
A green-light-emitting, spontaneously blinking fluorophore based on intramolecular spirocyclization for dual-colour super-resolution imaging†
Shin-nosuke
Uno
 a,
Mako
Kamiya
*bc,
Akihiko
Morozumi
a and
Yasuteru
Urano
*ab
a,
Mako
Kamiya
*bc,
Akihiko
Morozumi
a and
Yasuteru
Urano
*ab
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: uranokun@m.u-tokyo.ac.jp
bGraduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: mkamiya@m.u-tokyo.ac.jp
cPRESTO, Japan Science and Technology Agency, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 7th December 2017
Abstract
We have developed the first green-light-emitting, spontaneously blinking fluorophore (SBF), HEtetTFER. In combination with our near-infrared-light-emitting SBF (HMSiR), HEtetTFER allows dual-colour single-molecule localization microscopy (SMLM) in buffer solution without any additive and without photoactivation.
Super-resolution imaging has made it possible to visualize structures inside cells at unprecedented resolution.1,2 Single-molecule localization microscopy (SMLM) is one of the most frequently used methods,3–7 and utilizes repeated detection and high-precision localization of temporally resolved fluorescence signals to enable reconstruction of super-resolution images.8,9
Organic fluorophores are generally more suitable for SMLM than fluorescent proteins, since the spatial resolution depends on the detected photon number.10 To perform SMLM with conventional organic fluorophores, it is necessary to achieve a situation in which most of the fluorophores have been converted to the non-fluorescent dark state and only a small subset of them switch successively to the fluorescent state. So far, two major approaches have been developed: one employs caged fluorophores, and the other, known as dSTORM, requires initial conversion of the fluorophores to the non-emitting dark state by intense photoirradiation in a specific imaging buffer.8
Caged fluorophores exist in the non-emitting dark state and upon irradiation, a small subset of them is converted to the fluorescent state, providing high-contrast signals suitable for precise localization.11,12 However, the uncaged fluorophores need to be photobleached after obtaining a super-resolution image, and so repeated time-lapse imaging of the same target is virtually impossible. Moreover, an additional laser line is required for activation, and the phototoxic effects of UV irradiation are problematic for live-cell imaging.13
In contrast, dSTORM permits repeated time-lapse imaging, since it employs the reversible photoreaction of organic fluorophores with chemicals such as thiols to drive the fluorophores into the dark state.14 However, the composition of the imaging buffer has to be carefully optimized for each fluorophore used in the experiment.15–17 It is particularly difficult to find suitable conditions for inducing appropriate blinking states of plural fluorophores to visualize two different targets by dual-colour SMLM. Furthermore, to avoid crosstalk and to detect emitted photons as efficiently as possible, it is important that the absorption and fluorescence spectra are well separated. The pairs of fluorophores that meet these requirements are still few in number, although dual-colour SMLM has been reported.18–20
We have recently developed a spontaneously blinking fluorophore (SBF), HMSiR, as a first-in-class SMLM probe.21 In contrast to conventional fluorophores, HMSiR exists mostly in a colourless and non-fluorescent form in the ground state, and spontaneously blinks, reflecting the thermal equilibrium of intramolecular spirocyclization. By virtue of its spontaneous blinking property, HMSiR circumvents the need to use any chemical additive or photoactivation, so that SMLM can be conducted under flexible experimental conditions; in addition, HMSiR is suitable for time-lapse live-cell SMLM with minimum side effects.21,22 Since HMSiR emits fluorescence in the near-infrared (NIR) region, a green-light-emitting SBF (gSBF) that could easily be spectrally resolved from HMSiR would be valuable for dual-colour SMLM without the need for troublesome optimization.
In this work, we developed HEtetTFER as the first gSBF, based on our strategy of utilizing the thermal equilibrium of intramolecular spirocyclization of rhodamines (Fig. 1 and Fig. S1, ESI†). The new fluorophore shows bright fluorescence in the green region, and exhibits spontaneous blinking even in additive-free buffer solution. Furthermore, we confirmed that dual-colour SMLM can be achieved with the combination of HEtetTFER and HMSiR.
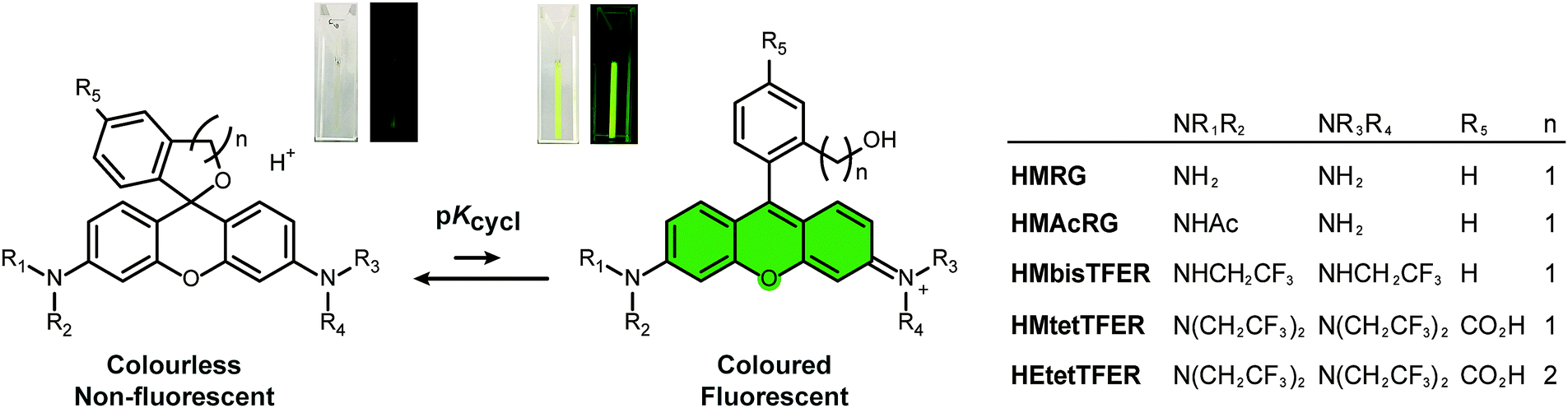 | ||
| Fig. 1 Chemical structures of green-light-emitting, spontaneously blinking fluorophores (gSBFs). gSBFs undergo thermally equilibrating intramolecular spirocyclization in aqueous solution (equilibrium constant: pKcycl). | ||
Our previous work demonstrated that rhodamine derivatives in which a carboxy group is replaced with a more nucleophilic group, such as a hydroxymethyl (HM) group, show thermally equilibrated intramolecular spirocyclization behaviour in aqueous solution.23–25 Specifically, they exist as a coloured and fluorescent open form under acidic conditions, but as a colourless and non-fluorescent closed form under basic conditions (Fig. 1). Aiming to develop SBFs suitable for SMLM, we established that the equilibrium constant of intramolecular spirocyclization (pKcycl, see the ESI†) and the duration until the fluorescent open form reverts to the non-fluorescent closed form are important factors to obtain spatially isolated signals and to accumulate sufficient photons in a millisecond-order camera frame. Here, we adopted the criterion that the pKcycl value should be less than 6, so that most molecules exist in the closed form at neutral pH, and we decided to focus on the HM group as an intramolecular nucleophile, as it tends to show a favourable duration of the fluorescent state (tens to hundreds of milliseconds).
First, we considered our previously reported green-light-emitting HM rhodamines, HMRG and HMAcRG. However, these were not satisfactory candidates for gSBFs (Table 1);21HMRG has a high pKcycl value of 8.1, which causes overlapping of fluorescence signals, while HMAcRG, though it has an appropriate pKcycl value of 5.3, is not bright enough to allow determination of its precise position, since amidation of rhodamine fluorophores tends not only to shift the equilibrium toward the closed form, but also to reduce the molar extinction coefficient (ε) and fluorescence quantum yield (Φfl).24,26 Thus, instead of amidation, we set out to install electron-withdrawing trifluoroethyl (TFE) groups on the nitrogen atom of HMRG to obtain the desired pKcycl value of less than 6 while maintaining the green fluorescence emission. N-Alkylation of rhodamines, such as N-methylation or N-ethylation, generally induces an increase of pKcycl value and a 15 nm shift in the absorption/emission wavelengths.27 However, we previously found that replacing an ethyl group of HM rhodol (HMRet) with a TFE group induces a decrease of the pKcycl value by 0.7, together with blue-shifts of the absorption and fluorescence maxima (λabs and λem) by 12 and 16 nm, respectively (Fig. S2, ESI†),28 pushing the absorption/emission wavelengths back to those of the non-alkylated analogue.29 Similar results were reported in rhodamine systems having a carboxylate group at the 2′-position; the introduction of two TFE groups onto the nitrogen of rhodamines30 or incorporation of fluorine at the 3-position of the azetidine rings in rhodamines31 shifted the equilibrium toward the closed spirolactone form in dioxane–water mixtures, and elicited hypsochromic shifts.
| λ abs [nm] | λ em [nm] | ε [M−1 cm−1] | Φ fl | pKcycl | Brightness (ε × Φfl) | |
|---|---|---|---|---|---|---|
| a Absorption maximum (λabs), fluorescence emission maximum (λem), molar extinction coefficient (ε), fluorescence quantum yield (Φfl), equilibrium constant for intramolecular spirocyclization (pKcycl), relative brightness (ε × Φfl). b Measured in 0.2 M sodium phosphate buffer (pH 2.0). c Measured in 0.2 M sodium phosphate buffer (pH 4.5). d Data from ref. 21. e The value for the fully open analogue. | ||||||
| HMRG , | 500 | 523 | 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.90 | 8.1 | 195 |
| HMAcRG , | 493 | 528 | 30200 |
0.39 | 5.3 | 35 |
| HMbisTFER | 502 | 526 | 81000 |
0.85 | 7.2 | 202 |
| HMtetTFER | 507 | 532 | 57000 |
0.89 | 5.0 | 149 |
| HEtetTFER | 507 | 530 | 80000 |
0.76 | 5.5 | 178 |
| HMSiR , | 650 | 671 | 110000e |
0.31 | 5.8 | 100 |
Thus, we designed and synthesized HMbisTFER and HMtetTFER, incorporating two and four TFE groups, respectively, on the nitrogen atoms (Fig. 1, Schemes S1 and S2, ESI†). As expected, HMbisTFER and HMtetTFER showed λabs/λem at 502/526 nm and 507/532 nm, respectively (Fig. S3, ESI†). These values are comparable with those of HMRG (Table 1). Although HMtetTFER showed two inflection points, as it is a diprotic acid, the pKcycl values were calculated to be 7.2 and 5.0 for HMbisTFER and HMtetTFER, respectively. Thus, the larger number of TFE groups resulted in a smaller pKcycl value, which can be explained in terms of lowering of the lowest unoccupied molecular orbital (LUMO) levels of the xanthene moiety (fluorophore) by the electron-withdrawing TFE groups (Fig. S4, ESI†).24,27 According to the criterion that pKcycl should be less than 6, we chose HMtetTFER as a candidate gSBF. To test the performance of HMtetTFER in SMLM, we prepared HMtetTEFR-labelled secondary antibody. Then, β-tubulin in Vero cells was immunostained, and SMLM was performed in oxygenated phosphate-buffered saline (PBS). However, the signals of fluorophores were blurred, with a low signal-to-background ratio, and clear super-resolution images could not be reconstructed. Possible reasons include aggregation during labelling, or a blinking rate faster than the acquisition rate.
To overcome this problem, we designed and synthesized HEtetTFER by replacing the HM group with a hydroxyethyl (HE) group as a similar alcoholic nucleophile (Fig. 1 and Scheme S3, ESI†). HEtetTFER showed λabs/λem of 507/530 nm, like HMtetTFER (Fig. 2a and b), and had sufficiently high values of ε and Φfl in aqueous buffer solution (80000 M−1 cm−1, 0.76, respectively) to achieve high localization accuracy (Table 1). The pKcycl value of HEtetTFER was 5.5, which is slightly shifted to the basic side compared to that of HMtetTFER, but the pKcycl value is still less than 6 (Fig. 2c).
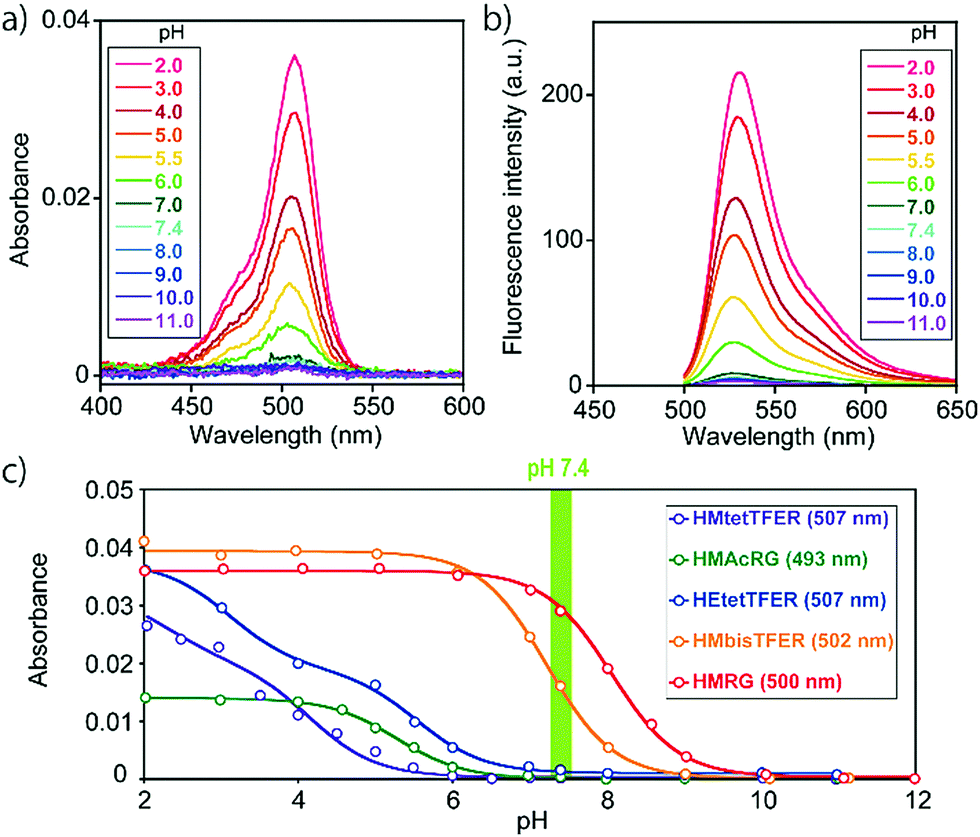 | ||
| Fig. 2 (a) Absorption and (b) fluorescence spectra of HEtetTFER at various pH values in a 0.2 M sodium phosphate buffer. The excitation wavelength was 480 nm. (c) pH profiles of gSBFs in 0.2 M sodium phosphate buffer at various pH values (dye concentration: 0.5 μM). | ||
Next, in order to evaluate the performance of HEtetTFER in SMLM under oxygenated imaging conditions, we synthesized HEtetTFER-sulfoNHS to label the secondary antibody. β-Tubulin was immunostained, and SMLM was performed in PBS with excitation at 488 nm. HEtetTFER showed spontaneous blinking immediately after starting acquisition (Movie S1, ESI†), and super-resolution images were successfully reconstructed (Fig. 3a–c). The photon number of each signal and the localization accuracy were 1145.6 ± 968.0 and 12.3 ± 5.7 nm (mean ± s.d., N = 566464), respectively (Fig. S5, ESI†). SMLM images showed fine structures of microtubules with a full-width at half-maximum (FWHM) of 70.9 ± 4.7 nm (mean ± s.e., N = 5), while conventional images had an FWHM of 389.7 ± 23.8 nm (mean ± s.e., N = 5) (Fig. 3d). Moreover, by using a cylindrical lens to perform three-dimensional (3D) SMLM, we could resolve the complex 3D microtubule network (Fig. 3e).
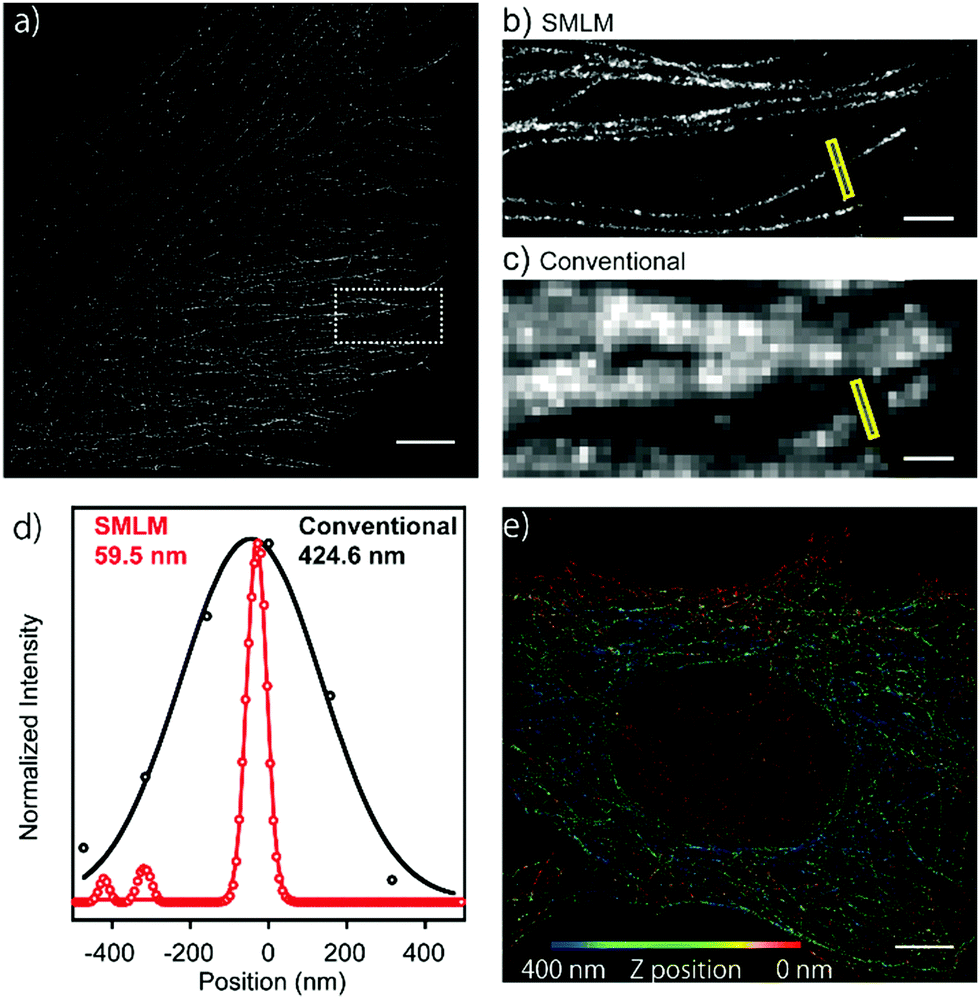 | ||
| Fig. 3 SMLM of microtubules (β-tubulin) in fixed Vero cells with HEtetTFER. (a) Super-resolution image reconstructed from 20000 frames (60 ms per frame). Measured in PBS with excitation at 488 nm (240 W cm−2). (b) SMLM and (c) conventional images of the region enclosed by the white box in (a). (d) Line profile of the microtubule in the yellow box in (b–c). Black, conventional image; red, SMLM image. The FWHM value was taken as the width. (e) Three-dimensional SMLM of microtubules (β-tubulin) with HEtetTFER. Scale bars, 5 μm for (a) and (e); 1 μm for (b) and (c). | ||
Since the absorption and fluorescence spectra of HEtetTFER are completely separable from those of NIR-light-emitting HMSiR (Fig. 4a), the combination of these different-coloured SBFs should be extremely effective for dual-colour SMLM without the need for imaging buffer optimization or photoactivation. Therefore, β-tubulin and Tom20 (a mitochondrial outer membrane marker) in Vero cells were immunostained with HEtetTFER and HMSiR, respectively, and sequential SMLM was performed. We observed little crosstalk between the two fluorophores, and super-resolution images of microtubules and mitochondria were successfully reconstructed (Fig. 4b).
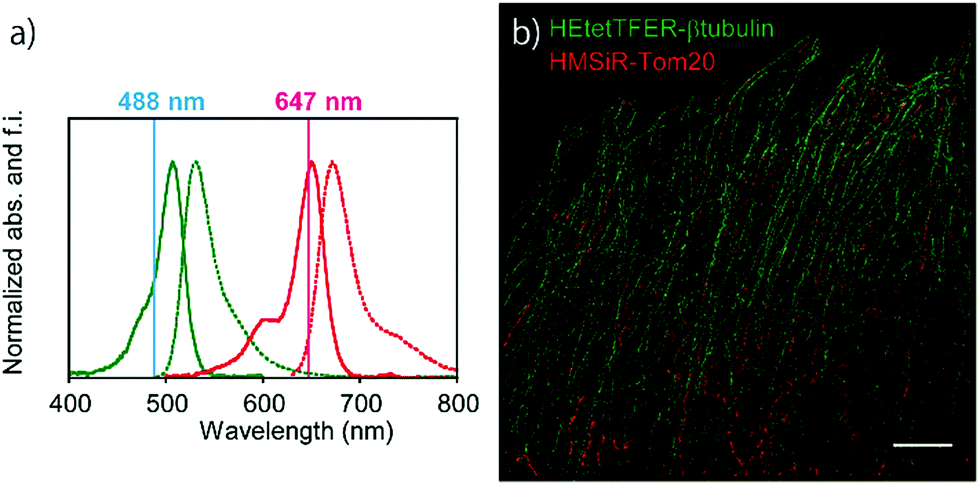 | ||
| Fig. 4 (a) Normalized absorption and fluorescence spectra of HEtetTFER (green) and HMSiR (red). Solid and dashed lines show absorption and fluorescence spectra, respectively. (b) Dual-colour SMLM of microtubules (HEtetTFER-β-tubulin) and mitochondria (HMSiR-Tom20) in fixed Vero cells. Scale bar, 5 μm. | ||
In conclusion, we developed a novel green-light-emitting SBF, HEtetTFER by exploiting a new rhodamine scaffold, and achieved dual-colour SMLM by using this agent in combination with our NIR-light-emitting SBF, HMSiR. This pair of SBFs exhibits spontaneous blinking due to thermal equilibration of spirocyclization, offering a simple procedure for dual-colour SMLM in additive-free buffer solution without optimization, and without an activation laser. Although it is not yet clear why we could not perform SMLM with HMtetTFER, the results suggest that it is important to optimize the combination of nucleophiles and fluorophores for successful SMLM. In principle, HEtetTFER could be adapted for live-cell imaging by preparing ligands for self-labelling tags such as SNAP-tag and HaloTag, and this approach should be useful for understanding in detail the interactions between two different proteins at sub-diffraction resolution.
This research was supported in part by JST/PRESTO grant JPMJPR14F8 (to M. K.), by MEXT/JSPS KAKENHI grants JP16H02606, JP26111012 (to Y. U.) and JP15H05951 “Resonance Bio” (to M. K.), by JSPS Core-to-Core Program, A. Advanced Research Networks, by grants from Hoansha Foundation (to Y. U.) and Japan Foundation for Applied Enzymology (to M. K.), by a JSPS postdoctoral fellowship (to S. U.), and by a JSPS stipend (to A. M.). Vero cells (JCRB0111) were obtained from the Japanese Collection of Research Bioresources (JCRB) cell bank.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 18, 685–701 CrossRef PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- S. T. Hess, T. P. K. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- J. Folling, M. Bossi, H. Bock, R. Medda, C. A. Wurm, B. Hein, S. Jakobs, C. Eggeling and S. W. Hell, Nat. Methods, 2008, 5, 943–945 CrossRef PubMed.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS PubMed.
- S. van de Linde, A. Loschberger, T. Klein, M. Heidbreder, S. Wolter, M. Heilemann and M. Sauer, Nat. Protoc., 2011, 6, 991–1009 CrossRef CAS PubMed.
- M. Sauer and M. Heilemann, Chem. Rev., 2017, 117, 7478–7509 CrossRef CAS PubMed.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS PubMed.
- J. B. Grimm, T. Klein, B. G. Kopek, G. Shtengel, H. F. Hess, M. Sauer and L. D. Lavis, Angew. Chem., Int. Ed., 2016, 55, 1723–1727 CrossRef CAS PubMed.
- J. C. Vaughan, S. Jia and X. Zhuang, Nat. Methods, 2012, 9, 1181–1184 CrossRef CAS PubMed.
- S. Wäldchen, J. Lehmann, T. Klein, S. van de Linde and M. Sauer, Sci. Rep., 2015, 5, 15348 CrossRef PubMed.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z.-G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed.
- J. Vogelsang, C. Steinhauer, C. Forthmann, I. H. Stein, B. Person-Skegro, T. Cordes and P. Tinnefeld, ChemPhysChem, 2010, 11, 2475–2490 CrossRef CAS PubMed.
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Nat. Methods, 2011, 8, 1027–1036 CrossRef CAS PubMed.
- L. Carlini and S. Manley, ACS Chem. Biol., 2013, 8, 2643–2648 CrossRef CAS PubMed.
- M. Bates, B. Huang, G. T. Dempsey and X. Zhuang, Science, 2007, 317, 1749–1753 CrossRef CAS PubMed.
- A. Löschberger, S. van de Linde, M.-C. Dabauvalle, B. Rieger, M. Heilemann, G. Krohne and M. Sauer, J. Cell Sci., 2012, 125, 570–575 CrossRef PubMed.
- W. Muranyi, S. Malkusch, B. Müller, M. Heilemann and H.-G. Kräusslich, PLoS Pathog., 2013, 9, e1003198 Search PubMed.
- S. Uno, M. Kamiya, T. Yoshihara, K. Sugawara, K. Okabe, M. C. Tarhan, H. Fujita, T. Funatsu, Y. Okada, S. Tobita and Y. Urano, Nat. Chem., 2014, 6, 681–689 CAS.
- H. Takakura, Y. Zhang, R. S. Erdmann, A. D. Thompson, Y. Lin, B. McNellis, F. Rivera-Molina, S. Uno, M. Kamiya, Y. Urano, J. E. Rothman, J. Bewersdorf, A. Schepartz and D. Toomre, Nat. Biotechnol., 2017, 35, 773–780 CrossRef CAS PubMed.
- S. Kenmoku, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 7313–7318 CrossRef CAS PubMed.
- M. Sakabe, D. Asanuma, M. Kamiya, R. J. Iwatate, K. Hanaoka, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2013, 135, 409–414 CrossRef CAS PubMed.
- Y. Urano, M. Sakabe, N. Kosaka, M. Ogawa, M. Mitsunaga, D. Asanuma, M. Kamiya, M. R. Young, T. Nagano, P. L. Choyke and H. Kobayashi, Sci. Transl. Med., 2011, 3, 110–119 Search PubMed.
- L. D. Lavis, T.-Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef CAS PubMed.
- R. J. Iwatate, M. Kamiya and Y. Urano, Chem. – Eur. J., 2016, 22, 1696–1703 CrossRef CAS PubMed.
- D. Asanuma, M. Sakabe, M. Kamiya, K. Yamamoto, J. Hiratake, M. Ogawa, N. Kosaka, P. L. Choyke, T. Nagano, H. Kobayashi and Y. Urano, Nat. Commun., 2015, 6, 6463 CrossRef CAS PubMed.
- Y.-H. Ahn, J.-S. Lee and Y.-T. Chang, J. Am. Chem. Soc., 2007, 129, 4510–4511 CrossRef CAS PubMed.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P.-T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290–3294 CrossRef CAS PubMed.
- J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987–994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization of new compounds, and super-resolution imaging details. See DOI: 10.1039/c7cc07783a |
| This journal is © The Royal Society of Chemistry 2018 |