
Photoresponsive spherical nucleic acid: spatiotemporal control of the assembly circuit and intracellular microRNA release†
Xiang
Zhou‡
a,
Hui
Li‡
ab,
Miao
He
a,
Xue
Yin
a,
Dongbao
Yao
a,
Shiyan
Xiao
a and
Haojun
Liang
 *ac
*ac
aCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: hjliang@ustc.edu.cn
bDepartment of Biology and Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China
cCollaborative Innovation Center of Chemistry for Energy Materials, Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 28th November 2017
Abstract
Herein, we designed and prepared a photoresponsive spherical nucleic acid (SNA) conjugate by inserting a photocleavable linker into the recognition sequence of SNA which can block the reactivity of the system temporally until specific UV light (∼365 nm) is introduced. The photoresponsive SNA realizes temporal regulation of a self-assembly reaction circuit and spatial selectivity of microRNA release in a cell population, respectively.
Over the past few decades, stimuli-responsive nanomaterials have attracted much attention and they play a pivotal role in many fields, including theranostic studies,1 biochemical sensing,2 self-healing materials,3 and energy research.4 These dynamic nanosystems can sensitively transform their physical or chemical properties, thereby realizing the on-demand “OFF/ON” switching upon application of external stimuli. In this regard, photoresponsiveness is highly attractive.5 Generally, the photoresponsiveness results from the light-switchable molecular group which can temporally block the reactivity of the nanomaterial until specific light is introduced. Light is a highly efficient and clean form of energy that enables remote and instantaneous manipulation. With the advantages of light irradiation, the photoresponsive system allows precise temporal and spatial control, such as gene detection and regulation in vivo at a designated location and time.6 Hence, photoresponsiveness is an ideal property for developing dynamic nanosystems.
Spherical nucleic acid (SNA) represents a fundamentally distinct class of nanomaterials for versatile biotechnological applications.7 The gold nanoparticle (AuNP)-based SNA conjugate is a typical form where the AuNP core acts as a scaffold and a dense oligonucleotide shell is covalently attached on the AuNP surface. One of the most studied properties of SNA is the programmable, DNA-mediated self-assembly which is widely applied in constructing higher-ordered materials8 and biosensors.9 Generally, SNA self-assembly is initiated by the complementary sequences (trigger strand), and the assembly dynamics also depends primarily on inherent factors of the trigger strand (e.g., concentration, length, and sequence composition). This restriction limits the accurate regulation of SNA nanoassembly. Hence, introducing a photoresponsive action to SNA could enable more accurate nanofabrication, such as providing temporal control of the assembly kinetics and a time-resolved aggregation level. In parallel, SNA conjugates also greatly facilitate intracellular assay,7 including biodetection, gene regulation and drug delivery and release (e.g., small molecule and small RNA).10 When incubated with cells, SNA undergoes rapid endocytosis by engaging a membrane-bound scavenger protein.11 This mechanism is common for many cell types, and hence SNA by itself cannot distinguish target cells from the healthy ones, which consequently weakens the specificity of SNA-assisted therapy. Fortunately, the photoresponsive molecular linker can offer a route to overcome this challenge, which can cage the molecular or RNA-based agent within the SNA nanocarriers until specific light is introduced at targeted locations. Accordingly, the spatial selectivity of SNA-assisted drug release can be achieved.
To address the two points, we developed a photoresponsive SNA conjugate via intercalating a photocleavable o-nitrobenzyl linker (PC linker) into the recognition sequence. The PC linker can block the reactivity of the SNA system temporally until specific UV irradiation (∼365 nm) is introduced. Herein, we show that photoresponsive SNA conjugates can achieve temporal and spatial control in a self-assembly reaction and intracellular microRNA release, respectively.
The synthesis of photoresponsive SNA and design of a SNA self-assembly circuit are illustrated in Fig. 1. The citrate-capped 13 ± 1 nm AuNPs were functionalized with thiolated DNA strands (capture DNA). Afterwards, photoresponsive SNA was formed by the hybridization of the hairpin precursor with capture DNA. The PC linker is intercalated at the junction between the loop region (blue) and the stem (green), and the loop sequence therefore is hidden by the PC linker. The details of the hairpin and the SNA conjugate are shown in Fig. S1 and S2 (ESI†), respectively. Upon UV irradiation, the PC linker is rapidly cleaved and the loop sequence is exposed, which results in a reactive single-stranded region called toehold (a). Hence, the photoactivated SNA conjugates (SNA-1) can perform the self-assembly network (Fig. 1b). Firstly, the trigger strand invades the toehold region (a) and displaces the protector (b* and c*) from the linker strand (b–d). After the migration of the protector, a new single-stranded region (c) is generated. Then the fuel strand on the surface of SNA-2 binds to the new lengthy toehold (c and d) and displaces the trigger strand from the intermediate. The trigger strand is released back into the solution and participates in the self-assembly process circularly, whereas SNA-1 and SNA-2 are linked, which forms the detectable aggregates. In contrast, without UV irradiation, the toehold remains concealed by the PC linker, and SNA remains in the “OFF” state, preventing the self-assembly reaction circuit.
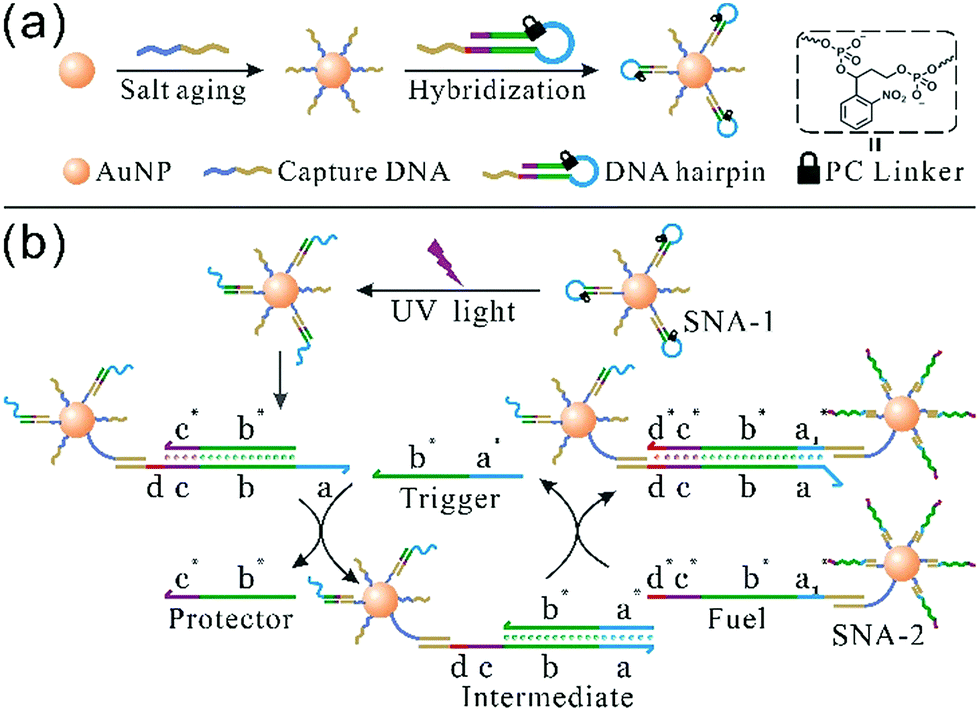 | ||
| Fig. 1 Schematic representation of (a) photoresponsive SNA synthesis and (b) a photocontrolled SNA self-assembly circuit. The hairpin precursor contains a loop (blue), a stem (green and purple) and an overhang end (red and dark yellow). After UV irradiation, the blue part forms a toehold region, and the double-stranded stem consists of a linker (b and c) and a protector (b* and c*). The red line (d) represents another toehold domain which drives the second step of the self-assembly reaction, while the sequence marked in dark yellow is hybridized with the capture strand. | ||
To monitor the assembly reaction, the real-time absorption value (Abs) of the SNA system at 520 nm was recorded. As shown in Fig. 2a, the Abs of the photoactivated system (irradiation duration, 5 min; trigger, 20 nM) sharply decreased within four hours, meaning the assembly dynamics is remarkably accelerated. Also, we employed transmission electron microscopy (TEM) to prove SNA aggregation (Fig. S3, ESI†). In the absence of UV light, the Abs of the inactivated system (trigger, 20 nM) almost remains constant, indicating that SNA cannot aggregate. On the other hand, when the system is subjected to UV irradiation but lacks a trigger strand, a detectable spontaneous aggregation, called “reaction leakage”, still occurs (Fig. S4, ESI†). Therefore, photoresponsiveness can effectively prevent reaction leakage for this assembly network. Despite the dominant influence of UV irradiation, we still need to optimize the amount of the trigger strand to achieve maximized sensitivity for the self-assembly reaction. After uniform UV irradiation for 5 min, the SNA aggregation rate is monitored at varying trigger concentrations (Fig. 2b). The self-assembly kinetics is remarkably expedited with the increase of trigger concentration from 1 nM to 20 nM. However, the acceleration becomes insignificant at higher concentrations of 20–40 nM. The trigger concentration of 20 nM therefore is an optimum choice to achieve the maximized sensitivity of the SNA self-assembly and subsequent experiments are performed using this parameter.
 | ||
| Fig. 2 The real-time kinetics of SNA self-assembly is monitored at 520 nm. (a) In the absence or presence of UV light; (b) the kinetic rate of SNA assembly at different trigger concentrations via a uniform 5 min UV treatment; (c) the influence of the UV irradiation period on the assembly rate; (d) time-resolved aggregation degree at different durations of UV irradiation. | ||
Next, we determined whether the photoresponsive SNA could ensure temporal regulation of SNA aggregation. As shown in Fig. 2c, prolonged exposure to UV light gradually accelerates the aggregation reaction until an exposure period of 5 min when the maximum rate is achieved. In addition, when the duration of UV irradiation exceeds 10 min, the aggregation rate becomes slow instead, which is attributed to the photodegradation of the DNA strands caused by excess UV light (Fig. S5, ESI†). Furthermore, we use the term “aggregation degree (AD)” to describe the assembly level (at ∼8 h). AD is calculated by dividing the change in Abs (ΔA) by the initial value (A0): AD = ΔA/A0. Within an irradiation time of 0–5 min, the AD is significantly elevated. More importantly, each time point results in a distinct AD value that represents a distinct aggregation level (Fig. 2d). Additionally, the hybridization-dependent optical property of the AuNPs indicates that the color of the solution will undergo a significant change after the aggregation reaction, and hence different aggregation levels can be observed by the naked eye (Fig. S6, ESI†). Initially, all samples show a homogeneous pure red color. After UV irradiation, only the non-illuminated colloid remains intact, whereas the other samples fade and sediments accumulate gradually with increasing UV treatment time. Taken together, the photoresponsive SNA conjugates are capable of achieving temporal regulation of assembly dynamics and providing a clearly time-resolved aggregation level at the reaction endpoint.
Parallelly, SNA also exhibits highly effective gene regulation and therapeutic outcome for cancer cells. In this regard, small RNA-based SNA conjugates have made great progress.12 As described previously, SNA can be internalized by many cell types, including cancer and normal cells. This property is disadvantageous to the specificity of SNA-aided intracellular assay. Nevertheless, this challenge could be attenuated via controlled release at a desired location. So, in this work, we prepared photocontrolled, microRNA-34a-functionalized SNA (miR-34a SNA) nanocarriers to achieve spatial selectivity of miR-34a release in MCF-7 breast cancer cells. Meanwhile, the released miR-34a strands can implement gene knockdown, reduce cell viability and induce apoptosis. As shown in Fig. 3, miR-34a SNA conjugates were fabricated by functionalizing AuNPs with DNA–RNA duplexes, which are formed by the hybridization of mature miR-34a mimics (blue) with the complementary DNA strands (purple). Noticeably, two PC linkers were intercalated into the DNA strand. Then the SNA nanocarriers were incubated with MCF-7 cells. To release miR-34a within the assigned region, the “white” area of the culture dish was selectively exposed to UV irradiation, and the other part (“dark”) was shielded. Under UV irradiation, the PC linker-labeled DNA strands are broken, thereby releasing miR-34a strands. In contrast, miR-34a still remains stably attached to the surface of the AuNPs in the “dark” area.
 | ||
| Fig. 3 The spatial selectivity of miR-34a release in MCF-7 population by means of photoresponsive SNA nanocarriers. | ||
To verify the design rationale, miR-34a strands are tagged with a Cy5 dye in order to examine the cellular fluorescence response. In the bound state, the fluorescence of the Cy5 dye is effectively quenched by the AuNP core. After incubation with miR-34a SNA and UV irradiation, the cells were imaged using scanning confocal microscopy (Fig. 4a and b). The cells exposed to UV light show a remarkable Cy5 (red) fluorescence signal, indicating that the PC linker-labeled DNA is cleaved and miR-34a strands are released. Then we selected a “dark” region, which is not illuminated and close to the exposed area. The cell image of the “dark” region shows a negligible Cy5 signal, suggesting that miR-34a still remains stably bound to the AuNP surface without UV irradiation. So, these photocontrolled SNA vehicles successfully achieve spatial control of miR-34a release. Meanwhile, this design provides an internal reference in the same culture dish without an additional blank group. Subsequently, flow cytometry analysis further confirms the cell imaging results (Fig. 4c). The cells treated with both miR-34a SNA and UV irradiation show a remarkable fluorescence intensity, but without UV light, the signal intensity is very weak. Additionally, we also measured the release rate of miR-34a strands in a buffer solution and in MCF-7 cells (Fig. S7, ESI†).
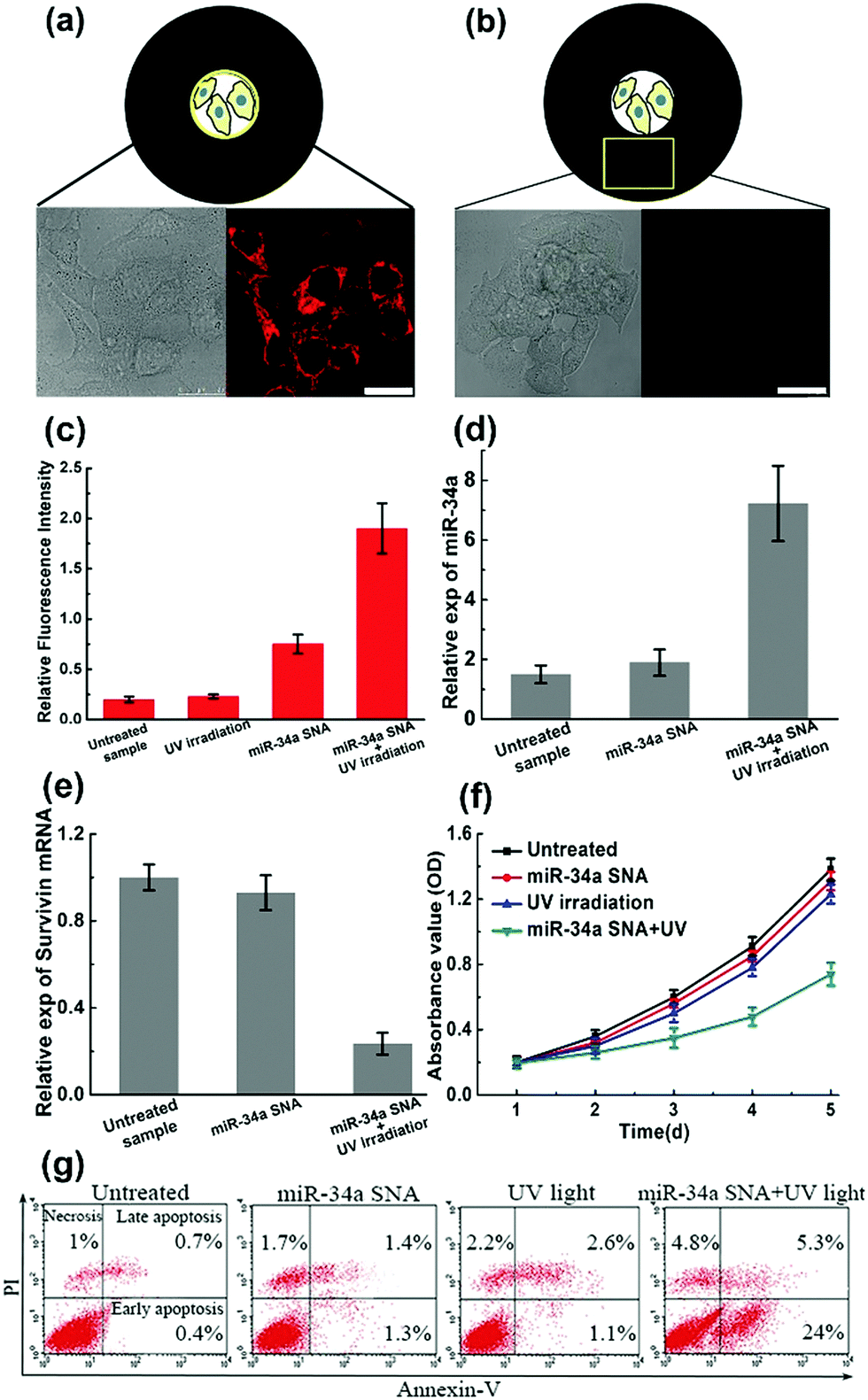 | ||
| Fig. 4 The performance of photoresponsive SNA nanocarriers. (a and b) The spatial control of miR-34a release. MiR-34a strands are labeled with Cy5 dye. (a) The cells exposed to UV irradiation show a remarkable fluorescence signal (red), indicating that miR-34a are released from SNA carriers. (b) In the unexposed area, the cells show a negligible signal, indicating that miR-34a is still caged within the SNA carriers. The scale bar is 25 μm; (c) flow cytometry analyses reinforce cell imaging data; (d and e) estimation of miR-34 expression and the survivin mRNA knockdown level; (f and g) cell growth curves and apoptosis percentages under different conditions. | ||
Numerous studies have shown that the overexpression of miR-34a can suppress proliferation, migration and invasion of the MCF-7 cells13 by targeting several key genes, including Notch, LMTK3, and survivin mRNA. To test the efficiency of the photocontrolled SNA nanocarriers, intracellular miR-34a and survivin mRNA levels are estimated by quantitative RT-PCR (qRT-PCR). When the MCF-7 population was treated with miR-34a SNA conjugates and exposed to UV light, the expression of miR-34a was significantly elevated (Fig. 4d), and the survivin mRNA level was reduced by ∼75% (Fig. 4e) compared with the untreated group. Next, a crucial assessment for this photoresponsive SNA nanosystem is the measurement of the cell viability. The cell growth curves for 5 days were determined by CCK-8 assay and the OD values at 450 nm indicate the viable cell percentage (Fig. 4f). When the cells were treated with SNA conjugates or UV irradiation alone, the OD values decreased slightly compared to that of the untreated sample. In contrast, when incubated with miR-34a SNA and exposed to UV light, the cell viability was significantly reduced from 72 to 120 h. Inducing apoptosis is identified as a major mechanism that miR-34a inhibits cell viability.13 Accordingly, we measured the apoptosis percentage of the cell population using fluorescein isothiocyanate (FITC)-Annexin V and counterstaining with propidium iodide (PI) using flow cytometry analysis (Fig. 4g). The separate treatment of miR-34a SNA or UV irradiation leads to inappreciable apoptosis rates (less than 5%). Strikingly, the MCF-7 population treated with both SNA conjugates and UV light exhibits a large percentage of apoptosis, especially early apoptosis reaching up to ∼24%. So, these measurements verify the high efficiency and availability of the proposed photoresponsive SNA nanocarriers. Furthermore, with the advantage of spatial selectivity of light irradiation, they exhibit great potential in precise cancer therapy.
In summary, we have reported a class of photoresponsive SNA conjugates formed via inserting light-switchable PC linkers into the DNA strands. The reactivity of the SNA nanosystem is initially blocked by the PC linker until specific UV light is applied which causes rapid dissociation of the PC linker. The SNA conjugate engineered through this design is capable of performing spatiotemporal regulation as expected. For SNA self-assembly, photoresponsiveness can effectively prevent spontaneous reaction leakage from the system. More importantly, photoresponsive SNA allows temporal regulation of the self-assembly dynamics and provides a time-resolved aggregation degree at the reaction endpoint at different durations of UV irradiation. Next, we used photoresponsive SNA nanocarriers to implement the spatial specificity of miR-34a release in the MCF-7 cell population. Initially, the miR-34a strands were caged within the SNA vehicles and were then released by subjecting them to UV light at designated locations. Subsequently, the released miR-34a strands demonstrate excellent performance in gene knockdown, inhibiting cell viability and inducing apoptosis, which indicates the high-efficiency and potential availability of the proposed strategy. Overall, endowing SNA conjugates with stimulus-responsive performance could provide a plethora of innovation for sensing methods, nanofabrication design, and diagnostic and therapeutic applications.
We acknowledge the National Natural Science Foundation of China (No. 21434007, 21574122, 91127046, 21404097, and 21404098), and the National Basic Research Program of China (No. 2012CB821500), and the Fundamental Research Funds for the Central Universities (Grant No. WK3450000002 and WK2060200017) for their financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Yi, H. Wang, X. Wang, Q. Liu, M. Ye and W. Tan, ACS Appl. Mater. Interfaces, 2017, 9, 5847 CAS.
- J. I. L. Chen, Y. Chen and D. S. Ginger, J. Am. Chem. Soc., 2010, 132, 9600 CrossRef CAS PubMed.
- Y. Chen and Z. Guan, Polym. Chem., 2013, 4, 4885 RSC.
- X. Guo, S. Xiao, M. Myers, Q. Miao, M. L. Steigerwald and C. Nuckolls, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 691 CrossRef CAS PubMed.
- (a) Z. Dai, H. M. Leung and P. K. Lo, Small, 2017, 13, 1602881 CrossRef PubMed; (b) L. Qiu, C. Wu, M. You, D. Han, T. Chen, G. Zhu, J. Jiang, R. Yu and W. Tan, J. Am. Chem. Soc., 2013, 135, 12952 CrossRef CAS PubMed.
- A. Deiters, R. A. Garner, H. Lusic, J. M. Govan, M. Dush, N. M. Nascone-Yoder and J. A. Yoder, J. Am. Chem. Soc., 2010, 132, 15644 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376 CrossRef CAS PubMed.
- (a) M. R. Jones, R. J. Macfarlane, B. Lee, J. Zhang, K. L. Young, A. J. Senesi and C. A. Mirkin, Nat. Mater., 2010, 9, 913 CrossRef CAS PubMed; (b) D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549 CrossRef CAS PubMed; (c) S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553 CrossRef CAS PubMed.
- (a) N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed; (b) H. Li, S. Xiao, D. Yao, M. H.-W. Lam and H. Liang, Chem. Commun., 2015, 51, 4670 RSC.
- (a) S. P. Narayan, C. H. J. Choi, L. Hao, C. M. Calabrese, E. Auyeung, C. Zhang, O. J. G. M. Goor and C. A. Mirkin, Small, 2015, 11, 4173 CrossRef CAS PubMed; (b) X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, J. Am. Chem. Soc., 2016, 138, 10834 CrossRef CAS PubMed; (c) F. M. Kouri, L. A. Hurley, W. L. Daniel, E. S. Day, Y. Hua, L. Hao, C.-Y. Peng, T. J. Merkel, M. A. Queisser, C. Ritner, H. Zhang, C. D. James, J. I. Sznajder, L. Chin, D. A. Giljohann, J. A. Kessler, M. E. Peter, C. A. Mirkin and A. H. Stegh, Genes Dev., 2015, 29, 732 CrossRef CAS PubMed.
- (a) C. H. J. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625 CrossRef CAS PubMed; (b) D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818 CrossRef CAS PubMed.
- (a) L. Zhang, W. Zheng, R. Tang, N. Wang, W. Zhang and X. Jiang, Biomaterials, 2016, 104, 269 CrossRef CAS PubMed; (b) H. Li, W. Shen, M. Hon-Wah Lam and H. Liang, New J. Chem., 2017, 41, 5255 RSC; (c) P. S. Randeria, M. A. Seeger, X.-Q. Wang, H. Wilson, D. Shipp, C. A. Mirkin and A. S. Paller, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5573 CrossRef CAS PubMed.
- (a) G. Misso, M. T. Di Martino, G. De Rosa, A. A. Farooqi, A. Lombardi, V. Campani, M. R. Zarone, A. Gullà, P. Tagliaferri, P. Tassone and M. Caraglia, Mol. Ther.–Nucleic Acids, 2014, 3, e194 CrossRef CAS PubMed; (b) X. J. Li, Z. J. Ren and J. H. Tang, Cell Death Discovery, 2014, 5, e1327 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and additional data. See DOI: 10.1039/c7cc07932j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |