
Chemical amplification accelerates reactive oxygen species triggered polymeric degradation†
Sangeun
Lee
ab,
Alexandra
Stubelius
ac,
Jason
Olejniczak
a,
Hongje
Jang
d,
Viet Anh Nguyen
Huu
a and
Adah
Almutairi
 *abc
*abc
aUCSD Center of Excellence in Nanomedicine and Engineering, University of California San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA. E-mail: aalmutairi@ucsd.edu
bDepartments of NanoEngineering, Jacobs School of Engineering, University of California San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA
cSkaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, 9500 Gilman Dr., La Jolla, California 92093, USA
dIII. Institute of Physics, Georg August University Goettingen, Friedrich-Hund-Platz 1, D-37077 Goettingen, Germany
First published on 28th November 2017
Abstract
Chemical amplification is a known strategy for improving the sensitivity of stimuli-responsive polymers. However, the chemical amplification effect has never been fully examined. Many questions remain about its mechanism and efficacy, obstructing its further implementation. Here, we design and demonstrate a reactive oxygen species (ROS) responsive polymer (ROS-ARP) with a chemical amplification strategy to dismiss these concerns. The ROS-ARP is designed to change the hydrophilicity by ROS, revealing a carboxylic acid, which also catalyzes ketal hydrolysis along the polymer backbone. The chemical amplification strategy of ROS-ARP accelerated the polymer degradation up to 17 fold compared to a previously reported ROS-responsive polymer. To investigate the mechanism behind this increased acceleration, we compared the degradation kinetics in various environments. Additionally, other effects such as hydrophilicity changes were excluded. The accelerated degradation of ROS-ARP is evaluated as a potential drug delivery system, demonstrating on-demand cargo release from the formulated polymeric particles.
Introduction
On-demand control of polymeric nanoparticle properties has been attractive in a broad range of fields such as cosmetics, environmental engineering, energy and electronics, and biomedical applications.1–3 Despite its significance, few polymers can degrade or change their properties by relevant stimuli concentrations.4–6 In particular, it is a challenge to develop polymers degrading by biological stimuli, since they need to be sensitive to subtle change of the triggers.7–11 For this reason, very few ROS (reactive oxygen species) responsive materials react to pathological ROS concentrations,12–15 although ROS are important biomarkers in inflammatory diseases.16–19One avenue to address this sensitivity challenge is by applying a chemical amplification strategy.20–23 However, the effect of the strategy and the mechanism are not fully understood. Recently, Song et al.24 reported a hydrogen peroxide sensitive, chemical amplifying polymer. A hydrophilicity switch occurred at the hydrophobic portion of the amphiphilic polymer when exposed to H2O2, which caused oxidation to a carboxylic acid, followed by ketal hydrolysis. Although the polymeric particle disassembly was accelerated successfully, it is difficult to evaluate a chemical amplification effect without considering the contribution from a hydrophilicity change by the carboxylic acid. Therefore, questions remain such as whether the revealed acid catalyzes ketal hydrolysis, and whether the chemical amplification strategy works directly on the polymer.
Here, we designed an H2O2-sensitive polymer incorporating a chemical amplification strategy for increased polymer degradation efficiency to dispel issues with the strategy (Scheme 1). The designed polymer is composed of three monomers each having different functionalities. The degradation of the polymer arises from the hydrolysis of ketal groups on the backbone. Ketal groups are great candidates because of their rapid catalytic degradation.25–29 However, they suffer from poor stability and a short shelf life. One way to overcome the stability issue is to add a logic gate system. The logic gate system is based on different ketal hydrolysis rates depending on the hydrophilicity of the polymers the ketal groups are incorporated in.30–34 In this study, we incorporated two ROS-sensitive functional groups into the polymer to control the hydrolysis rate of the ketals.
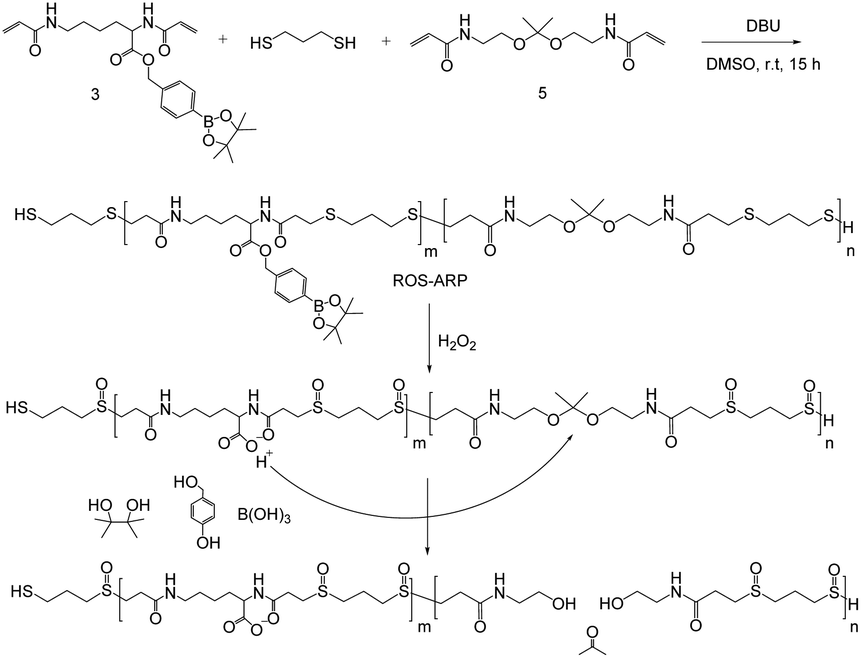 | ||
| Scheme 1 Polymerization scheme and H2O2-responsive ROS-ARP degradation mechanism. | ||
First, a sulfide group is incorporated, not only as a Michael addition donor for polymerization but also as an ROS sensitive hydrophilicity switch. Sulfide groups are oxidized to sulfoxides or sulfonyl groups by reacting with various ROS, such as H2O2 and hypochlorous acid.35–37 This oxidation of sulfides changes the polymer's polarity and eventually induces hydrophilicity change of sulfide containing polymers.38–42
Another ROS-sensitive functional group is hydroxymethyl-phenyl boronic acid pinacol ester (BE). It is well known that BE can be quantitatively cleaved by H2O2 with high selectivity and sensitivity.43–50 This functional group is conjugated with a carboxylic acid of an acryloyl modified lysine to yield an H2O2-responsive group masking the acid. Acid release by H2O2 amplifies the ROS signal and catalyzes the hydrolysis of the ketals in the polymer backbone.
By polymerizing the monomers with three different functionalities, a chemically amplified highly efficient polymer degradation was expected. To prove that the chemical amplification accelerated the polymer degradation, we compared the degradation kinetics between a newly designed polymer and a control polymer without an amplification strategy. Additionally, a hydrophilicity switch effect was excluded by suppressing ketal hydrolysis following increased water accessibility. Based on the investigated degradation mechanism, on-demand polymeric particle degradation was demonstrated after nanoparticle (NP) formulation. The polymeric NPs were also evaluated for potential use as a drug delivery material by studying H2O2-responsive particle degradation, controlled cargo release and cell viability.
