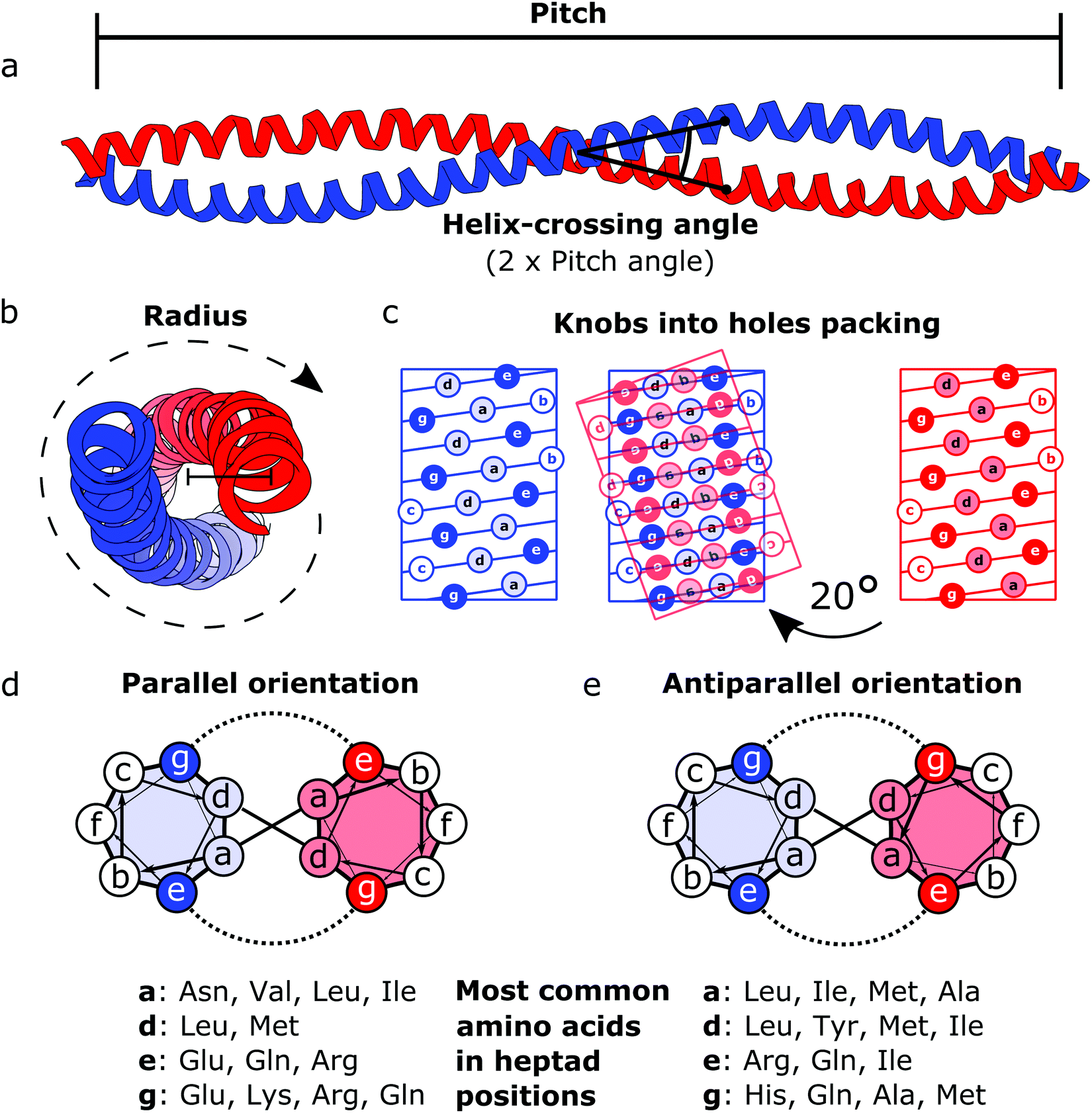 | ||
| Fig. 1 Schematic representation of the coiled coil structure and interaction pattern. (a and b) Side (a) and top (b) view of a representative coiled coil structure (PDB code 1C1G) depicting the characteristic parameters defining the final supercoiled structures. (c) Knobs into holes packing in dimeric coiled coils. (d) Heptad wheel representation of a parallel coiled coil with a list of most frequently observed amino acids on a, d, e and g position.29 (e) Heptad wheel representation of an antiparallel coiled coil with a list of most frequently observed amino acids in a, d, e and g positions.29 | ||
After the crystal structure of GCN4,30 a parallel homodimeric CC transcription factor in yeast, was solved at high resolution, several peptides were designed and characterized starting from GCN4, giving a more accurate description of the roles of core residues and of the relation between sequence and oligomerization propensity in CCs.31 For instance, one of the first examples of CC engineering, based on the GCN4 sequence, was the pairing system proposed for the design of the Peptide Velcro heterodimer.32 This synthetic heterodimer formed a stable complex consisting of two helices designed to have e and g positions occupied respectively by either lysine or glutamate residues, highlighting the importance of electrostatic interactions in these positions. Similarly, the peptides called EE and KK, also designed by exploiting electrostatic interactions between residues at e and g positions, showed a high degree of specificity33 and are still widely used as a model for CC interaction and for applications that require the heterodimerization complementarity.34,35 Charges can also be utilized to regulate the orientation of the two helices by matching complementary charges along the peptides. When designing the synthetic CC APH, Gurnon et al. enforced an antiparallel orientation to the homodimer by placing the appropriate amino acids in e and g positions in order to ensure an interaction between the two opposite termini of the peptides.36 Therefore, at least in principle, it is possible to look at CCs as a simplified study case in which protein–protein interaction surfaces can be engineered by the rule of thumb, resembling the simplicity of pairwise interactions that characterizes DNA. Although the correct formation of salt bridges provides a large thermodynamic contribution towards stable complexes, also van der Waals interactions and steric repulsion are involved in defining the CC specificity. As elegantly shown by systematically replacing residues in a and d positions with either leucine, isoleucine or valine residues in GCN4, Harbury et al. observed the formation of different oligomerization states and provided the insight regarding the influence of these buried residues on the assembly of CCs.31 Notably, polar amino acids can be found in buried positions along the hydrophobic patches of CCs.37,38 Taking as example the Peptide Velcro again, it has been shown how changing the position of a couple of buried asparagine residues could determine a change in the orientation from parallel to antiparallel.39 The ability to control the oligomerization state of CCs has been displayed by the successful design of a series of CC assemblies, which span from dimers to tetramers, via modification of the core residues in a and d positions.40 However, moving beyond dimers, helical bundles having higher oligomerization state can be built by extending the hydrophobic surface, engaging also e and g positions. A series of bundles, from natural pentamers all the way up to de novo designed hexameric41 and heptameric assemblies,42 were investigated,43 and indexed in a large set of CC structures.44 Although in classic dimeric CCs, b, c and f positions are not involved in protein–protein interactions, they play a role in determining the stability of CC dimers. Increasing the local helical propensity by formation of salt bridges via pairwise (i, i + 3) and (i, i + 4) interactions allows modulating the stability of the dimer without modifying the specificity of the interaction.45
2.2 Orthogonal coiled coil sets
The ability to construct complex modular protein assemblies depends on the availability of required building blocks. Whereas nature offers large sets of specific CCs,46 the design of toolsets of CC elements that bind their target with high specificity remains a challenge. Sets of CC elements that bind solely to their designated partner peptide and do not cross-interact, also called orthogonal sets, designed so far possess only a limited size. This is mostly due to the small free energy differences between the desired and off-target associations. To facilitate the design of CC nanostructures several orthogonal CC sets have been developed in the last decade, differing in size, length, and orientation of constituent peptides.One of the first examples were the α-helical tectons designed by Bromley et al.47 The set was composed of 6 three-heptad-long peptides that specifically formed 3 parallel CC heterodimers. Gradišar et al.48 reported the design of a set of 4 parallel CC heterodimers based on the combinations of patterns of charge interactions and a pattern of asparagine residues at heptad position a and evaluated by the energy scoring function introduced by Hagemann.49 The peptides comprised 4 heptads and contained a N-terminal capping sequence intended to stabilize the α-helical sequence. Orthogonal sets have been constructed from subgroups of synthetic peptides called SYNZIP.50 It was discovered that these peptides, initially designed to specifically bind the leucine zipper region of bZIP transcription factors,51 also exhibit strong hetero-association within the set. In vitro biophysical characterization of 14 SNYZIP peptides revealed that they were capable of assembling into 22 different heterodimeric CCs with groups of up to 4 CC pairs in each orthogonal set. Here, the peptides were of varying length, spanning 5–7 heptad repeats, which was reflected in the high stability of CC dimers, with the majority of the measured KD values in the nM range. Recently, Crooks et al. developed the largest CC set so far.52 Using the bCIPA algorithm53 a set of 8 parallel heterodimers was constructed comprising 4 heptad repeats. However, Tm measurements revealed that only 7 pairs behaved as designed with Tm > 70 °C and at least a 10 °C gap before the most stable off-target interaction.
Negron et al.54 reported the design of the only orthogonal set of antiparallel CC dimers found in the literature. Two sets of three homodimeric antiparallel CCs consisting of 6 heptads were designed. Three of the designed peptides preferentially formed antiparallel homodimers and were furthermore orthogonal to a previously designed antiparallel CC dimer,36 while higher order structures were observed for two of the designs. The availability of antiparallel orthogonal CCs is of high significance in the design of CC based protein origami, since it was shown that certain CCPO structures can be achieved only by inclusion of both parallel and antiparallel CC pairs.
The design of the above described orthogonal sets was achieved by a mixture of rational and computational approaches. Although different computational algorithms were used, they share similar core features. Firstly, the explored sequence space is restrained in accordance with previous rules discovered to govern CC oligomerization and pairing specificity. In the previously mentioned examples concerning the design of parallel CC sets,47,48,52 only lysine and glutamic acid were allowed at e:g positions, while the a heptad positions could be occupied either by asparagine or isoleucine and at d positions only leucine was allowed. Only a, d, e and g heptad positions were subjected to design, while b, c, and f positions were occupied by helicity promoting amino acids. Secondly, the interaction energy between peptide pairs is evaluated using sequence-based scoring functions as a weighted sum of terms for hydrophobic and electrostatic interactions. The scoring functions differ in weights used and inclusion of certain terms (e.g. helicity53).
In most cases,47,48,52 the above described sequence rules were used to construct a library of possible sequences based on the limited amino acid variability at interacting positions. Scores were then assigned to all possible peptide pairs and orthogonally interacting sets were selected from the library. Since this approach comes at a high computational cost, algorithms have been developed that optimize the energy gap between the on-target and off-target interactions already at the sequence design level.51,54
Interestingly, while the above described orthogonal CC sets are synthetic in nature, considerable degree of specificity was observed also for human bZIP transcription factors despite their high sequence homology.55
First successes of modular protein design exploiting orthogonally interacting CC sets highlighted the importance of such sets for the field of protein design. Strategically linking non-associating α-helical tectons via glycine–glycine linkers resulted in 6–9 nm long helical nanorods as revealed by CD, DLS and AUC measurements.47 Using the same strategy that yielded α-helical tectons an additional set of 3 heterodimeric CC was designed with two pairs interacting in a parallel fashion, while helix orientation in the third pair was antiparallel.56 The latter comprised 3 heptads, while parallel dimers were composed of 4 heptad repeats. The developed set allowed successful construction of a three-stranded chassis intended to function as a hub in synthetic molecular motors (Fig. 2a and c). The resulting assembly was verified via CD, AUC and MALDI-TOF measurements (Fig. 2a). SYNZIP peptides served as the basis for the design of a two-dimensional nano-triangle composed of three polypeptide chains. Similarly to nanorods, the triangle shape was specified by cleverly linking non-interacting peptide pairs by a 10 amino acid linker (Fig. 2b and d).57 A combination of structure characterization techniques (DLS, SAXS, AFM) confirmed the fusion proteins assembled into a triangular shape as intended with a characteristic particle dimension of 10 nm (Fig. 2b).
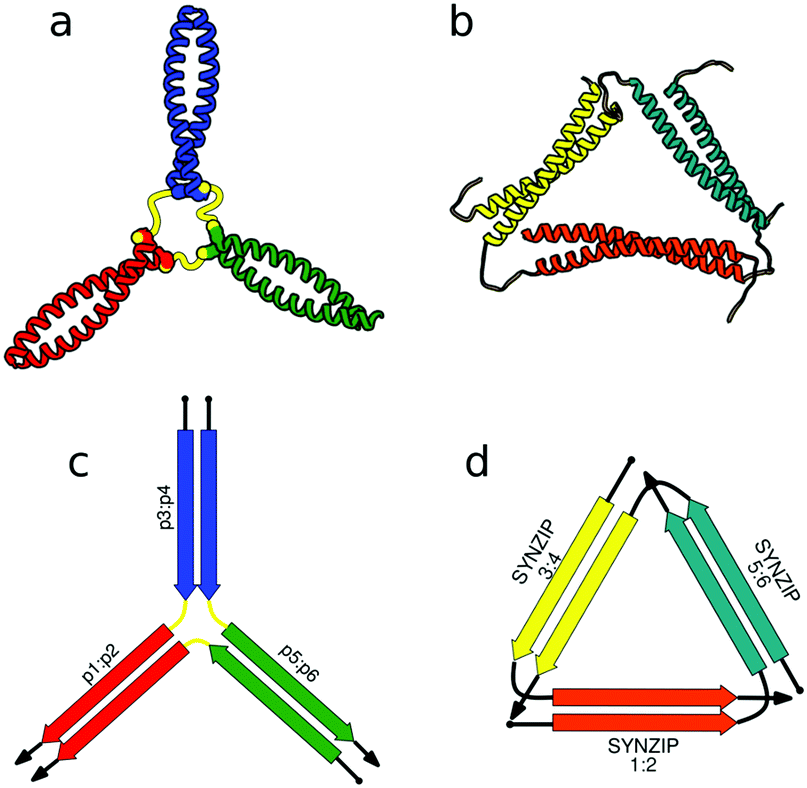 | ||
| Fig. 2 Models of nanostructures based on orthogonally interacting CC peptides. (a) Tripartite chassis formed by linking three CC dimers through disulfide bonds; the cysteine residues are visible as spheres.56 (b) Nano-triangle composed from three interacting peptide chains confirmed by SAXS analysis.57 (c and d) Respective schemes; CC segments are represented as colored arrows. | ||
In the above described studies, the final assemblies were achieved as a result of correct pairing between multiple polypeptide chains, which depended on concentration and equilibrium determined by the affinity of CC segments. However, discrete protein structures could be realized more accurately by connecting orthogonally interacting CC dimers into a single polypeptide chain.58 This strategy was utilized to design the CCPO structures, i.e. polyhedron-shaped protein cages (Fig. 3),20,21 from building blocks provided by the CC set introduced by Gradišar et al.,45,48 the APH toolkit54 and modified naturally occurring CC dimers.59,60 Since finding the right sequential order of peptide modules for more complex polyhedral shapes becomes quickly intractable using back-of-the-envelope approaches, this design strategy relies on the foundations established by the mathematical graph theory.
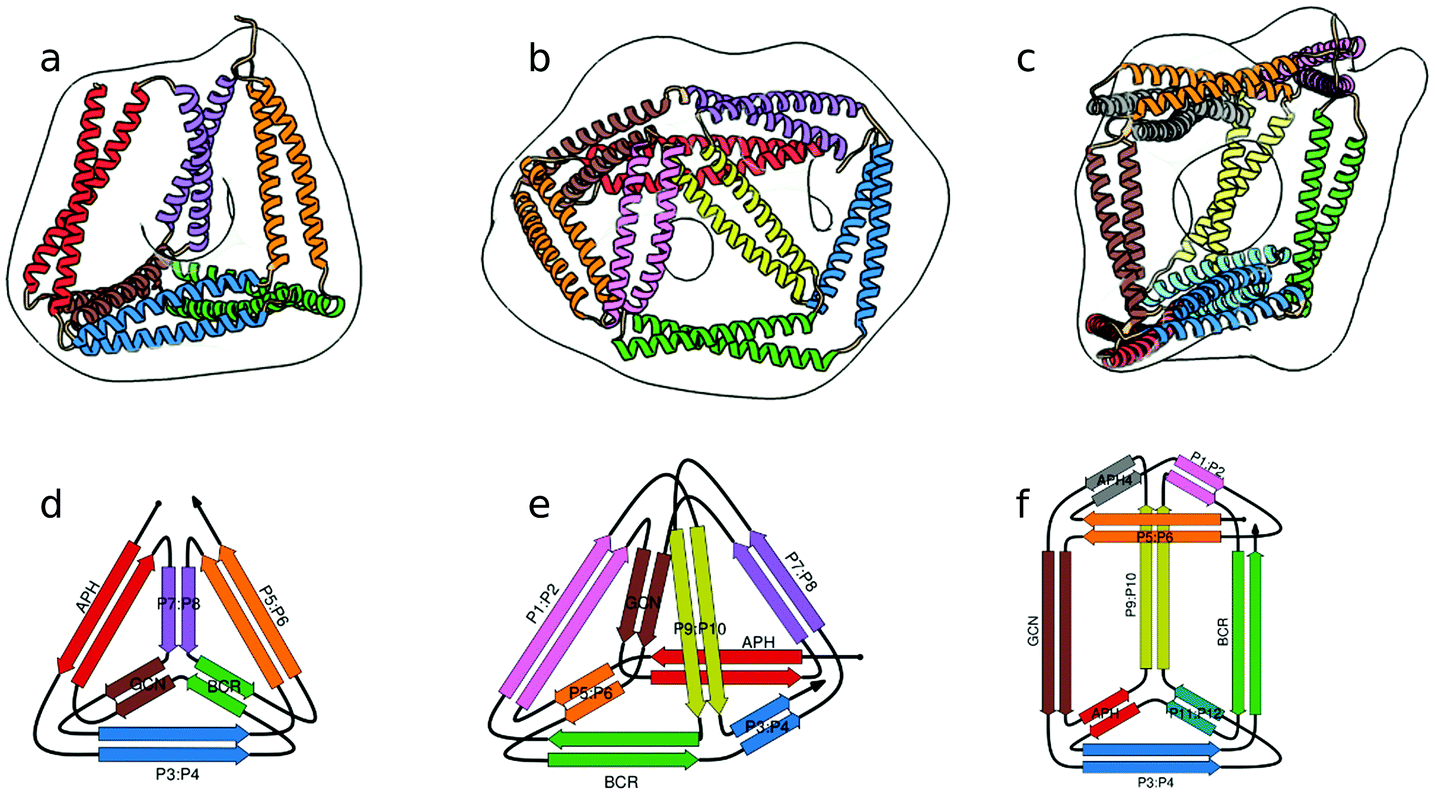 | ||
| Fig. 3 Model structure of representative CCPO cages in the shape of a (a) tetrahedron, (b) square pyramid and (c) trigonal prism, respectively composed of 12, 16 or 18 CC dimers, obtained from SAXS analysis and superimposed to volumes obtained by EM negative stain single-particle reconstruction.21 (d–f) Respective schemes; CC segments are represented as colored arrows. | ||
3 Coiled coil protein origami structures
3.1 Modelling of CCPO designs
At its core, the design of CCPO structures consists of connecting orthogonal CC peptides into a single-chain that will guide the polypeptide chain to fold into a polyhedron-shaped protein cage as the peptide modules self-assemble into intramolecular CC dimers forming the edges of the polyhedron. The task of finding the right arrangements of peptide segments is equivalent to the mathematical problem of finding a strong trace, a subset of double Eulerian paths, i.e. an oriented path that traverses each edge of the graph object exactly twice and interlocks the path into a stable structure, which means that all edges are connected with others in vertices.61,62 While the principles of designing CCPO structures have been described in detail,21 here we provide a brief overview and underline some important considerations.CCPO design can be divided into multiple steps (Fig. 4):
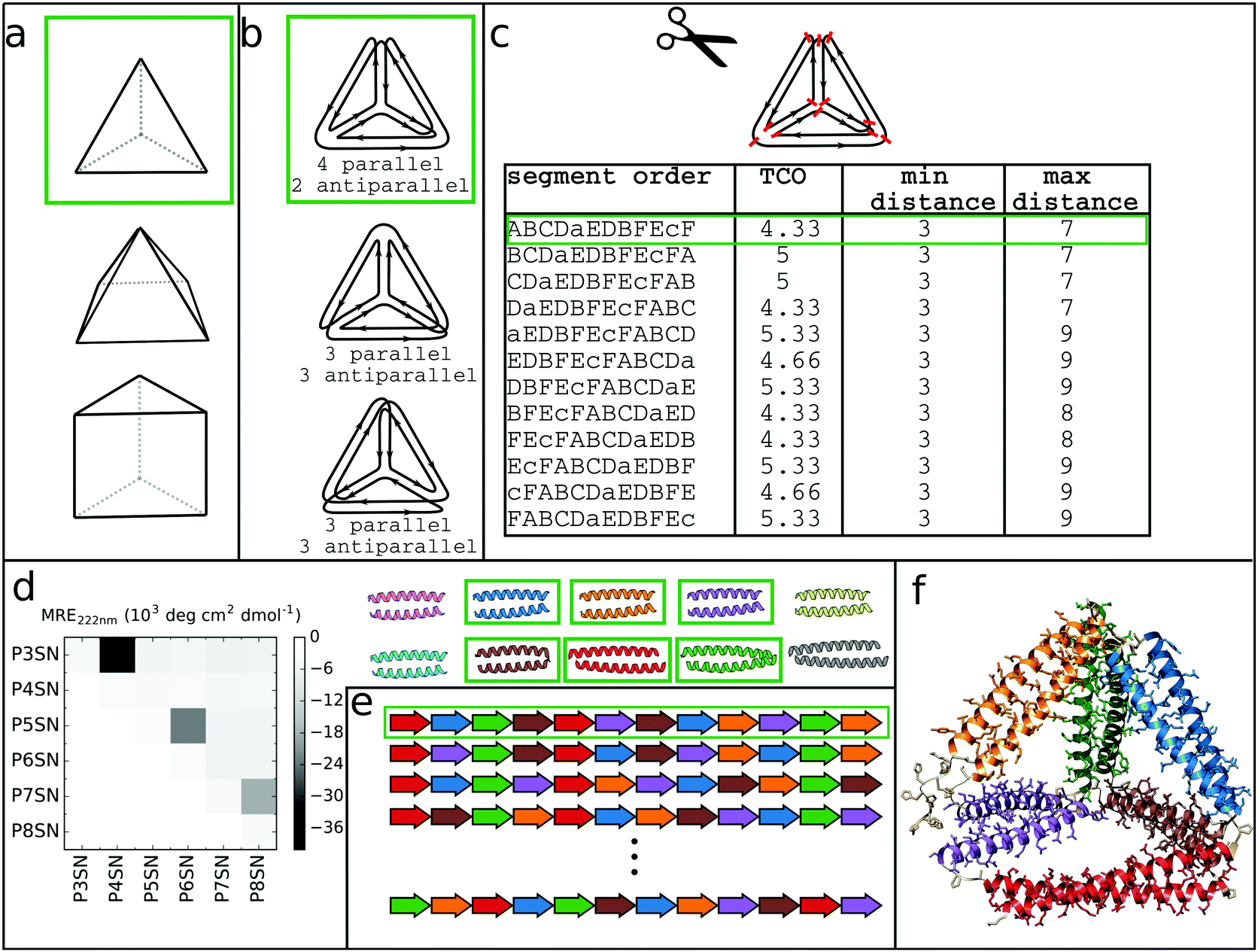 | ||
| Fig. 4 Steps involved in the design of CCPO structures. (a) Selection of the target shape. The target shape dictates the number of necessary orthogonal segments, a potential limiting factor for designing higher-order structures. (b) Construction and selection of a double Eulerian path. Different paths require the same total number of CC dimers; however the number of parallel vs. antiparallel pairs differs between paths. (c) Linearization of the double chain. TCO is the guiding parameter for selecting the best circular permutation. (d) Selection of needed CC building blocks. The heatmap shows the protein–protein interaction pattern for the orthogonal peptide set reported in ref. 21. (e) Placing CC-forming peptides into a single polypeptide chain according to the selected circular permutation. (f) Construction of the atomistic model for the designed polyhedral protein cage. | ||
3.2 De novo design of CCPO cages
The CCPO folds are based on a highly modular design strategy, based on long range, designable native contacts defined at the level of dimeric CC units to form the edges and guide the assembly of the cage. This strategy therefore bypasses the complexity of the design of cooperative protein core interactions. The affinity and specificity of CC segments to their partner modules underlies the formation of the CCPO protein fold. Therefore, the abovementioned importance of developing orthogonal CC sets assumes particular relevance for the construction of high order CCPO structures. CCPO cages form an internal cavity, whose shape and volume are determined by the geometry of the chosen polyhedron and the length of the edges. In a recent publication,21 the boundaries of CCPO design have been further extended to high order polyhedra with experimentally confirmed construction of cages possessing the shape of a square pyramid and a triangular prism in addition to alternative tetrahedral topologies. The first generation CCPO structures had to be refolded in vitro from the produced protein, as in most designs of DNA nanostructures, which limited the potential technological and therapeutic applications. One of the elements pivotal for the success of the second generation CCPOs was the design and usage of supercharged CC elements that ushered the correct in vivo self-assembly of CCPO under the physiological conditions, without the requirement of in vitro refolding steps.20 The design platform CoCoPOD provided a suitable environment for the design of different polyhedra, showcasing the utility of the developed software.Three representative structures, a tetrahedron, a square pyramid and a triangular prism, formed by 12, 16 and 18 CC segments respectively, were confirmed by both small angle X-ray scattering (SAXS) and single particle TEM reconstruction (Fig. 3). As an indication of the flexibility of these nanostructures, SAXS experiments revealed that the trigonal prism is present in solution in both rectangular and oblique conformation. Furthermore, Kratky plots of other CCPO structures also indicated partial flexibility. Since CC elements represent rigid modules, conformational changes are due to the flexibility of the loops, which allows angles of non-constrained faces a certain degree of freedom, resulting in cages with a limited conformational variability. In addition, the tetrahedron TET12SN was structurally characterized by application of chemical cross-linking coupled with proteolytic digestion and mass spectrometry,21 which can be employed to investigate the fold of modular CCPOs. Crosslinking was performed with three different reagents, DSS, BS(PEG)5, and BS(PEG)9, that can bridge Cα–Cα distances up to 2.4, 3.4 and 4.8 nm respectively, covering the range of distances relevant for the tetrahedral protein cage. After cross-linking, the protein was subjected to proteolytic cleavage resulting in crosslinked peptide fragments. The latter were analyzed using mass spectrometry. In the case of the shorter cross-linker, several connections between pairing CC segments were detected, confirming that in the context of TET12SN peptides assembled into CC dimers as expected. With the longer cross-linkers, BS(PEG)5 and BS(PEG)9, long-range crosslinks between non-neighboring pairs of peptides (in terms of sequence) were detected. These connections were consistent with distances observed for the corresponding peptide fragments during MD simulations of the model TET12SN cage, indicating that the polypeptide chain folded according to the design.
The increase in the complexity of CCPO structures is also reflected in the increase of TCO values (4.3, 5, and 5.6 for the CCPO tetrahedron, square pyramid and trigonal prism). Although CCPO structures are defined by long-range interactions between CC modules and not a tightly packed hydrophobic core, the kinetics of folding for the CCPO tetrahedron, square pyramid and trigonal prism, obtained via stopped-flow CD and stopped-flow FRET experiments,21 were discovered to be comparable to that of natural proteins of similar length.63 The experimentally determined secondary structure folding rates (17 s−1, 14 s−1 and 7.7 s−1, respectively for the tetrahedron, square pyramid and trigonal prism) were in agreement with the overall folding rates observed via the FRET effect of fluorescently labeled N- and C-terminal ends (31 s−1, 15 s−1 and 10 s−1 respectively for the tetrahedron, square pyramid and trigonal prism). Interestingly, the increase in TCO values was correlated with a decrease in folding rates as expected from theoretical considerations.64 However, this correlation, as well as the complete characterization of the folding pathways of CCPO cages (e.g. potential kinetic traps, folding rates after annealing), still requires additional studies that would offer a better understanding and a means of controlling the CCPO folding process. A further increase of the CC module length will likely introduce knotted CCPOs, which will represent strong kinetic barriers that will need to be considered but also exploited as demonstrated before for single chain DNA nanostructures.19 The current state of the art CCPO cages undergo a reversible unfolding process, retaining their monomeric state after temperature unfolding followed by rapid cooling, while at 4 °C they are stable for weeks. Successful testing of approx. 20 CCPO cages reflected the robustness of this strategy. Additionally, biophysical characterization and SAXS analysis of 10 tetrahedral variants and another four-squared pyramid confirmed the applicability of this strategy to differently composed polyhedra and helped to understand the rules governing the formation of these non-natural folds.
The novelty of this modular design strategy is in the atypical fold assumed by these cages, whose robust designability and formation of an internal cavity can, in turn, be used for different applications. Besides self-assembly in an in vitro transcription–translation reaction and bacterial production, CCPO structures also self-assembled in mammalian cells as well as in living animals. The correct folding was confirmed by reconstitution of protein reporters, fluorescent proteins and luciferase catalytic activity fused to the termini of the CCPO structures. Biocompatibility of the designed CCPO structures with mammalian cells was proved by monitoring inflammasome activation and unfolded protein response. In addition, the absence of inflammation and liver damage markers confirmed that the tested CCPO structures are not sensed by mammalian cells as foreign and adopt the correct native structure in vivo. Therefore, due to the lack of observable adverse effects in vivo, CCPO cages show considerable promise for biological applications.
4 Prospects of modular CCPO protein design for applications
The ability to accurately manipulate objects at the nano-scale level is advantageous for various applications from biomedicine, materials science to chemical technology and beyond. While a wide spectrum of different nanomaterials is already available,65–67 polypeptide-based materials own a specific combination of features such as programmability, ability to accommodate functional chemical moieties with nano-scale accuracy, self-assembly, biocompatibility, biodegradability and sustainable technological production that make them a highly suitable material for biomedicine and other technological applications.Designed proteins have already been used for production of nano-vaccines and drug delivery systems.68–70 Protein-based technologies are increasingly used to address the problem of producing new safer nano-vaccines.71 Subunit vaccines, composed of discrete molecular effectors, provide a safer and viable alternative to inactivated- or attenuated-pathogen based vaccines.72 The advantages of designed subunit vaccines reside in the controllability over molecular composition, the higher safety offered by the system and the control over size, shape and geometry. The close arrangement of epitopes in the crystalline lattice of Gp23, the major capsid protein of bacteriophage T4, provided a substrate with 7–10 nm spacing between epitopes that increased the antibody titer.73 Although polymeric nanoparticles such as PLGA or PGA and lipid nanoparticles have been used as carriers for subunit vaccines and drug delivery,65,74 protein-based nanoparticles feature strong controllability and high biodegradability in comparison to polymer-based particles, whose long-term effects on health are still not known.68
Self-assembling peptide nanoparticles (SAPNs) are based on a bottom-up approach, where the intrinsic affinity between the components leads to the formation of structurally defined nanocarriers that present multiple copies of antigens. Epitopes are usually fused to short peptides that assemble into fibers or compact particles. Protein self-assembling nanofibers conjugated to different antigens produced auto-adjuvant effects and elicited activation of antigen-specific T cell differentiation,75,76 for treatment of major diseases such as HIV, malaria, SARS and avian influenza.77–81 Parallel presentation of immuno-stimulatory compounds and antigens also represents an attractive strategy, as demonstrated by the use of the innate immunity TLR ligand in combination with antigenic protein.82–86
On the other hand, what protein engineering and de novo protein design brings to the table is the power to build novel and well-defined architectures at atomic accuracy.6 The computational protein design software such as Rosetta provides a platform for the design of customized immunomodulatory proteins, which mimic specific structural epitopes87–90 or function as peptide-based inhibitors.91,92 Epitope focused design and backbone grafting permit to move epitopes into structural scaffolds. This strategy allowed grafting HIV and RSV epitopes in protein scaffolds, which was shown to elicit neutralizing activity in animals.88,90 Protein design was also used to generate novel biocompatible inhibitors.89 This approach yielded extremely tight HA binders with IC50 < 150 pM that protected mice from viral infection91 and small interactors that effectively provided protection against viral infection in animals.92
Matching both correct size and presentation of antigens is a critical parameter in vaccine development.93 The size of modular CC assemblies can be regulated via modulation of CC length. Utilization of extended CC units or implementation of higher order structures could allow fine-tuning of the final size of CCPO structures. The other advantage offered by CCPO designs is the possibility to precisely engineer and design fusion partners, either chemically or genetically encoded, as for instance the fusion of natural protein reporters and fluorescent dyes.21
Manipulation of self-assembling protein modules is the key to achieve highly controllable protein cages for encapsulation and drug delivery. As for multimeric protein cages, subunits can be modified via different chemistries, fused to protein domains pointing either inwards or outwards and self-assembled in order to encapsulate diverse compounds.94 Protein cages showed promising results as drug delivery systems.94 Naturally occurring proteins, such as ferritin, vault proteins or viral capsids, have already been successfully developed into drug delivery systems.95–98 Notably, Hilvert and coworkers reengineered natural proteins such as lumazine and ferritin for encapsulation of small proteins into natural capsids via electrostatic interactions.99–101
However, designing protein cages from scratch can yield more versatile scaffolds for the purposes of direct applications such as drug delivery. Self-assembling natural domains have been employed to construct de novo oligomeric polyhedral cages as in the studies initiated by Yeates,102–104 that led to the development of computational procedures for the de novo design of multicomponent large cages.105–108
Cages can be constructed by employing a wide range of different building blocks, from bulky protein domains to smaller, rigid units, as in the case of CC elements. In particular, CCs were used as modular units either for direct self-assembly of multimeric hollow ∼100 nm large spheres109,110 or for the design of monomeric protein cages as in the case of CCPO structures. In comparison to other de novo cages, CCPO structures are the only example of assemblies that accommodate cavities within a single polypeptide chain.
Monomeric natural proteins possess internal cavities with an average volume of 0.25 nm3; larger cavities reach around 2.5 nm3 but often assume irregular shapes and typically occur at the protein–protein interaction surfaces (Fig. 5a).111 In contrast, multimeric protein cages either natural or de novo designed can accommodate large internal cavities, measuring from 40 nm3 to more than 4000 nm3 (Fig. 5b–d and f–g). Due to their peculiar fold, CCPO structures exhibit a large and hydrophilic cavity of approximately 40 nm3 (in the case of a tetrahedral fold) within a single polypeptide chain (Fig. 5e). In comparison to symmetry-based protein self-assemblies, where the cavity is tightly enclosed, CCPO cages offer a much more exposed cavity. The formation of such extensive cavities in a single polypeptide assembly makes CCPO design an attractive tool for targeted drug delivery and for molecular cages.
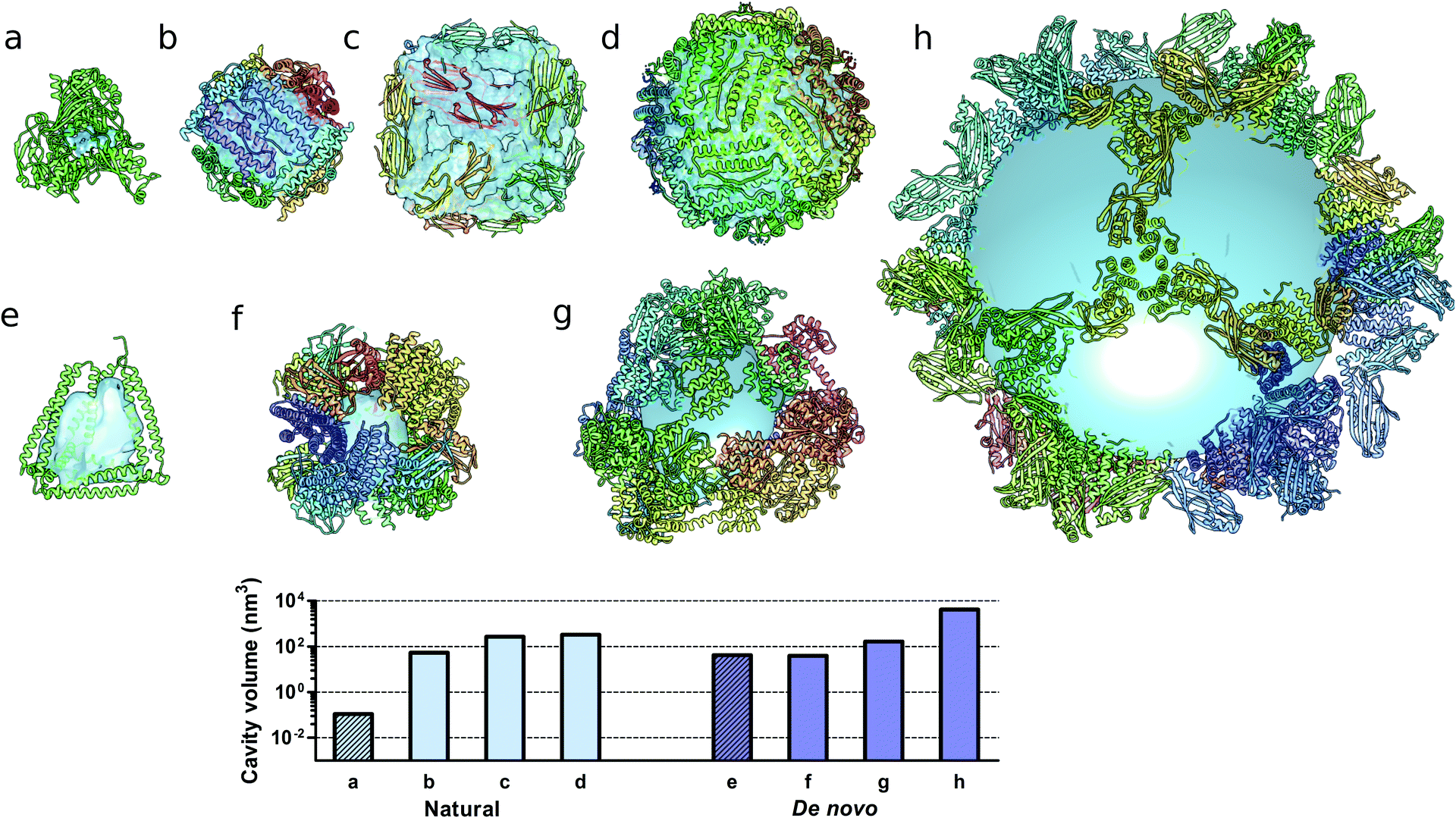 | ||
| Fig. 5 Molecular structures of protein cages and internal volumes. Each chain composing the final structure has a different color on a rainbow scale, and the internal cavities are represented as light blue surfaces. (a) Aldehyde oxidoreductase (PDB code 1VLB), a large monomeric protein having a small internal cavity. Multimeric natural assemblies: (b) DNA-binding protein from Starving cells (DPS) (PDB code 2YJJ); (c) small heat shock protein (PDB code 1SHS); (d) human ferritin (PDB code 2CEI). De novo designed proteins: (e) single-chain CCPO tetrahedron21 (f) 24-subunit assembly (PDB code 4NWP); (g) 12-subunit tetrahedron (PDB code 4ITV); (h) 120-subunit icosahedron (PDB code 5IM6). The plot at the bottom shows the volumes of the internal cavities for different proteins, the bars in light blue correspond to natural proteins and bars in dark blue correspond to de novo designed proteins. The internal volume of monomeric proteins is represented by diagonal lines patterned bars. The cavities were generated by using either Computed Atlas of Surface Topography of proteins (CASTp),123 Voss Volume Voxelator (3V)124 or a 10 nm radius sphere (h), and volumes were then calculated using UCSF Chimera.125 | ||
5 Conclusion and outlook
Diverse strategies to obtain polypeptide sequences that fold into a designed three dimensional structure are available and are continuously being improved.4,6,112,113 Due to the intrinsic adaptability of CC elements, design strategies that use sets of orthogonal CCs offer a solid and designable platform for the development of functional nanostructures. Examples of CC modules used to sense pH changes and drug release from liposomes114 as well as CC elements able to rearrange their structure upon binding of metal ions115,116 suggest that protein designs based on these modules could serve as scaffolds for developing conformationally flexible nanostructures. In comparison to design strategies based on single-state energy minimization of large folds, precise control over structural rearrangements is a major advantage offered by small and well-studied protein modules as exemplified by CCs based on azobenzene crosslinkers which trigger light-induced conformational rearrangements of CC helices.117,118Control over proteins' three dimensional rearrangement can readily be achieved by grafting flexible conformational hinges within rigid structural elements. In the context of CCPO structures, CC formation involves a conformational transition from unfolded monomers to structurally rigid dimeric units, an example of folding-upon-binding behavior, resembling some intrinsically disordered proteins.119 In the context of CCPO, the reversibility of the folding transition offers the possibility to further engineer CCPO folds into dynamic assemblies able to assume different conformations upon interaction.
Additionally, the flexible linkers intersecting CC units in CCPO cages offer an additional degree of freedom to polyhedral cages (as experimentally observed in the case of a trigonal prism), which in turn allows considerable movement of CC dimers affecting the volume of the internal cavity, leading to a large breathing capacity of the whole structure. CCPO structures represent an interesting example of modular yet monomeric protein assembly. Foremost, these folds do not rely on symmetric oligomerization and, therefore, permit the assembly of cages with addressable unique sites. Secondly, these cages possess a large cavity that could accommodate chemically linked compounds. Currently, the largest designed CCPOs comprise 700 amino acid residues resulting in one of the largest single chain protein designs. However, it is likely that construction of larger CC nanostructures will require assembly from several partially assembled subunits. Additionally, the expansion of CC orthogonal sets, in terms of both their number and their size, will facilitate the design of more complex modular folds. Besides, discrete multi-chain coiled coil protein assemblies can also be achieved based on the control of the angle between building blocks via linker length120 or charge repulsion.109,110
Modular CC-based designs exhibit several properties that make them appealing for therapeutic purposes. Such properties are also gaining importance in biotechnology, in particular in controlled catalysis, where the precise stoichiometric and spatial clustering of catalytic elements is important.121,122 In this regard, CCPO nanostructures may offer high customization. Furthermore, the biocompatibility of these novel folds, already demonstrated by in vivo studies,21 provides solid foundations for further development. In particular, understanding these novel folds and repurposing them for geometrical rearrangement of grafted moieties and molecular shielding represent interesting perspectives towards biotechnological applications, and while these challenges may require substantial efforts, recent advances in designing CC-based nanostructures offer all the reasons to be optimistic.
List of abbreviations
| CC | Coiled coil |
| CCPO | Coiled coil protein origami |
| CD | Circular dichroism |
| DLS | Dynamic light scattering |
| AUC | Analytical ultracentrifugation |
| TCO | Total contact order |
| RCO | Relative contact order |
| MALDI-TOF | Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry |
| SAXS | Small angle X-ray scattering |
| AFM | Atomic force microscopy |
| TEM | Transmission electron microscopy |
| DSS | Disuccinimidyl suberate |
| BS(PEG)5 | Bis-N-succinimidyl-(pentaethylene glycol)ester |
| BS(PEG)9 | Bis-N-succinimidyl-(nonaethylene glycol)ester |
| FRET | Förster resonance energy transfer |
| PLGA | Poly(lactic-co-glycolic acid) |
| PGA | Poly(glycolic acid) |
| SAPN | Self-assembling peptide nanoparticles |
| HIV | Human immunodeficiency virus |
| SARS | Severe acute respiratory syndrome |
| RSV | Respiratory syncytial virus |
| HA | Human influenza hemagglutinin |
Conflicts of interest
RJ is the author of the patent application on the design of self-assembling polypeptide polyhedra.Acknowledgements
The authors acknowledge the support of Slovenian Research Agency (program no. P4-0176, project N4-0037), MSC-RTN 642157 TOLLerant H2020 (R. J. and F. L.) and the ERANET SynBio project Bioorigami (ERASYNBIO1-006) and COST action CM1306. Dr Prof. B. Bromley, Dr L. Small and Dr Prof. A. Keating kindly provided the models used in Fig. 2a and b.Notes and references
- S. Govindarajan, R. Recabarren and R. A. Goldstein, Proteins: Struct., Funct., Genet., 1999, 35, 408–414 CrossRef CAS.
- R. D. Schaeffer and V. Daggett, Protein Eng., Des. Sel., 2011, 24, 11–19 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
- W. R. Taylor, V. Chelliah, S. M. Hollup, J. T. MacDonald and I. Jonassen, Structure, 2009, 17, 1244–1252 CrossRef CAS PubMed.
- M. Dorn, M. B. E. Silva, L. S. Buriol and L. C. Lamb, Comput. Biol. Chem., 2014, 53, 251–276 CrossRef CAS PubMed.
- P. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS PubMed.
- F. Parmeggiani, P. Huang, S. Vorobiev, R. Xiao, K. Park, S. Caprari, M. Su, J. Seetharaman, L. Mao, H. Janjua, G. T. Montelione, J. Hunt and D. Baker, J. Mol. Biol., 2015, 427, 563–575 CrossRef CAS PubMed.
- K. M. Weeks, Biopolymers, 2015, 103, 438–448 CrossRef CAS PubMed.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- H. F. Noller, Science, 2005, 309, 1508–1514 CrossRef CAS PubMed.
- C. Lin, Y. Liu, S. Rinker and H. Yan, ChemPhysChem, 2006, 7, 1641–1647 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- A. A. Greschner, V. Toader and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 14382–14389 CrossRef CAS PubMed.
- J. S. Shin and N. A. Pierce, J. Am. Chem. Soc., 2004, 126, 10834–10835 CrossRef CAS PubMed.
- R. N. Grass, R. Heckel, M. Puddu, D. Paunescu and W. J. Stark, Angew. Chem., Int. Ed., 2015, 54, 2552–2555 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- R. Jungmann, T. Liedl, T. L. Sobey, W. Shih and F. C. Simmel, J. Am. Chem. Soc., 2008, 130, 10062–10063 CrossRef CAS PubMed.
- V. Kočar, J. S. Schreck, S. Čeru, H. Gradišar, N. Bašić, T. Pisanski, J. P. K. Doye and R. Jerala, Nat. Commun., 2016, 7, 10803 CrossRef PubMed.
- H. Gradišar, S. Božič, T. Doles, D. Vengust, I. Hafner-Bratkovič, A. Mertelj, B. Webb, A. Šali, S. Klavžar and R. Jerala, Nat. Chem. Biol., 2013, 9, 362–366 CrossRef PubMed.
- A. Ljubetič, F. Lapenta, H. Gradišar, I. Drobnak, J. Aupič, Ž. Strmšek, D. Lainšček, I. Hafner-Bratkovič, A. Majerle, N. Krivec, M. Benčina, T. Pisanski, T. Ć. Veličković, A. Round, J. M. Carazo, R. Melero and R. Jerala, Nat. Biotechnol., 2017, 35, 1094–1101 Search PubMed.
- J. Liu and B. Rost, Protein Sci., 2001, 10, 1970–1979 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37–78 CrossRef CAS PubMed.
- A. N. Lupas, Trends Biochem. Sci., 1996, 21, 375–382 CrossRef CAS PubMed.
- F. H. C. Crick, Acta Crystallogr., 1953, 6, 689–697 CrossRef CAS.
- J. Walshaw and D. N. Woolfson, J. Struct. Biol., 2003, 144, 349–361 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37–78 CrossRef CAS PubMed.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS PubMed.
- J. Walshaw and D. N. Woolfson, J. Mol. Biol., 2001, 307, 1427–1450 CrossRef CAS PubMed.
- E. K. O’Shea, J. D. Klemm, P. S. Kim and T. Alber, Science, 1991, 254, 539–544 Search PubMed.
- P. Harbury, T. Zhang, P. Kim and T. Alber, Science, 1993, 262, 1401–1407 CAS.
- E. K. O’Shea, K. J. Lumb and P. S. Kim, Curr. Biol., 1993, 3, 658–667 CrossRef.
- N. E. Zhou, C. M. Kay and R. S. Hodges, J. Mol. Biol., 1994, 237, 500–512 CrossRef CAS PubMed.
- J. Yang, A. Bahreman, G. Daudey, J. Bussmann, R. C. L. Olsthoorn and A. Kros, ACS Cent. Sci., 2016, 2, 621–630 CrossRef CAS PubMed.
- M. Rabe, C. Aisenbrey, K. Pluhackova, V. de Wert, A. L. Boyle, D. F. Bruggeman, S. A. Kirsch, R. A. Böckmann, A. Kros, J. Raap and B. Bechinger, Biophys. J., 2016, 111, 2162–2175 CrossRef CAS PubMed.
- D. G. Gurnon, J. A. Whitaker and M. G. Oakley, J. Am. Chem. Soc., 2003, 125, 7518–7519 CrossRef CAS PubMed.
- X. Zeng, A. M. Herndon and J. C. Hu, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3673–3678 CrossRef CAS.
- D. L. Akey, V. N. Malashkevich and P. S. Kim, Biochemistry, 2001, 40, 6352–6360 CrossRef CAS PubMed.
- M. G. Oakley and P. S. Kim, Biochemistry, 1998, 37, 12603–12610 CrossRef CAS PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- N. R. Zaccai, B. Chi, A. R. Thomson, A. L. Boyle, G. J. Bartlett, M. Bruning, N. Linden, R. B. Sessions, P. J. Booth, R. L. Brady and D. N. Woolfson, Nat. Chem. Biol., 2011, 7, 935–941 CrossRef CAS PubMed.
- J. Liu, Q. Zheng, Y. Deng, C.-S. Cheng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15457–15462 CrossRef CAS PubMed.
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- O. D. Testa, E. Moutevelis and D. N. Woolfson, Nucleic Acids Res., 2009, 37, D315–D322 CrossRef CAS PubMed.
- I. Drobnak, H. Gradišar, A. Ljubetič, E. Merljak and R. Jerala, J. Am. Chem. Soc., 2017, 139, 8229–8236 CrossRef CAS PubMed.
- Y. Wang, X. Zhang, H. Zhang, Y. Lu, H. Huang, X. Dong, J. Chen, J. Dong, X. Yang, H. Hang and T. Jiang, Mol. Biol. Cell, 2012, 23, 3911–3922 CrossRef CAS PubMed.
- E. H. C. Bromley, R. B. Sessions, A. R. Thomson and D. N. Woolfson, J. Am. Chem. Soc., 2009, 131, 928–930 CrossRef CAS PubMed.
- H. Gradišar and R. Jerala, J. Pept. Sci., 2011, 17, 100–106 CrossRef PubMed.
- U. B. Hagemann, J. M. Mason, K. M. Müller and K. M. Arndt, J. Mol. Biol., 2008, 381, 73–88 CrossRef CAS PubMed.
- K. E. Thompson, C. J. Bashor, W. A. Lim and A. E. Keating, ACS Synth. Biol., 2012, 1, 118–129 CrossRef CAS PubMed.
- G. Grigoryan, A. W. Reinke and A. E. Keating, Nature, 2009, 458, 859–864 CrossRef CAS PubMed.
- R. O. Crooks, A. Lathbridge, A. S. Panek and J. M. Mason, Biochemistry, 2017, 56, 1573–1584 CrossRef CAS PubMed.
- J. M. Mason, M. A. Schmitz, K. M. Muller and K. M. Arndt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8989–8994 CrossRef CAS PubMed.
- C. Negron and A. E. Keating, J. Am. Chem. Soc., 2014, 136, 16544–16556 CrossRef CAS PubMed.
- J. R. S. Newman and A. E. Keating, Science, 2003, 300, 2097–2101 CrossRef CAS PubMed.
- L. S. R. Small, M. Bruning, A. R. Thomson, A. L. Boyle, R. B. Davies, P. M. G. Curmi, N. R. Forde, H. Linke, D. N. Woolfson and E. H. C. Bromley, ACS Synth. Biol., 2017, 6, 1096–1102 CrossRef CAS PubMed.
- W. M. Park, M. Bedewy, K. K. Berggren and A. E. Keating, Sci. Rep., 2017, 7, 10577 CrossRef PubMed.
- V. Kočar, S. Božič Abram, T. Doles, N. Bašić, H. Gradišar, T. Pisanski and R. Jerala, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 218–237 CrossRef PubMed.
- C. M. Taylor and A. E. Keating, Biochemistry, 2005, 44, 16246–16256 CrossRef CAS PubMed.
- K. J. Lumb, C. M. Carr and P. S. Kim, Biochemistry, 1994, 33, 7361–7367 CrossRef CAS PubMed.
- S. Klavzar and J. Rus, MATCH Commun. Math. Comput. Chem., 2013, 70, 317–330 CAS.
- G. Fijavž, T. Pisanski and J. Rus, MATCH Commun. Math. Comput. Chem., 2014, 71, 199–212 Search PubMed.
- K. W. Plaxco, K. T. Simons and D. Baker, J. Mol. Biol., 1998, 277, 985–994 CrossRef CAS PubMed.
- N. Koga and S. Takada, J. Mol. Biol., 2001, 313, 171–180 CrossRef CAS PubMed.
- T. A. P. F. Doll, S. Raman, R. Dey and P. Burkhard, J. R. Soc., Interface, 2013, 10, 20120740 CrossRef PubMed.
- S. Mitragotri, D. G. Anderson, X. Chen, E. K. Chow, D. Ho, A. V. Kabanov, J. M. Karp, K. Kataoka, C. A. Mirkin, S. H. Petrosko, J. Shi, M. M. Stevens, S. Sun, S. Teoh, S. S. Venkatraman, Y. Xia, S. Wang, Z. Gu and C. Xu, ACS Nano, 2015, 9, 6644–6654 CrossRef CAS PubMed.
- O. V. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef PubMed.
- C. P. Karch and P. Burkhard, Biochem. Pharmacol., 2016, 120, 1–14 CrossRef CAS PubMed.
- G. A. Khoury, J. Smadbeck, C. A. Kieslich and C. A. Floudas, Trends Biotechnol., 2014, 32, 99–109 CrossRef CAS PubMed.
- A. W. Purcell, J. McCluskey and J. Rossjohn, Nat. Rev. Drug Discovery, 2007, 6, 404–414 CrossRef CAS PubMed.
- M. Skwarczynski and I. Toth, Nanomedicine, 2014, 9, 2657–2669 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2005, 23, 1359–1366 CrossRef CAS PubMed.
- W. Baschong, L. Hasler, M. Häner, J. Kistler and U. Aebi, J. Struct. Biol., 2003, 143, 258–262 CrossRef CAS PubMed.
- K. A. Ghaffar, A. K. Giddam, M. Zaman, M. Skwarczynski and I. Toth, Curr. Top. Med. Chem., 2014, 14, 1194–1208 CrossRef PubMed.
- J. Chen, R. R. Pompano, F. W. Santiago, L. Maillat, R. Sciammas, T. Sun, H. Han, D. J. Topham, A. S. Chong and J. H. Collier, Biomaterials, 2013, 34, 8776–8785 CrossRef CAS PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- N. Wahome, T. Pfeiffer, I. Ambiel, Y. Yang, O. T. Keppler, V. Bosch and P. Burkhard, Chem. Biol. Drug Des., 2012, 80, 349–357 CAS.
- S. A. Kaba, C. Brando, Q. Guo, C. Mittelholzer, S. Raman, D. Tropel, U. Aebi, P. Burkhard and D. E. Lanar, J. Immunol., 2009, 183, 7268–7277 CrossRef CAS PubMed.
- S. A. Kaba, M. E. McCoy, T. A. P. F. Doll, C. Brando, Q. Guo, D. Dasgupta, Y. Yang, C. Mittelholzer, R. Spaccapelo, A. Crisanti, P. Burkhard and D. E. Lanar, PLoS One, 2012, 7, e48304 CAS.
- T. A. P. F. Pimentel, Z. Yan, S. A. Jeffers, K. V. Holmes, R. S. Hodges and P. Burkhard, Chem. Biol. Drug Des., 2009, 73, 53–61 CAS.
- S. Babapoor, T. Neef, C. Mittelholzer, T. Girshick, A. Garmendia, H. Shang, M. I. Khan and P. Burkhard, Influenza Res. Treat., 2011, 2011, 1–12 CrossRef PubMed.
- Y. Ding, J. Liu, S. Lu, J. Igweze, W. Xu, D. Kuang, C. Zealey, D. Liu, A. Gregor, A. Bozorgzad, L. Zhang, E. Yue, S. Mujib, M. Ostrowski and P. Chen, J. Controlled Release, 2016, 236, 22–30 CrossRef CAS PubMed.
- K. El Bissati, Y. Zhou, S. M. Paulillo, S. K. Raman, C. P. Karch, C. W. Roberts, D. E. Lanar, S. Reed, C. Fox, D. Carter, J. Alexander, A. Sette, J. Sidney, H. Lorenzi, I. J. Begeman, P. Burkhard and R. McLeod, npj Vaccines, 2017, 2, 24 CrossRef PubMed.
- D. J. Dowling, E. A. Scott, A. Scheid, I. Bergelson, S. Joshi, C. Pietrasanta, S. Brightman, G. Sanchez-Schmitz, S. D. Van Haren, J. Ninković, D. Kats, C. Guiducci, A. de Titta, D. K. Bonner, S. Hirosue, M. A. Swartz, J. A. Hubbell and O. Levy, J. Allergy Clin. Immunol., 2015, 140, 1339–1350 CrossRef PubMed.
- S. Burgdorf, C. Schölz, A. Kautz, R. Tampé and C. Kurts, Nat. Immunol., 2008, 9, 558–566 CrossRef CAS PubMed.
- K. J. Ishii and S. Akira, J. Clin. Immunol., 2007, 27, 363–371 CrossRef CAS PubMed.
- G. Ofek, F. J. Guenaga, W. R. Schief, J. Skinner, D. Baker, R. Wyatt and P. D. Kwong, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17880–17887 CrossRef CAS PubMed.
- B. E. Correia, Y. E. A. Ban, M. A. Holmes, H. Xu, K. Ellingson, Z. Kraft, C. Carrico, E. Boni, D. N. Sather, C. Zenobia, K. Y. Burke, T. Bradley-Hewitt, J. F. Bruhn-Johannsen, O. Kalyuzhniy, D. Baker, R. K. Strong, L. Stamatatos and W. R. Schief, Structure, 2010, 18, 1116–1126 CrossRef CAS PubMed.
- M. L. Azoitei, Y. E. A. Ban, J. P. Julien, S. Bryson, A. Schroeter, O. Kalyuzhniy, J. R. Porter, Y. Adachi, D. Baker, E. F. Pai and W. R. Schief, J. Mol. Biol., 2012, 415, 175–192 CrossRef CAS PubMed.
- B. E. Correia, J. T. Bates, R. J. Loomis, G. Baneyx, C. Carrico, J. G. Jardine, P. Rupert, C. Correnti, O. Kalyuzhniy, V. Vittal, M. J. Connell, E. Stevens, A. Schroeter, M. Chen, S. MacPherson, A. M. Serra, Y. Adachi, M. A. Holmes, Y. Li, R. E. Klevit, B. S. Graham, R. T. Wyatt, D. Baker, R. K. Strong, J. E. Crowe, P. R. Johnson and W. R. Schief, Nature, 2014, 507, 201–206 CrossRef CAS PubMed.
- E.-M. Strauch, S. M. Bernard, D. La, A. J. Bohn, P. S. Lee, C. E. Anderson, T. Nieusma, C. A. Holstein, N. K. Garcia, K. A. Hooper, R. Ravichandran, J. W. Nelson, W. Sheffler, J. D. Bloom, K. K. Lee, A. B. Ward, P. Yager, D. H. Fuller, I. A. Wilson and D. Baker, Nat. Biotechnol., 2017, 35, 667–671 CrossRef CAS PubMed.
- A. Chevalier, D.-A. Silva, G. J. Rocklin, D. R. Hicks, R. Vergara, P. Murapa, S. M. Bernard, L. Zhang, K.-H. Lam, G. Yao, C. D. Bahl, S.-I. Miyashita, I. Goreshnik, J. T. Fuller, M. T. Koday, C. M. Jenkins, T. Colvin, L. Carter, A. Bohn, C. M. Bryan, D. A. Fernández-Velasco, L. Stewart, M. Dong, X. Huang, R. Jin, I. A. Wilson, D. H. Fuller and D. Baker, Nature, 2017, 550, 74–79 CAS.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- N. M. Molino and S. W. Wang, Curr. Opin. Biotechnol., 2014, 28, 75–82 CrossRef CAS PubMed.
- Z. Yang, X. Wang, H. Diao, J. Zhang, H. Li, H. Sun and Z. Guo, Chem. Commun., 2007, 3453–3455 RSC.
- X. T. Ji, L. Huang and H. Q. Huang, J. Proteomics, 2012, 75, 3145–3157 CrossRef CAS PubMed.
- Z. Zhen, W. Tang, H. Chen, X. Lin, T. Todd, G. Wang, T. Cowger, X. Chen and J. Xie, ACS Nano, 2013, 7, 4830–4837 CrossRef CAS PubMed.
- T. Douglas and M. Young, Science, 2006, 312, 873–875 CrossRef CAS PubMed.
- T. Beck, S. Tetter, M. Künzle and D. Hilvert, Angew. Chem., Int. Ed., 2015, 54, 937–940 CrossRef CAS PubMed.
- E. Sasaki, D. Böhringer, M. van de Waterbeemd, M. Leibundgut, R. Zschoche, A. J. R. Heck, N. Ban and D. Hilvert, Nat. Commun., 2017, 8, 14663 CrossRef PubMed.
- S. Tetter and D. Hilvert, Angew. Chem., Int. Ed., 2017, 56, 14933–14936 CrossRef CAS PubMed.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 CrossRef CAS PubMed.
- Y. T. Lai, D. Cascio and T. O. Yeates, Science, 2012, 336, 1129 CrossRef CAS PubMed.
- Y. T. Lai, E. Reading, G. L. Hura, K. L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 6, 1065–1071 CrossRef CAS PubMed.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. André, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- Y. Hsia, J. B. Bale, S. Gonen, D. Shi, W. Sheffler, K. K. Fong, U. Nattermann, C. Xu, P. Huang, R. Ravichandran, S. Yi, T. N. Davis, T. Gonen, N. P. King and D. Baker, Nature, 2016, 535, 136–139 CrossRef CAS PubMed.
- J. B. Bale, S. Gonen, Y. Liu, W. Sheffler, D. Ellis, C. Thomas, D. Cascio, T. O. Yeates, T. Gonen, N. P. King and D. Baker, Science, 2016, 353, 389–394 CrossRef CAS PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- J. F. Ross, A. Bridges, J. M. Fletcher, D. Shoemark, D. Alibhai, H. E. V. Bray, J. L. Beesley, W. M. Dawson, L. R. Hodgson, J. Mantell, P. Verkade, C. M. Edge, R. B. Sessions, D. Tew and D. N. Woolfson, ACS Nano, 2017, 11, 7901–7914 CrossRef CAS PubMed.
- S. Sonavane and P. Chakrabarti, PLoS Comput. Biol., 2008, 4, e1000188 Search PubMed.
- J. Aupič, F. Lapenta, Ž. Strmšek and R. Jerala, Essays Biochem., 2016, 60, 315–324 CrossRef PubMed.
- L. Regan, D. Caballero, M. R. Hinrichsen, A. Virrueta, D. M. Williams and C. S. O’Hern, Biopolymers, 2015, 104, 334–350 CrossRef CAS PubMed.
- R. M. Reja, M. Khan, S. K. Singh, R. Misra, A. Shiras and H. N. Gopi, Nanoscale, 2016, 8, 5139–5145 RSC.
- X. I. Ambroggio and B. Kuhlman, J. Am. Chem. Soc., 2006, 128, 1154–1161 CrossRef CAS PubMed.
- E. Cerasoli, B. K. Sharpe and D. N. Woolfson, J. Am. Chem. Soc., 2005, 127, 15008–15009 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- A. M. Ali, M. W. Forbes and G. A. Woolley, ChemBioChem, 2015, 16, 1757–1763 CrossRef CAS PubMed.
- V. N. Uversky, Chem. Soc. Rev., 2011, 40, 1623–1634 RSC.
- A. L. Boyle, E. H. C. Bromley, G. J. Bartlett, R. B. Sessions, T. H. Sharp, C. L. Williams, P. M. G. Curmi, N. R. Forde, H. Linke and D. N. Woolfson, J. Am. Chem. Soc., 2012, 134, 15457–15467 CrossRef CAS PubMed.
- A. Heyman, Y. Barak, J. Caspi, D. B. Wilson, A. Altman, E. A. Bayer and O. Shoseyov, J. Biotechnol., 2007, 131, 433–439 CrossRef CAS PubMed.
- A. R. Chandrasekaran, Nanoscale, 2016, 8, 4436–4446 RSC.
- J. Dundas, Z. Ouyang, J. Tseng, A. Binkowski, Y. Turpaz and J. Liang, Nucleic Acids Res., 2006, 34, W116–W118 CrossRef CAS PubMed.
- N. R. Voss and M. Gerstein, Nucleic Acids Res., 2010, 38, W555–W562 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |