
Synthesis and characterization of AgGaS2 nanoparticles: a study of growth and fluorescence†
Sky
Paderick
,
Matthew
Kessler‡
,
Tyler J.
Hurlburt§
and
Steven M.
Hughes
 *
*
Department of Chemistry, Roanoke College, 221 College Lane, Salem, Virginia 24153, USA. E-mail: shughes@roanoke.edu
First published on 30th November 2017
Abstract
Silver gallium sulfide nanocrystals were synthesized and characterized by fluorescence, TEM, EDS, and XRD to better understand the material system with an ideal band gap of 2.7 eV. The crystals were found to nucleate in the monoclinic structure, and develop two fluorescence peaks (650, 460 nm) influenced by stoichiometry.
Semiconductor nanocrystals are finally beginning to live up to their promise, and we are seeing these materials make the transition from research lab to industrial applications more and more frequently, driving all new areas of research.1–5 One area these materials seem to have found a foothold in is the display and lighting market, where cadmium based semiconductor nanocrystals in particular are being used for their ability to downshift ultraviolet light to a wide spectrum of colors.6–8 These materials have many advantages in this market, including narrow line widths and strong absorption efficiencies, but one significant drawback is the innate use of cadmium.9–11
In the search for a material system that displays similar high quantum yield efficiencies to cadmium based materials, one of the most popularly studied material groups is the I–III–VI2 system including CuInS2 (CIS). CIS has achieved extremely high quantum yields when alloyed with zinc, with the most efficient fluorescence typically shown in the red-orange part of the visible spectrum.12–15 Unlike the II–VI material systems, the I–III–VI2's typically control emission wavelength through compositional variation.15,16 While effective, this can be more challenging than working with a material that has the ideal band gap for the desired emission, especially at shorter wavelengths. As a result, there is interest in finding a new material system with band gap emission in the blue and green regions of the visible spectrum. Additionally, as demonstrated by the CdSe/CdS pair, there are many advantages to having a complementary set of materials with nested band gaps, suggesting a wider band gap material with similar crystal structure to CIS could be very beneficial.
In this study we look at the synthesis of uniform AgGaS2 (AGS) nanocrystals. This highly overlooked material has a direct band gap of approximately 2.7 eV, and a reported crystal structure similar to CIS.17,18 There has been some recent interest in this material system, with groups researching a variety of possibilities including: tuning the composition of AgInS2 by the inclusion of Ga; growing orthorhombic AGS crystals as previously reported in this journal; or even the growth of structurally interesting nanoflowers.19–22 Much of the renewed interest in AGS has stemmed from its potential as a water-splitting photocatalyst as identified in the bulk phase.23–25 Given the advantages of nanomaterials, there is hope that AGS nanocrystals will work in this capacity as well. However, the optical emission properties of AGS have been less well characterized, in part due to a lack of an easily reproducible synthesis for uniform particles. Given its band gap energy, the corresponding fluorescence at 460 nm would make it well suited as a blue-green emitter for a variety of applications, as well as a shelling material on smaller band gap systems such as CIS (1.53 eV). In this study we present a robust synthesis, and carefully analyze the fluorescence behavior of AGS nanocrystals throughout their growth in order to better understand how they develop and their potential for optical applications.
The formation of our AGS nanoparticles was initiated by the rapid injection of elemental sulfur in 1-dodecanethiol (DDT) to a reaction vessel containing AgNO3 and Ga(acac)3 in octadecene at 170 °C. Nucleation and growth of the nanoparticles was monitored both by eye and fluorescence, Fig. 1. Immediately upon the addition of the sulfur the solution appeared opaque black, and as time progressed the reaction mixture transitioned to a translucent red-brown. An earlier publication on a synthetic approach for nanocrystalline AGS in the tetragonal phase reported by Huang et al. in Nanoscale suggests that the particles nucleate initially as Ag2S, and subsequent incorporation of gallium transforms the particles to AGS.22 While the color transition of our reaction does suggest this same behavior, analysis by XRD shows only the smallest influence from the expected silver sulfide structure of acanthite at times as early as 5 seconds after injection, Fig. 2. Despite the large change in the appearance of the colloidal suspension, structurally there is little change during growth.
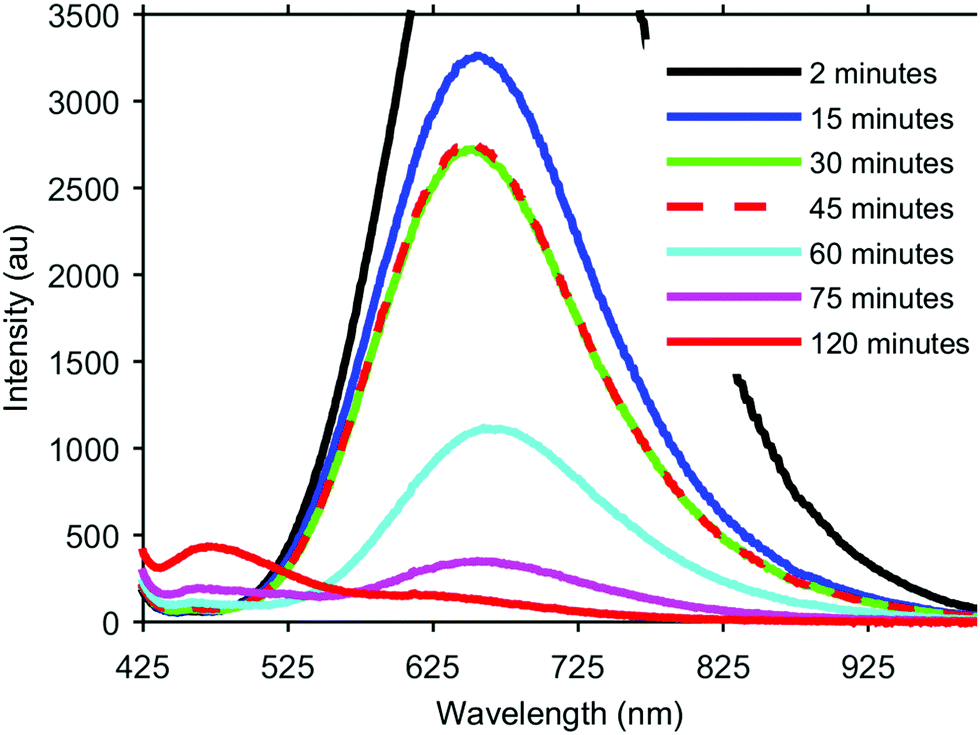 | ||
| Fig. 1 Fluorescence evolution in time during the growth of AGS nanocrystals. Times 2–45 min are at 170 °C, while subsequent times had additional growth at 240 °C. The tail of the excitation source at 405 nm can be seen at the short wavelength end of the spectra. | ||
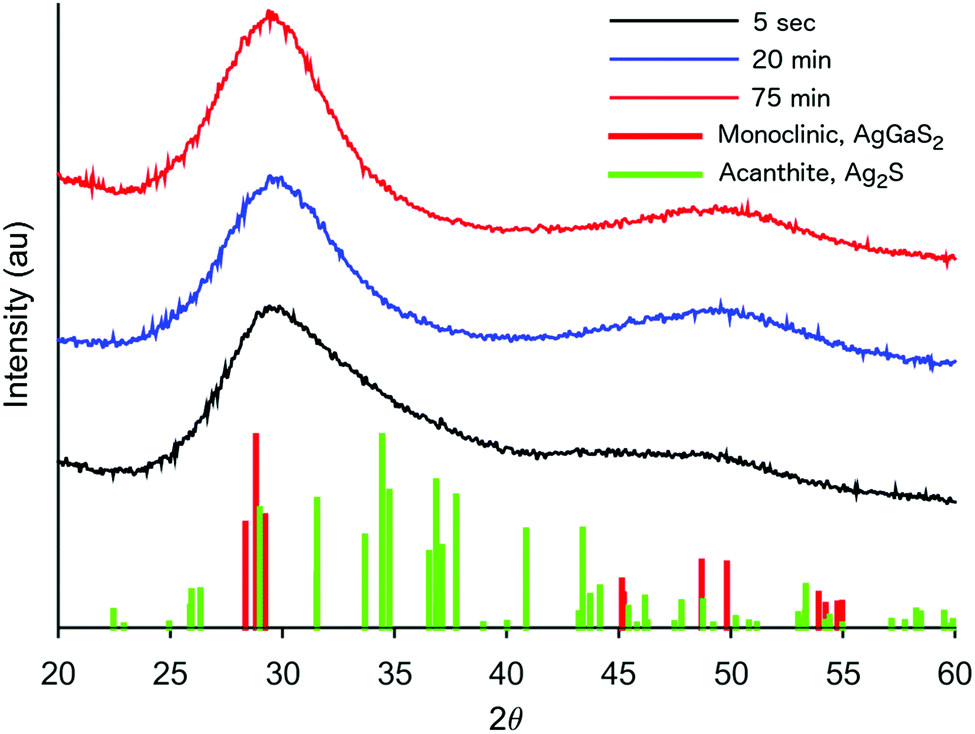 | ||
| Fig. 2 XRD patterns of AGS nanocrystals taken by stopping the growth at various times during a standard reaction. | ||
Of particular interest, is the assignment of our XRD pattern to the monoclinic phase of AgGaS2. As mentioned earlier, AGS was chosen in particular for its chalcopyrite structure, a tertiary system based on a tetragonal unit cell. However, despite the broadness of the peaks, our system appears to best match the pattern shown for the monoclinic structure, which was first measured by synchrotron for AGS at high pressures in a diamond anvil cell at 5.5 GPa.26 As the authors of that original work discuss, their assignment of the atomic positions is essentially a distorted chalcopyrite unit cell. It is therefore not too surprising to find this pattern for the nanocrystals described here. Nanocrystals are by their nature a strained system due to the high fraction of surface to interior atoms. Additionally, as shown in Table 1, the odd stoichiometry of the material during growth also likely contributes to distortions in the lattice.
| Time (min) | Ag (%) | Ga (%) | S (%) | Formula |
|---|---|---|---|---|
| 0.5 | 32.0 ± 1.2 | 25.9 ± 1.1 | 42.1 ± 2.2 | AgGa0.808S1.32 |
| 2 | 29.7 ± 2.8 | 25.8 ± 1.8 | 44.4 ± 4.6 | AgGa0.869S1.50 |
| 30 | 33.1 ± 3.0 | 37.6 ± 1.7 | 29.3 ± 3.0 | AgGa1.14S0.886 |
| 60 | 28.6 ± 1.8 | 25.0 ± 1.9 | 46.4 ± 3.6 | AgGa0.872S1.62 |
| 120 | 19.1 ± 1.9 | 22.3 ± 2.0 | 58.6 ± 2.0 | AgGa1.17S3.06 |
During this initial low temperature nucleation and growth period however, there is a dramatic change in the fluorescence of the nanocrystals. As a semiconductor with a direct band gap of 2.7 eV, it was hoped that the nanocrystals would exhibit strong fluorescence corresponding to this energy at 460 nm. However, aliquots taken at very early times show dramatic fluorescence centered typically around 650 nm with a FWHM of approximately 300 nm. As the crystals continue to grow at this temperature, the 650 nm peak rapidly decreases in intensity, and the expected peak at 460 nm begins to emerge. If the reaction is maintained at a low temperature this transition between peaks eventually stalls, as can be seen in Fig. 1 between the aliquots taken at 30 min and 45 min. In order to continue the transition and intensify the higher energy fluorescence, the reaction is raised to 240 °C. At this elevated temperature the long wavelength peak continues to diminish and the short wavelength peak continues to increase. The low intensity of the 460 nm peak is expected since our final material is unshelled, and likely has a poorly passivated surface. While the intensity of this emission can be further improved with additional growth time at 240 °C for complete conversion of the 650 nm peak, careful monitoring is necessary as too long of growth can lead to rapid aggregation and have the opposite affect of quenched fluorescence.
We believe the origins of these two peaks rise from the overall stoichiometry and crystallinity of the nanoparticles as they grow. The key element in the development of this material appears to be the incorporation of sulfur. In order to establish the role of the elemental sulfur and DDT, the two sources of sulfur present, reactions were run with varying amounts of elemental sulfur present in the injection. It was observed that the reaction would stall as it does in Fig. 1 (between 30 min and 45 min) at different times based on how much elemental sulfur was provided, despite always being in stoichiometric abundance. By adding more elemental sulfur, the nanocrystals would continue to develop longer at the lower temperature, and the 650 nm peak would continue to decrease. Unfortunately, if enough sulfur was included to completely eliminate the 650 nm peak through growth at 170 °C, the particles were found to be highly aggregated through what was likely the formation of disulfide bonds bridging individual nanocrystals. By limiting the sulfur precursor at this stage of growth the crystals form with improved dispersion in solution, but growth at this temperature is ultimately limited, Fig. 3. Because there is an excess of elemental sulfur provided to the reaction even when limited as described above, it is evident that the formation of the sulfur precursor is a complex process involving multiple components in the reaction. By raising the reaction temperature to 240 °C more sulfur becomes available in the system, and the particles begin to grow.
 | ||
| Fig. 3 AGS nanocrystals at various times during growth: 30 s (A), 2 min (B), 30 min (C), 60 min (D), and 120 min (E). Samples (A–C) were grown at 170 °C, while (D and E) were subsequently grown for additional time at 240 °C. | ||
This behavior can also be observed in the compositional analysis performed using EDS, shown in Table 1. During the first two points of growth (0.5 min, 2 min) the ratios of the three elements remain fairly constant. By 30 min though, the sulfur fraction of the particles has clearly dropped, suggesting that while sulphur addition slowed, silver and gallium continued to be added to the crystal. Once the temperature is elevated and growth is allowed to continue (60 min, 120 min) the sulfur fraction begins to increase again, and the final particle stoichiometry is approximately AgGaS3, suggesting we have formed the intended nanocrystals. The higher than ideal sulfur fraction most likely arises from a sulfur rich surface, due to dodecanethiol passivating the nanocrystals.
In addition to improving stoichiometry at the elevated temperatures of the reaction, the particle size increases significantly as well. Fig. 3A–E, shows the development of the nanocrystals at the same points in time as the compositional analysis. At the earliest time, 30 s, the particles are roughly 3.0 ± 0.7 nm (148 particles). When the reaction has stalled at the low growth temperature, 30 min, the particles are effectively the same diameter though the size distribution has narrowed slightly to 3.0 ± 0.6 nm (117 particles). The stoichiometry is changing, leading to a reduction in the 650 nm fluorescence, but the overall size of the particles is effectively the same. After completing growth at the elevated temperature, the nanocrystals in Fig. 3E have a size distribution of 5.6 ± 0.9 nm (139 particles). During this elevated temperature growth period, there appears to be a small shift in the fluorescence peak to longer wavelengths, best observed in the 60 min sample, but as the particles continue to grow this peak rapidly diminishes, and it is difficult to discern any significant shifts. Additionally, it may be that only the smaller particles with the greatest deviations in stoichiometry contribute to the 650 nm fluorescence, and as the particles grow and the stoichiometry approaches the ideal ratios, this fluorescence is suppressed and the band gap fluorescence emerges.
Also worth note is the lack of significant quantum confinement effects. The exciton Bohr radius has been reported in literature to be 3.3 nm for AGS,22 which may explain this absence. The particles very rapidly grow to near the size of the Bohr radius within 30 s, and subsequent growth at the elevated temperatures moves them well beyond this size. Interestingly, between 2 min to 30 min, there is a slight blue-shift in the fluorescence, followed by the noted red shift as the peak diminishes at higher temperatures. It is unclear what the origins of these two shifts are, but it seems more likely that this behavior is a result of the stoichiometry changes than quantum confinement.
