
Facile formation of mesoporous structured mixed-phase (anatase/rutile) TiO2 with enhanced visible light photocatalytic activity†
Weiwei
Fu
 ab,
Guode
Li
*a,
Yu
Wang
b,
Shangjing
Zeng
c,
Zhuojun
Yan
d,
Junwei
Wang
a,
Shigang
Xin
a,
Lei
Zhang
a,
Shiwei
Wu
a and
Zongtao
Zhang
*b
ab,
Guode
Li
*a,
Yu
Wang
b,
Shangjing
Zeng
c,
Zhuojun
Yan
d,
Junwei
Wang
a,
Shigang
Xin
a,
Lei
Zhang
a,
Shiwei
Wu
a and
Zongtao
Zhang
*b
aExperimental Center, Shenyang Normal University, Shenyang 110034, China. E-mail: fuweiwei1984@126.com; Fax: +86 24 86506356; Tel: +86 24 86506356
bState Key Laboratory of Inorganic Synthesis & Preparative Chemistry, Jilin University, Changchun 130012, China
cKey Laboratory of Synthetic Rubber, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130012, Jilin, China
dCollege of Chemistry, Liaoning University, Shenyang 110036, P. R. China
First published on 24th November 2017
Abstract
A new mixed-phase (anatase/rutile) TiO2 with mesoporous structures and smaller crystal size (3–5 nm) was successfully synthesized by a facile sol–gel method at a lower calcination temperature (100 °C). Rhodamine B can be completely decomposed in the presence of the as-synthesized nanocomposite after only 60 minutes under visible light. Therefore it is believed to be a promising candidate for wastewater treatment.
Water pollution, resulting from the organic dyes in industries and residential use, is becoming an overwhelming problem all over the world. In recent years, more and more people focus on heterogeneous photocatalysts for the decomposition of organic pollutants under light irradiation.1 To date, numerous interests have been focused on the titanium dioxide material due to its strong redox ability, inexpensiveness, nontoxic nature and long-term stability.2 It is reported3 that the mixed-phase TiO2 (anatase/rutile) shows more favourable photocatalytic properties than the pure anatase TiO2 due to the transfer of electrons from the anatase to the rutile phase, which reduces the recombination rate of anatase, leading to a more efficient electron–hole separation and a greater catalytic activity. Commercial TiO2 (Degussa P25) is widely accepted as the benchmark because of its excellent photocatalytic activity.4 However, its practical application is limited due to its nonporous nature, low surface area, complicated preparation and low visible light activity. The porous structure of the photocatalyst is beneficial for the adsorption of the reactants, and can significantly improve the photocatalytic efficiency.5 So the high surface area and nanocrystallinity of the mesoporous materials qualify them as excellent candidates as active photocatalysts.
In this work, we report a new mesoporous anatase/rutile mixed-phase TiO2 material with smaller nanocrystal size. The mixed-phase TiO2 was synthesized through a simple sol–gel route at a relatively low temperature (100 °C). Compared to P25, the obtained mixed-phase TiO2 (anatase/rutile) exhibited significant photocatalytic activity.
Fig. 1 shows the X-ray diffraction (XRD) patterns of all the as-prepared samples. The samples are mixed phase crystalline. The observed peaks marked with blue can be attributed to the anatase-type TiO2 (JCPDS no. 21-1272)6 and the others marked with pink can be assigned to the rutile-type TiO2 (JCPDS no. 21-1276).7 All diffraction lines can be assigned as either anatase or rutile phases, and no other titania related peaks were observed. Another obvious observation is that most broadening peaks become sharper with higher temperature. Compared with TiO2-100, TiO2-500 showed the best crystallinity, which indicates a potential influence of temperature on the TiO2 lattice (the grain size of the obtained nanoparticles may increase with the temperature). Meanwhile, the intensity of the (101) peaks of the anatase and the (110) peaks of the rutile TiO2 also changed with the calcination temperature. The polymorph composition of the products is characterized by the anatase content fA, which is derived from
| fA = (1 + 1.265IR/IA)−1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ) will be smaller. Fig. S2 (ESI†) shows the TEM images of the samples, which show that the obtained products are nanocrystals. It also shows that the increasing calcination temperature resulted in an increase of the nanocrystal size (∼3 nm to ∼20 nm), which is consistent with the above XRD analysis.
sinθ) will be smaller. Fig. S2 (ESI†) shows the TEM images of the samples, which show that the obtained products are nanocrystals. It also shows that the increasing calcination temperature resulted in an increase of the nanocrystal size (∼3 nm to ∼20 nm), which is consistent with the above XRD analysis.
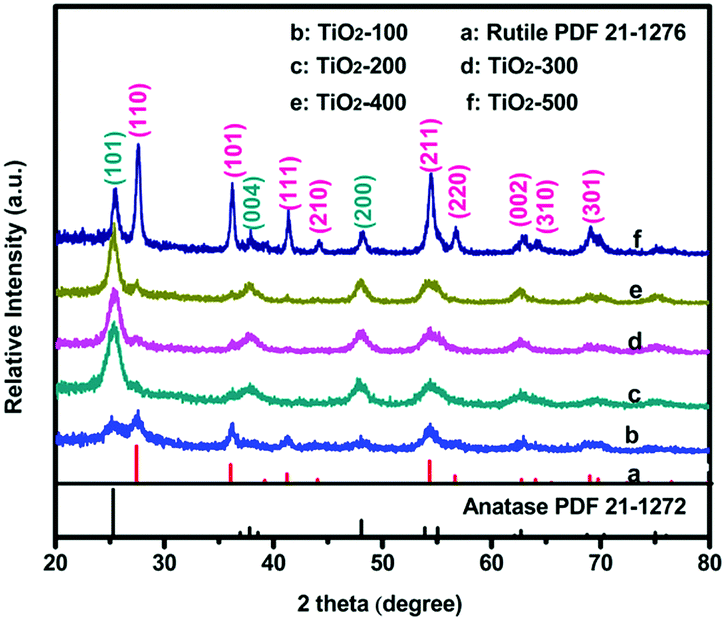 | ||
| Fig. 1 XRD patterns of the as-prepared anatase–rutile TiO2 powders. | ||
The surface chemical states of the samples were studied with XPS. Fig. S3a (ESI†) shows the survey spectra of TiO2-100. We can see that there are only C, Ti and O related peaks, indicating that the powders possess a high chemical purity. Fig. 2a displays the Ti2p spectrum of TiO2-100. As shown, the Ti2p1/2 and Ti2p3/2 peaks are observed at binding energies of 464.5 and 458.85 eV, demonstrating the Ti4+ nature of the titanium in the samples.9 The high-resolution O1s spectra of TiO2-100 are shown in Fig. 2b. The spectra show two components: Olatt and Ox−. Olatt represents the O atom in the crystal lattice, and Ox− is assigned to the absorbed oxygen ion which results from kinds of oxygen-related defect such as an oxygen vacancy, oxygen interstitial site and oxygen antisite. The O1s spectra of all samples consist exclusively of Olatt and Ox− (Fig. S3b (ESI†)). So the Ox− ratios of TiO2-100, TiO2-200, TiO2-300, TiO2-400 and TiO2-500 can be analyzed by EDX measurement (Table S2, ESI†). They are 39.1%, 38.6%, 6.1%, 22.5% and 25.7%, respectively, which indicates that there is an appreciable quantity of defects in the as-prepared TiO2. These defects may further lead to a certain amount of utilization of visible light and consequently the photocatalytic activity under visible light.10
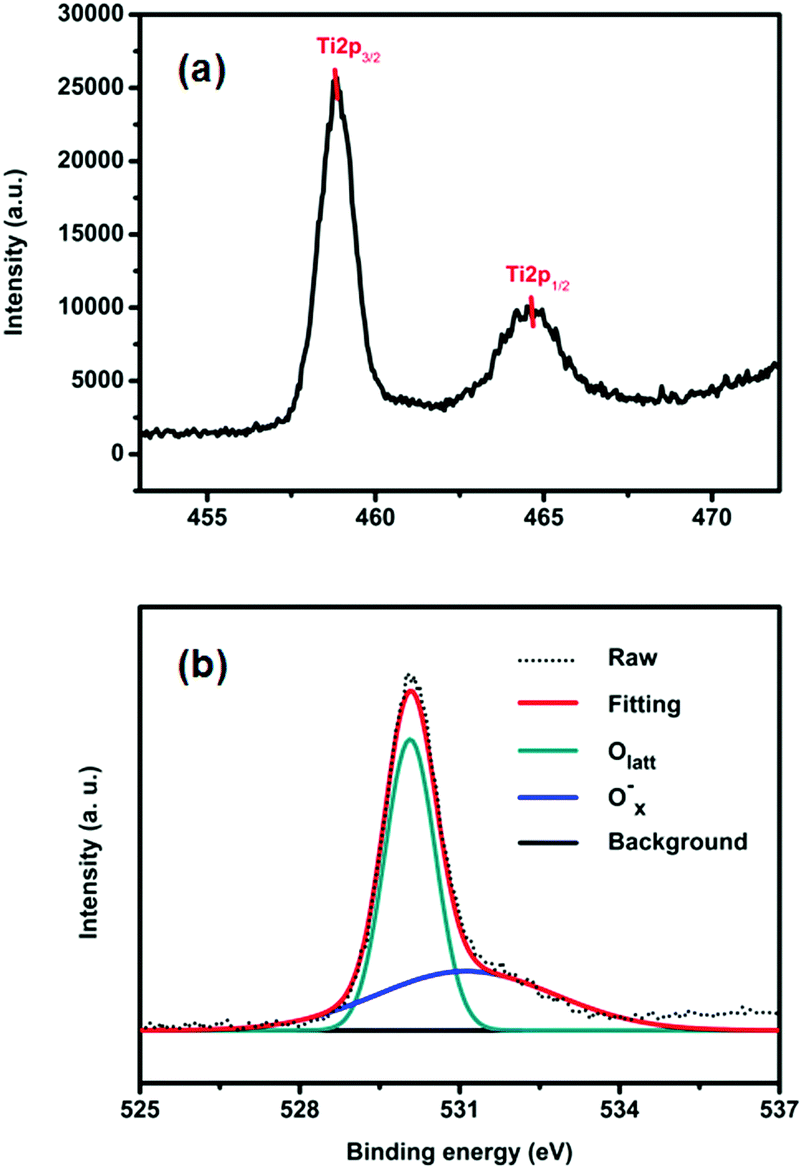 | ||
| Fig. 2 XPS spectra of TiO2-100: (a) the Ti2p spectrum and (b) the O1s spectra. | ||
Fig. 3a shows the UV-vis DRS of P25 and TiO2-100. Compared with that of P25, the absorption edge of TiO2-100 and other samples shifted toward longer wavelengths. This may be attributed to the shallow trap states created by oxygen-related defects,11 which lead to the narrowing of the band gap, which play a crucial role in photocatalysis. In addition, TiO2-200 demonstrates strong photo-absorption in the wavelength range of 350–800 nm (Fig. S4, ESI†), which is mainly attributed to the remaining organics carbonized at 200 °C (Table S3, ESI†). Fig. 3b displays the Tauc plots of (αhν)2vs. photon energy (hν) for P25 and TiO2-100, which are derived using the Kubelka–Munk method,12 and the corresponding band gap energies of 3.45 and 3.10 eV, respectively. With the same method, the band gaps of TiO2-200, TiO2-300, TiO2-400, and TiO2-500 were determined to be 1.70, 3.22, 3.07, and 3.11 eV, respectively, which are mainly attributed to the oxygen-related defects, the synergistic effect of the anatase–rutile phase and the quantum-size effect which results in a larger band gap. The PL emission peak is an intrinsic characteristic of the radiative recombination of the charge carriers for a material. Fig. 3c shows the PL spectra of P25 and TiO2-100 excited at 325 nm. The sample of TiO2-100 shows a much weaker emission intensity than P25, which indicates that the efficient separation of the charge carriers is accelerated and the recombination is retarded at the interface between the anatase and the rutile phase of TiO2-100. This causes many more free electrons to participate in the photocatalytic reactions and enables the photocatalytic process.
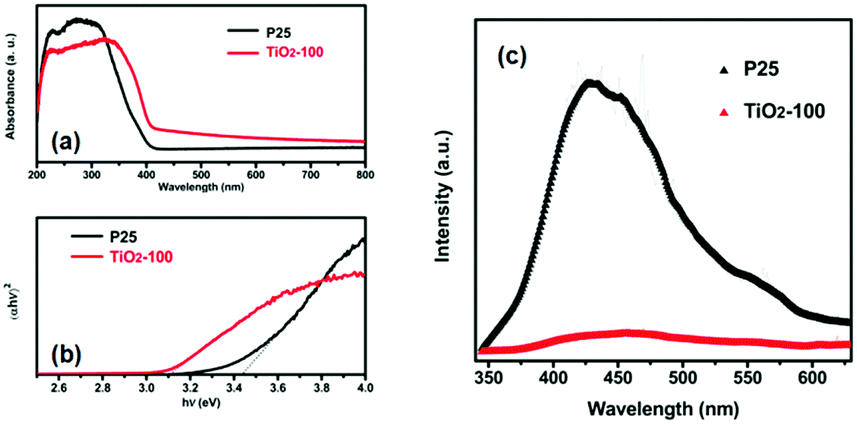 | ||
| Fig. 3 (a) UV-vis DRS of P25 and TiO2-100. (b) Tauc plots of (αhν)2versus hν. (c) PL spectra of P25 and TiO2-100. | ||
The coexistence of the two TiO2 phases in the as-prepared samples was further proven by Raman spectroscopic analysis. In Fig. S5 (ESI†), the peaks centered at 143 (Eg), 199 (Eg), 396 (B1g), 514 (A1g) and 636 cm−1 (Eg) are attributed to the anatase phase,13 while the other peaks are characteristic of the rutile phase.14 The Raman spectrum consisted of the characteristic peaks of the anatase and the rutile phase, which is consistent with the XRD analysis. Compared with TiO2-100, the principal peaks at 142 cm−1 of all others have a hypsochromic shift accompanied by the calcination temperatures increasing. This change in the Raman spectrum is associated with the disorder in the TiO2 crystal, which originates from the defects of the oxygen vacancies.15
The porosities of the products were examined by N2 sorption. Fig. 4 shows the N2 sorption isotherms along with the BJH pore size distributions. The obtained isotherm of the sample is a typical IUPAC type-IV isotherm, which indicates the presence of mesoporous structures in the sample. The BJH desorption pore size distributions are 3.0 nm for TiO2-100, 3.3 nm for TiO2-200, 3.1 nm for TiO2-300, 4.1 nm for TiO2-400 and 6.0 nm for TiO2-500. The changes in pore size may be due to the grain size of the mixed-phase TiO2 nanoparticles. Table S4 (ESI†) shows that most samples have a higher BET surface area. When the calcination temperature was increased to 500 °C, the surface area decreased to 33.5 m2 g−1. This may be due to the high proportion of rutile. Rutile has a density of 4.23 g cm−3, while anatase has a smaller density of 3.78 g cm−3. The other possibility is the increase of the grain size of the nanoparticles and the partial loss of mesoporosity. The adsorption ability tests of the samples were conducted by performing all the degradation reactions in the dark (Fig. 5). P25 did not show any obvious adsorption ability (5%) after 30 min. Compared to P25, most of the as-prepared samples showed higher adsorption abilities (∼20%) of the RhB dye due to their larger surface areas and mesoporous structures, which is beneficial for improving the photocatalytic efficiency.
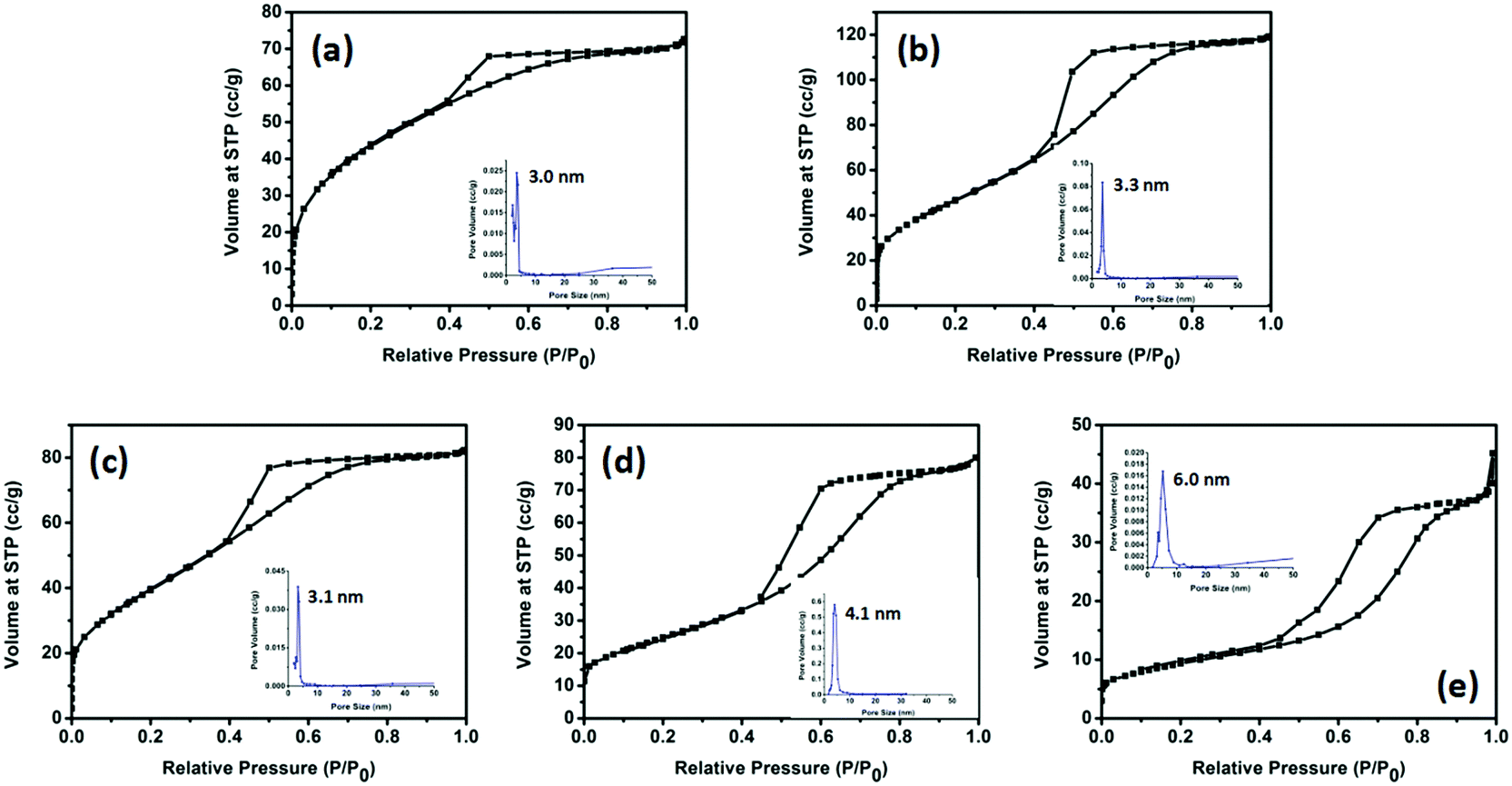 | ||
| Fig. 4 N2 sorption isotherms for the (a) TiO2-100, (b) TiO2-200, (c) TiO2-300, (d) TiO2-400 and (e) TiO2-500 samples. The inset figures are the BJH desorption pore-size distributions for each sample: (a) 3.0 nm, (b) 3.3 nm, (c) 3.1 nm, (d) 4.1 nm and (e) 6.0 nm. | ||
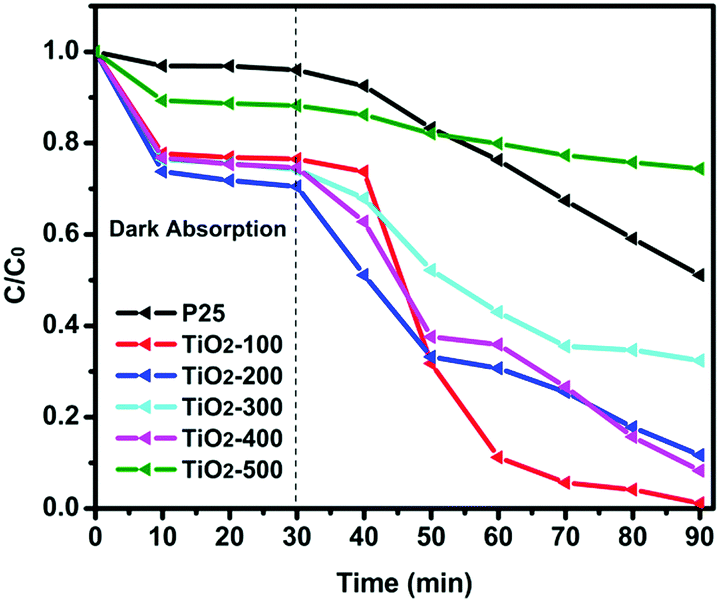 | ||
| Fig. 5 Photocatalytic degradation of RhB over the as-prepared catalysts and P25 under visible light irradiation. | ||
The photocatalytic activity of the as-prepared samples and P25 was studied by degradation of RhB in aqueous solution under visible light at room temperature. It is clearly seen in Fig. 5 that the dye is almost decomposed completely in the test of the TiO2-100 nanocomposite after 60 min of visible light irradiation, which indicates the high photocatalytic ability of the TiO2-100 nanocrystals. Table S5 (ESI†) shows the degradation rate constant of the catalysts for RhB under visible lights irradiation. It is observed that most as-prepared samples show better degradation efficiency than P25. TiO2-100 has a 7 times higher photoactivity compared to P25 under visible light radiation. Fig. S6(2) (ESI†) shows that all as-prepared catalysts exhibited higher UV-vis light photoactivity than P25. Compared with other samples, TiO2-300 and TiO2-400 exhibited the best photoactivity, which also confirmed that 77% anatase and 23% rutile is the optimal ratio for the synergic effect (Table S1, ESI†).16 To further understand the photocatalytic performance of the mixed phase mesoporous TiO2, TiO2-100 was used to degrade solutions of phenol and MB (Fig. S6(3) and (4), ESI†). After visible light was turned on, TiO2-100 was able to remove around 90% MB (Fig. S6(3), ESI†) in 2 h and 50% phenol (Fig. S6(4), ESI†) in 1 h. P25 shows no significant degradation for phenol in 1 h. The above observations confirmed the high reactivity of the synthesized mixed-phase mesoporous materials.
Based on the results, we may ask why the mixed-phase (anatase/rutile) TiO2 exhibits a higher photodegradation capacity under visible light. It can be explained by considering several factors: (1) the large shift of the absorption band extended to the visible light region (narrowed the band gap). In the mixed phase, the reduction in the band gap is because of the presence of the oxygen-related defects. Some Ti–O bonds of anatase or rutile are ruptured and new Ti–O bonds are formed and in the process oxygen defects are created at the interface, which increases the non-radiative transition of the charge carriers hindering their recombination17 and leads to a certain amount of utilization of visible light.10 (2) The mesopore structure was preserved in the mixed-phase TiO2, which had a higher BET surface area. Larger surface areas and pore structures can offer more active sites to adsorb bulky dye molecules and facilitate the diffusion of dye molecules inside the pores. The presence of the mesopores also favors the rapid diffusion of quanta, which form during the photocatalytic reaction and further promote photogenerated charge transport to the surface and improve the separation rate.5a,18 (3) The result of the smaller average crystal size implies more powerful redox ability due to the quantum-size effect.19 Reaction rate enhancement can be due to the increased migration of electrons and holes to the semiconductor surfaces, which is possible in smaller particles, allowing their participation in the reaction, thereby reducing the net electron–hole recombinations.20 The synergetic effect between anatase and rutile is another reason for the high photocatalytic activity. When these two phases combined, a staggered band gap formed and the synergistic effect causes an efficient charge separation across the phase junctions.21 Combining all these advantages, the mesoporous mixed-phase TiO2 exhibited high visible light photocatalytic activity.
In summary, a new mixed-phase (anatase/rutile) TiO2 nanocomposite with mesoporous structures and a smaller crystal size was synthesized by a simple sol–gel method at a relatively low temperature. It exhibited a higher photocatalytic activity than P25 under visible light because of the oxygen defects produced in the process of the mixed-phase being formed, the mesoporous structure and large surface areas, the smaller average crystal size, the synergistic effect of the anatase–rutile mixed phases, and so on. This work presents a simple and economical method to synthesize efficient and stable visible-light-responsive TiO2 photocatalysts for wide applications in environmental remediation.
This work was supported by the Doctoral Research Foundation of Liaoning Province (Grant No. 201501058), the Natural Science Foundation of Liaoning Province (Grant No. 201602683), the Science and Technology Planning Project of Shenyang (Grant No. F15-199-1-21), the Instrument Sharing Service Foundation of Liaoning Province (Grant No. 2015), and the Doctoral Research Foundation and Intramural Research Project (Grant No. L201509) from Shenyang Normal University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. Basnet, G. K. Larsen, R. P. Jadeja, Y.-C. Hung and Y. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 2085 CrossRef CAS PubMed; (b) P. Basnet and Y. Zhao, Catal. Sci. Technol., 2016, 6, 2228 RSC; (c) K. Yao, P. Basnet, H. Sessions, G. K. Larsen, S. E. Hunyadi Murph and Y. Zhao, Catal. Today, 2016, 270, 51 CrossRef CAS; (d) Y. He, P. Basnet, S. E. Hunyadi Murph and Y. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 11818 CrossRef CAS PubMed; (e) H. Yao, F. Li, J. Lutkenhaus, M. Kotaki and H.-J. Sue, AIMS Mater. Sci., 2016, 3, 1410 CrossRef.
- (a) X. Chen and S. S. Mao, Chem. Rev., 2006, 107, 2891 CrossRef PubMed; (b) W. B. Cross, I. P. Parkin, S. A. O’Neill, P. A. Williams, M. F. Mahon and K. C. Molloy, Chem. Mater., 2003, 15, 2786 CrossRef CAS.
- (a) D. C. Hurum, A. G. Agrios and K. A. Gray, J. Phys. Chem. B, 2003, 107, 4545 CrossRef CAS; (b) J. Ovenstone and K. Yanagisawa, Chem. Mater., 1999, 11, 2770 CrossRef CAS.
- H. R. Jafry, M. V. Liga, Q. Li and A. R. Barron, Environ. Sci. Technol., 2011, 45, 1563 CrossRef CAS PubMed.
- (a) Z. Luo, A. S. Poyraz, C.-H. Kuo, R. Miao, Y. Meng, S.-Y. Chen, T. Jiang, C. Wenos and S. L. Suib, Chem. Mater., 2015, 27, 6 CrossRef CAS; (b) E. Masolo, M. Meloni, S. Garroni, G. Mulas, S. Enzo, M. D. Baró, E. Rossinyol, A. Rzeszutek, I. Herrmann-Geppert and M. Pilo, Nanomaterials, 2014, 4, 583 CrossRef PubMed; (c) H. Hou, M. Shang, L. Wang, W. Li, B. Tang and W. Yang, Sci. Rep., 2015, 5, 15228 CrossRef CAS PubMed.
- Z. G. Xiong and X. S. Zhao, J. Am. Chem. Soc., 2012, 134, 5754 CrossRef CAS PubMed.
- Y. Xie, K. L. Ding, Z. M. Liu, R. T. Tao, Z. Y. Sun, H. Y. Zhang and G. M. An, J. Am. Chem. Soc., 2009, 131, 6648 CrossRef CAS PubMed.
- R. A. Spurr and H. Myers, Anal. Chem., 1957, 29, 760 CrossRef CAS.
- G. Song, F. Xin and X. Yin, J. Colloid Interface Sci., 2015, 442, 60 CrossRef CAS PubMed.
- Y. Wang, C. Feng, M. Zhang, J. Yang and Z. Zhang, Appl. Catal., B, 2011, 104, 268 CrossRef CAS.
- B. Santara, P. K. Giri, K. Imakita and M. Fujii, Nanoscale, 2013, 5, 5476 RSC.
- M. Nowak, B. Kauch and P. Szperlich, Rev. Sci. Instrum., 2009, 80, 046107 CrossRef CAS PubMed.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321 CrossRef.
- J. Jhang, M. Li, Z. Feng, J. Chen and C. Li, J. Phys. Chem. B, 2006, 110, 927 CrossRef PubMed.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600 CrossRef CAS PubMed.
- H. Li, X. Shen, Y. Liu, L. Wang, J. Lei and J. Zhang, J. Alloys Compd., 2015, 646, 380 CrossRef CAS.
- R. Verma and S. K. Samdarshi, J. Alloys Compd., 2015, 629, 105 CrossRef CAS.
- M. Gurulakshmi, M. Selvaraj, A. Selvamani, P. Vijayan, N. R. Sasi Rekha and K. Shanthi, Appl. Catal., A, 2012, 449, 31 CrossRef CAS.
- Z. Y. Liu, D. D. Sun, P. Guo and J. O. Leckie, Chem. – Eur. J., 2007, 13, 1851 CrossRef CAS PubMed.
- (a) M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS; (b) P. V. Kamat, Chem. Rev., 1993, 93, 267 CrossRef CAS; (c) N. I. Al-Salim, S. A. Bagshaw, A. Bittar, T. Kemmitt, A. J. McQuillan, A. M. Mills and M. J. Ryan, J. Mater. Chem., 2000, 10, 2358 RSC.
- Y. K. Kho, A. Iwase, W. Y. Teoh, L. Mädler, A. Kudo and R. Amal, J. Phys. Chem. C, 2010, 114, 2821 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc05750d |
| This journal is © The Royal Society of Chemistry 2018 |