
Spontaneous protein desorption from self-assembled monolayer (SAM)-coated gold nanoparticles†
Ranran
Tian
,
Mengbo
Luo
* and
Jingyuan
Li
 *
*
Department of Physics, Zhejiang University, Hangzhou, 310027, China. E-mail: jingyuanli@zju.edu.cn; luomengbo@zju.edu.cn
First published on 27th November 2017
Abstract
When nanoparticles enter blood or other biological fluids, they tend to contact with a variety of proteins which may hamper their application and even bring adverse impacts. Such nonspecific protein binding is usually weak and proteins reach dynamic equilibrium between adsorption and desorption. Studies on the spontaneous desorption of weakly bound proteins should not only be helpful to enrich our understanding about such nonspecific interactions but also shed light on the strategy to avoid nonspecific binding of proteins. Here, we use molecular dynamics simulations to investigate the interactions of human serum albumin with the self-assembled monolayer (SAM)-coated gold(111) surface, a typical facet of various gold nanoparticles. The response of the protein interfacial behavior to solution pH, especially the spontaneous desorption, is studied. When the solution pH is relatively low, the protein can adsorb to the SAM surface. Such adhesion is attributed to several salt bridges between acidic residues and SAM's protonated groups, and there are water molecules distributed under the adsorbed protein, i.e. interlayer water. Interestingly, the increase of the solution pH reduces the binding affinity of the protein, which engenders the lateral diffusion of the protein and the increase of interlayer water. When the solution pH is further increased, the enhanced lateral diffusion of the protein and the growth of interlayer water disrupt the formation of salt bridges between the protein and the SAM, and the protein progressively dissociates from the SAM. The spontaneous desorption process of the protein and the corresponding mechanisms illustrated in this work may be helpful to develop an antifouling surface and enhance the biosafety of nanomaterials.
Introduction
Nanoparticles experience growing applications in the biomedicine field, including disease diagnosis, drug delivery and gene therapy.1–11 When nanoparticles enter blood or other biological fluids, they tend to contact with a variety of proteins (e.g., human serum albumin, fibrinogen and gamma globulin).12–16 Nonspecific interactions with proteins will hamper the targeting ability and delivery efficiency of the nanoparticles,14,17 and even bring adverse impacts.18–21 Therefore, it is highly desired to avoid the nonspecific binding of proteins. It has been recognized that such binding is reversible with a finite residence time.22 Moreover, the dynamic equilibrium between adsorption and desorption is vulnerable to the changes in the environment, such as temperature, solution pH and ionic strength.23–25 Studies on the desorption process of weakly bound proteins and the underlying mechanisms should be helpful to better understand their interfacial behavior and promote the desorption of non-specifically bound proteins.Extensive experimental works have investigated the effects of environmental factors on the interactions between proteins and nanoparticles, especially the impacts of solution pH.26–33 In most cases, efforts have been focused on the change of protein adsorption amounts. Proteins tend to aggregate on the nanomaterial surface at pH close to the isoelectric point of proteins, leading to the increase of their adsorption.26–28 Besides, the solution pH also influences the zeta potential of nanomaterials, which affects the interaction with proteins and the resulting protein adsorption. For example, at acidic pH (e.g., pH = 1) the cerium oxide nanoparticles adsorb much BSA because of electrostatic attraction with proteins, the adsorption decreases as pH increases.29 In short, protein adsorption can be regulated by the solution pH. However, detailed information about the response of the interactions between proteins and nanoparticles, such as the interaction mode, kinetic processes of protein adsorption and desorption as well as the underlying mechanism, still remains elusive.
Molecular dynamics (MD) simulations have been widely used to study the interactions between proteins and nanomaterials, such as graphene,16,34–36 carbon nanotubes,13,37 gold nanorods,15 gold nanospheres38 and monolayer-protected gold nanoparticles.39 Useful information about the protein adsorption, such as the adsorption process, binding stability and the influencing factors (e.g. the size and surface properties of nanoparticles), has been illustrated in these studies. A comprehensive understanding about the interactions between proteins and nanoparticles requires full description of the protein interfacial behavior. However, there is still limited work about other aspects of the interfacial behavior of proteins, e.g., the evolution of the adsorbed protein and the process of spontaneous desorption.
In the present study, we use MD simulations to investigate the protein adsorption, as well as the desorption of weakly bound proteins on the self-assembled monolayer (SAM)-coated gold(111) surface, a typical facet of various gold nanomaterials, including nanorods and prisms.15,40–43 The most abundant plasma protein, human serum albumin (HSA), is selected as the model protein, and the interfacial behavior of the protein in response to the change in the solution pH is systematically investigated. When the solution pH is relatively low, the protein can stably bind to the SAM surface. Such binding is largely attributed to several salt bridges formed by acidic residues with SAM's protonated groups, and there are water molecules distributed under the adsorbed protein (i.e. interlayer water). As the solution pH increases and the protonation degree of the SAM reduces, the adsorption stability of the protein dramatically decreases. The protein undergoes substantial lateral diffusion and the number of interlayer water increases. When the solution pH is further increased, the enhanced lateral diffusion of the protein, the growth of interlayer water and the sparse population of protonated groups disrupt the formation of salt bridges between the protein and the SAM, and the protein progressively dissociates from the SAM surface. In short, our results illustrate the kinetic process and the underlying mechanism of protein desorption which may be helpful for the development of an antifouling surface by enhancing protein desorption.
System and methods
Self-assembled monolayers (SAMs) are widely used to protect gold nanomaterials, such as gold nanorods, spheres, cages, and prisms, and such coatings profoundly affect the interactions with proteins.15,39–49 In the present work, a SAM is constructed using 10-amino-1-decanethiol (–S(CH2)10NH2, Fig. 1a), a typical alkanethiol used for the surface coating of gold nanomaterials.50 Moreover, the molecules are packed on the gold(111) surface with a √3 × √3 lattice structure.41,47,51–53 There are 1360 thiol chains, and the dimension of the SAM is 17.3 nm × 17.0 nm. According to Matulis's work, the pKa of –(CH2)10NH2 in aggregation is 10.23.54 The protonation degree of SAMs is 50%, 25% and 6.3% when the solution pH is 10.2, 10.7 and 11.4, respectively. There are a wide variety of functional groups whose protonation state can also be regulated by the solution pH,55 and they can similarly be exploited to construct pH-stimuli nanoparticles with different response intervals.45,46 For example, the pKa of N-ethylethylenediamine, diethyltoluine and N-acetyl-L-histidine monohydrate is around 7 and their protonation state may change during the circulation in blood. | ||
| Fig. 1 (a) Optimized structures of –S(CH2)10NH3+ (left) and –S(CH2)10NH2 (right). (b) The sketch of the 50%-protonated SAM. The amine (–NH2) and protonated amine (–NH3+) groups are shown as light blue and blue spheres, respectively. (c and d) Two initial configurations used in the simulation of protein adsorption, wherein HSA is placed with planes A and B orientated toward the SAM surface, respectively. Domains, I, II and III are colored in blue, green and magenta, respectively. | ||
The structure of human serum albumin (HSA) is obtained from the Protein Data Bank (PDB entry 1N5U).56 The corresponding protonation states of HSA residues at different pHs are determined by PDB2PQR 2.0.0 (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0).57 HSA is a heart-shaped protein composed of 583 residues with three domains (named I, II and III). A previous study has shown that HSA tends to adopt side-on orientations when it adsorbs on the hydrophilic surface.58 Therefore, there are two possible binding surfaces that are denoted by plane A and plane B (Fig. S1, ESI†).
First, SAMs with 50% and 25% protonation degree are prepared respectively (Fig. 1b and Fig. S2, ESI†). The SAMs and HSA are then equilibrated separately. Subsequently, HSA is placed at ∼5 Å above the SAMs with side-on orientations. The adsorption processes with both plane A and plane B orientating toward the SAMs are studied (Fig. 1c and d). Chlorine and sodium ions are added to maintain the system as neutral with an ionic strength of 0.2 M. After initial steepest descent minimization, 10 ns simulation is performed with all heavy atoms of both HSA and SAMs restrained to equilibrate the water molecules and ions. Next, 80 ns simulation is conducted to study the adsorption behavior of HSA on the 50% (i.e. adsorption-50%) and 25% (i.e. adsorption-25%) protonated SAMs.
To study the response of the adsorbed protein to the changes in the solution pH, we construct the other two systems on the basis of the final configuration of the system adsorption-50%, wherein the protonation degree of SAMs is switched to 25% (i.e. adjustment-25%) and 6.3% (i.e. adjustment-6.3%), respectively.59 The behaviors of adsorbed HSA in these two systems are investigated by 30 ns simulation.
All MD simulations are performed in the NPT ensemble (1 atm and 300 K). The pressure is controlled using a semi-isotropic Parrinello–Rahman barostat,60,61 and only the fluctuation in the z-axis is allowed. The temperature is maintained using a Nose–Hoover thermostat62 with a relaxation time of 0.5 ps. Parameters of SAMs are taken from the CHARMM General Force Field (CGENFF);63 and the CHARMM36 Force Field64 is adopted to describe protein molecules, sodium and chloride ions. The TIP3P model65 which is consistent with the chosen force fields is employed for water. The particle-mesh Ewald (PME) method66 is employed to calculate long-range electrostatic interactions, and a typical cutoff distance of 12 Å is applied to compute short-range electrostatic and van der Waals energies. Standard periodic boundary conditions are applied to all simulations, and the use of the LINCS algorithm allows a time step of 2 fs.67 All simulations are carried out using the Gromacs 4.6.5 package,68 and snapshots are rendered by the visual molecular dynamics (VMD) program.69
Results and discussion
The interactions between human serum albumin (HSA) and a series of self-assembled monolayer (SAM)-protected gold nanoparticles are systematically investigated. We first study the adsorption behavior of HSA onto the SAMs with 50% protonation degree (i.e. adsorption-50%, Fig. S3, ESI†). HSA can approach the SAMs and firmly adsorb on both plane A and plane B. And the binding stability is largely attributed to the salt-bridge interaction between the acidic residues of HSA and the protonated amine groups of SAMs. The acidic Asp or Glu residues are considered to form salt bridges with SAMs when their side-chain carbonyl oxygen atoms are found to be within 0.4 nm from the SAM protonated groups’ nitrogen atoms. The average number of salt bridges in both systems are calculated based on the last 10 ns trajectory. And the protein forms more salt bridges via plane A (12.5) than plane B (8.0). Therefore, the binding with plane A should be more stable and dominate the adsorption behavior of the protein.Fig. 2 shows that all three domains contribute to the adsorption of HSA with plane A, while only a fraction of the surface region directly contacts with the SAM (i.e. binding regions, Fig. 2b). And about 92.8% of the salt bridges are attributed to nine acidic residues, i.e. E167 and D173 (domain I), E277, E280, E297 and D301 (domain II), and E396, E442 and D563 (domain III), in these binding regions. In other words, the protein is essentially anchored by several salt bridges formed by these key residues. And there are water molecules distributed under the binding regions of the protein (denoted as interlayer water) after the adsorption is accomplished (Fig. 3a). Even though the protein adsorption does not fully exclude the hydration layer of SAMs, the approaching protein still needs to displace the hydration water. We compute the number of interlayer water (i.e. water molecules under the protein binding regions), and the number considerably decreases from ∼500 to ∼140 (Fig. 3b). Similar to many cases of the protein adsorbed onto hydrophilic surface,70–73 the interfacial water mediates the adsorption process of HSA onto the 10-amino-1-decanethiol (–S(CH2)10NH2)-protected gold nanoparticles.
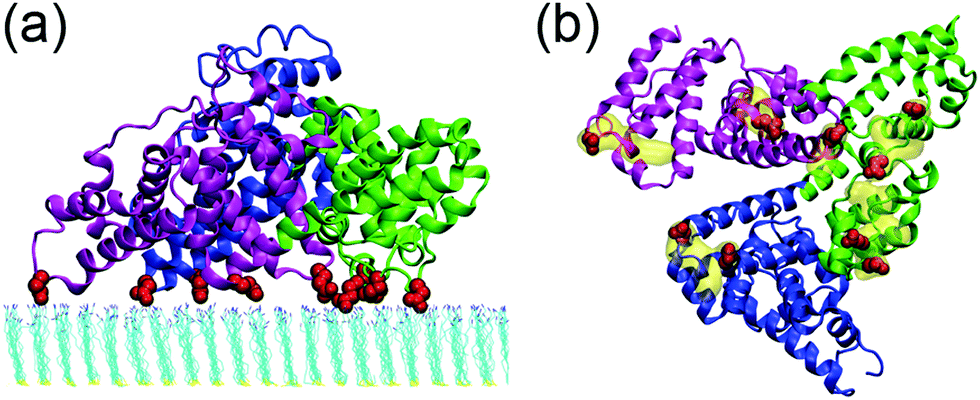 | ||
| Fig. 2 Snapshots of HSA adsorbed on the 50%-protonated SAM (i.e. adsorption-50%): side view (a); bottom view (b). Domains I, II and III are colored in blue, green and magenta, respectively. Acidic residues that form salt bridges with the SAM are colored in red, and the binding regions are highlighted in yellow. | ||
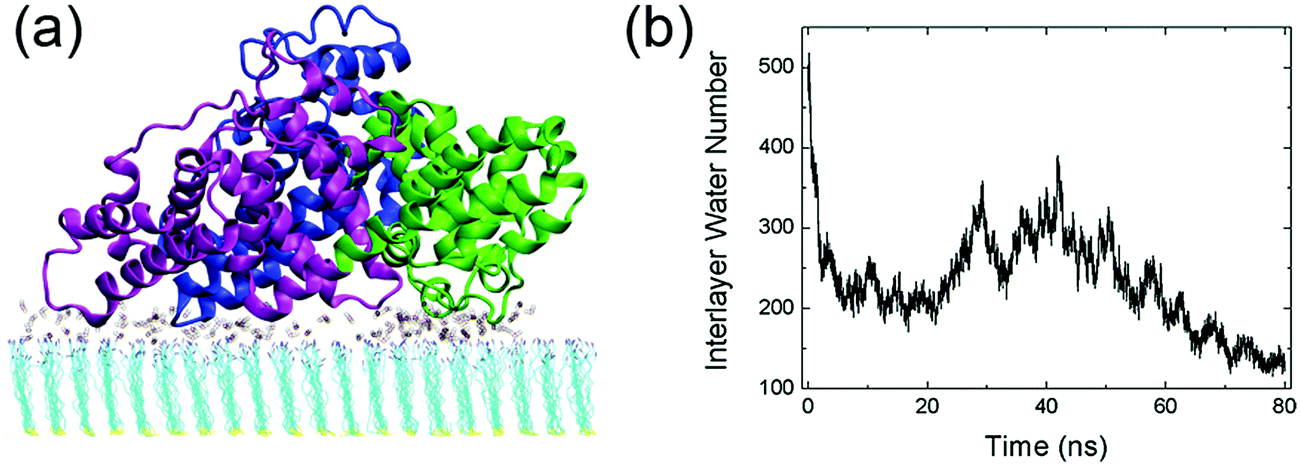 | ||
| Fig. 3 (a) The water molecules located under the binding regions of the protein, i.e. interlayer water. (b) The number of interlayer water during the adsorption process of the protein to the 50%-protonated SAM. | ||
The response of adsorbed HSA to the change of the solution pH is then investigated. As the solution pH increases from 10.2 to 10.7, the protonation degree decreases to 25% correspondingly. HSA remains bound to the SAM (Fig. 4a, adjustment-25%), while the interaction between the protein and SAM is substantially weakened. More interestingly, the protein undergoes considerable lateral diffusion on the SAM surface. The self-diffusion coefficient of HSA in both adsorption-50% and adjustment-25% is then determined by measuring its in-plane mean square displacement (based on the last 10 ns trajectory, Fig. 4b). The calculated self-diffusion coefficient of HSA in adjustment-25% is 2.6 × 10−7 cm2 s−1, which is comparable to the other weakly adsorbed proteins (e.g., Aβ trimer on the COOH-SAM, 2.3 × 10−7 cm2 s−1)74 and one order of magnitude larger than the self-diffusion coefficient of the protein in adsorption-50% (2.8 × 10−8 cm2 s−1). Such enhanced lateral diffusion of HSA should reciprocally contribute to the reduction of adsorption stability. On top of it, there are more water molecules distributed under the binding regions of the protein. The average number of interlayer water molecules increases to ∼370 (based on the last 10 ns trajectory, Fig. S4a, ESI†), much higher than the case of adsorption-50% (∼150). It should be noted that some water molecules are in the vicinity of salt bridges formed by acidic residues with protonated amine groups. Some of these water molecules form hydrogen bonds with the salt-bridge groups (Fig. S5, ESI†), and the increase of interlayer water should also disturb the interactions between HSA and the SAM. The average number of salt bridges (i.e. the last 10 ns of the simulation, Fig. S4b, ESI†) is only 1.9, which is one-sixth of the corresponding number in adsorption-50%. That is, the extent of salt bridge reduction is more dramatic than the reduction of the SAM's protonation degree. Besides, only two domains (domain II and III) participate in the protein adsorption, and the salt-bridge interactions are mainly attributed to three residues: D301 of domain II, E396 and D563 of domain III. The distance between the mass center of HSA and the SAM surface is about 3.1 nm, which is considerably higher than that in adsorption-50% (2.5 nm, see Fig. S6, ESI†). Taken together, the increase of the solution pH and the concomitantly decreased protonation degree of the SAM considerably affect the interfacial behavior of the adsorbed protein.
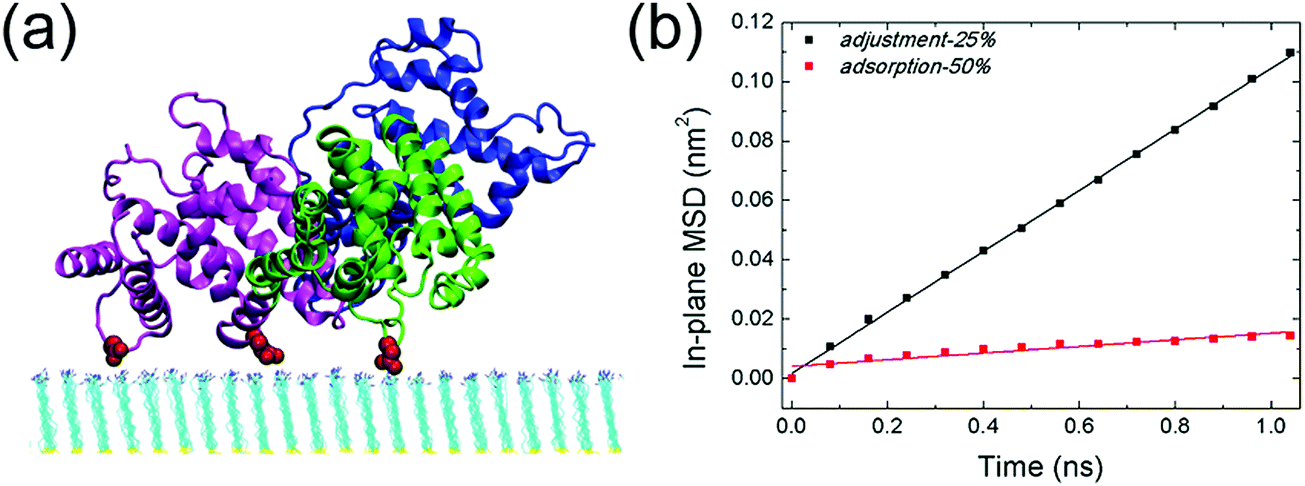 | ||
| Fig. 4 (a) Final configuration of adsorbed HSA when the protonation degree of the SAM is decreased to 25% (i.e. adjustment-25%). Domains I, II and III are colored in blue, green and magenta, respectively. Acidic residues that form salt bridges with the SAM surface are highlighted in red. (b) The in-plane mean square displacement (MSD) of HSA in adsorption-50% (red) and adjustment-25% (black). | ||
To further characterize the behaviors of HSA on the 25%-protonated SAM surface, we also assess its adsorption behavior (i.e. adsorption-25%). The protein can approach and bind to the SAM. The adsorbed protein similarly undergoes substantial lateral diffusion on the SAM surface, and the corresponding diffusion coefficient is 4.1 × 10−7 cm2 s−1 (based on the last 10 ns trajectory, Fig. S7, ESI†), comparable to the value of adjustment-25%. The average number of salt bridges (Fig. S8, ESI†) is 2.0, which is also close to the case of adjustment-25%. Besides, there are also two domains (domains II and III) involved in the adsorption of HSA (Fig. S9, ESI†), and water molecules are similarly distributed in the region between the protein and SAM. Moreover, the distance between the mass center of HSA and the SAM surface (∼3.0 nm, Fig. S10, ESI†) is still similar to the case of adjustment-25%. In short, the interfacial behavior of the protein in adsorption-25% converges with the adjusted behavior of the pre-adsorbed protein in adjustment-25% and is distinct to the case of adsorption-50%. Such an interfacial behavior can thus be denoted as the weakly-bound state. In addition, our results suggest that HSA can still adsorb on the surface as long as there are two salt bridges.
In addition, we examine the response of the adsorbed protein when the protonation degree of the SAM is further decreased to 6.3% (i.e. adjustment-6.3%). In this case, the adsorption stability of HSA continuously decreases and the protein essentially desorbs from the surface after t = 21 ns (Fig. 5): HSA no longer forms salt bridges with the SAM, and the average number of heavy atoms in contact with the SAM (within 0.5 nm from nitrogen atoms, the uppermost heavy atoms, of the SAM) becomes merely 0.4. Moreover, the protein progressively moves away from the SAM: the mass center distance, i.e. the distance between the mass center of HSA and the SAM surface, increases from 2.4 nm to 4.1 nm.
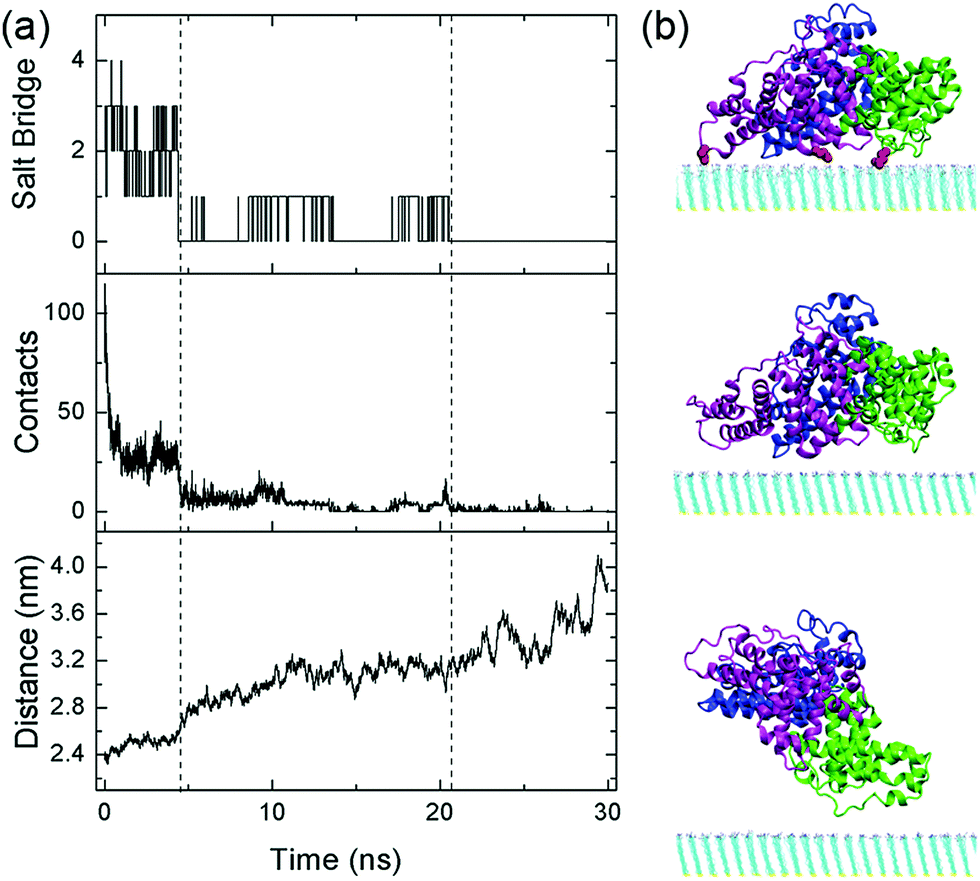 | ||
| Fig. 5 Desorption process of HSA when the protonation degree of the SAM is decreased to 6.3% (i.e. adjustment-6.3%). (a) The salt bridge number, heavy atom contact number as well as the mass center distance between HSA and the SAM (from top to bottom). (b) Representative snapshots at t = 0, 4.5 and 30 ns (top, middle, bottom). Domains I, II and III are colored in blue, green and magenta, respectively. Acidic residues that form salt bridges with the SAM are highlighted in red. | ||
Even in the early stage of desorption (the first 4.5 ns), the protein undergoes considerable lateral diffusion with a diffusion coefficient of 1.4 × 10−7 cm2 s−1. Such diffusion indeed disrupts the interaction with the SAM. The number of salt bridge decreases to 0, and the heavy atom contact number decreases dramatically to about 10. Meanwhile, there are more water molecules distributed in the region between the protein and the SAM. The number of interlayer water increases from ∼140 to ∼240 at t = 4.5 ns (Fig. S11, ESI†). As mentioned above, the growth of interlayer water disturbs the interaction between the protein and SAM. Especially, the surrounding water molecules facilitate the breakage and prohibit the re-formation of salt-bridge interactions. For example, among the water molecules in the vicinity of the salt bridge between E277 and the protonated amine group, there are at least five water molecules that form hydrogen bonds with the salt-bridge groups (Fig. 6a). When the salt bridge breaks due to the lateral diffusion of the protein or the fluctuation of residue side chains, one water molecule nimbly inserts into the broken salt bridges, forming a water bridge between E227 and the amine group (Fig. 6b). The salt bridge is then replaced by the water bridge. In addition, the invasion of water molecules also enhances the hydration of salt-bridge groups, and the re-formation of the salt bridge is thus suppressed by the dehydration penalty. Similar phenomena are found during the breakage of salt bridges of E396 and D563 (Fig. 6c, d and Fig. S12, ESI†). In short, the invasion of interlayer water along with the lateral diffusion of protein disturbs the interactions between the protein and SAM.
 | ||
| Fig. 6 Water-mediated breakage of salt bridges. The salt bridge formed between Glu277 and protonated amine group is surrounded by multiple water molecules (a). Once the salt bridge is broken, the water molecule inserts in-between the salt-bridge groups, forming a water bridge (b). Similar phenomena are found in the breakage of salt bridge formed by Glu396 and the protonated amine group (c and d). | ||
After t = 4.5 ns, the lateral diffusion of the protein is further enhanced and the diffusion coefficient increases up to 5.3 × 10−7 cm2 s−1 (Fig. S13, ESI†), even larger than the case of the weakly-bound protein. Due to the limited number of protonated amine groups, the probability of acidic residues moving to the vicinity of these groups is very low. Besides, relatively rapid lateral diffusion of the protein further prevents the formation of the salt bridge with the SAM even when the acidic residues are close to the protonated amine groups. In addition, along with the dissociation of the protein from the SAM, the number of interlayer water further increases to ∼480 (Fig. S11, ESI†). An extended hydration layer forms in-between the protein and the SAM, and both protein's acidic residues and SAM's amine groups are well solvated. Therefore, the re-formation of salt bridges with the SAM needs to remove multiple water molecules and surmount considerable desolvation penalty. In short, the enhanced lateral diffusion of the protein, the existence of interlayer water and the sparse population of protonated amine groups substantially prevent the protein from forming the stable salt bridge with the SAM. After t = 21 ns, there are no salt-bridge interactions with the SAM. The protein diffuses away from the SAM surface even though there is still weak attraction with the surface.
During the desorption process, three domains sequentially depart from the SAM surface. The heavy atom contact number of each domain is calculated separately (Fig. 7). At t = 7 ns, the contact number of domain I decreases to 0, and this domain is then considered to be desorbed. The following dissociation of domain III occurs at t = 11 ns. In addition, domain II finally desorbs from the SAM surface at t = 21 ns, leading to the departure of the whole protein. It should be noted that the dissociation of each domain is triggered by the elimination of the salt bridges with the SAM. For example, domain III no longer forms the salt bridge with the SAM after t = 4.5 ns (Fig. S14, ESI†), the contact number of domain III progressively decreases to 0, and the domain desorbs at t = 11 ns. Similarly, there is no longer the salt bridge between domain II and the SAM after t = 21 ns (Fig. S14, ESI†). As the number of salt bridges decreases to ∼0, it is hard for the domain to contact with the SAM and thus the protein diffuses away from the surface. Hence, the breakage of salt bridges plays a critical role for the desorption of each domain.
 | ||
| Fig. 7 The heavy atom contact number of domains I, II and III during the desorption process. | ||
Conclusions
In the present work, we investigate the interfacial behavior of HSA, especially the spontaneous desorption due to the change of the solution pH. When the solution pH is 10.2, the protein can stably adhere to the SAM, and such binding is largely attributed to the salt bridge interactions with the SAM. As the solution pH increases and the ratio of the protonated amine groups reduces, the number of salt bridges dramatically decreases. Meanwhile, the protein undergoes substantial lateral diffusion and the behavior of such a weakly-bound protein is distinct from the case of stable adsorption. It should be noted that there are water molecules under the adsorbed protein (i.e. interlayer water). When the salt bridge incidentally breaks due to the thermal fluctuation of the protein, water molecule nimbly insert into the broken salt bridge, forming a water bridge. When the solution pH increases to 11.4, the formation of a salt bridge with sparsely distributed protonated groups becomes very difficult. Besides, the formation of salt bridges is hindered by the enhanced lateral diffusion of the protein as well as the increase of interlayer water. The number of salt bridges gradually decreases to zero, resulting in the dissociation of the protein from the SAM surface. Studies on the process of spontaneous desorption of the weakly adsorbed protein will enrich our understanding about the nonspecific interactions between proteins and nanomaterials. It should be noted that the protein adsorption is also affected by the temperature and ionic strength.24,31,33 Further works are highly desired to study the protein desorption in these systems as well as the other nanoparticles with different pH response intervals, especially the importance of interfacial water and protein lateral diffusion. Moreover, detailed information about the spontaneous desorption process can also be obtained by a carefully designed experimental work which can validate our findings and provide insights into the rational design of antifouling materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (NSFC) grants (11722434, 21473207, 11374255 and 11674277). We acknowledge the support of the Tianhe-2 Supercomputer Center.References
- V. Holzapfel, M. Lorenz, C. K. Weiss, H. Schrezenmeier, K. Landfester and V. Mailaender, J. Phys.: Condens. Matter, 2006, 18, S2581–S2594 CrossRef CAS.
- W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- S. Zhang, Z. Chu, C. Yin, C. Zhang, G. Lin and Q. Li, J. Am. Chem. Soc., 2013, 135, 5709–5716 CrossRef CAS PubMed.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- S. Nimesh, S. Halappanavar, N. K. Kaushik and P. Kumar, Biomed. Res. Int., 2015, 2015, 610342 Search PubMed.
- L. Zhang, Q. Feng, J. Wang, S. Zhang, B. Ding, Y. Wei, M. Dong, J.-Y. Ryu, T.-Y. Yoon, X. Shi, J. Sun and X. Jiang, ACS Nano, 2015, 9, 9912–9921 CrossRef CAS PubMed.
- D. M. Shin, Drug Metab. Rev., 2010, 42, 13 Search PubMed.
- D. An, J. Su, J. K. Weber, X. Gao, R. Zhou and J. Li, J. Am. Chem. Soc., 2015, 137, 8412–8418 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- L. Wang, J. Li, J. Pan, X. Jiang, Y. Ji, Y. Li, Y. Qu, Y. Zhao, X. Wu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- Y. Chong, C. Ge, Z. Yang, J. A. Garate, Z. Gu, J. K. Weber, J. Liu and R. Zhou, ACS Nano, 2015, 9, 5713–5724 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- M. Dong, S. Xu, C. L. P. Oliveira, J. S. Pedersen, S. Thiel, F. Besenbacher and T. Vorup-Jensen, J. Immunol., 2007, 178, 3016–3022 CrossRef CAS.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- N. Gilbert, Nature, 2009, 460, 937 CrossRef CAS PubMed.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- C. C. Fuller and J. A. Davis, Nature, 1989, 340, 52–54 CrossRef CAS.
- G. Yu, J. Liu and J. Zhou, J. Phys. Chem. B, 2014, 118, 4451–4460 CrossRef CAS PubMed.
- P. Szabelski, A. Cavazzini, K. Kaczmarski, X. Liu, J. Van Horn and G. Guiochon, J. Chromatogr. A, 2002, 950, 41–53 CrossRef CAS PubMed.
- B. Bharti, J. Meissner and G. H. Findenegg, Langmuir, 2011, 27, 9823–9833 CrossRef CAS PubMed.
- J. Meissner, A. Prause, B. Bharti and G. H. Findenegg, Colloid Polym. Sci., 2015, 293, 3381–3391 CAS.
- M. Veen, W. Norde and M. C. Stuart, Colloids Surf., B, 2004, 35, 33–40 CrossRef PubMed.
- S. Patil, A. Sandberg, E. Heckert, W. Self and S. Seal, Biomaterials, 2007, 28, 4600–4607 CrossRef CAS PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- M. Mohsen-Nia, M. M. Bidgoli, M. Behrashi and A. M. Nia, Protein J., 2012, 31, 150–157 CrossRef CAS PubMed.
- R. Huang, R. P. Carney, F. Stellacci and B. L. Lau, Nanoscale, 2013, 5, 6928–6935 RSC.
- B. Bharti, J. Meissner, S. H. Klapp and G. H. Findenegg, Soft Matter, 2014, 10, 718–728 RSC.
- Z. Gu, Z. Yang, L. Wang, H. Zhou, C. A. Jimenez-Cruz and R. Zhou, Sci. Rep., 2015, 5, 10873–10884 CrossRef CAS PubMed.
- X. Wang, J. K. Weber, L. Liu, M. Dong, R. Zhou and J. Li, Nanoscale, 2015, 7, 15341–15348 RSC.
- C. Muecksch and H. M. Urbassek, Langmuir, 2011, 27, 12938–12943 CrossRef PubMed.
- Z. Gu, Z. Yang, Y. Chong, C. Ge, J. K. Weber, D. R. Bell and R. Zhou, Sci. Rep., 2015, 5, 10886–10895 CrossRef CAS PubMed.
- F. Ramezani and H. Rafii-Tabar, Mol. BioSyst., 2015, 11, 454–462 RSC.
- A. Hung, S. Mwenifumbo, M. Mager, J. J. Kuna, F. Stellacci, I. Yarovsky and M. M. Stevens, J. Am. Chem. Soc., 2011, 133, 1438–1450 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Metraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- G. E. Poirier, Chem. Rev., 1997, 97, 1117–1127 CrossRef CAS PubMed.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- H. Hakkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, 15, 3–30 CrossRef CAS.
- A. C. Templeton, M. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS PubMed.
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943–946 CrossRef CAS PubMed.
- R. Huang, R. P. Carney, K. Ikuma, F. Stellacci and B. L. Lau, ACS Nano, 2014, 8, 5402–5412 CrossRef CAS PubMed.
- J. Chen, M. Yang, Q. Zhang, E. C. Cho, C. M. Cobley, C. Kim, C. Glaus, L. V. Wang, M. J. Welch and Y. Xia, Adv. Funct. Mater., 2010, 20, 3684–3694 CrossRef CAS.
- M. Ozboyaci, D. B. Kokh, S. Corni and R. C. Wade, Q. Rev. Biophys., 2016, 49, 1–45 CrossRef PubMed.
- C. E. D. Chidsey and D. N. Loiacono, Langmuir, 1990, 6, 682–691 CrossRef CAS.
- L. Strong and G. M. Whitesides, Langmuir, 1988, 4, 546–558 CrossRef CAS.
- C. Vericat, M. E. Vela, G. A. Benitez, J. A. M. Gago, X. Torrelles and R. C. Salvarezza, J. Phys.: Condens. Matter, 2006, 18, R867–R900 CrossRef CAS.
- D. Matulis and V. A. Bloomfield, Biophys. Chem., 2001, 93, 37–51 CrossRef CAS PubMed.
- J. A. Dean, Lange's handbook of chemistry, McGraw-Hill, New York, 1999 Search PubMed.
- M. Wardell, Z. M. Wang, J. X. Ho, J. Robert, F. Ruker, J. Ruble and D. C. Carter, Biochem. Biophys. Res. Commun., 2002, 291, 813–819 CrossRef CAS PubMed.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- H. J. Hsu, S. Y. Sheu and R. Y. Tsay, Colloids Surf., B, 2008, 67, 183–191 CrossRef CAS PubMed.
- C. R. Sondergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nose and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef CAS.
- S. Nose, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. MacKerell, Jr., J. Comput. Chem., 2010, 31, 671–690 CAS.
- J. Huang and A. D. MacKerell, Jr., J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- M. J. Penna, M. Mijajlovic and M. J. Biggs, J. Am. Chem. Soc., 2014, 136, 5323–5331 CrossRef CAS PubMed.
- T. Utesch, G. Daminelli and M. A. Mroginski, Langmuir, 2011, 27, 13144–13153 CrossRef CAS PubMed.
- Y. Kang, X. Li, Y. Tu, Q. Wang and H. Agren, J. Phys. Chem. C, 2010, 114, 14496–14502 CAS.
- D. Zhao, C. Peng and J. Zhou, Phys. Chem. Chem. Phys., 2015, 17, 840–850 RSC.
- J. Zhao, Q. Wang, G. Liang and J. Zheng, Langmuir, 2011, 27, 14876–14887 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp05515c |
| This journal is © the Owner Societies 2018 |