The broad 650 nm peak is particularly interesting due to its similarity to the typical emission observed in CIS. This broad emission in CIS is still poorly understood, and there is considerable debate regarding its origins, which has been well covered by Leach and Macdonald in a recent JPC Letters perspective, as well as in recent publications from the Gamelin group.27–32 Because of the similarities between CIS and AGS, both ternary I–III–VI2 materials with ideally chalcopyrite crystal structures, one might expect the mechanisms behind the emission to possibly be related. In an interesting investigation by Nelson et al., Cu+ and Ag+ doped CdSe nanocrystals were found to exhibit many of the same spectroscopic behaviors despite possessing clearly different electronic structures.32 Similarly, the ability to intentionally eliminate the long-wavelength fluorescence of AGS and promote the band gap emission through additional growth and annealing at elevated temperatures suggests some unique differences between AGS and CIS nanoparticles. This behavior however, does support the hypothesis summarized in Leach and Macdonald's perspective that this is trap state emission arising from a transition between a quantized level and an internal defect since the intensity of this emission drops as our stoichiometry improves.
In some instances, the broad red emission may be desirable, in which case this fluorescence can be intentionally promoted. Interestingly, the most intense emission was observed at very short times, with significantly deviant stoichiometry and the poorest crystal structure. Because of this, the long wavelength emission can be promoted by growing the crystals with sub-stoichiometric amounts of reagents present. Unfortunately, this may lead to complications in determining whether the sample has been modified by an additional dopant, or simply poorly formed. For instance in Huang's AGS research, they were intentionally doping the AGS with Mn2+ ions, in order to generate manganese's characteristic broad emission.22 Unfortunately, their methods for doping the crystals involved running their reaction with a gallium deficit, which we would expect to lead to the broad red emission with or without the presence of the manganese. It is clearly important that in future doping studies the fluorescence be monitored throughout the crystal growth, or chemically comparable control reactions be run, in order to show that the observed fluorescence is due to the presence of the dopant.
In summary we have demonstrated that AGS has the potential to become a useful medium-gap, ternary semiconductor for those seeking an alternative to cadmium based nanomaterials such as CdS. It is a particularly complementary system to CIS given the similar crystal structures and larger band gap. The nanocrystals grown using the synthesis outlined in this work were found to grow in the monoclinic form, a distorted chalcopyrite lattice previously only observed at high pressures for the bulk form. Unlike CIS, AGS offers the ability to selectively promote either trap-state or band gap emission, which we hope may ultimately help elucidate the mechanism of the long-wavelength emission observed in both this system and others.
This work was supported by Roanoke College, and by the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), supported by NSF (ECCS 1542100).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef CAS PubMed.
- C. B. Williamson, D. R. Nevers, T. Hanrath and R. D. Robinson, J. Am. Chem. Soc., 2015, 137, 15843–15851 CrossRef CAS PubMed.
- P. K. Santra, P. V. Nair, K. George Thomas and P. V. Kamat, J. Phys. Chem. Lett., 2013, 4, 722–729 CrossRef CAS PubMed.
- S. Deka, A. Quarta, M. G. Lupo, A. Falqui, S. Boninelli, C. Giannini, G. Morello, M. De Giorgi, G. Lanzani, C. Spinella, R. Cingolani, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2009, 131, 2948–2958 CrossRef CAS PubMed.
- M. Bruchez Jr, Science, 1998, 281, 2013–2016 CrossRef CAS.
- J. Kwak, J. Lim, M. Park, S. Lee, K. Char and C. Lee, Nano Lett., 2015, 15, 3793–3799 CrossRef CAS PubMed.
- X. Fang, M. Roushan, R. Zhang, J. Peng, H. Zeng and J. Li, Chem. Mater., 2012, 24, 1710–1717 CrossRef CAS.
- M. J. Anc, N. L. Pickett, N. C. Gresty, J. A. Harris and K. C. Mishra, ECS J. Solid State Sci. Technol., 2012, 2, R3071–R3082 CrossRef.
- K. M. Tsoi, Q. Dai, B. A. Alman and W. C. W. Chan, Acc. Chem. Res., 2013, 46, 662–671 CrossRef CAS PubMed.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039–7050 RSC.
- W. Zhang, K. Lin, Y. Miao, Q. Dong, C. Huang, H. Wang, M. Guo and X. Cui, J. Hazard. Mater., 2012, 213–214, 413–420 CrossRef CAS PubMed.
- R. Xie, M. Rutherford and X. Peng, J. Am. Chem. Soc., 2009, 131, 5691–5697 CrossRef CAS PubMed.
- L. Li, T. J. Daou, I. Texier, T. T. Kim Chi, N. Q. Liem and P. Reiss, Chem. Mater., 2009, 21, 2422–2429 CrossRef CAS.
- L. Li, A. Pandey, D. J. Werder, B. P. Khanal, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2011, 133, 1176–1179 CrossRef CAS PubMed.
- W. Zhang, Q. Lou, W. Ji, J. Zhao and X. Zhong, Chem Mater., 2014, 26, 1204–1212 CrossRef CAS.
- X. Tang, W. B. A. Ho and J. M. Xue, J. Phys. Chem. C, 2012, 116, 9769–9773 CAS.
- B. Tell, J. L. Shay and H. M. Kasper, J. Appl. Phys., 1972, 43, 2469–2470 CrossRef CAS.
- G. Brandt and V. Krämer, Mater. Res. Bull., 1976, 11, 1381–1388 CrossRef CAS.
- T. Uematsu, T. Doi, T. Torimoto and S. Kuwabata, J. Phys. Chem. Lett., 2010, 1, 3283–3287 CrossRef CAS.
- Y. Yuan, J. Zai, Y. Su and X. Qian, J. Solid State Chem., 2011, 184, 1227–1235 CrossRef CAS.
- C.-M. Fan, M. D. Regulacio, C. Ye, S. H. Lim, Y. Zheng, Q.-H. Xu, A.-W. Xu and M.-Y. Han, Chem. Commun., 2014, 50, 7128 RSC.
- F. Huang, J. Zhou, J. Xu and Y. Wang, Nanoscale, 2014, 6, 2340 RSC.
- J. S. Jang, S. H. Choi, N. Shin, C. Yu and J. S. Lee, J. Solid State Chem., 2007, 180, 1110–1118 CrossRef CAS.
- J. S. Jang, S. J. Hong, J. Y. Kim and J. S. Lee, Chem. Phys. Lett., 2009, 475, 78–81 CrossRef CAS.
- N. Liang, Q. He, S. Huang, M. Wang, W. Chen, M. Xu, Y. Yuan, J. Zai, N. Fang and X. Qian, CrystEngComm, 2014, 16, 10123–10130 RSC.
- E. H. Kitahara, N. Ishizawa, F. Marumo and Y. Noda, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3310–3316 CrossRef.
- A. D. P. Leach and J. E. Macdonald, J. Phys. Chem. Lett., 2016, 7, 572–583 CrossRef CAS PubMed.
- A. D. P. Leach, X. Shen, A. Faust, M. C. Cleveland, A. D. La Croix, U. Banin, S. T. Pantelides and J. E. Macdonald, J. Phys. Chem. C, 2016, 120, 5207–5212 CAS.
- K. E. Knowles, H. D. Nelson, T. B. Kilburn and D. R. Gamelin, J. Am. Chem. Soc., 2015, 137, 13138–13147 CrossRef CAS PubMed.
- W. D. Rice, H. McDaniel, V. I. Klimov and S. A. Crooker, J. Phys. Chem. Lett., 2014, 5, 4105–4109 CrossRef CAS PubMed.
- P. J. Whitham, A. Marchioro, K. E. Knowles, T. B. Kilburn, P. J. Reid and D. R. Gamelin, J. Phys. Chem. C, 2016, 120, 17136–17142 CAS.
- H. D. Nelson, S. O. M. Hinterding, R. Fainblat, S. E. Creutz, X. Li and D. R. Gamelin, J. Am. Chem. Soc., 2017, 139, 6411–6421 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, absorption curves for samples shown in Fig. 1, expanded and un-cropped fluorescence spectra from Fig. 1, XRD patterns with chalcopyrite reference lines. See DOI: 10.1039/c7cc08070k |
| ‡ Present address: Department of Chemistry, University of Maine, Orono, ME 04469, USA. |
| § Present address: College of Chemistry, University of California, Berkeley, CA 94720, USA. |
| This journal is © The Royal Society of Chemistry 2018 |