
Advanced manufacturing of microdisk vaccines for uniform control of material properties and immune cell function
Qin
Zeng
a,
Peipei
Zhang
a,
Xiangbin
Zeng
a,
Lisa H.
Tostanoski
a and
Christopher M.
Jewell
 *abcd
*abcd
aFischell Department of Bioengineering, University of Maryland, 2212 Jeong H. Kim Engineering Building, 8228 Paint Branch Drive, College Park, Maryland 20742, USA. E-mail: qzeng@umd.edu; pzhang14@umd.edu; xzeng@umd.edu; ltostano@umd.edu; cmjewell@umd.edu; Web: http://jewell.umd.edu Tel: +1 301-405-9628
bDepartment of Microbiology and Immunology, University of Maryland School of Medicine, 685 West Baltimore Street, HSF-I Suite 380, Baltimore, Maryland 21201, USA
cMarlene and Stewart Greenebaum Cancer Center, Executive Office, Suite N9E17, 22 S. Greene Street, Baltimore, Maryland 21201, USA
dUnited States Department of Veterans Affairs, 10 North Greene Street, Baltimore, Maryland 21201, USA
First published on 13th November 2017
Abstract
The continued challenges facing vaccines in infectious disease and cancer highlight a need for better control over the features of vaccines and the responses they generate. Biomaterials offer unique advantages to achieve this goal through features such as controlled release and co-delivery of antigens and adjuvants. However, many synthesis strategies lead to particles with heterogeneity in diameter, shape, loading level, or other properties. In contrast, advanced manufacturing techniques allow precision control of material properties at the micro- and nano-scale. These capabilities in vaccines and immunotherapies could allow more rational design to speed efficient design and clinical translation. Here we employed soft lithography to generate polymer microdisk vaccines with uniform structures and tunable compositions of vaccine antigens and toll like receptor agonists (TLRas) that serve as molecular adjuvants. Compared to conventional PLGA particles formed by emulsion, microdisks provided a dramatic improvement in the consistency of properties such as diameter. During culture with primary dendritic cells (DCs) from mice, microdisks were internalized by the cells without toxicity, while promoting co-delivery of antigen and TLRa to the same cell. Analysis of DC surface activation markers by flow cytometry revealed microdisk vaccines activated dendritic cells in a manner that depended on the level of TLRa, while antigen processing and presentation depended on the amount of antigen in the microdisks. Together, this work demonstrates the use of advanced manufacturing techniques to produce uniform vaccines that direct DC function depending on the composition in the disks.
Introduction
Biomaterials offer unique advantages for vaccine design by providing features such as loading of multiple cargos, controlled release, and increased stability.1–4 However, one of the translational challenges associated with biomaterials is the complexity and heterogeneity of many common vaccine synthesis technologies. For example, PLGA nanoparticles and microparticles prepared by emulsion, one of the most common approaches, exhibit polydispersed sizes and loadings.5,6 Highlighting the challenges of this diversity, a number of studies demonstrate that particle size and shape greatly impact the function of the immune cells that process and present antigens, as well as the T cells responsible for eliminating pathogens.7–12 More broadly in the vaccine and adjuvant development field, the need for potent and tailored immune responses is driving increasingly complex vaccines. Much of the work continues to draw on empirical research models that, while important, are inefficient.13 Thus, there is great opportunity for new technologies that generate vaccines with programmable features – such as precision control over the loading of multiple cargos – and highly uniform properties, such as diameter and aspect ratio. This control could support rational vaccine design that enables creation and testing of effective vaccines more quickly.Advanced manufacturing techniques such as photolithography, soft lithography, and 3D printing are distinguished by the ability to generate materials with uniform, precise features.14–16 For example, photolithography techniques have been employed to design asymmetrical structures with silicon and polymer materials exhibiting little deviation in size, and these techniques also allow direct control over material shape.17–20 Imprint lithography has also been used to produce particles with uniform structures. In this technique, liquid precursors are filled in the surface cavities of a mold, then cured to produce replicates from the mold template.21 The DeSimone lab has developed a platform called Particle Replication In Nonwetting Templates (PRINT), which allows production of monodispersed particles with simultaneous control over particle size, shape, and composition.22,23 Additionally, techniques are being combined to enable more advanced capabilities. For example, photolithography has been combined with microfluidics to produce particles with controllable features. In this approach, photosensitive materials flow in a microfluidic channel, and upon exposing to UV light, are shaped into particles with controllable structures that replicate the photomask filtering the UV light.24 Further, soft lithography has also been integrated with layer-by-layer assembly techniques to generate multilayered microparticles with well-defined shape, size and spatial composition.25
For vaccine design, advanced manufacturing techniques offer several unique opportunities. Producing particles with precisely-defined features (i.e. size, aspect ratio, relative cargo ratio), allows isolation of the role of these properties in driving specific types of immune function. Further, these approaches maximize the uniformity of particle features, reducing experimental variability in cell culture, and in the pre-clinical animal models of infection and cancer that vaccines and immunotherapies are studied in. DeSimone and co-workers have illustrated such advantages using PRINT as a tool to study how nanoparticle charge and ligand display impacts biodistribution in tumor models,26,27 and to enhance response to clinically-relevant vaccines.28,29 Beyond conventional injectable vaccine applications, others have used advanced manufacturing techniques to direct immune response by modifying immune cells. For example, the Mitragotri lab used photolithography to prepare disk-shaped “backpack” that could be attached to macrophages, important immune cells involved in phagocytosing pathogens and in antigen presentation.30 Compared to conventional polymeric materials, these backpacks were resistant to endocytosis and sustained the release of model drug cargo while attached to the macrophages.
An important area of vaccine design where advanced manufacturing has not been applied is precision delivery of antigens – components of vaccines that confer specificity to a given target, and adjuvants – vaccine components that enhance the magnitude and type of immune response. Here we used soft lithography to build highly uniform vaccines containing a model antigen from ovalbumin (SIINFEKL), and CpG, an important molecular adjuvant that activates evolutionary immune pathways called toll-like receptors (TLRs). Photolithography was used to create a PDMS array that could be coated with poly(lactic-co-glycolic acid) (PLGA) particles incorporating defined levels of SIINFEKL (SIIN) and CpG (Fig. 1A). By printing these disks onto a water soluble polyvinyl alcohol (PVA) coating, free PLGA microdisks were obtained. Compared to conventional particles generated by emulsion techniques, these microdisks were more uniform in size and shape, and could be prepared with tunable levels of antigen (i.e., SIIN) and adjuvant (i.e., CpG) to control the function of immune cells (Fig. 1B). In culture with dendritic cells (DCs) – antigen presenting cells that play vital roles in both innate and adaptive immunity, microdisks increase expression of DC surface activation markers in a manner that depends on the level of CpG in microdisks. The presentation of SIIN peptide antigen by DCs is also tunable by controlling the level of peptide in the microdisks. The approach reported here allows generation of vaccines with tunable, highly uniform features to support more rational design and faster testing and characterization.
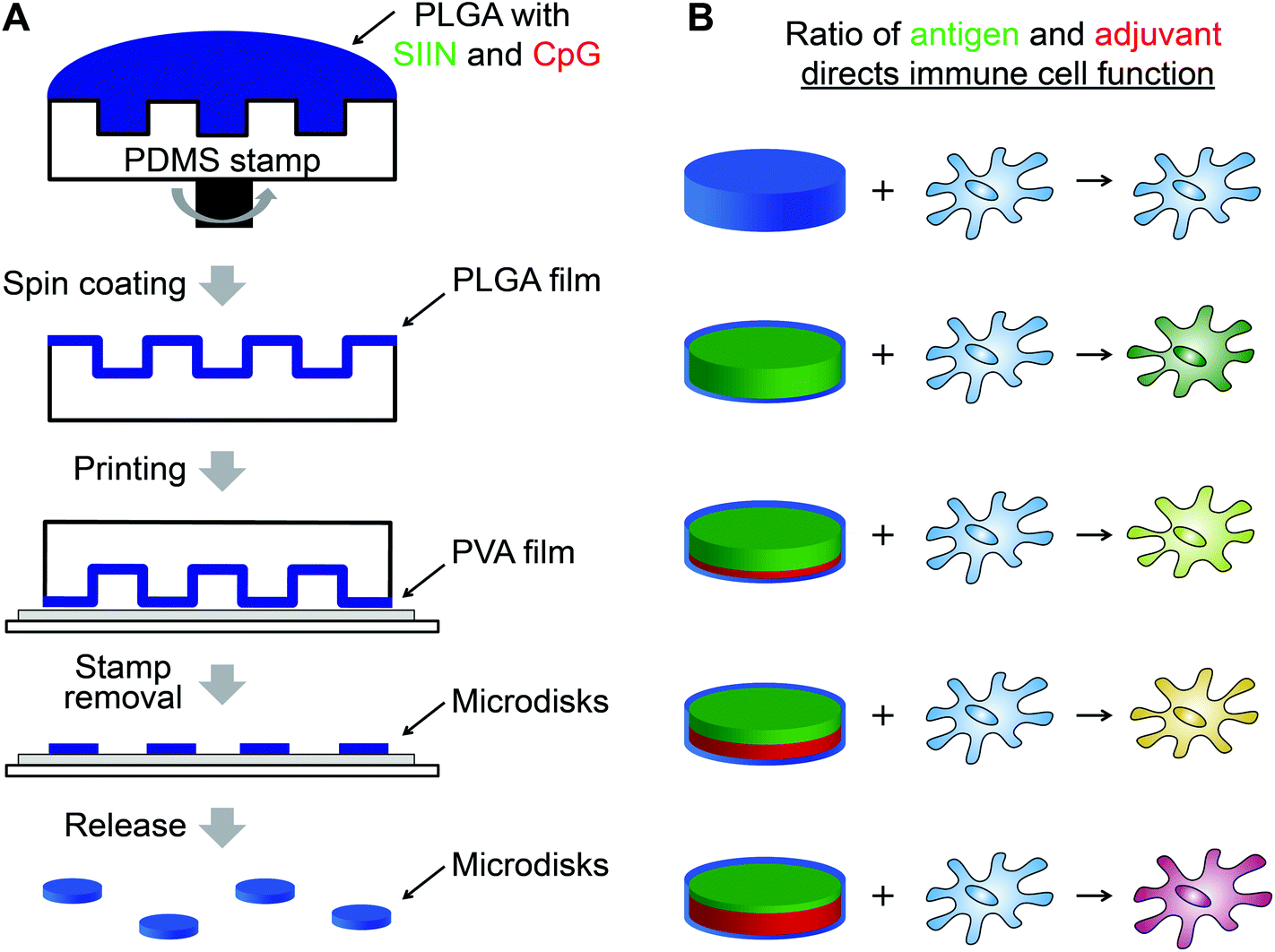 | ||
| Fig. 1 Schematic view of the microdisk strategy for tunable, uniform vaccines. (A) Design scheme showing the process to generate microdisk by lithography and contact printing. PLGA containing antigens and adjuvants at defined ratios are spin coated onto a PDMS stamp, then the resulting PLGA disks are printed onto a PVA-coated glass substrate. The disks are released by exposure to water to dissolve the PVA layer. (B) Microdisks with different levels of antigen (SIIN, green) and adjuvant (CpG, red) can be produced, with the relative composition of each defining the function of immune cells interacting with these vaccines. | ||
Materials and methods
CpG DNA (5′-TCC ATG ACG TTC CTG ACG TT-3′) was purchased from IDT (Coralville, IA). SIINFEKL (SIIN) was synthesized by Genscript (Piscataway, NJ) with or without a FITC tag. TE buffer were purchased from Amresco (Solon, OH). RPMI-1640 media was purchased from Lonza (Allendale, NJ) and fetal bovine serum (FBS) was supplied by Corning (Tewksbury, MA). 2-Mercaptoethanol was purchased from Sigma Aldrich (St Louis, MO). HEPES and non-essential amino acids were purchased from VWR (Radnor, PA). L-Glutamine, penicillin–streptomycin, and DAPI were purchased from Thermo Fisher Scientific (Grand Island, NY). Spleen Dissociation Medium and CD8 negative selection kits were from STEMCELL Technologies (Vancouver, British Columbia, Canada). CD11c microbeads were purchased from Miltenyi Biotec (Cambridge, MA). Rhodamine isocyanate (RD) nucleic acid labelling kits for fluorescently-labeling CpG were from Mirus (Madison, WI). Fluorescent antibody conjugates were purchased from BD (San Jose, CA). Poly(vinyl alcohol) (PVA) with MW of 3000 Da was purchased from Scientific Polymer Products, Inc. Poly(D-, L-lactide-co-glycolide) (PLGA) with a lactide-to-glycolide ratio of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 and molecular weight (Mw) of 4000–15000 Da was from Sigma Aldrich (St Louis, MO). PDMS prepolymer the associated curing agent was from Dow Corning.
:25 and molecular weight (Mw) of 4000–15000 Da was from Sigma Aldrich (St Louis, MO). PDMS prepolymer the associated curing agent was from Dow Corning.
PDMS stamp preparation
PDMS prepolymer and the curing agent were mixed completely at a ratio of 10 to 1. The mixture was poured onto a silicon master and degassed using a vacuum pump. After 4 h at 65 °C, the cured PDMS substrate was peeled from the master and sectioned in 1 cm × 1 cm substrates. The micropillar features on the PDMS substrates were 7 μm in diameter, 5 μm in height, and had a center-to-center spacing of 20 μm in a square lattice.Microdisk production
PLGA microdisks were prepared using a PLGA solution (300 μL; 10 wt%) in acetone that was spin coated on the PDMS substrates at 2500 rpm for 45 s. PVA-coated glass slides were prepared by spin coating 2 mL PVA (10 wt%) at a speed of 3000 rpm for 1 min. A PDMS stamp was then printed on a PVA coated glass substrate for 1 min, followed by removal of the PDMS substrate. To prepare cargo-loaded microdisks, defined levels of CpG and SIIN were added to PLGA solutions prior to the spin coating procedure described above. The level of CpG and SIIN was controlled by fixing the total input cargo dose at 30 μg, then varying the relative ratio of SIIN input to CpG input across the range of inputs indicated in the figures. The microdisks were then released by adding 500 μL of H2O to the disk-coated slides for 1 min to dissolve the PVA coating. The solution containing the microdisks was then collected and centrifuged at 5000g for 10 min. The disks were then washed twice by cycles of resuspension in water and centrifugation.Microdisk characterization
To characterize the cargo loading in microdisks, FITC-labeled SIIN and RD-labeled CpG were used in the microdisk synthesis procedure described above. Solutions containing released microdisks were then deposited in pre-weighed vials and lyophilized overnight. After recording the mass, the dried microdisks were dissolved in DMSO containing 5% SDS. The fluorescence was then measured by fluorimetry in microtiter plates for SIIN-FITC (Ex: 488 nm, Em: 519 nm) and CpG-RD (Ex: 546, Em: 576). Mass concentrations and loading levels were calculated by comparison to a fluorescence standard curve for each of the labeled cargos.Kinetic release studies were conducted in a similar manner to the loading studies using known masses of microdisks containing labeled cargo. The disks were placed in PBS buffer and incubated at 37 °C. At defined points, the concentration of each component in the supernatant was determined by fluorimetry. The values were converted to a release percent by comparison to the total cargo measured in an equivalent aliquot after dissolution in DMSO/SDS.
Atomic force microscopy (AFM) characterization of microdisks was obtained using a Digital Instruments (Veeco) multimode atomic force microscope in tapping mode in air. Microdisks were printed onto PVA coated glass substrate for AFM characterization. For fluorescence imaging, microdisks were loaded with (rhodamine isocyanate, RD) and imaged under fluorescence microscopy.
Microparticle synthesis and characterization
PLGA microparticles were synthesized using a double emulsion method. An aqueous phase of 500 μl ultra-pure water and an organic phase composed of 80 mg PLGA dissolved in 5 mL dichloromethane were freshly prepared. The aqueous phase was added to the organic phase to form a water/oil emulsion by sonication on ice for 30 s. The primary emulsion was added to 40 mL PVA (2%, w/v) and homogenized at 16000 rpm for 3 min to form the secondary emulsion. The particles were stirred at 600 rpm for 24 h to evaporate the solvent, then collected by centrifugation (5 min, 6000g, and 4 °C). Microparticles were then washed three times with 5 mL water, and the supernatants were decanted. 200 μL of the particle suspension was then dried on a glass substrate under vacuum for SEM imaging (Hitachi SU-70 FEG SEM). Microparticle diameters was measured using a Partica LA-950 laser diffraction particle size distribution analyzer (HORIBA) and automated image analysis of SEM images with ImageJ.
Cells and animals
Primary DCs were isolated from female C57BL6 mice (4–12 weeks, stock #000664) purchased from Jackson Laboratories (Bar Harbor, ME). All studies involving primary cells isolated from animals were carried out in compliance with Federal, State, and local guidelines, and using protocols reviewed and approved by the University of Maryland's Institutional Animal Care and Use Committee (IACUC).DC uptake and activation studies
CD11c+ DCs were isolated from spleens by positive selection as directed by the manufacturer using Spleen Dissociation Media and CD11c Microbeads. DCs were cultured in a 96 well plate at a density of 100000 cells per well in culture medium at 37 °C and 5% CO2. The culture medium was RPMI 1640 media supplemented with 10% FBS, 2 mM L-glutamine, 1× non-essential amino acids, 10 mM HEPES buffer, 1× penicillin and streptomycin, and 55 μM β-mercaptoethanol. For confocal microscopy imaging, cells were treated with microdisks containing SIIN-FITC and CpG-RD for 4 h, then fixed and stained using DAPI (nucleus) and a fluorescent wheat germ agglutinin conjugate (thermofisher) for the membrane. Similarly, for uptake studies by flow cytometry, DCs were treated with microdisk loaded with each fluorescent cargo for 24 hours, then assessed.
For activation studies, cells were treated with each of the samples specified in the text for 24 hours, then stained with DAPI as an indication or viability (i.e., DAPI− cells). Expression of DC surface activation markers (CD40+, CD80+, CD86+) was assessed by staining the cells with fluorescent antibody conjugates for each marker. To quantify presentation of SIINFEKL via the MHC-I pathway, cells were stained with a PE-Cy7-labeled antibody (BioLegend) against anti-mouse H-2Kb bound to OVA257-264 (SIINFEKL). Cells were then assessed by flow cytometry (BD Cantoll, San Jose, CA) and data was analyzed with FlowJo v.10 (TreeStar, Ashland, OR).
Statistics
Statistical analysis was performed using one-way ANOVA with a Dunnett's test post-test in Graphpad Prism (version 6.02). P values of <0.05 (*), <0.01 (**), <0.001 (***), and <0.0001 (****) were used to indicate statistical significance. Data are expressed as mean ± standard error of the mean (SEM). All experiments were conducted by using 3 or 4 replicates. Data shown in all figures are representative of results obtained in 2–3 similar experiments for each study type.Results
Microdisk vaccines exhibit uniform, tunable features
To produce microdisks, a PDMS stamp with a micropillar structure was first prepared by soft lithography (Fig. 2A). Each micropillar exhibited a diameter of 7 μm and a center-to-center distance of 30 μm. PLGA solution were then spun on the PDMS stamp to generate PLGA microdisks at the top of the pillars (Fig. 2B, left). The microdisks were printed on a PVA film coated on a glass substrate, resulting in transfer of the microdisks to the PVA-coated substrate (Fig. 2B, right). AFM was used to characterize the microdisks, revealing a thickness of 683 ± 11 nm at the center of the microdisks (Fig. 2C). Microdisks could be directly visualized by including a fluorescence dye (RD) in the PLGA solution during synthesis (Fig. 2D, left). Analysis of the fluorescence intensity of these microdisks revealed diameters in good agreement with the SEM measurements (Fig. 2D, right), and also in agreement with measurements from AFM analysis.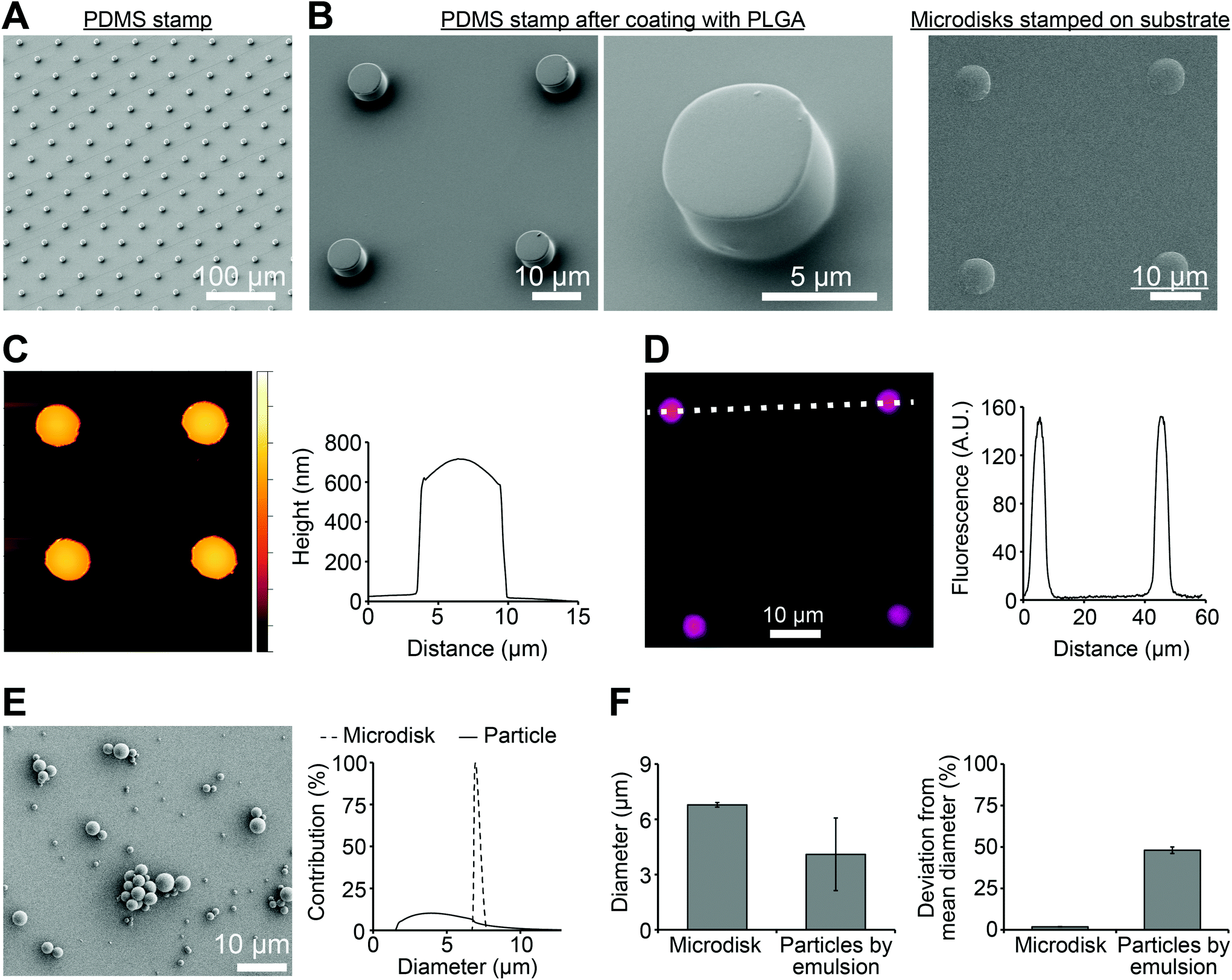 | ||
| Fig. 2 Microdisks exhibit uniform diameters. (A) SEM image of the PDMS stamp, (B) low and high magnification SEM images of the PDMS stamp after coating with PLGA (left) and an SEM image of microdisks after printing onto the PVA-coated glass substrate (right). (C) AFM characterization of microdisks printed on the PVA film (left) and height profile across an individual microdisk (right; dashed line). (D) Fluorescent microscopy image of a fluorescent dye (RD) loaded in microdisks printed on the PVA film (left), and the corresponding intensity profiles along the dashed line (right). (E) SEM image of PLGA particles prepared by emulsion (left) and the diameter distribution of the emulsion particles compared with the diameter distribution of microdisks (right). (F) Comparison of diameters of microdisks produced by lithography and microparticles prepared by emulsion (left), and the corresponding deviations from the mean diameters using each technique (right). | ||
One of the advantage of this technology is the uniformity of microdisks resulting from the precision of the PDMS master. Thus we next compared the variation in diameter of PLGA microdisks with those of PLGA microparticles prepared by a common technique used in preparing nanoparticle and microparticle vaccines. Spherical PLGA particles synthesized using a conventional double emulsion protocol exhibited diameters of 4.10 ± 1.97 μm (Fig. 2E). Compared to the microparticles prepare by emulsion, microdisks exhibited a striking reduction in the width of the diameter distribution (Fig. 2E, right). These distributions corresponded to microdisk diameters of 6.97 ± 0.13 μm, compared to 4.10 ± 1.97 μm for the emulsion particles (Fig. 2F, left). While the diameters were not identical, importantly, the deviation from the mean for microdisks was 1.8% compared with 48% for the spherical microparticles (Fig. 2F, right). Together, these data indicated that these advanced manufacturing techniques can generate more uniform materials than conventional emulsion method.
Microdisks enable programmable loading of multiple vaccine cargos
Using the approach above, microdisks were synthesized using specific compositions of antigen (SIIN) and adjuvant (CpG). Each cargo was fluorescently labeled, allowing visualization and measurement of cargo loading for both CpG and SIIN (Fig. 3A). Importantly, the signals for each of these components revealed exclusive co-localization of the antigen and the adjuvant, as indicated by color mixing in overlay images. Intensity profiles also supported a high degree of uniformity in the spatial localization of signal from disk to disk, both in x–y spacing, and in intensity (Fig. 3A). Similar results were obtained from analysis of higher magnification fluorescent images (Fig. 3B). Microdisks loaded with SIIN and CpG on the PVA-coated glass substrate were subsequently released by dissolution of the PVA in water. The released microdisks also exhibited localized signal for each component, which were co-localized with one another (Fig. 3C). A comparison of microdisk diameter before and after release revealed a diameter of 6.79 ± 0.13 μm and 6.71 ± 0.18 μm, respectively, indicating microdisk diameter remained unchanged and tightly distributed (Fig. 3D).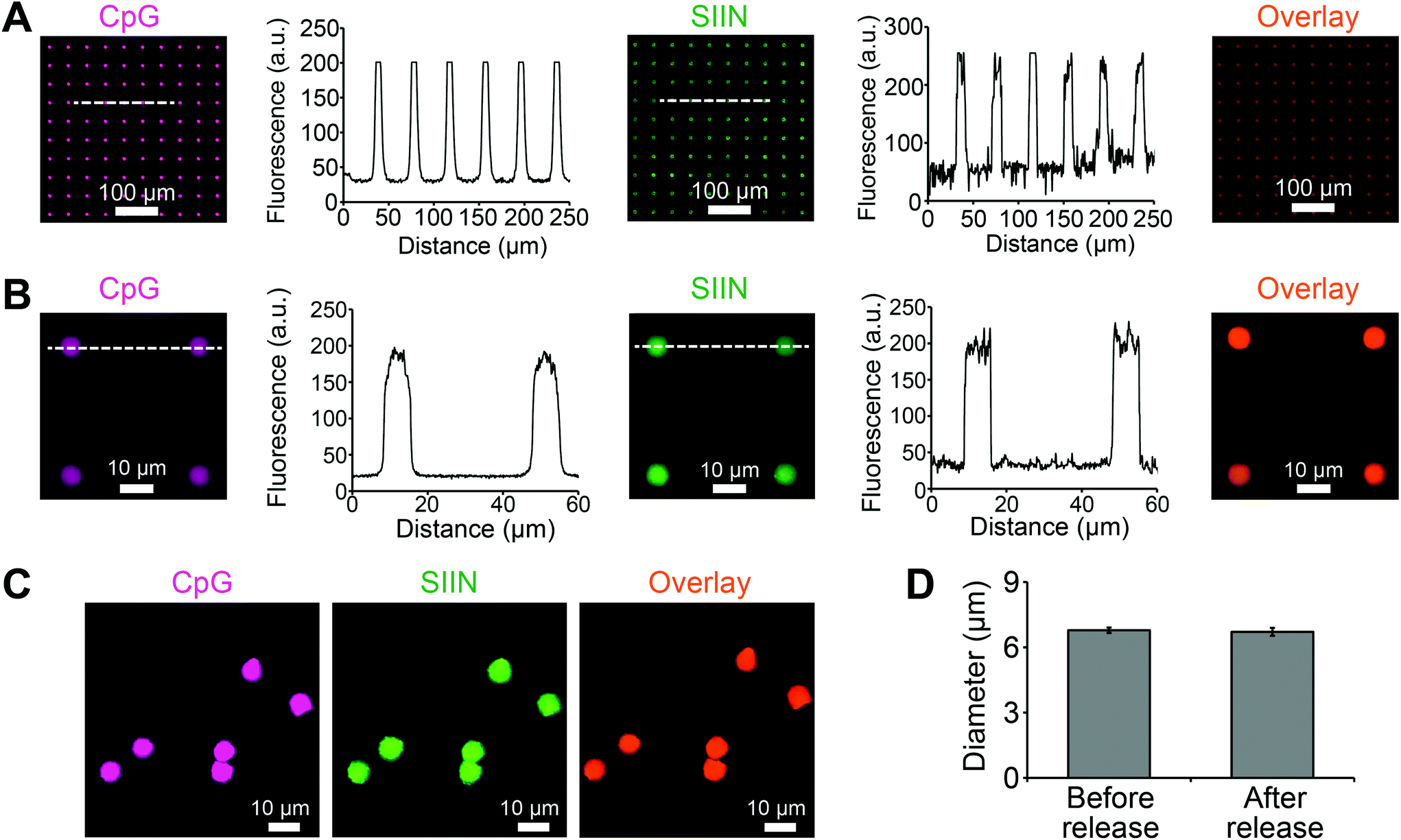 | ||
| Fig. 3 Characterization of geometries of microdisks loaded with CpG and SIIN. (A) Fluorescence images of adjuvant (CpG-RD, purple) and antigen (SIIN-FITC, green) in microdisks. The intensity profiles correspond to the dashed lines for each channel, and the color mixing (orange) in the overlay image indicates exclusive co-localization of the antigen and adjuvant in the microdisks. (B) Higher magnification images, overlays, and intensity profiles of microdisks loaded with adjuvant (CpG-RD, purple) and antigen (SIIN-FITC, green). (C) Fluorescence images of microdisks demonstrating signals are still co-localized after release of microdisks into solution by dissolving the sacrificial PVA core in water. (D) Diameters of microdisks before and after release do not change. | ||
Next we tested if the composition of vaccine signals in microdisks could be controlled by fixing the total input cargo concentration, but altering the relative cargo input levels of SIIN and CpG during microdisk synthesis. Measurement of each cargo loading level by fluorimetry after synthesis revealed programmable control over both SIIN and CpG. As SIIN input decreased, the SIIN loading level decreased (Fig. 4A), while over the same range, CpG loading increased as the amount of CpG input was increased (Fig. 4B). Thus, this strategy allowed control over the absolute and relative composition (Fig. 4C) across a range of microdisks containing from 100% SIIN to microdisks containing 100% CpG. The cargo loading efficiencies for microdisks ranged from more than 80% to 25%, with higher efficiencies generally observed at higher SIIN:CpG ratios (Fig. 4D). Further, the SIIN component generally exhibited more efficient incorporation. The combination of uniformity and tunability could support modular vaccines that can be designed more rationally.
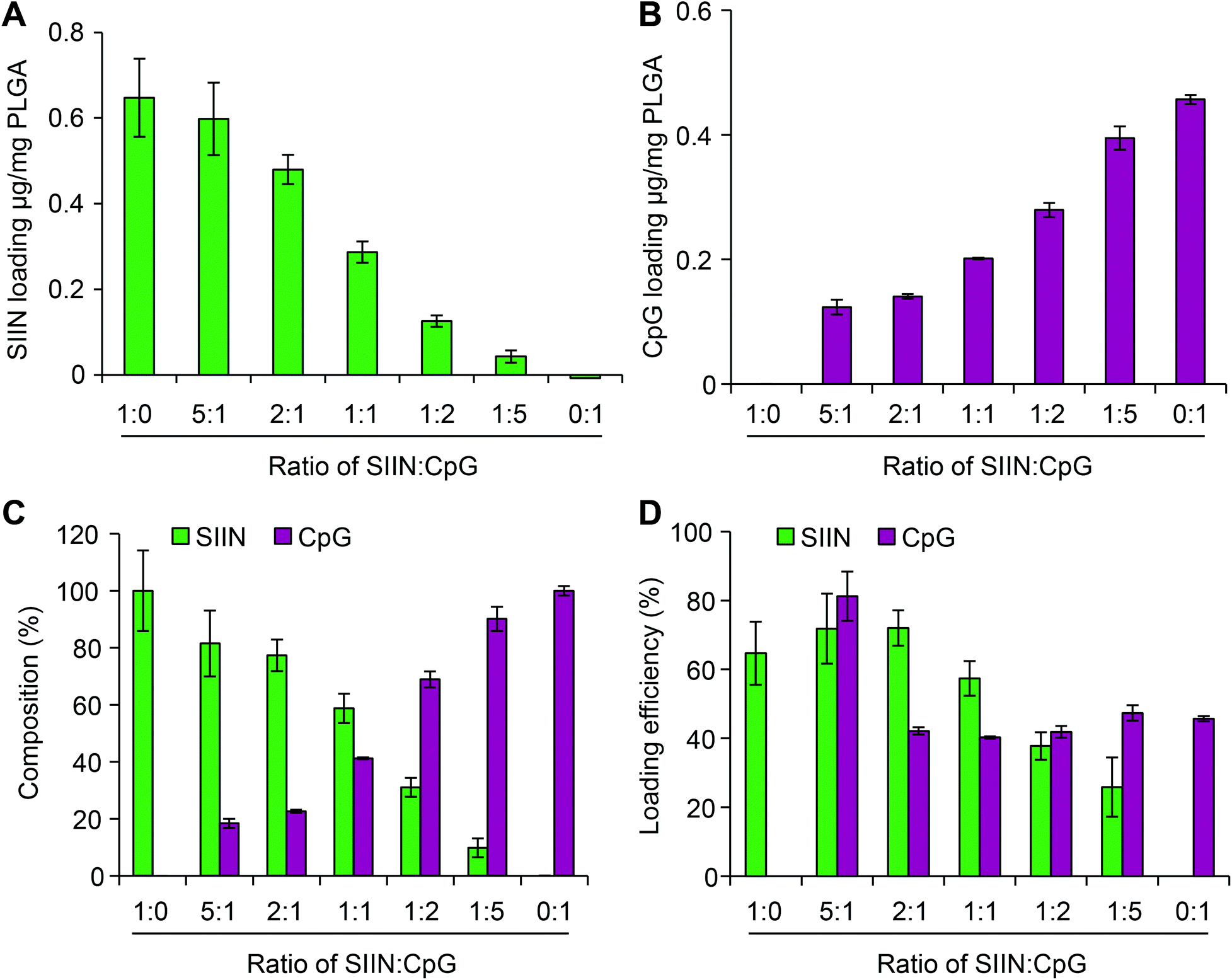 | ||
| Fig. 4 Characterization of microdisks loaded with defined compositions of SIIN and CpG. Control over the loading of microdisks is achieved by altering the input ratios of CpG:SIIN in the PLGA solutions, resulting in microdisks with tunable absolute loadings of (A) SIIN and (B) CpG. Also shown are the corresponding (C) cargo compositions and (D) cargo loading efficiencies of the microdisks. | ||
Microdisks are efficiently internalized by DCs without toxicity
To prepare for cell uptake studies, microdisks loaded with fluorescence cargo were incubated in buffer at 37 °C to assess release kinetics. Over 5 days, cargo release was observed. A higher percentage of release was measured for CpG, but neither cargo was fully released from the microdisks (Fig. 5A). SIIN/CpG microdisks were next incubated with DCs isolated from mouse spleens to assess uptake of each component and any toxicity of the vaccines. For these studies, microdisks with a final composition of 47:53 (SIIN:CpG) were used to treat DCs with serial-dilutions. Flow cytometry revealed dose-dependent uptake with up to 75% and 77% cells of cells positive for CpG and SIIN, respectively (Fig. 5B). These levels then decreased to near-baseline, but still detectable, levels during dilution over two orders of magnitude (i.e., 128 dilution). Confocal microscopy revealed that cells internalized both cargos, as indicated by the presence of both SIIN and CpG within the cytosolic region (Fig. 5C). Some level of co-localization was observed in these studies, but punctate red and green regions were also observed, indicating some of the cargo components were not associated with each other. Across the dosing range, no toxicity was observed (Fig. 5D) in the cells.
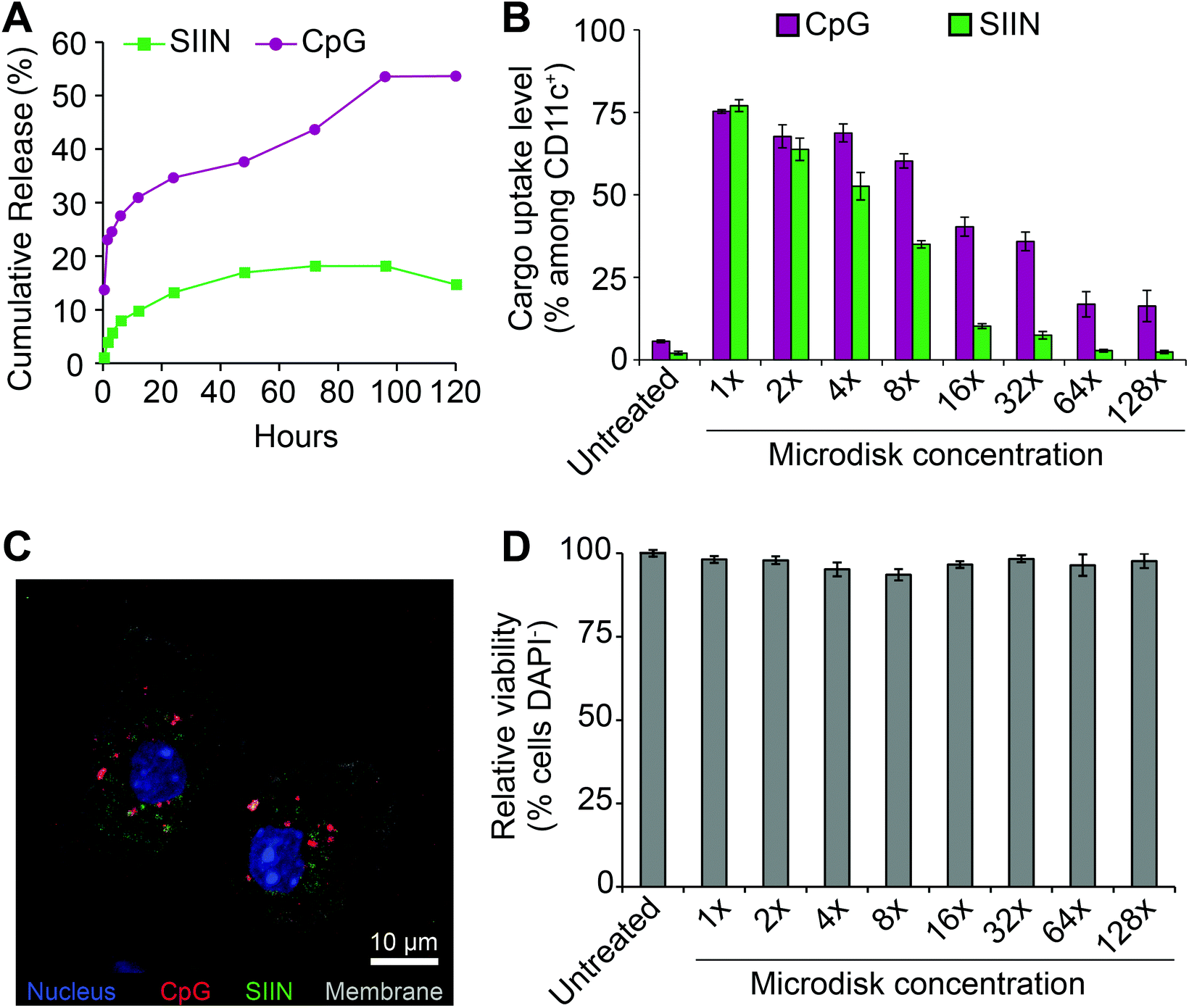 | ||
| Fig. 5 Microdisks release vaccine cargo and are internalized by DCs without toxicity. (A) Release of SIIN and CpG from microdisks in PBS. DC uptake of CpG and SIIN following incubation with microdisks across a range of serial dilutions as measured by (B) flow cytometry and (C) confocal microscopy. (D) Relative viability of DCs treated with microdisks across a range of serial dilutions. | ||
Microdisks activate DC surface markers in a composition-dependent manner
We next tested if altering the relative levels of antigen and adjuvant in microdisks can be used to control DC activation. We first assessed DC activation by measuring expression of common DC surface activation markers: CD86, CD80, CD40. As above, DCs were treated with microdisk vaccines loaded with defined levels of CpG and SIIN, then the impact on DC surface activation markers and SIIN peptide display was assessed. Untreated DCs served as a negative control, while free CpG, free SIIN, and free CpG plus free SIIN were employed as additional controls. Flow cytometry analysis of DCs revealed 51.6%, 47.6%, 42.0%, 24.9% and 24.5% DCs were activated, as assessed by CD86+ expression, for treatment with vaccines at 100%, 91%, 53%, 12% and 0% CpG, respectively (Fig. 6A). The values measured for the 12% and 0% samples were at baseline levels measured in untreated cells, reflecting the low level (12%) and absence (0%) of adjuvant (CpG) in these samples. Similarly, free SIIN and empty PLGA microdisks did not activate DCs relative to untreated cells (Fig. 6A). Analogous trends were observed for additional activation makers, CD80+ (Fig. 6B) and CD40+ (Fig. 6C). Together, these data indicate microdisk adjuvant loading correlates with the level of DC activation.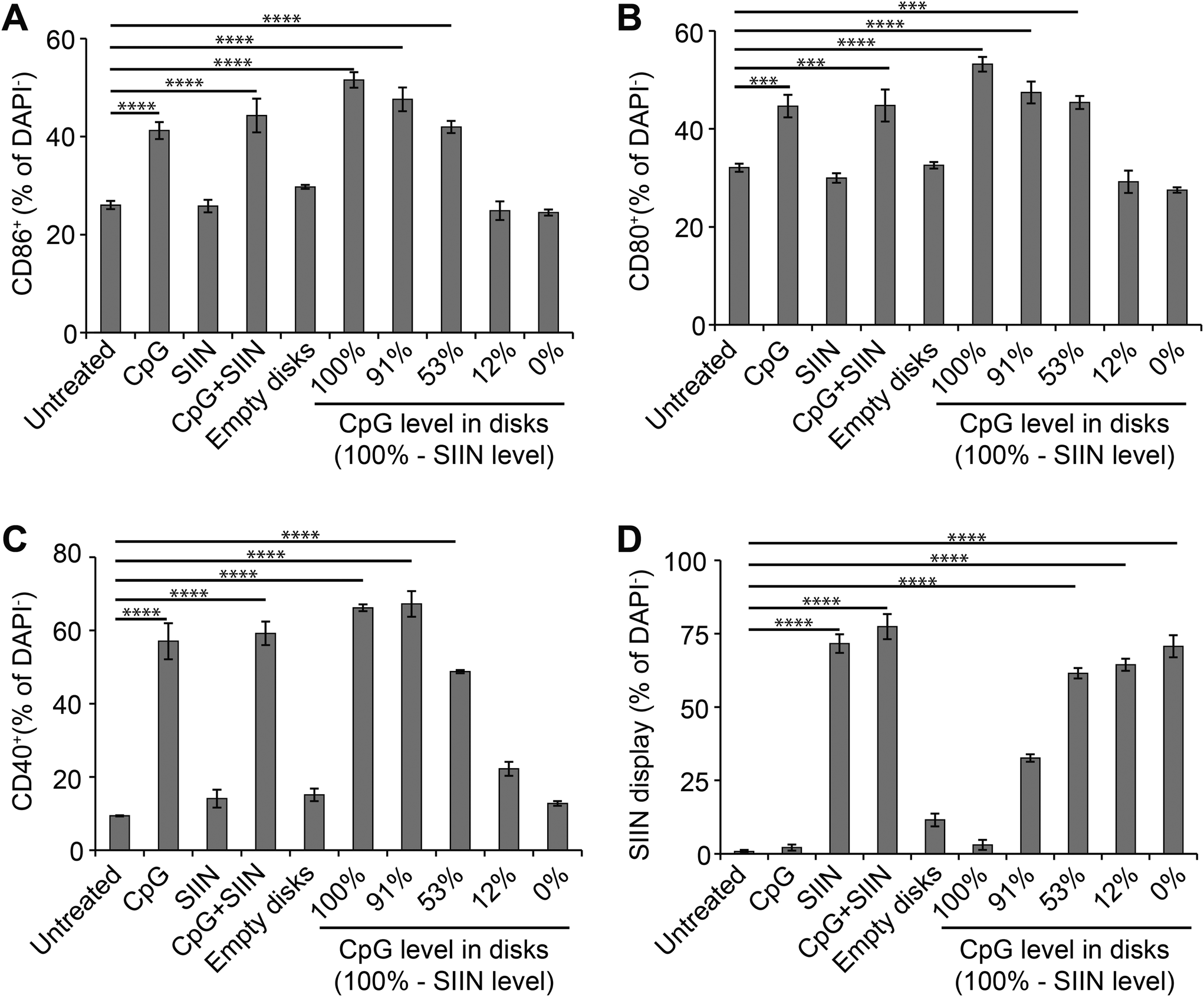 | ||
| Fig. 6 Microdisks activate DCs and promote presentation of SIIN antigen. Activation of DCs incubated with microdisks loaded with defined levels of CpG and SIIN, as indicated by expression of the surface activation markers (A) CD86, (B) CD80, and (C) CD40. (D) Presentation of SIIN peptide on DCs in the MHC-I complex as measured by flow cytometry. ***, p < 0.001; ****, p < 0.0001. | ||
Microdisk antigen composition controls SIIN display by DCs
Processing and presentation of SIIN antigen by DCs incubated with microdisks was next assessed as a function of microdisk antigen and adjuvant composition. SIIN display increased with the level of SIIN in the microdisks (Fig. 6D), corresponding to values of 3.0%, 32.7%, 61.5%, 64.4% and 70.7% of cells displaying SIIN in MHC-I for the same compositions studied above in Fig. 6A–C. Interestingly, microdisks without CpG or containing low levels of CpG, also exhibited increased SIIN display, while DCs treated with microdisks containing low levels of SIIN (and high levels of CpG) exhibited low levels of SIIN (i.e., compositions “100%” and “91%”). In contrast, untreated DCs, DCs treated with free CpG, and DCs treated with empty microdisks caused display by 0.8%, 2.1% and 11.3% of cells. The results of these antigen display studies demonstrate microdisk composition controls not only the level of DC activation, but also the level of antigen display. This is an important step in enabling DCs to prime and expand functional T cells able to fight infection or tumors as part of adaptive immune response.Discussion
One of the broad challenges in vaccine design is that most vaccines are still developed empirically. More rationale design approaches would greatly improve the efficiency of vaccine development. The complexity of the immune system and the interaction with vaccines is one factor hindering this transition, while another is the relatively large variation in properties of many pre-clinical vaccines seeking to exploit the unique features of biomaterials. For example, the most common micro and nanoparticle synthesis approach used in vaccines, emulsion synthesis, generates particles with polydisperse diameters and rely on complex stabilizers.5,6 More uniform and programmable properties would reduce the challenges of immunologically characterizing and testing these vaccines in the setting of higher order animal models or in humans.31 Other studies using advanced manufacturing in the context of vaccines28,29 have not investigated vaccines comprised of antigens and adjuvants, but rather, focused on adsorbing preformed vaccines such as influenza. Thus the knowledge generated in our studies helps broaden the understanding of how modular design of vaccines using lithographic tools or other precision design modalities can be exploited for more uniform vaccines that also enable more tunable control over immune cell function.In this study we employed advanced manufacturing techniques to produce microdisk vaccines with precision features – geometries and loading of multiple cargos. The vaccines have defined diameters controlled by master template materials that can be designed with features of interest. This is in contrast to microparticles synthesized by emulsion techniques. For example, microdisks exhibited a low level of deviation in diameter compared to microparticles prepared by emulsion (Fig. 2E). While the performance of an individual microdisk may not exceed that of an individual particle, the uniformity is important in vaccine design because particulate materials reach lymph nodes – tissues that orchestrate immune function – either through free drainage, if small enough, or through active transport by DCs.32 Materials with uniform diameters mean the mechanism of transport to draining lymph nodes is more consistent, and thus, easier to study and characterize, demonstrating the high level of control in vaccine structures. Along related lines, several studies demonstrate that aspect ratio and shape of biomaterial carriers can impact the magnitude and nature of immune response by changing, for example, the surface area for interaction with DCs, T cells, or other populations.10 While we used a disk design, this advanced manufacturing approach creates future opportunities to explore shape and other properties in a precision manner.
In addition to diameter, this microdisk platform allowed tuning of the absolute and relative amounts of antigen and adjuvant loaded in the microdisks. This resulted in co-localization in primary immune cells. However, the low level of release observed with SIIN in Fig. 5A may indicate a stronger affinity of the peptide with the microdisk compared to CpG. The slight decrease in concentration of SIIN at the final time point may indicate degradation of the released cargo. With respect to uptake, the current data only reveal that SIIN and CpG were both in DCs (Fig. 5B and C), not if the cargo were still incorporated in the microdisks, or if the cargo entered before or after release from the microdisks. Future studies elucidating the answers to these questions could reveal opportunities to further tune the responses to microdisk vaccines by altering the kinetics of cargo release.
Microdisks promoted co-localization in DCs, and this uptake led to several interesting observations with respect to the specific impact of the antigen and adjuvant composition on DC and T cell function. First, during the activation studies of Fig. 6, as adjuvant composition decreased, activation decreased. These cells were activated even at the highest CpG compositions where SIIN was completely or nearly absent. While this is expected, SIIN was processed and displayed at the highest levels when CpG was nearly or completely absent, even though the DCs were not activated at these doses (e.g., 0%, 12% comparing Fig. 6D to other panels). This reveals the specific function of each vaccine component, as DCs preferentially internalize particulate materials (such as microdisks incorporating SIIN), but do not become activated unless they also receive stimulatory signals, such as CpG. These different profiles of activation and antigen presentation indicate the control that can be achieved with different ratio of adjuvant and antigen. This is an important step in generating vaccines exhibiting consistent features that can tune immune responses in a controlled manner.
Conclusion
This work investigated the use of advanced manufacturing techniques for generating microdisk vaccines with uniform features. We show that this approach enables consistent, programmable designs that control both physicochemical features and the interactions with primary immune cells. There are a variety of open questions suggested by this work. For example, how can release kinetics, along with particle aspect ratio or shape, be used to achieve greater levels of control. Additionally, our studies were conducted ex vivo, but future studies should investigate how this platform, as a function of design parameters or adjuvant classes, impact the magnitude and nature of immune response in in vivo.Conflicts of interest
The authors have no conflict of interest.Acknowledgements
This work was supported in part by NSF CAREER Award # 1351688. L. H. T. is a National Science Foundation Graduate Fellow no. DGE1322106. C. M. J. is an employee of the United States Department of Veterans Affairs, a Damon Runyon-Rachleff Innovator supported by the Damon Runyon Foundation (# DRR3415), and a Young Investigator of the Alliance for Cancer Gene Therapy (# 15051543) and the Melanoma Research Alliance (# 348963).References
- M. A. Swartz, S. Hirosue and J. A. Hubbell, Sci. Transl. Med., 2012, 4(148), 148rv9.
- L. H. Tostanoski and C. M. Jewell, Adv. Drug Delivery Rev., 2017, 114, 60–78.
- L. H. Tostanoski, Y. C. Chiu, J. M. Gammon, T. Simon, J. I. Andorko, J. S. Bromberg and C. M. Jewell, Cell Rep., 2016, 16, 2940–2952 CrossRef CAS PubMed.
- J. M. Gammon, E. A. Gosselin, L. H. Tostanoski, Y. C. Chiu, X. Zeng, Q. Zeng and C. M. Jewell, J. Controlled Release, 2017, 263, 151–161.
- S. P. Kasturi, I. Skountzou, R. A. Albrecht, D. Koutsonanos, T. Hua, H. I. Nakaya, R. Ravindran, S. Stewart, M. Alam, M. Kwissa, F. Villinger, N. Murthy, J. Steel, J. Jacob, R. J. Hogan, A. Garcia-Sastre, R. Compans and B. Pulendran, Nature, 2011, 470, 543–547 CrossRef CAS PubMed.
- M. C. Hanson, A. Bershteyn, M. P. Crespo and D. J. Irvine, Biomacromolecules, 2014, 15, 2475–2481 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- P. Kolhar and S. Mitragotri, Adv. Funct. Mater., 2012, 22, 3759–3764 CrossRef CAS.
- S. Barua, J. W. Yoo, P. Kolhar, A. Wakankar, Y. R. Gokarn and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3270–3275 CrossRef CAS PubMed.
- J. C. Sunshine, K. Perica, J. P. Schneck and J. J. Green, Biomaterials, 2014, 35, 269–277 CrossRef CAS PubMed.
- Y. Z. He and K. Park, Mol. Pharmaceutics, 2016, 13, 2164–2171 CrossRef CAS PubMed.
- J. I. Andorko, K. L. Hess, K. G. Pineault and C. M. Jewell, Acta Biomater., 2016, 32, 24–34 CrossRef CAS PubMed.
- T. Y. H. Wu, M. Singh, A. T. Miller, E. De Gregorio, F. Doro, U. D'Oro, D. A. G. Skibinski, M. L. Mbow, S. Bufali, A. E. Herman, A. Cortez, Y. Li, B. P. Nayak, E. Tritto, C. M. Filippi, G. R. Otten, L. A. Brito, E. Monaci, C. Li, S. Aprea, S. Valentini, S. Calabro, D. Laera, B. Brunelli, E. Caproni, P. Malyala, R. G. Panchal, T. K. Warren, S. Bavari, D. T. O'Hagan, M. P. Cooke and N. M. Valiante, Sci. Transl. Med., 2014, 6(263), 263ra160.
- N. A. Peppas, Adv. Drug Delivery Rev., 2013, 65, 5–9 CrossRef CAS PubMed.
- Y. Zhang, H. F. Chan and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS PubMed.
- N. Bhattacharjee, A. Urrios, S. Kang and A. Folch, Lab Chip, 2016, 16, 1720–1742 RSC.
- L. Bogunovic, D. Anselmetti and J. Regtmeier, J. Micromech. Microeng., 2011, 21, 027003.
- R. K. Pal, N. E. Kurland, C. Y. Jiang, S. C. Kundu, N. Zhang and V. K. Yadavalli, Eur. Polym. J., 2016, 85, 421–430 CrossRef CAS.
- P. Zorlutuna, N. Annabi, G. Camci-Unal, M. Nikkhah, J. M. Cha, J. W. Nichol, A. Manbachi, H. Bae, S. Chen and A. Khademhosseini, Adv. Mater., 2012, 24, 1782–1804 CrossRef CAS PubMed.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed.
- M. C. Traub, W. Longsine and V. N. Truskett, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 583–604.
- J. Xu, D. H. C. Wong, J. D. Byrne, K. Chen, C. Bowerman and J. M. DeSimone, Angew. Chem., Int. Ed., 2013, 52, 6580–6589 CrossRef CAS PubMed.
- J. M. DeSimone, J. Controlled Release, 2016, 240, 541–543 CrossRef CAS PubMed.
- D. Dendukuri, D. C. Pregibon, J. Collins, T. A. Hatton and P. S. Doyle, Nat. Mater., 2006, 5, 365–369 CrossRef CAS PubMed.
- P. Zhang and J. Guan, Small, 2011, 7, 2998–3004 CrossRef CAS PubMed.
- C. A. Fromen, G. R. Robbins, T. W. Shen, M. P. Kai, J. P. Ting and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 488–493 CrossRef CAS PubMed.
- K. G. Reuter, J. L. Perry, D. Kim, J. C. Luft, R. H. Liu and J. M. DeSimone, Nano Lett., 2015, 15, 6371–6378 CrossRef CAS PubMed.
- A. L. Galloway, A. Murphy, J. M. DeSimone, J. Di, J. P. Herrmann, M. E. Hunter, J. P. Kindig, F. J. Malinoski, M. A. Rumley, D. M. Stoltz, T. S. Templeman and B. Hubby, Nanomedicine, 2013, 9, 523–531 CrossRef CAS PubMed.
- S. N. Mueller, S. Tian and J. M. DeSimone, Mol. Pharm., 2015, 12, 1356–1365 CrossRef CAS PubMed.
- N. Doshi, A. J. Swiston, J. B. Gilbert, M. L. Alcaraz, R. E. Cohen, M. F. Rubner and S. Mitragotri, Adv. Mater., 2011, 23, H105–H109 CrossRef CAS PubMed.
- Q. Zeng, J. M. Gammon, L. H. Tostanoski, Y. C. Chiu and C. M. Jewell, ACS Biomater. Sci. Eng., 2017, 3, 195–205 CrossRef CAS PubMed.
- D. J. Irvine, M. A. Swartz and G. L. Szeto, Nat. Mater., 2013, 12, 978–990 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |