
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expeditious synthesis of polyacetylenic water hemlock toxins and their effects on the major GABAA receptor isoform†
Martin
Berger‡
a,
Yong
Chen‡
a,
Konstantina
Bampali
b,
Margot
Ernst
b and
Nuno
Maulide
 *a
*a
aInstitute of Organic Chemistry, University of Vienna, Währinger Straße 38, 1090 Vienna, Austria. E-mail: nuno.maulide@univie.ac.at
bDepartment of Molecular Neurosciences, Center for Brain Research/Medical University of Vienna, Spitalgasse 4, 1090 Vienna, Austria
First published on 7th February 2018
Abstract
Classical synthetic approaches to highly unsaturated polyene/yne natural products rely on iterative cross-coupling of linear fragments. Herein, we present an expeditious and unified approach to the unsaturated backbone of polyacetylenes via domino cuprate addition/4π-electrocyclic ring opening of a stereodefined cyclobutene intermediate. This sets the stage for a detailed biological assessment of the role of Virol A and Cicutoxin as inhibitors of GABA induced chloride currents, providing further insight into the interaction of these highly potent toxins towards the GABAA receptor, including the structure–activity relationship of the derivatives.
Water hemlock (Cicuta species) is regarded as one of the most poisonous plants in Northern America and Central Europe.1 The reason for this unusual toxicity is conjugated, polyunsaturated diols, such as Virol A (1) and Cicutoxin (2) (Fig. 1). The ingestion of even small parts of the plant can cause seizures and lead to death, and numerous cases with lethal outcome have been reported. Due to the high risk of confusion with different plant species, water hemlock still poses a serious danger to all forms of livestock.2
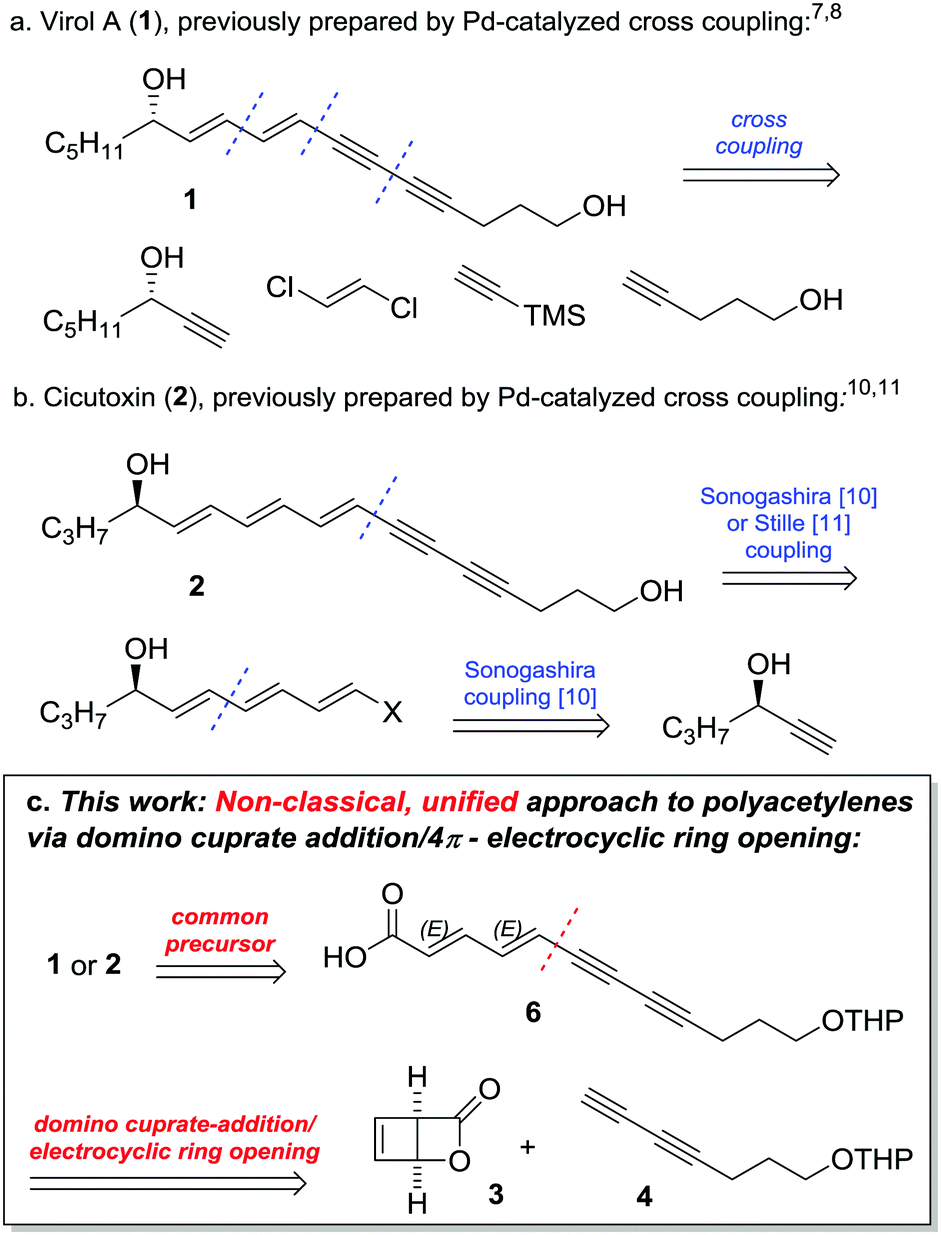 | ||
| Fig. 1 (a) Structure of Virol A (1) and previous synthesis via Sonogashira cross-coupling; (b) Cicutoxin (2) and previous synthesis by cross-coupling and (c) our approach via a cuprate-addition/4π-electrocyclic ring opening cascade. | ||
The highly conjugated backbone of these compounds imparts on them considerable light- and air-sensitivity. This notwithstanding, the isolation of Cicutoxin (2) was reported as early as 19153 and its structural determination along with the first (low-yielding) racemic total synthesis waited until 1953.4,5 The stereochemical assignment of Cicutoxin (2) and Virol A (1) along with their relative toxicity was first reported much later in 1999,6 followed by the first asymmetric total synthesis of Virol A (1) by Uwai in the same year. This approach made extensive use of palladium-catalyzed cross coupling (Fig. 1a).7–9 However, a convergent cross coupling using the diyne building block 4 (vide infra) failed to give a useful yield due to polymerization. As a result, the unsaturated core of Virol A was assembled by the cross coupling of four smaller units, including an enantiopure alcohol as an early-stage building block. The first total synthesis of Cicutoxin was reported by Gung in 2009, using a very similar cross-coupling strategy (Fig. 1b).10 The previously unsuccessful building block diyne 4 was a viable partner in this specific case and, as before, the synthesis started from an enantiopure alcohol building block. In 2011, Brückner reported an elegant approach using Stille cross-coupling of an advanced stannane intermediate.11 This use of iterative transition metal-catalyzed cross-coupling reactions of linear fragments reflects the general trend in the field of polyene natural product synthesis.12
Herein, we present a concise total synthesis of the water hemlock toxins Virol A (1) and Cicutoxin (2). Our strategy provides a unified approach to both natural products through a concise route.13 We also investigate the influence of the absolute configuration and the number of –OH groups on the GABAA inhibitory effects of Virol A (1) and Cicutoxin (2).
Synthesis: Our group has contributed to the chemistry of strained bicyclo[2.2.0]lactones such as 3 in recent years,14,15 and we envisaged an assembly of the key unsaturated core structure of the toxins by domino ring opening/4π-electrocyclic ring opening of bicyclo lactone 3 to stereoselectively afford the all-trans polyacetylenic carboxylic acid 6 (Fig. 1c). In this context, the entire unsaturated core of Virol A 1 could be constructed in a single step utilizing the logical building block 4 – a diyne which was not applicable in prior synthesis of 1. Furthermore, key intermediate 6 would also afford an advanced intermediate en route to the higher homologue Cicutoxin 2, uniting both syntheses.
In the synthetic direction, cupration16 of known diyne 47 followed by the addition of lactone 3 at −78 °C afforded the desired trans-cyclobutene 5, which was found to be metastable at room temperature. In this event, the main pathway for the decomposition of 5 appeared to be its desired ring opening, which already took place in trace amounts during the reaction itself (Scheme 1, top). Pleasingly, the crude product underwent full conversion into 6 upon standing for 12 h at room temperature. Diene–diyne 6, isolated in 82% yield, was formed exclusively as the (E,E)-isomer as expected for the conrotatory ring opening of the trans-cyclobutene precursor 5. Straightforward amidation of 6 led to Weinreb amide 7, the pivotal common intermediate for both toxins 1 and 2. Amide 7 can be conveniently stored at room temperature for several weeks without noticeable degradation. In order to obtain Virol A 1, enone 8 was prepared by simple treatment of 7 with pentyllithium. The addition of LiCl was essential in order to obtain a reasonable yield of 67%. Presumably owing to its lowered LUMO, 8 was prone to facile visible light-mediated photoisomerization of the central olefin from (E) to (Z).13 Enantioselective reduction of 8 using (R)-CBS afforded the desired THP-protected (S)-Virol A 9 in 76% yield and an (S)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(R) ratio of >95:5.§17 Final deprotection with pyridinium para-toluenesulfonate (PPTS) afforded Virol A 1, which was found to be stable upon storage at −20 °C.
:(R) ratio of >95:5.§17 Final deprotection with pyridinium para-toluenesulfonate (PPTS) afforded Virol A 1, which was found to be stable upon storage at −20 °C.
 | ||
| Scheme 1 Top: Synthesis of Virol A (1). Legend: (a) 4 (2.0 equiv.), MeLi (2.0 equiv.), CuCN (2.0 equiv.), THF, −78 °C to r.t., 4 h. (b) Weinreb salt (1.5 equiv.), EDCI (1.2 equiv.), HOBt (1.2 equiv.), Et3N (2.0 equiv.), DCM, r.t., 12 h. (c) Pentyllithium (3.0 equiv.), LiCl (5.0 equiv.), Et2O, −40 °C, 40 min. (d) (R)-(+)-CBS (2.0 equiv.), BH3 * Me2S (2.0 equiv.), THF, −50 °C, 1.5 h. (e) PPTS (1.0 equiv.), MeOH, 35 °C, 2 h. Bottom: Synthesis of Cicutoxin 2 and condition-dependent formation of methoxy analog 12 during THP-deprotection. Legend: (a) DIBAL-H (1.5 equiv.), toluene, −78 °C, 1 h. (b) C3H7COCH2PO(OEt)2 (2.0 equiv.), Ba(OH)2 (4.0 equiv.), THF/H2O, r.t., 1 h. (c) (S)-(−)-CBS (2.0 equiv.), BH3 * Me2S (2.0 equiv.), THF, −50 °C, 1.5 h. (d) PTSA (0.5 equiv.), MeOH, r.t., 1.5 h. (e) PPTS (1.0 equiv.), MeOH, 45 °C, 2 h. | ||
With both racemic and enantioenriched samples of 1 in hand, we then focused on the synthesis of the more chemically labile Cicutoxin 2 (Scheme 1, bottom). Taking into account the photolability of 8, reduction of the Weinreb amide 7 to the corresponding enal with DIBAL and immediate homologation of the unsaturated system via HWE olefination (performed directly on the crude mixture) were carried out under strict exclusion of oxygen and light to yield enone 10 in reproducible 52% yield over two steps, as a single isomer. Enantioselective reduction of 10 using (S)-CBS afforded the desired THP-protected (R)-Cicutoxin 11 in 76% yield and an (R):(S) ratio of >95:5. Deprotection was achieved with p-toluene sulfonic acid (PTSA) at room temperature yielding Cicutoxin 2. The attempted unravelling using PPTS at elevated temperature led to the isolation of a considerable amount of the methoxy derivative 12. We found that 11 can be quantitatively converted to the methoxy analog 12 simply by extending the reaction time. As such a substitution was not observed during the unmasking of 9en route to Virol A, it can be ultimately ascribed to extended delocalization of a putative heptadienyl carbenium ion.
Owing to the late-stage chirality introduction in our synthetic plan, racemic samples of 1 and 2 can be prepared by simple reduction with NaBH4 instead of CBS-reduction from advanced intermediates 8 and 10.§
Evaluation of compound effects on the major GABA A receptor isoform: It is interesting to note that the toxicity of these diols is reported to increase with the number of conjugated double bonds.13 A study by Uwai et al. suggests a correlation of the inhibition of GABA-induced chloride currents with the acute toxicity of Cicutoxin13 and Virol A.18
We carried out a full characterization of α1β3γ2 GABAA receptors, which are the major GABAA receptor isoforms in the adult mammalian brain.19 Two-Electrode Voltage Clamp Electrophysiology was employed to test the compound effects on currents elicited by an EC20 concentration of GABA on the receptors expressed in Xenopus laevis oocytes. Subsequently, their half maximal inhibitory concentration values (IC50 values) were determined.
As shown in Fig. 2, co-application of racemic Virol A or Cicutoxin inhibits GABA-evoked currents in α1β3γ2 GABAA receptors.
 | ||
| Fig. 2 Inhibition of the control chloride current (100%) by racemic (a and c) and enantioenriched (b and d) Virol A (1) and Cicutoxin (2) suggesting similar inhibitory efficacy independent of the configuration. Concentration-dependent inhibition of the GABA EC20 current at α1β3γ2 receptors for racemic (a and c) and enantioenriched (b and d) Virol A and Cicutoxin, with IC50 values of 0.93 μM and 1.4 μM for Virol A and 2.4 μM and 1.7 μM for Cicutoxin, respectively. (e) shows concentration-dependent effects of 14-O-Me-Cicutoxin rac-12 on the GABA EC20 current at α1β3γ2 receptors. The compound was measured one day after synthesis. The dotted line is used to visualize the baseline of 100% of control current, which corresponds to no modulatory activity. Data are depicted as mean ± SEM (n = 4). | ||
A similar inhibition for the enantioenriched compounds was observed, suggesting that the detected activity is independent of the enantiopurity of the diol (Fig. 2). Chiral recognition, therefore, does not appear to affect the interaction of these compounds with the receptors.
Interestingly, our 14-O-methylated Cicutoxin analogue 12 did not inhibit the GABA-induced chloride current (Fig. 2e). Our data imply the secondary alcohol of the toxins 1 and 2 to be absolutely essential for the inhibitory effect on the tested receptor. Thus, the sole mode of action by the primary alcohol can be ruled out. In agreement with this, Cicutol, the 14-deoxyanalog of 2, is reported to lack the capability of prolonging action potentials.20 The simple presence of an oxygen atom as a hydrogen atom acceptor at the allylic position 14 seems to be, in the case of a methoxy-group, insufficient to cause inhibition. As for the case of Virol A (1) and Cicutoxin (2), it is tempting to propose that capability for hydrogen bond donation is required for the interaction with the GABAA receptor complex.21
We have presented an approach to the common unsaturated backbone of water hemlock polyacetylene natural products Virol A and Cicutoxin by a domino cuprate addition/electrocyclic ring opening of a stereodefined cyclobutene intermediate. Our strategy stands in contrast to classical paradigms of polyene synthesis that rely on extensive cross-coupling of small building blocks and offers a more general solution to the preparation of structurally related polyenic natural products. We generated the targets in racemic and enantioenriched forms along a novel analogue, enabling a detailed biological characterization of their role as inhibitors of GABA-induced chloride currents on the major GABAA receptor isoform in the adult mammalian brain. The observation that both –OH groups are mandatory for the inhibitory effect is in line with the idea that the effect on GABAA receptors contributes to the compounds’ toxicity.
This research was funded by the Austrian Research Fund (FWF) Doctoral Program “Molecular Drug Targets” (W1232) and Stand-alone grant P27194. We are grateful to the University of Vienna for continued support of our research. Dr H. Kählig (U. Vienna) is acknowledged for expert NMR analysis. We are grateful to R. Holzinger (Med. U. Vienna) for help with the Oocyte work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. J. Schep, R. J. Slaughter, G. Becket and D. M. G. Beasley, Clin. Toxicol., 2009, 47, 270 CrossRef CAS PubMed.
- For case reports of Water Hemlock poisoning, see: (a) K. Sweeney, K. F. Gensheimer, J. Knowlton-Field and R. A. Smith, Morb. Mortal. Wkly. Rep., 1994, 43, 229 Search PubMed; (b) L. Mutter, Can. J. Public Health, 1976, 67, 386 CAS; (c) D. Landers, K. Seppi and W. Blauer, West. J. Med., 1985, 142, 637 CAS; (d) K. E. Panter, R. F. Keeler and D. C. Baker, J. Anim. Sci., 1988, 66, 2407 CrossRef CAS PubMed.
- C. A. Jacobson, J. Am. Chem. Soc., 1915, 37, 916 CrossRef CAS.
- E. L. F. J. Anet, B. Lythgoe, M. H. Silk and S. Trippett, J. Chem. Soc., 1953, 309 RSC.
- B. E. Hill, B. Lythgoe, S. Mirvish and S. Trippett, J. Chem. Soc., 1955, 1770 RSC.
- T. Ohta, K. Uwai, R. Kikuchi, S. Nozoe, Y. Oshima, K. Sasaki and F. Yoshizaki, Tetrahedron, 1999, 55, 12087 CrossRef CAS.
- K. Uwai, Y. Oshima, T. Sugihara and T. Ohta, Tetrahedron, 1999, 55, 9469 CrossRef CAS.
- More recently, Virol A was prepared by Yu et al. via similar disconnections as ref. 7, see: J. Liu, H.-L. Li, X.-R. Guo, L. Zhou, Y. Wang, Y.-N. Duan, M.-Z. Wang, R.-S. Na and B. Yu, Tetrahedron, 2016, 72, 6603 CrossRef CAS.
- For a synthesis of the (1)-dehydroxyanalog of 1, see: V. Fiandanese, D. Bottalico, C. Cardellicchio, G. Marchese and A. Punzi, Tetrahedron, 2005, 61, 4551 CrossRef CAS.
- B. W. Gung and A. O. Omollo, Eur. J. Org. Chem., 2009, 1136 CrossRef CAS PubMed.
- J. Burghart and R. Brückner, Eur. J. Org. Chem., 2011, 150 CrossRef CAS.
- For a review on the synthesis of naturally occurring poly–ynes, see: A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034 CrossRef PubMed.
- K. Uwai, K. Ohashi, Y. Takaya, T. Ohta, T. Tadano, K. Kisara, K. Shibusawa, R. Sakakibara and Y. Oshima, J. Med. Chem., 2000, 43, 4508 CrossRef CAS PubMed.
- For a recent overview of our efforts in cyclobutene chemistry, see: A. Misale, S. Niyomchon and N. Maulide, Acc. Chem. Res., 2016, 49, 2444 CrossRef CAS PubMed.
- (a) F. Frebault, M. Luparia, M. T. Oliveira, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 5672 CrossRef CAS PubMed; (b) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed; (c) C. Souris, F. Frébault, D. Audisio, C. Farès and N. Maulide, Synlett, 2013, 1286 CAS; (d) C. Souris, M. Luparia, F. Frébault, D. Audisio, C. Farès, R. Goddard and N. Maulide, Chem. – Eur. J., 2013, 19, 6566 CrossRef CAS PubMed; (e) C. Souris, F. Frébault, A. Patel, D. Audisio, K. N. Houk and N. Maulide, Org. Lett., 2013, 15, 3242 CrossRef CAS PubMed; (f) C. Souris, A. Misale, Y. Chen, M. Luparia and N. Maulide, Org. Lett., 2015, 17, 4486 CrossRef CAS PubMed.
- B. H. Lipshutz, R. S. Wilhelm and D. M. Floyd, J. Am. Chem. Soc., 1981, 103, 7672 CrossRef CAS.
- (a) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS; (b) T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451 CrossRef CAS PubMed; (c) D. E. Ward and C. K. Rhee, Tetrahedron Lett., 1991, 32, 7165 CrossRef CAS.
- K. Uwai, K. Ohashi, Y. Takaya, Y. Oshima, K. Furukawa, K. Yamagata, T. Omura and S. Okuyama, Brain Res., 2001, 889, 174 CrossRef CAS PubMed.
- R. W. Olsen and W. Sieghart, Pharmacol. Rev., 2008, 60, 243 CrossRef CAS PubMed.
- U. Wittstock, K.-H. Lichtnow and E. Teuscher, Planta Med., 1997, 63, 120 CrossRef CAS PubMed.
- In contrast, a 14-O-acetyl-protected analog of Cicutoxin 2 was reported to be as toxic as the unprotected natural product (cf.ref. 13 for details). However, toxic effects mediated by another receptor isoform or acetolysis in vivo cannot be excluded (cf.ref. 20 for details).
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and spectral data. See DOI: 10.1039/c7cc09801d |
| ‡ These authors contributed equally. |
| § See the ESI† for details. |
| This journal is © The Royal Society of Chemistry 2018 |