Results and discussion
Synthesis of monomers and polymerization
Monomers were synthesized by following Scheme 2. For H2O2 sensitive BE monomer synthesis, 4-(hydroxymethyl)phenyl boronic acid pinacol ester was conjugated onto N,N′-di-boc-L-lysine by DCC coupling. By following tert-butyloxycarbonyl (boc) de-protection, compound 2 was synthesized. The ROS sensitive, BE modified monomer, compound 3, was prepared by reacting acryloyl chlorides with compound 2 (Fig. S1†). A pH sensitive ketal monomer was synthesized by following previously reported methods.32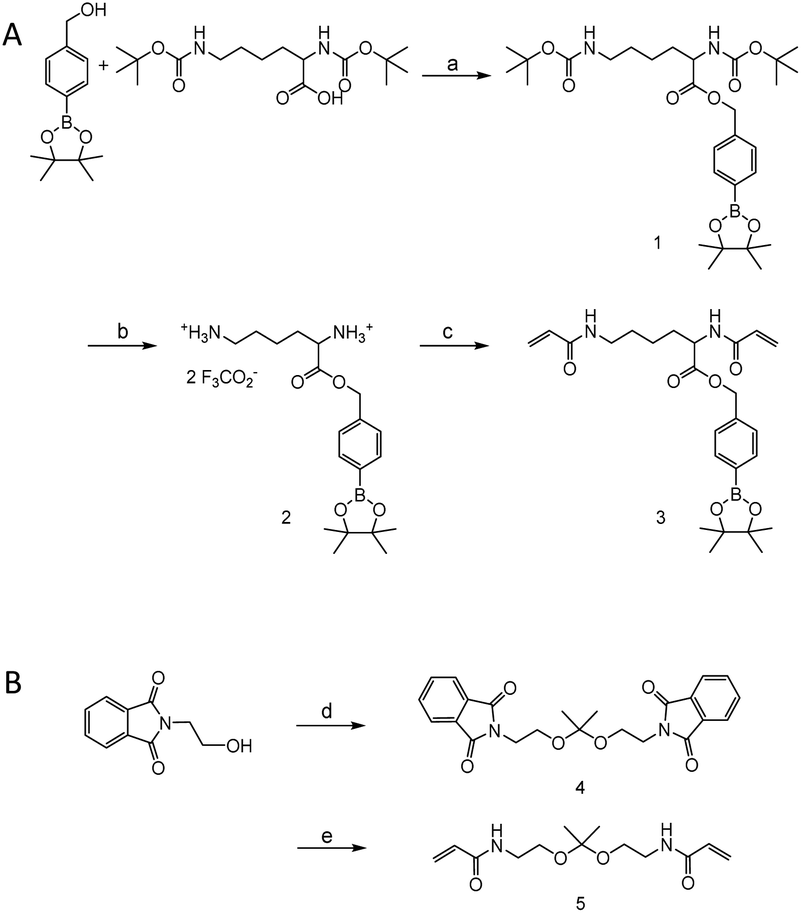 | ||
| Scheme 2 Synthesis scheme of H2O2-sensitive boronic ester monomer (compound 3) (A) and acid-sensitive ketal monomer (compound 5) (B). Reagents and conditions: (a) DCC, DMAP, DCM, r.t., 15 h, 49%; (b) TFA/DCM, r.t., 1 h, 96%; (c) acryloyl chloride, TEA, DCM, 0 °C, 1 h, 33%; (d) (i) 2-methoxypropene, PTSA, benzene, r.t., 1 h, (ii) TEA, acetic anhydride, benzene, r.t., 15 h, 66% over 2 steps; (e) (i) NaOH/H2O, reflux, 15 h, (ii) acryloyl chloride, TEA, H2O/dioxane, −5 °C, 1 h, 90% over 2 steps. | ||
Compounds 3 and 5 were polymerized with 1,3-propandithiol in d6-DMSO under argon. DBU is used as a catalyst. After 2 hours of reaction time, the crude solution became viscous. Proton peaks of the acryloyl group were completely flattened in the NMR spectrum after overnight incubation. This implies that all the acryloyl groups are consumed and the polymerization completed. This reaction time is remarkably shorter than TEA catalyzed Michael addition polymerization.20,31 DBU induced faster polymerization since DBU has higher basicity and also supports the reaction by acting as a nucleophile.51–53 After purification, a pale yellow oily product was obtained and characterized by gel permeation chromatography (GPC) and 1H-NMR (Fig. S2†). The molecular weight of the polymer was calculated based on a PMMA standard curve. The calculated average molecular weight of the polymer was 10 kDa and the polydispersity index was 1.25. The monomer incorporation ratio was calculated based on H-NMR peaks, and the ratio between BE monomers and ketal monomers was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3.
:1.3.
Stimuli triggered polymer degradation
After obtaining the polymer, H2O2 triggered polymer degradation was evaluated by GPC. The degradation of the polymer backbone is triggered by H2O2 but ultimately leads to the hydrolysis of ketals on the polymers. Thus, after each time point, polymer solutions in two different H2O2 concentrations (0 mM and 10 mM) were compared at different pH values (5.0, 7.4, and 10.0) (Fig. S3 and S4†).First, the polymer degradation at neutral pH, pH 7.4, with and without H2O2 was compared. Fig. 1A is the RI signal of the ROS-ARP obtained by GPC. Using the signal, we calculated and analyzed the molecular weight changes of the ROS-ARP in two different H2O2 concentrations at each time point (Fig. 1B). Under 10 mM H2O2, 59% of the calculated molecular weight of the polymer decreased in 24 hours, while the polymer in 0 mM H2O2 at pH 7.4 degraded only 8% during the same period. Also, UV absorption in the low molecular weight range remarkably increased with incubation time (Fig. S5†). These are considered as peaks of products from the H2O2 and BE reaction. To support this, a representative product, 4-hydroxybenzyl alcohol, was injected into the GPC under the same conditions as the polymers. The retention time of 4-hydroxybenzyl alcohol had a similar UV absorption to the aforementioned peaks (Fig. S5†).
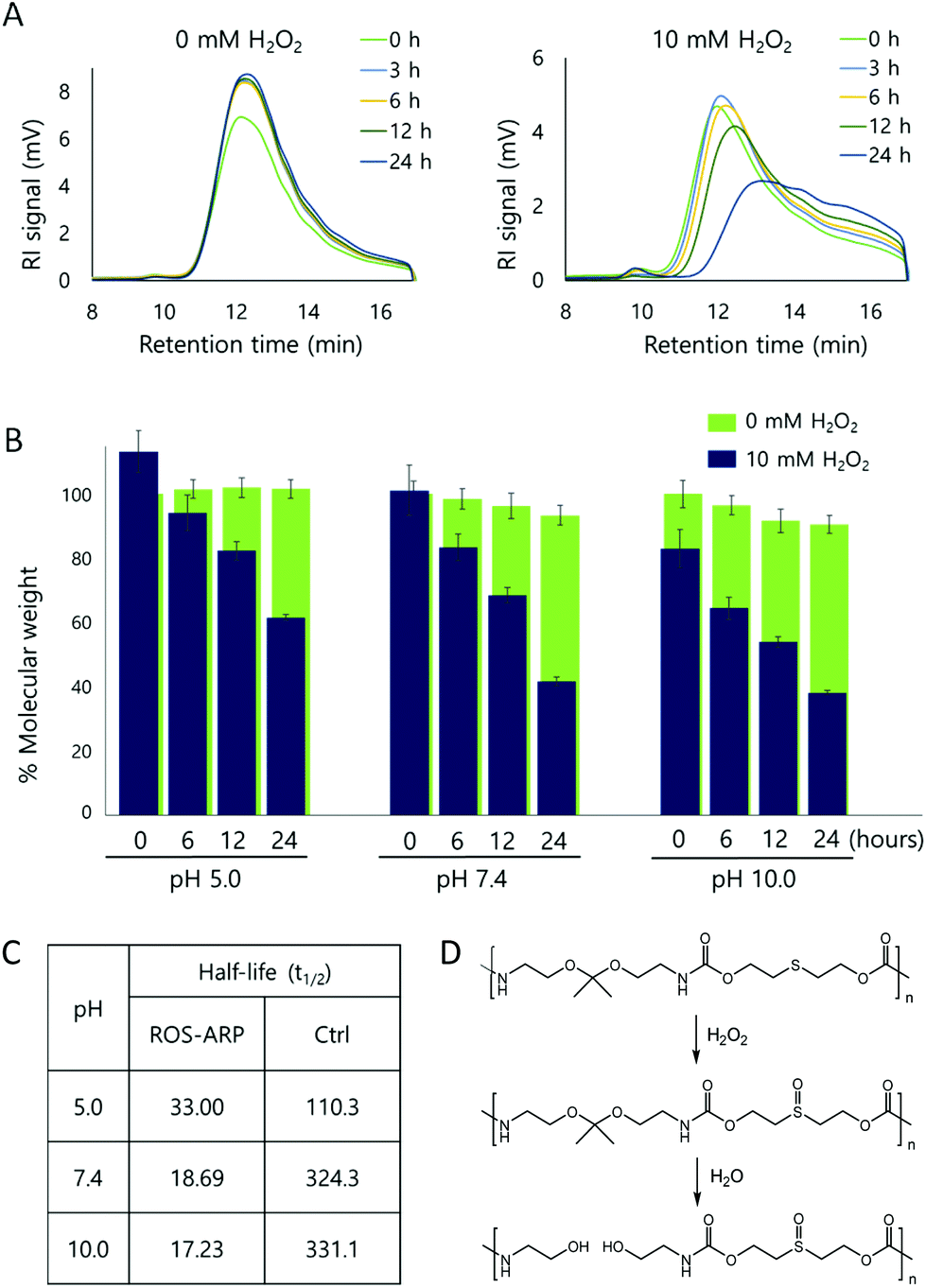 | ||
| Fig. 1 (A) GPC chromatogram of ROS-ARP in 0 mM H2O2 (left) and 10 mM H2O2 (right) at pH 7.4 (detector; RI, 20% buffer and 80% DMF, 24 hours incubation at 37 °C). (B) Molecular weight change of the polymer with and without H2O2 at pH 5.0, pH 7.4 and pH 10.0. Polymer MW was calculated by GPC with the PMMA standard (n = 3). (C) Half-lives of polymer degradations by 10 mM H2O2 in a different pH buffer. (D) Polymer structure and degradation mechanism of the control polymer. | ||
The half-life (t1/2) of the polymer degradation by 10 mM H2O2 was calculated and compared with a control polymer which has been previously published by our group30 (Fig. 1C). This control polymer has sulfide groups and ketal groups in the polymer backbone as ROS-ARP (Fig. 1D). However, the BE group, the ROS sensitive carboxylic acid protecting group, is not included. Therefore, the ketal hydrolysis rate is assumed to rely only on hydrophilicity changes by sulfide oxidation. The t1/2 of the ROS-ARP at pH 7.4 with 10 mM H2O2 was 18.69 hours and it was 17.35 times shorter than the t1/2 of the control polymer which does not contain BE groups for chemical amplification.
Investigation of the degradation mechanism
The ROS-ARP degraded more efficiently and faster than the previously reported control polymer. Several factors contribute to the accelerated ROS-ARP degradation.Firstly, the H2O2 sensitivity of ROS-ARP is higher than that of the control polymer. Compared with the control polymer, which only has sulfides as an ROS sensitive group, the ROS-ARP additionally has BE groups. The BE group is more sensitive to H2O2 than the sulfide.44–46 Therefore, the hydrophilicity change of the ROS-ARP is faster, and contributes to faster polymer degradation.
Secondly, upon reaction with ROS, the ROS-ARP transforms into a more hydrophilic polymer than its control. The ROS oxidizes the sulfide groups in the control polymer, and the produced sulfoxides induce a hydrophilicity change of the entire polymer. Since the ROS-ARP has two of these sulfide groups in a repeating unit while the control has one, the hydrophilicity change of ROS-ARP by H2O2 is greater than the control. Moreover, in the ROS-ARP, H2O2 unmasks carboxylic acids, further enhancing its hydrophilicity in addition to the change done by the sulfoxides. Thus, the ROS-ARP has two switches that cause increased hydrophilicity. Furthermore, since a carboxylic acid is more polar than a sulfoxide, the hydrophilicity increase of the ROS-ARP is more drastic than the control. The increased hydrophilicity promotes water access to the polymer, and it accelerates the polymer degradation by the ketal hydrolysis.
Lastly, a local pH change by unmasked carboxylic acids accelerates the ROS-ARP degradation. In the ROS-ARP, the carboxylic acid group is revealed by ROS after the BE group detachment. We hypothesize a possibility that the revealed carboxylic acid catalyzes the ketal hydrolysis in the ROS-ARP backbone in a chemical amplification process. This hypothesis is based on several previous studies applying the strategy.20,23 However, it has not been clarified whether a hydrophilicity change or a chemical amplification is the primary contributor to polymer degradation. Consequently, the efficiency of chemical amplification is still questionable.
To investigate which effect dominates between hydrophilicity change and local pH change by acid release for accelerating ketal hydrolysis, polymer degradation was evaluated at basic pH (pH 10.0). Under basic conditions, the ketal hydrolysis is extremely suppressed.25,54 Therefore, the ketal hydrolysis caused by a hydrophilicity change of the polymer is inhibited as no protons are available for catalyzing hydrolysis. In other words, although the hydrophilicity of the polymer is largely increased by ROS, the ROS-ARP degradation is not attributed to increased water accessibility under basic conditions. Note that the hydrolysis rate of amide bonds, the other functional group in the polymer backbone, has negligible difference between pH 5.0 and 10.0.55
The molecular weight changes of the ROS-ARP in basic solution are shown in Fig. 1B. Compared to the initial molecular weight, it decreased by 20% right after the H2O2 addition and decreased by 62% in 24 hours with 10 mM H2O2 at pH 10.0. This confirms that the ketal groups were hydrolyzed even under basic conditions. This means that the polymer was degraded by the unmasked acid, and not by a hydrophilicity change. Moreover, at higher pH, the t1/2 of ROS-ARP was shorter (Fig. 1C). This is a reverse tendency to the pH dependent ketal hydrolysis rate, but corresponds well to the BE reactivity to H2O2. Since the BE and H2O2 reaction is faster at a higher pH,56–59 carboxylate unmasking is also faster under basic conditions. On the other hand, the control polymer's half-life was shorter at lower pH, because the polymer degradation by ketal hydrolysis fully relies on the ROS-triggered hydrophilicity change. These two results support that the BE groups and unmasked carboxylic acids are the main factors in the degradation kinetics of ROS-ARP.
Additionally, ketal hydrolysis is more influenced by carboxylic acids in the same molecule than the surrounding acids, because of the conformational proximity.60–63 We assume that a similar effect occurs in a polymer, so that carboxylic acids in a polymer catalyze ketal groups in the same polymer backbone more efficiently than the surrounding protons in a solution. Therefore, we conclude that the accelerated ROS-ARP degradation depends more on chemical amplification than hydrophilicity change, due to the H2O2-sensitive acid releasing followed by the acid catalyzed ketal hydrolysis.
Nanoparticle formulation and H2O2 triggered degradation
After verification of H2O2-responsive polymer degradation, nanoparticle formulation and degradation under varying H2O2 concentrations were demonstrated. Nanoparticles were formed via nanoprecipitation, and DLS estimated the hydrodynamic nanoparticle size as 208 nm (polydispersity index (PDI) = 0.188) (Fig. S6†). Based on TEM observations, the morphology of the nanoparticles was spherical and the size corresponded to the DLS measurements (Fig. S6†).The nanoparticles were treated with various H2O2 concentrations at pH 7.4 and degradation was observed based on the light scattering intensity measured by DLS (Fig. 2). In 10 mM H2O2, the scattering intensity of the nanoparticle solution decreased by 30% immediately after H2O2 addition, and by 93% in 24 hours. At 100 μM H2O2, the intensity decreased by 25% after 24 hours. At the physiological H2O2 concentration in inflammation, 50 μM, 9% of the scattering intensity decreased in the same period.
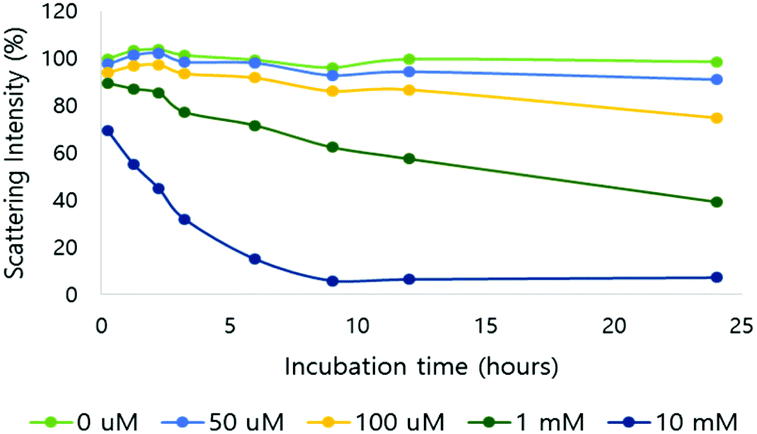 | ||
| Fig. 2 Light scattering intensity change of NPs in different H2O2 concentrations (0, 50, 100 μM and 1 and 10 mM; pH 7.4, 10 mM phosphate buffer). | ||
The morphology and size population change of NPs in a 10 mM H2O2 environment were also evaluated by TEM (Fig. 3A and B). Three representative images of NPs, treated with 0 mM or 10 mM H2O2 for 24 hours at 37 °C, were compared for size (Fig. S8†). The size of the nanoparticles was analyzed from the TEM images. In 0 mM H2O2, the ROS-ARP NPs maintained their initial mean size of 150 nm (Fig. 3B, left). In 10 mM H2O2 solution, the mean size of the NPs was 62 nm based on the images (Fig. 3B, right). The size change of the NPs with H2O2 suggests that NPs are degraded by polymer degradation. This size change corresponds to the DLS scattering light intensity change by H2O2 (Fig. 2). Furthermore, in the presence of H2O2, larger particles (400–700 nm) were observed with a different morphology compared to the NPs in 0 mM H2O2 (Fig. 3A). These NPs had lower electron density believed to be caused by swelling of the NPs and polymer degradation.
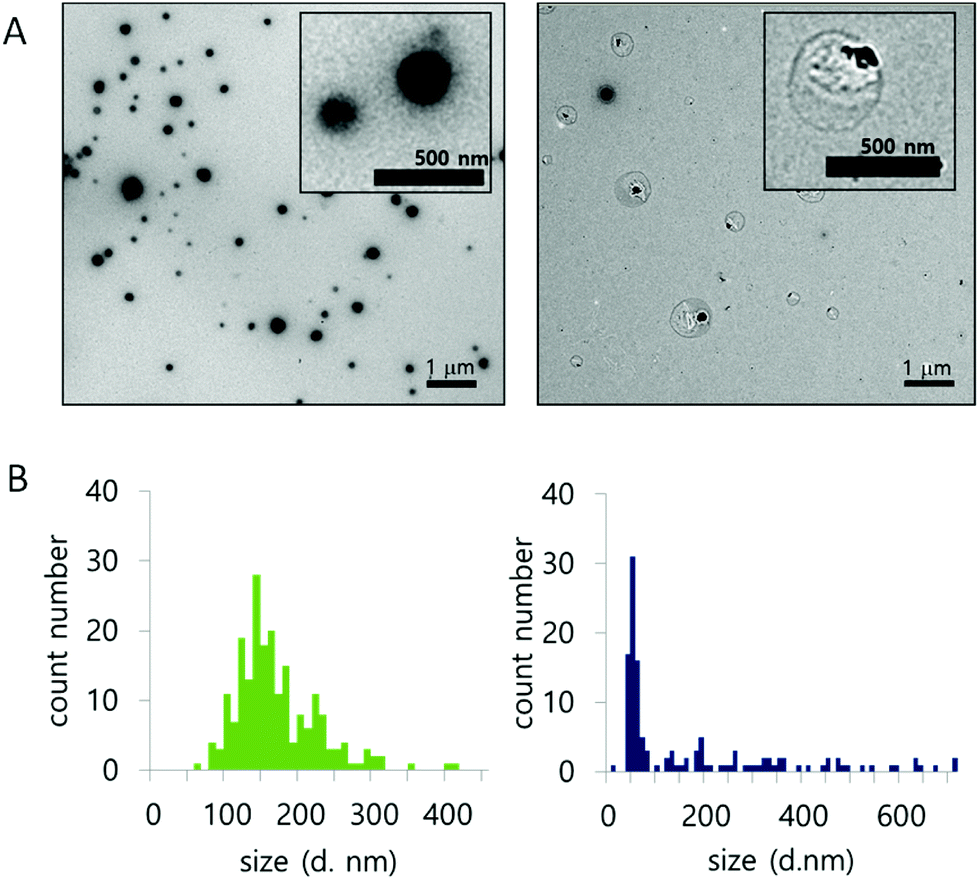 | ||
| Fig. 3 (A) Morphology of NPs, with 0 mM (left) and 10 mM H2O2 (right). (B) Size population histogram based on TEM images (left; 0 mM H2O2, right; 10 mM H2O2) (after 24 h incubation at 37 °C). | ||
ROS-responsive payload release
To demonstrate the ROS-dependent payload release from the NPs, IR-780 iodide was loaded into the NPs (IR780-NPs). The IR780-NP solutions were treated with varying concentrations of H2O2 and the fluorescence was measured at different time points. Free IR-780 has emission at λmax = 798 nm in pH 7.4 10 mM phosphate buffer, but the λmax shifts (λmax = 823 nm) due to encapsulation.64,65 Nonetheless, the area under the curve (AUC) comparison of the fluorescence intensity of the loaded and released dye is still difficult as the peaks are in close proximity. Thus, Gaussian fitting of the raw fluorescence intensity signal was used to separate released vs. encapsulated IR-780. The details are described in the ESI (Fig. S9†). The fitting method itself was verified by mixing various amounts (0, 10, 20, 30, 40 and 50 μl) of 0.3 μg ml−1 IR-780 solution with 100 μl of 0.1 mg ml−1 IR780-NP solution. Consequently, the calculated AUC of the free dye increased linearly (R2 = 0.9971) while the AUC of the loaded dye (IR780-NPs) signal remained constant (Fig. S10†).Using the signal separation method, the fluorescence signal from the loaded IR-780 was compared at different incubation concentrations of H2O2 and time in 10 mM phosphate buffer (Fig. S11†). Note that the signals of the released IR-780 are not comparable due to the hydrophobic, unstable nature of IR-780 in the buffer. Fig. 4A shows the separated fluorescence signal of the encapsulated IR-780 with 0 mM (Fig. 4A, left) and 10 mM H2O2 (Fig. 4A, right). In the presence of H2O2, the loaded dye signal clearly decreased. Furthermore, the signal decrease was faster when increasing the H2O2 concentration (Fig. 4B and Fig. S11†).
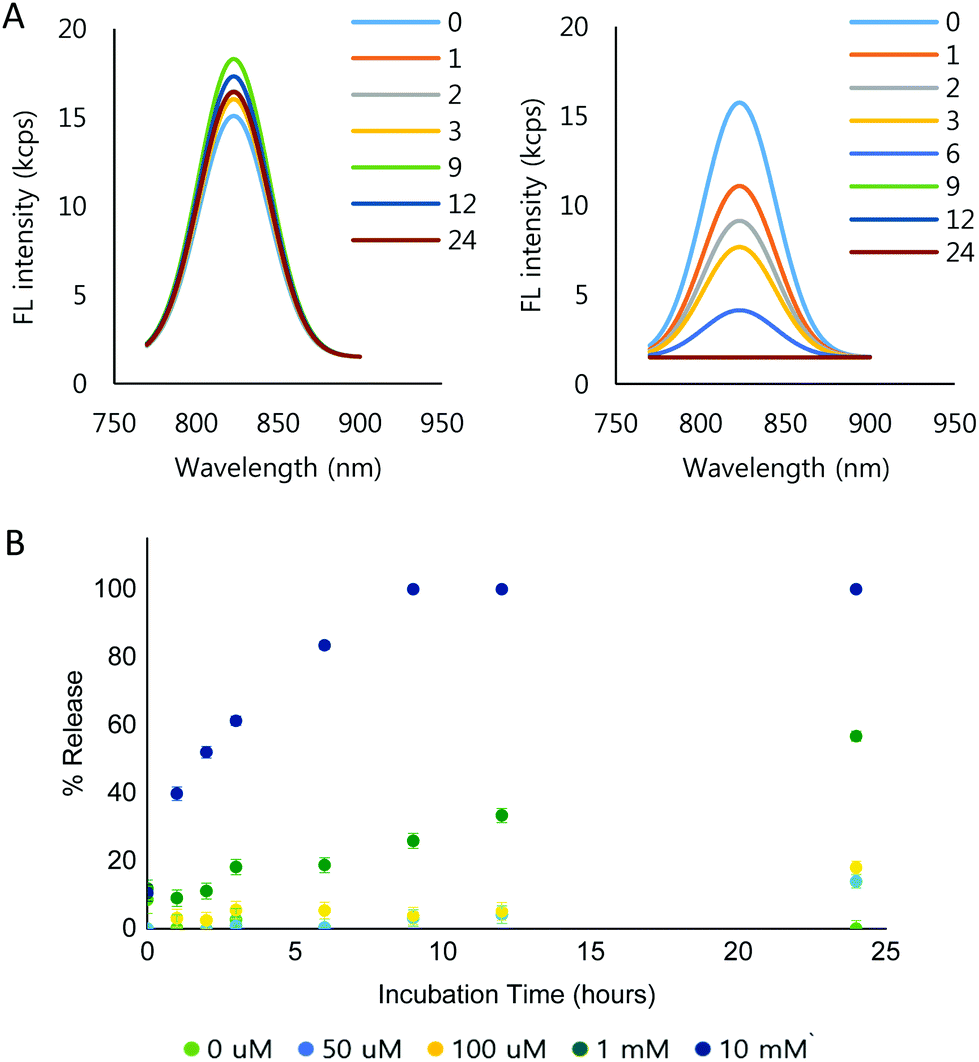 | ||
| Fig. 4 (A) Fluorescence intensity of IR780-NPs in 0 mM (left) and 10 mM H2O2 (right) at pH 7.4, r.t. (B) percent release of dye, based on the AUC of IR780-NP fluorescence intensity (Standard deviation was obtained from Gaussian fitting). | ||
Cell viability
To test the applicability of ROS-ARP nanoparticles as drug delivery vehicles, we assessed the cellular compatibility in Raw 264.7 mouse macrophages (Fig. S12†). The cells tolerated the NPs well up to the maximum tested concentration of 300 μg mL−1, suggesting their potential for drug delivery.Conclusions
Herein we designed an ROS sensitive degradable polymer applying a chemical amplification strategy for fast and efficient polymer degradation. We successfully synthesized the polymer and verified the chemical amplification effect on its degradation. The ROS-ARP degraded over 17 times faster than its control polymer without an amplification strategy. The accelerated degradation arises from the unmasked carboxylic acid which catalyzes a ketal hydrolysis. As suggested here, this strategy and degradation mechanism may be applied in future biomedical applications such as for drug delivery.Experimental
General methods and instrumentation
All chemicals and solvents were purchased from Sigma-Aldrich unless specified. Silica gel column chromatography for purification was conducted with an automated CombiFlash® Rf 200 system. The synthesized compounds were confirmed by nuclear magnetic resonance (NMR) and high-resolution mass spectroscopy. Acquired 1H NMR and 13C NMR spectra were based on a Varian spectrometer working at 600 MHz and 150 MHz, respectively. An Agilent 6230 ESI-TOFMS was used for high-resolution mass spectra in positive ion mode. The synthesized polymers and degradation were characterized by size exclusion gel chromatography, an Agilent 1100 Series HPLC system equipped with RI, Agilent 1260 Light Scattering and PDA detectors and a Waters Styragel HR 2 size-exclusion 22 column with 0.1% LiBr/DMF as the eluent and a flow rate of 1 mL min−1 at 37 °C. The molecular weight and polydispersity of polymers were calculated based on monodisperse poly(methylmethacrylate) (PMMA) standards. For particle washing, tangential flow filtration was conducted (Pellicon XL, 500 kDa, Millipore) with 300 mL cell-culture grade water (HyClone) at 50 RPM. Dynamic light scattering (DLS), Malvern Instruments Zetasizer ZS, was utilized for particle characterization and degradation study, and size of the particles was acquired based on culmulants analysis using the scattering of a 4 mW 633 nm laser. The morphology of the nanoparticles was observed by transmission electron microscopy (TEM, Tecnai FEI Spirit). An Agilent Technologies 1260/1290 Infinity HPLC-MS equipped with a ZORBAX 1.8 μm (2.1 × 50 mm) SB-C18 column and a 6120 Quadrapole detector was used for HPLC-MS chromatograms, and it was run in a gradient of water and acetonitrile with 0.1% formic acid.Synthesis of compound 1
4-(Hydroxymethyl)phenyl boronic acid pinacol ester (1.0 g, 4.27 mmol), N,N′-di-boc-L-lysine (1.48 g, 4.27 mmol) and 4-(dimethylamino)pyridine (DMAP) (260 mg, 2.14 mmol) were dissolved in 20 ml of anhydrous dichloromethane (DCM) under an argon flow. The mixture was cooled down using an ice bath and followed by dropwise addition of 1.32 g (6.4 mmol) of N,N′-dicyclohexylcarbodiimide (DCC) in 3 ml of DCM. After the addition, the resulting solution was stirred overnight at r.t. The crude solution was filtered to remove side products (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU), and then purified with a column. A final product was obtained after purification using gel chromatography with 20% ethyl acetate in hexane (1.04 g, 43.3%).1H NMR (600 MHz, CDCl3) δ 7.80 (d, J = 7.7 Hz), 7.34 (d, J = 7.7 Hz), 5.17 (dd, J = 45.6, 12.7 Hz), 4.32 (dd, J = 11.9, 7.6 Hz), 3.06 (dd, J = 11.2, 5.7 Hz), 1.60 (bs), 1.44 (s), 1.34 (s).
13C NMR (151 MHz, d6-CDCl3) δ 172.6, 156.1, 155.49, 138.4, 135.0, 127.4, 83.9, 79.9, 79.1, 66.9, 53.3, 40.1, 32.3, 29.5, 22.4, 28.5, 28.4, 24.9.
MS (ESI) m/z: [M + Na]+ calcd for C29H47BN2O8Na, 585.33; found 585.2.
Synthesis of compound 2
Compound 1 (885 mg, 1.57 mmol) was dissolved in 4 ml of DCM and 4 ml of trifluoroacetic acid (TFA) and stirred for 1 h at r.t. The reaction was monitored by thin-layer chromatography (TLC) and LCMS. When the reaction was completed, a crude solution was dried using a rota-vap to remove TFA. (1.19 g, 93%).1H NMR (600 MHz, d6-DMSO) δ 8.48 (bs, 3H), 7.78 (bs, 3H), 7.70 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 5.27 (dd, J = 15.2, 12.8 Hz, 2H), 5.15 (bs, 2 equiv. of TFA), 4.10 (dd, J = 11.3, 6.1 Hz, 1H), 2.74 (m, 2H), 1.80 (m, 2H), 1.53 (m, 2H), 1.43–1.31 (m, 2H), 1.29 (s, 12H).
13C NMR (151 MHz, d6-DMSO) δ 169.4, 138.5, 127.5, 83.8, 67.0, 51.8, 38.4, 29.5, 26.5, 21.3.
MS (ESI) m/z: [M + H]+ calcd for C19H31BN2O4, 363.24; found 363.1.
Synthesis of compound 3
Compound 2 (521 mg, 0.93 mmol) was dissolved in 8 ml of anhydrous DCM and 1 ml (7.44 mmol) of TEA. The solution was cooled down using an ice-bath and followed by acryloyl chloride (165.7 μl, 2.05 mmol) addition. After 30 min of stirring at r.t., the product was washed with 800 ml of 50 mM HCl (aq) and 200 ml of additional DCM. The organic layer was collected and dried using a rota-vap, after treating with MgSO4 to remove the remaining H2O. The product was purified again using column chromatography with 95% DCM and 5% MeOH (136 mg, 33%) (Fig. S1†).1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 6.38 (d, J = 8.1 Hz, 1H), 6.33–6.06 (m, 4H), 5.82 (t, J = 5.5 Hz, 1H), 5.67 (d, J = 10.4 Hz, 1H), 5.60 (d, J = 10.4 Hz, 1H), 5.17 (dd, J = 42.8, 12.5 Hz, 2H), 4.67 (m, 1H), 3.34–3.20 (m, 2H), 1.87 (m, 1H), 1.74 (m, 1H), 1.61–1.45 (m, 2H), 1.39–1.33 (m, 1H), 1.32 (s, 12H).
13C NMR (151 MHz, CDCl3) δ 172.2, 165.9, 138.2, 135.1, 130.6, 127.5, 127.4, 126.4, 84.0, 67.1, 51.8, 38.7, 31.9, 28.5, 24.9, 22.1.
MS (ESI) m/z: [M + Na]+ calcd for C25H35BN2O6Na, 493.25; found 493.1.
Synthesis of compound 5
Compound 4 was obtained by following the same procedure as in ref. 32. Compound 4 (3.0 g, 7.1 mmol) was dissolved in 6 N NaOH (aq) (9 ml) and refluxed overnight. After the overnight reaction, 9 ml of 1,4-dioxane was added into the resulting solution and cooled down using an ice-bath. TEA (11.9 ml, 85.2 mmol) and acryloyl chloride (1.15 ml, 14.2 mmol) were added dropwise into the mixture, while monitoring the temperature and pH of the solution. The temperature of the solution was kept under 10 °C, and the pH was maintained above pH 13. After the addition, the solution was stirred for 1 h, and extracted with ethyl acetate (EA) and column chromatography followed. After the gel column purification, a light yellow compound was obtained (500 mg, 20%).1H NMR (600 MHz, d6-DMSO) δ 8.15 (t, J = 5.5 Hz, 2H), 6.24 (dd, J = 17.10, 10.10 Hz, 2H), 6.07 (dd, J = 16.9, 2.0 Hz, 2H), 5.56 (dd, J = 10.2, 2.0 Hz, 2H), 5.27 (dd, J = 15.2, 12.8 Hz, 2H), 3.39 (t, J = 5.9 Hz, 4H), 3.23 (dd, J = 11.7, 5.80 Hz, 4H), 1.26 (s, 6H).
13C NMR (151 MHz, d6-DMSO) δ 164.7, 131.7, 125.1, 99.6, 66.4, 59.0, 24.7.
Synthesis of ROS-ARP
102 mg of compound 3 (1 equiv.) and 58.5 mg of compound 5 (1 equiv.) were put together under Ar and dissolved in 500 μl of d6-DMSO. 1,3-Propanedithiol (43.5 μl, 2 equiv.) and DBU (195 μl, 6 equiv.) were added into the reaction mixture. The mixture was stirred at r.t. overnight. The completion of polymerization was confirmed by NMR. The resulting viscous polymer solution was diluted with DCM and precipitated in diethyl ether, and this was repeated 3 times. The purified polymer was evaluated by NMR and GPC (Fig. S2†).The molecular weight was 10280 Da and the PDI was 1.25 with the PMMA standard.
1H NMR (600 MHz, d6-DMSO, and 5% CD3OD) δ 8.15 (t, J = 5.5 Hz, 2H), 6.24 (dd, J = 17.10, 10.10 Hz, 2H), 6.07 (dd, J = 16.9, 2.0 Hz, 2H), 5.56 (dd, J = 10.2, 2.0 Hz, 2H), 5.27 (dd, J = 15.2, 12.8 Hz, 2H), 3.39 (t, J = 5.9 Hz, 4H), 3.23 (dd, J = 11.7, 5.80 Hz, 4H), 1.26 (s, 6H).
H2O2 sensitive polymer degradation
The ROS-ARP was dissolved in 0.1% LiBr DMF to 5 mg ml−1. 400 μl of the polymer solution was mixed with 100 μl of aqueous solution (10 mM acetate buffer for pH 5.0, 10 mM phosphate buffer for pH 7.4, and 10 mM tris buffer for pH 10.0) and the final H2O2 concentration was set to 0 mM and 10 mM. Both samples were incubated for 24 h at 37 °C. Each sample was filtered using a 0.22 μm PVDF syringe filter before the GPC injection (Fig. S3†). The molecular weight of the polymers was calculated by analyzing the peaks between 10 and 16 min of retention time (Fig. S4†). 4-Hydroxybenzyl alcohol, a representative side product of the BE reaction with H2O2, was injected into the GPC under the same conditions to compare the retention time (Fig. S5†).Particle formulation
The ROS-ARP (10 mg) was dissolved in 1 ml of DMF. The resulting polymer solution was added dropwise into 9.0 ml of 1 wt% polyvinyl alcohol (PVA) aqueous solution and stirred for 2 h at r.t. The crude solution was washed using tangential flow filtration to remove excess PVA and DMF. For IR-780 or rhodamine 6G encapsulated particles, 2 mg of encapsulates were dissolved in 1 ml DMF with ROS-ARP. Loading was calculated by dividing the amount of cargo by the weight of particles. The loading of IR-780 was 2 wt%.The formulated particles were characterized by DLS and TEM (Fig. S6†).
Stimuli triggered nanoparticle degradation
The size distribution of nanoparticles (NPs) with different H2O2 concentrations was compared by DLS. 50 μg ml−1 of nanoparticle solution with 2 mM phosphate buffer (pH 7.4) and 0.2% PVA aqueous solution was prepared. Each sample was mixed with a H2O2 solution and the final H2O2 concentrations were adjusted to 0 μM, 50 μM, 100 μM, 1 mM and 10 mM. All the samples were monitored by DLS for 24 h at r.t. Size distribution was measured at given time points (0, 1, 2, 3, 6, 9, 12 and 24 h). The nanoparticles were also incubated under acidic (pH 5.0) and basic conditions (pH 10.0) by mixing 10 mM acetate buffer or 10 mM of tris buffer.To observe the morphological change of nanoparticles, 5 μl of 0 mM and 10 mM of H2O2 treated nanoparticle solutions were put on a TEM copper grid. The TEM grids were dried under vacuum and imaged by TEM. Size distribution was calculated from three representative images of each sample (Fig. S8†).
Release study
IR-780 was loaded into the ROS-ARP NPs, and 1 mg ml−1 of NP solution was prepared. The NP solution was diluted 10 times with 0, 50, 100 μM and 1, 10 mM H2O2, 10 mM phosphate solution. Fluorescence was measured by using a spectrofluorometer (Horiba, FL-1000). The fluorescence intensity of the loaded IR-780 was separated from the released dye Gaussian fitting of the raw signal. All the input parameters are shown in Fig. S9.† The fitting method was verified using various amounts (0, 10, 20, 30, 40 and 50 μl) of 0.3 μg ml−1 IR-780 solution mixed with 100 μl of 0.1 mg ml−1 IR-780 loaded ROS-ARP NP solution. The loaded dye signal was separated out using the above method. Consequently, the calculated AUC of the free dye increased linearly (R2 = 0.9971) while the AUC of the loaded dye (IR-780 loaded ROS-ARP NPs) was kept constant (Fig. S10†). These values were converted into AUC changes by free dye concentrations, and a linear calibration curve was obtained (R2 = 0.9966, Fig. S10†). The separated IR-780 NP signals by H2O2 concentration are shown in Fig. S11.†Cell viability
Empty ROS-ARP nanoparticles were diluted in sterile DMEM/FBS media and further diluted to appropriate concentrations. Raw 264.7 cells (American Type Culture Collection, ATCC® TIB-71™) were seeded in a 96-well plate (Corning) at a density of 20000 cells per well before NP addition. The cells were incubated with particle suspensions in quadruplicates for 24 h at 37 °C, in 5% CO2. Cells were then washed twice in warm PBS and AlamarBlue reagent was added to each well (according to manufacturer's instructions) at a 1:10 DMEM concentration and incubated for 4 hours. To quantify the cell viability, the fluorescence excitation/emission was set to 530/590 nm and read using a plate reader (Molecular Devices SpectraMax M5).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Air Force office of Scientific Research (AFOSR), FA9550-15-1-0273, for funding. NMR data were acquired at the UCSD Skaggs School of Pharmacy and Pharmaceutical Sciences NMR Facility.References
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- G. R. Martin and R. K. Jain, Cancer Res., 1994, 54, 5670–5674 CAS.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS.
- P. Niethammer, C. Grabher, A. T. Look and T. J. Mitchison, Nature, 2009, 459, 996–U123 CrossRef CAS PubMed.
- S. Yang, Z. Tang, D. Zhang, M. Deng and X. Chen, Biomater. Sci., 2017, 5, 2169–2178 RSC.
- C. D. Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef PubMed.
- D. L. Zhang, Y. L. Wei, K. Chen, X. J. Zhang, X. Q. Xu, Q. Shi, S. L. Han, X. Chen, H. Gong, X. H. Li and J. X. Zhang, Adv. Healthcare Mater., 2015, 4(1), 69–76.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed.
- Q. R. Deng, X. D. Li, L. P. Zhu, H. He, D. L. Chen, Y. B. Chen and L. C. Yin, Biomater. Sci., 2017, 5, 1174–1182 RSC.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- I. L. C. Chapple, J. Clin. Periodontol, 1997, 24, 287–296 CrossRef CAS PubMed.
- H. Wiseman and B. Halliwell, Biochem. J., 1996, 313, 17–29 CrossRef CAS PubMed.
- S. Joshi-Barr, C. D. Lux, E. Mahmoud and A. Almutairi, Antioxid. Redox Signaling, 2014, 21, 730–754 CrossRef CAS PubMed.
- J. Olejniczak, V. A. N. Huu, J. Lux, M. Grossman, S. He and A. Almutairi, Chem. Commun., 2015, 51, 16980–16983 RSC.
- E. Han, B. Kwon, D. Yoo, C. Kang, G. Khang and D. Lee, Bioconjugate Chem., 2017, 28, 968–978 CrossRef CAS PubMed.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2016, 116, 1309–1352 CrossRef CAS PubMed.
- E. Sella and D. Shabat, Org. Biomol. Chem., 2013, 11, 5074–5078 CAS.
- C. C. Song, R. Ji, F. S. Du, D. H. Liang and Z. C. Li, ACS Macro Lett., 2013, 2, 273–277 CrossRef CAS.
- E. H. Cordes and H. G. Bull, Chem. Rev., 1974, 74, 581–603 CrossRef CAS.
- T. H. Fife, Acc. Chem. Res., 1972, 5, 264–272 CrossRef CAS.
- T. H. Fife and L. K. Jao, J. Org. Chem., 1965, 30, 1492–1495 CrossRef CAS.
- A. J. Kirby, Acc. Chem. Res., 1984, 17, 305–311 CrossRef CAS.
- W. Chen, F. H. Meng, R. Cheng and Z. Y. Zhong, J. Controlled Release, 2010, 142, 40–46 CrossRef CAS PubMed.
- E. A. Mahmoud, J. Sankaranarayanan, J. M. Morachis, G. Kim and A. Almutairi, Bioconjugate Chem., 2011, 22, 1416–1421 CrossRef CAS PubMed.
- J. Sankaranarayanan, E. A. Mahmoud, G. Kim, J. M. Morachis and A. Almutairi, ACS Nano, 2010, 4, 5930–5936 CrossRef CAS PubMed.
- S. E. Paramonov, E. M. Bachelder, T. T. Beaudette, S. M. Standley, C. C. Lee, J. Dashe and J. M. J. Frechet, Bioconjugate Chem., 2008, 19, 911–919 CrossRef CAS PubMed.
- M. J. Heffernan and N. Murthy, Bioconjugate Chem., 2005, 16, 1340–1342 CrossRef CAS PubMed.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306–2317 CrossRef CAS PubMed.
- C. Schoneich, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 111–119 CrossRef PubMed.
- X. H. Fu, Y. A. Ma, Y. Shen, W. X. Fu and Z. B. Li, Biomacromolecules, 2014, 15, 1055–1061 CrossRef CAS PubMed.
- B. L. Allen, J. D. Johnson and J. P. Walker, ACS Nano, 2011, 5, 5263–5272 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- Y. T. Chiang, Y. W. Yen and C. L. Lo, Biomaterials, 2015, 61, 150–161 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- B. Liu, D. L. Wang, Y. K. Liu, Q. Zhang, L. L. Meng, H. R. Chi, J. N. Shi, G. L. Li, J. C. Li and X. Y. Zhu, Polym. Chem., 2015, 6, 3460–3471 RSC.
- M. S. Shim and Y. N. Xia, Angew. Chem., Int. Ed., 2013, 52, 6926–6929 CrossRef CAS PubMed.
- C. C. Song, R. Ji, F. S. Du and Z. C. Li, Macromolecules, 2013, 46, 8416–8425 CrossRef CAS.
- E. Lallana and N. Tirelli, Macromol. Chem. Phys., 2013, 214, 143–158 CrossRef CAS.
- C. C. Song, F. S. Du and Z. C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- W. Q. Yang, X. M. Gao and B. H. Wang, Med. Res. Rev., 2003, 23, 346–368 CrossRef CAS PubMed.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553–5585 CrossRef CAS PubMed.
- M. C. Y. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392–15393 CrossRef CAS PubMed.
- E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652–16659 CrossRef CAS PubMed.
- D. Srikun, A. E. Albers, C. I. Nam, A. T. Iavaron and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 4455–4465 CrossRef CAS PubMed.
- C. E. Yeom, M. J. Kim and B. M. Kim, Tetrahedron, 2007, 63, 904–909 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. X. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Org. Chem., 2009, 74, 58–63 CrossRef CAS PubMed.
- R. M. Smith and D. E. Hansen, J. Am. Chem. Soc., 1998, 120, 8910–8913 CrossRef CAS.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 2544–2511 CrossRef CAS.
- H. G. Kuivila, J. Am. Chem. Soc., 1954, 76, 870–874 CrossRef CAS.
- H. G. Kuivila and R. A. Wiles, J. Am. Chem. Soc., 1955, 77, 4830–4834 CrossRef CAS.
- H. G. Kuivila and A. G. Armour, J. Am. Chem. Soc., 1957, 79, 5659–5662 CrossRef CAS.
- B. Capon and M. C. Smith, Chem. Commun., 1965, 523–524 RSC.
- T. H. Fife and E. Anderson, J. Am. Chem. Soc., 1971, 93, 6610–6614 CrossRef CAS.
- G. A. Craze and A. J. Kirby, J. Chem. Soc., Perkin Trans. 2, 1974, 61–66, 10.1039/p29740000061.
- C. J. Brown and A. J. Kirby, J. Chem. Soc., Perkin Trans. 2, 1997, 1081–1093, 10.1039/a700155j.
- C. X. Yue, P. Liu, M. B. Zheng, P. F. Zhao, Y. Q. Wang, Y. F. Ma and L. T. Cai, Biomaterials, 2013, 34, 6853–6861 CrossRef CAS PubMed.
- M. L. Viger, G. Collet, J. Lux, V. A. N. Huu, M. Guma, A. Foucault-Collet, J. Olejniczak, S. Joshi-Barr, G. S. Firestein and A. Almutairi, Biomaterials, 2017, 133, 119–131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7bm00758b |
| This journal is © The Royal Society of Chemistry 2018 |