
Combined determination of copper ions and β-amyloid peptide by a single ratiometric electrochemical biosensor†
Yanyan
Yu‡
*a,
Peng
Wang‡
b,
Xiaodan
Zhu‡
b,
Qiwen
Peng
a,
Yi
Zhou
b,
Tianxiao
Yin
a,
Yixin
Liang
c and
Xiaoxing
Yin
 *a
*a
aJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical University, 209 Tongshan Road, Xuzhou 221004, Jiangsu, P.R. China. E-mail: yinxx@xzhmu.edu.cn
bDepartment of Pharmaceutical Analysis, School of Pharmacy, Xuzhou Medical University, 209 Tongshan Road, Xuzhou 221004, Jiangsu, P.R. China
cSchool of Liberal Arts and Science, Shanghai University of Medicine & Health Sciences, 279 Zhouzhu Highway, Shanghai 201318, P.R. China
First published on 15th November 2017
Abstract
Copper ions (Cu2+) play a critical role in biological processes and are directly involved in β-amyloid peptide (Aβ) aggregation, which is responsible for the occurrence and development of Alzheimer's disease (AD). Therefore, combined determination of Cu2+ and Aβ in one analytical system is of great significance to understand the exact nature of the AD event. This work presents a novel ratiometric electrochemical biosensor for the dual determination of Cu2+ and Aβ1–42. This unique sensor is based on a 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulphonate) (ABTS) and poly(diallyldimethylammonium chloride) (PDDA)-bi functionalized single-walled carbon nanotubes (ABTS-PDDA/CNTs) composite. The inclusion of ABTS not only enhanced the sensitivity, but it also acted as an inner reference molecule to improve detection accuracy. The specific recognition of Cu2+ was realized by neurokinin B (NKB) coatings on the ABTS-PDDA/CNTs surface to form a [CuII(NKB)2] complex with Cu2+. The ABTS-PDDA/CNTs-NKB modified electrode also displayed an excellent electrochemical response toward the Aβ1–42 monomer, when a certain amount of the Aβ1–42 monomer was added to Cu2+-contained PBS buffer, which was due to the release of Cu2+ from the [CuII(NKB)2] complex through Aβ binding to Cu2+. Meanwhile, our work showed that Cu2+ bound Aβ1–42 was concentration-dependent. Consequently, the presented electrochemical approach was capable of quantifying two important biological species associated with AD by one single biosensor, with the detection limits of 0.04 μM for Cu2+ and 0.5 ng mL−1 for Aβ1–42, respectively. Finally, the ratiometric electrode was successfully applied for monitoring Cu2+ and Aβ1–42 variations in plasma and hippocampus of normal and AD rats.
Introduction
The formation of neutric plaques in the brain has been considered as one of the primary hallmarks of Alzheimer's disease (AD).1 In this process, β-amyloid peptide (Aβ) monomer aggregations by way of oligomerization and fibrillization are central to the formation of these plaques.2,3 More studies have indicated that the Aβ monomer aggregations self-assemble into higher order structures in vitro and in vivo (e.g., dimers, trimers and tetramers) and eventually form the elongated fibrils that are observed in the late-stage AD patients.4 Both the soluble oligomers and mature fibrils from Aβ are more neurotoxic compared with the monomer and can cause death of brain cells.5–8 Consequently, the development of efficient techniques to observe variations in the level of Aβ monomers could provide a clue to unveil the extent of aggregation as well as a definitive molecular basis for understanding the disease mechanism and diagnosing AD. Fluorescence, colorimetry, electrochemistry, surface plasmon resonance (SPR), mass spectrometry (MS) and capillary electrophoresis (CE) have been employed to monitor Aβ species from body fluids and cell media.9–15However, more and more research has indicated that the hypothesis of self-aggregation of Aβ alone is inadequate to explain the accumulation of Aβ in brain regions. Transition metals (Cu2+, Fe3+ and Zn2+) have been directly involved in modulating Aβ aggregation owing to the fact that abnormally high concentrations of metal ions exist within senile plaques.16–18 This evidence has been further confirmed when Aβ aggregates were treated with metal chelators, which produced soluble Aβ peptides.19,20 The redox active transition metals generate toxic reactive oxygen species (ROS), which cause oxidative stress and precede the formation of the amyloid aggregates that are associated with AD.21–23 Nowadays, prevention of Aβ–metal interactions to inhibit oligomer formation has already been proposed as a disease-modifying strategy for AD. Among various metals, the contribution of Cu2+ to amyloid deposition has been proposed as a crucial step in the amyloid cascade.24 Although Cu2+ induced Aβ aggregation is still questionable, its complexation with peptides might be responsible for enhancing the formation of the α-helix conformation of the alanine-based peptides.25,26 Considering the tight correlations between Aβ and Cu2+ in the pathological process of AD, a quantitative understanding of simultaneous variations in their levels to uncover the detailed mechanism is essential. However, most of the published work focused only on one specific substance (e.g., Aβ or Cu2+), which is insufficient to explain the exact role of Aβ–metal interactions played in the AD progress. More importantly, up to now, a combined determination of two analytes associated with one disease in a single biological system has been a great challenge due to the unsatisfactory sensitivity.
Electrochemical biosensors have received more and more attention because of their striking advantages such as simplicity, selectivity, low instrumental cost, and capability in real-time, even in vivo detection.27–30 For their practical applications, signal amplification is crucial for obtaining a low detection limit and high sensitivity. Nowadays, the general strategy is nanomaterial involved electrocatalytic amplification.31–33 The unique chemical and physical properties of the nanoscaled materials, such as metal/semiconductor nanoparticles, can be used to prepare advanced electrocatalytic materials, which thus enable electrochemical monitoring with nanoscale spatial resolution, yielding unique information for better understanding heterogeneous electrode/solution interfaces.34 Owing to their unique properties, such as high specific surface areas, excellent electrical conductivities and chemical stability,35 carbon nanotubes (CNTs) have demonstrated a wide availability in many aspects, including supporting noble-metal nanoparticles to prepare functional nanohybrids36–38 and fabricating biosensors to accelerate the electron transfer rate of redox reactions.39,40 However, insufficient binding sites on CNTs for supporting molecules are a well-known drawback, resulting in poor stability and catalytic activity.38 Consequently, surface functionalization of CNTs is commonly required by chemical or electrochemical oxidation, wrapping with polymer or grafting of tether approaches.41–43
In this work, we report a facile but effective strategy for combined determination of Cu2+ and Aβ in a single analytical system. To achieve this goal, poly(diallyldimethylammonium chloride) (PDDA) and 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulphonate) (ABTS)-functionalized CNTs were firstly prepared for loading a Cu2+-specific recognition element, neurokinin B (NKB), and then a ratiometric ABTS-PDDA/CNTs-NKB electrode was constructed. The resulting bioelectrocatalytic film was utilized for effective electroreduction of Cu2+ with a high sensitivity and accuracy. Then the peak current decreased after a certain amount of Aβ1–42 monomer was added to a Cu2+-contained PBS solution, which originated from the stronger coordination between Cu2+ and Aβ peptide. It was found that by adjusting the concentration of Cu2+ in the detection electrolyte (0.38, 0.95 and 5.7 μM), the ABTS-PDDA/CNTs-NKB electrode was capable of detecting down to 0.001 and up to 3.8 μg mL−1 Aβ1–42. Noticeably, metal ions, amino acids and other endogenous substances in vivo, even Aβ oligomers, could not produce any response changes under the same conditions, indicating a satisfactory selectivity. Compared with the common strategy that utilized two independent electrodes to construct a two-channel system for the dual determination, this method only involved one single electrode, which was much easier to operate and provided a possible route for better understanding the close relationship between two analytes in physiological and pathological events of the brain.
Experimental
Materials and reagents
Pristine single-walled CNTs (ϕ = 10–30 nm) were bought from Chengdu Institute of Organic Chemistry Nanotech Port Co., Ltd (Chengdu, China). Purified synthetic Aβ1–42 was obtained from ChinaPeptides Co., Ltd (Shanghai, China). Stimulated aggregation of Aβ1–42 samples was achieved by incubation at 37 °C in PBS with shaking. NKB, ABTS, PDDA, glutaraldehyde solution (Glu, 25%), glucose (Glu), lactic acid (Lac), uric acid (UA), ascorbic acid (AA), dopamine (DA), all the amino acids and copper sulfate (CuSO4) were all purchased from Sigma-Aldrich (USA) and used without further purification. Phosphate buffered saline (PBS, 0.1 M, pH 7.4) containing 8.6 mM Na2HPO4·12H2O, 1.88 mM NaH2PO4, 50.9 mM NaCl and 2.94 mM KCl was employed as the detection electrolyte. A stock solution of Aβ1–42 (1 mg mL−1) was prepared by independently dissolving a desired amount in 25 mM Tris-HCl buffer solution (pH 7.4) and diluted to appropriate concentrations using 10 mM NaOH and freshly prepared daily before experiments. Water (≥18 M) used throughout the whole experiment was purified with a Millipore system. All other reagents were of at least analytical grade and commercially available. The plasma and hippocampus homogenate samples from normal and AD rats were kindly provided by L. Zhang from another group.Apparatus
Scanning electron microscope images (SEM, Hitachi Co. Ltd, Tokyo, Japan) were taken using a field emission gun Hitachi S-4800 scanning electron microscope (operating at 1 kV) for morphological analysis of electrodes. The changes in the diameter of CNT bundles before and after functionalization were measured by using a FEI Tecnai G2 T12 transmission electron microscope (TEM, USA) operating at 120 kV. The TEM specimens were prepared by dropping the sample solutions onto 50 Å carbon coated copper grids with the excess solution being immediately wicked away. X-ray photoelectron spectroscopy (XPS) investigation was carried out on an ESCALab MKII X-ray photoelectron spectrometer using Mg Kα radiation. Fourier transform infrared spectroscopy (FT-IR) was performed on a Nicolet model 460 Fourier transform-infrared spectrometer. Raman measurements were carried out on a confocal microscope Raman spectrometer system (Renishaw, inVia Reflex Raman) with an excitation wavelength of 785 nm.All the electrochemical experiments were performed on a CHI 832D electrochemical workstation (Shanghai Chenhua Instrument, CHI, China). A conventional three-electrode system was used throughout the experiments composed of a platinum wire as a counter electrode, a saturated calomel electrode (SCE) as a reference electrode, and a bare or a modified glassy carbon electrode (GC) as a working electrode. Unless otherwise indicated, all electrochemical measurements were performed under an oxygen atmosphere.
Preparation of ABTS-PDDA/CNTs hybrids
Before ABTS hybridization, CNTs were firstly functionalized by PDDA according to the previous report.44 Briefly, 0.5 mg mL−1 CNTs was dispersed in a 0.5% PDDA aqueous solution containing 0.5 M NaCl by 50 min sonication to give a homogeneous black suspension. Then, the solution was filtered and washed several times with distilled water to remove the free PDDA and dried under vacuum at 70 °C for 24 h. After that, 10 mg of the prepared PDDA/CNTs was added to 1 mL of 1.8 mM ABTS aqueous solution. The formed suspension was stirred continuously for two days. Then, it was centrifuged. The supernatant was removed and replaced with water. This process was repeated at least two times and finally, a stable colloidal suspension of ABTS-PDDA/CNTs was obtained.Electrode modification
The electrode modification is as shown in Scheme 1. Before fabrication, bare GC was firstly polished with 0.05 μm alumina paste on a micro-cloth and then cleaned using acetone, ethanol and distilled water with ultrasonication for 10 min, respectively and dried at room temperature (25 ± 1 °C). 0.2 mg mL−1 ABTS-PDDA/CNTs or PDDA/CNTs black suspension solutions were obtained by adding 0.2 mg ABTS-PDDA/CNTs or PDDA/CNTs to 1 mL of N,N-dimethylformamide (DMF). A 5 μL of ABTS-PDDA/CNTs or PDDA/CNTs suspension was dropped onto the cleaned electrode surface, which was denoted as ABTS-PDDA/CNTs-GC or PDDA/CNTs-GC. To realize the specific determination of Cu2+, 5 μL of NKB solution in PBS (0.1 M, pH 7.4) (0.5 mg mL−1) was introduced onto the ABTS-PDDA/CNTs or PDDA/CNTs layer and allowed to dry at ambient temperature. The obtained electrode was named ABTS-PDDA/CNTs-NKB-GC or PDDA/CNTs-NKB-GC. Finally, 2 μL of glutaraldehyde (0.1%) was drop-cast to complete the cross-linking of NKB with the ABTS-PDDA/CNTs or PDDA/CNTs hybrid.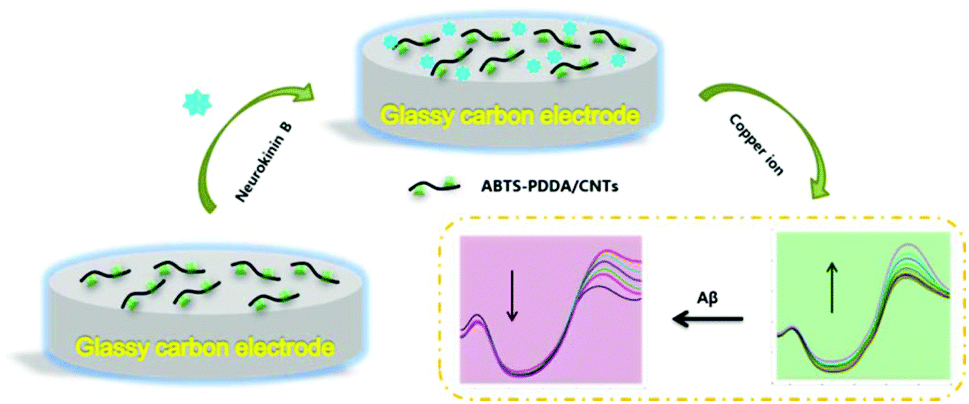 | ||
| Scheme 1 Combined determination of Cu2+ and β-amyloid peptide based on the ratiometric ABTS-PDDA/CNTs-NKB biosensor. | ||
Electrochemical determination of Cu2+ and Aβ1–42 in one analytical system
The electrochemical sensing of Cu2+ and Aβ1–42 was performed at ambient temperature in a PBS (0.1 M, pH 7.4) buffer solution. As shown in Scheme 1, in a typical run, firstly, different concentrations of a Cu2+ standard solution or real samples from rats were added to 5 mL PBS buffer, followed by gentle stirring for a certain time. The differential pulse voltammetry (DPV) signals were recorded by the potential scan in the range between −0.4 and 0.7 V vs. SCE. After that, additions of certain amounts of Aβ1–42 standard solutions or real samples from rats to Cu2+-contained PBS were performed to observe the signal change at the original potential of −0.12 V belonging to Cu2+. The quantifications of Cu2+ and Aβ1–42 were respectively estimated by calculating the ratios of the respective current intensity between the reduction peaks at −0.12 V for Cu2+ and Aβ1–42 and 0.58 V for the reference substance ABTS.Animal experiments
The induction of AD rats was performed according to our previous work.14 Briefly, male Wistar rats, weighing 100–120 g at the beginning of experiments, were obtained from Shanghai Yisen Biotechnology Co., Ltd. Rats were housed in plastic cages, with food and water available ad libitum, and kept under standard environmental conditions (12 h light/dark cycle, 22 °C). All animal experiments were conducted with approval of the Animal Ethics Committee in Xuzhou Medical University, China. All efforts were made to minimize the number of animals used and their suffering.D-Galactose (D-gal, Aladdin Reagent Co., Ltd) and ibotenic acid (IBO, Enzo Life Science Inc.) were dissolved in sterile saline and ice PBS (pH 7.4) at their final concentrations of 10 g L−1 and 8 g L−1, respectively. Rats were randomly divided into two groups: normal group and D-gal + IBO group (AD model group) (n = 15 for each group). The latter group was daily administered D-gal (50 mg kg−1 in 0.5 mL saline, i.p.) for six weeks, which has been demonstrated to be able to produce aging rats.
After D-gal administration, rats in the AD group were anesthetized with sodium pentobarbital (50 mg kg−1, i.p.) and placed on a stereotaxic apparatus (Shenzhen RWD Life Science Co.), with the incisor bar set at 5 mm above the interaural line and appropriately placed holes were drilled through the skull. Stereotaxic coordinates of nucleus basalis magnocellularis (NBM) were set at −1.0 mm posterior and −2.6 mm lateral to bregma and 7.8 mm below from the top of the skull. Bilateral infusions of 1 μL volume of IBO into NBM using a 5 μL Hamilton syringe lasted over a period of 5 min and the needle was left in place for 5 min after completing the infusion. After the surgery, rats were allowed to recover for 45 days before experiments. Hippocampal homogenates were centrifuged at 4000 rpm for 15 min and supernatant of the homogenates was used for the following determination.
Results and discussion
Characterization of electrodes
CNTs have always been used as carriers to load numerous enzymes or proteins in the preparation of an integrated bio-electrocatalytic system. To promote the electron transfer and stability of pristine CNTs, PDDA and ABTS were successively functionalized with CNTs to produce a more stable colloidal suspension. On the one hand, the non-covalent functionalization by PDDA led to a high density and homogeneous distribution of surface functional groups, such as the amine group, which was favorable for the self-assembling of negatively-charged ABTS and NKB. On the other hand, the inclusion of ABTS not only enhanced the sensitivity, but it also acted as an inner reference molecule, and thus provided a built-in correction for environmental effects and improved detection accuracy. The functionalization of CNTs was confirmed by Raman and FT-IR characterization techniques. As presented in Fig. S1,† both CNTs and ABTS-PDDA/CNTs showed similar Raman scattering patterns. The peaks near 1350 and 1580 cm−1 could be ascribed to the A1g breathing mode of a disordered graphite structure (D band) and the E2g structure mode of graphite (G band).45 The extent of the modification or defects in CNTs can be evaluated by the intensity ratio of D and G bands (ID/IG). This ratio was found to be higher at the ABTS-PDDA/CNTs surface (0.28) than that of CNTs (0.08), which was caused by the surface modification process.38 The FT-IR spectrum of CNTs showed absorption bands at 1165 and 1705 cm−1, corresponding to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of CNTs (Fig. S2†). The band at 1624 cm−1 was attributed to the aromatic skeletal CC stretching vibration of CNTs.46 The intensity of bands at 1165 and 1705 cm−1 decreased in ABTS-PDDA/CNTs, suggesting the interaction between CNTs and PDDA. These above results clearly illustrated that ABTS-PDDA/CNTs have been successfully synthesized.
O stretching vibrations of CNTs (Fig. S2†). The band at 1624 cm−1 was attributed to the aromatic skeletal CC stretching vibration of CNTs.46 The intensity of bands at 1165 and 1705 cm−1 decreased in ABTS-PDDA/CNTs, suggesting the interaction between CNTs and PDDA. These above results clearly illustrated that ABTS-PDDA/CNTs have been successfully synthesized.
The surface morphologies of ABTS-PDDA/CNTs and ABTS-PDDA/CNTs-NKB films on ITO were characterized by the SEM technique as shown in Fig. 1. The functionalized CNTs showed a homogeneous surface and good dispersion (Fig. 1A), indicating that the modifications of PDDA and ABTS preserved the intrinsic properties of CNTs. After NKB was attached to the ABTS-PDDA/CNTs part and cross-linked with glutaraldehyde, surface topography significantly changed with increased degree of cross-linking on the whole ITO, due to the massive loading of proteins throughout the three-dimensional structure of CNTs (Fig. 1B). A comparison of TEM images of ABTS-PDDA/CNTs and ABTS-PDDA/CNTs-NKB composites at the same magnification showed that the surface of ABTS-PDDA/CNTs became rough and bundles of CNTs became thicker following NKB modification. Such results confirmed the successful fabrication of ABTS-PDDA/CNTs-NKB-GC. The changes in chemical compositions with the step-by-step modification were also tracked by XPS analysis (see Fig. S3 in the ESI†).
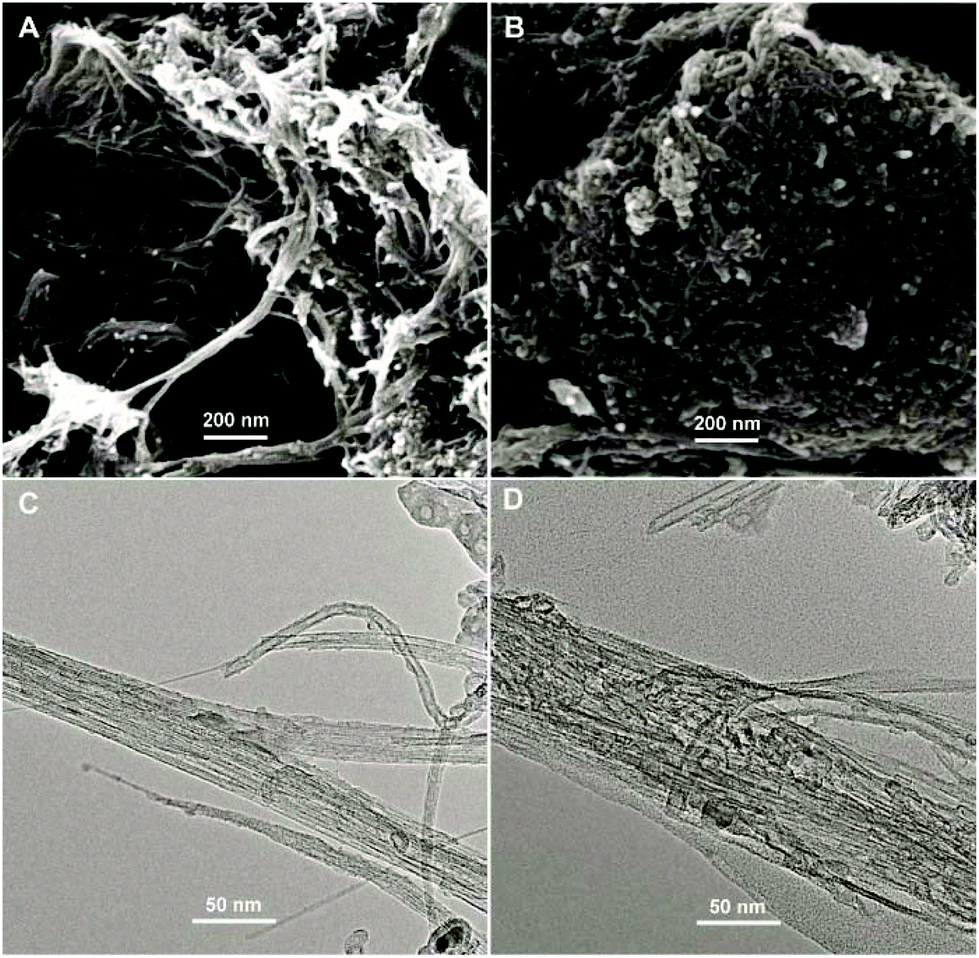 | ||
| Fig. 1 SEM (A, B) and TEM (C, D) images of ABTS-PDDA/CNTs (A, C) and ABTS-PDDA/CNTs-NKB (B, D) composites. | ||
Cyclic voltammetry (CV) was also employed to investigate the electrochemistry of the stepwise fabricated electrode interfaces in a Fe(CN)63−/Fe(CN)64− redox couple in pH 7.4 PBS buffer. As seen from Fig. S4,† the pristine bare GC exhibited a well-defined couple of redox peaks in the range between −0.4 and 0.7 V. The attachment of the conductive PDDA/CNTs or ABTS-PDDA/CNTs hybrid appreciably enhanced the redox currents by improving the electronic coupling to the underlying electrode, which was beneficial for the following analyte detection with increased sensitivity. After further immobilization of NKB onto the prepared ABTS-PDDA/CNTs interface, as expected, the peak current significantly decreased due to the impeding essence of native proteins for the electron transfer on electrodes. Such a result showed that PDDA/CNTs or ABTS-PDDA/CNTs and NKB have been fabricated onto the electrode step by step, which coincided with TEM, SEM and XPS results.
Electrochemical performance of the ABTS-PDDA/CNTs-NKB electrode toward Cu2+ determination
Our previous work had employed NKB as a unique biomolecular recognition agent for Cu2+ to form a [CuII(NKB)2] complex, which ensured the selectivity of Cu2+ determination against a series of potential interferences in vivo.42 In the present contribution, after the ABTS-PDDA/CNTs-NKB electrode was successfully fabricated, the Cu2+-sensitive behavior was investigated in 100 mM PBS buffer (pH 7.4) by DPV. The electrochemical responses of Cu2+ with the same concentration (8.0 μM) were compared among four electrodes, e.g., bare GC (I), PDDA/CNTs-GC (II), ABTS-PDDA/CNTs-GC (III) and ABTS-PDDA/CNTs-NKB-GC (IV). As illustrated in Fig. 2, only charge current was observed on bare GC in an 8.0 μM Cu2+-contained PBS buffer solution (curve I), while an obvious reduction peak at around −0.1 V vs. SCE appeared for PDDA/CNTs-GC, which can be ascribed to the electrochemical reduction of PDDA/CNTs (curve II). Afterwards, a new peak at 0.58 V belonging to the reduced state of ABTS (ABTS2−) on the ABTS-PDDA/CNTs film appeared in the 8.0 μM Cu2+-contained PBS buffer (curve III). After a certain amount of NKB solution was cast onto the ABTS-PDDA/CNT-GC and determined in the 8.0 μM Cu2+ solution, as expected, the electrochemical signal at −0.12 V obviously increased, whereas the current at 0.58 V remained unchanged (curve IV), indicating that Cu2+ was recognized by NKB and an electron transfer between the electrode and Cu2+ ion took place. Consequently, the quantification of Cu2+ in this ratiometric assay can be realized on the basis of the current ratios (IP/IP(R)) between the two peaks at −0.12 and 0.58 V, in which the current at −0.12 V for Cu2+ was denoted as IP, and the IP(R) represents the current of ABTS at 0.58 V.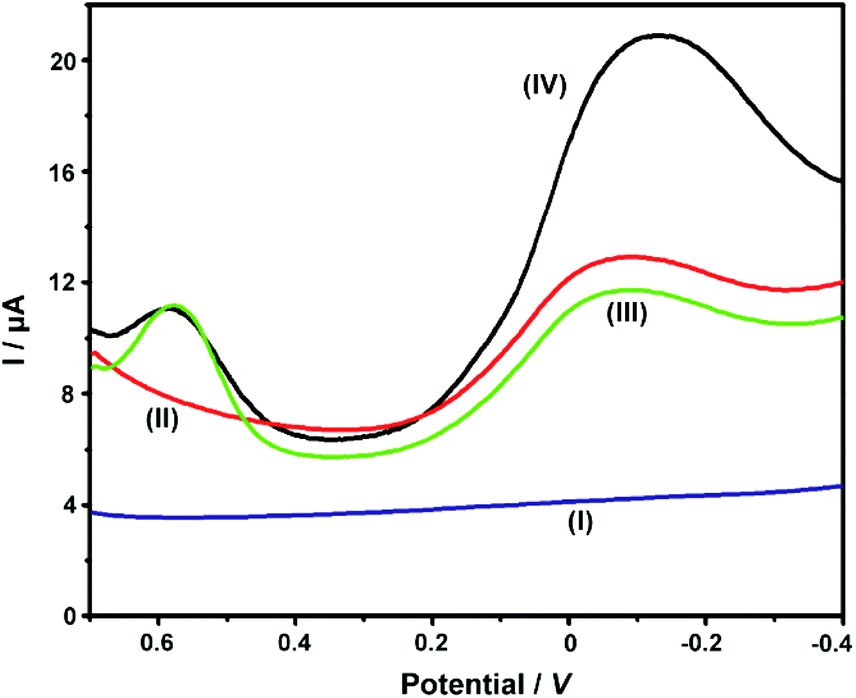 | ||
| Fig. 2 DPV curves obtained on bare GC (I), PDDA/CNTs-GC (II), ABTS-PDDA/CNTs-GC (III) and ABTS-PDDA/CNTs-NKB-GC (IV) in the 100 mM PBS buffer solution containing 8.0 μM Cu2+. | ||
Different concentrations of Cu2+ standard solutions were added to PBS buffer and the corresponding DPV responses obtained on ABTS-PDDA/CNTs-NKB-GC were recorded. As shown in Fig. 3, the peak current at −0.12 V increased gradually with the addition of Cu2+, while the peak current at 0.58 V remained constant. The calculated IP/IP(R) ratios showed a good linear range toward Cu2+ within a wide concentration range of 0.1 to 10 μM. In consideration of the wide span of Cu2+ levels between normal and AD rats, it would be more ideal for practical applications to have a broader range of dynamic responses. The linear dependencies of Cu2+ concentrations yielded the regression equation of IP/IP(R) = 0.03C(μM) + 1.63, with a correlation coefficient R of 0.9913. The detection limit of such a detection strategy was 0.04 μM, estimated from 3σ of the baseline signals. This value was much lower than our previously published work that utilized functionalized polymerized ionic liquids as the substrate for the loading and permeation of NKB.47 Moreover, it was also equal to or better than that of other reported Cu2+ detection assays,48–52 which indicated that the established electrochemical strategy held great potential for Cu2+ determination in a biological matrix.
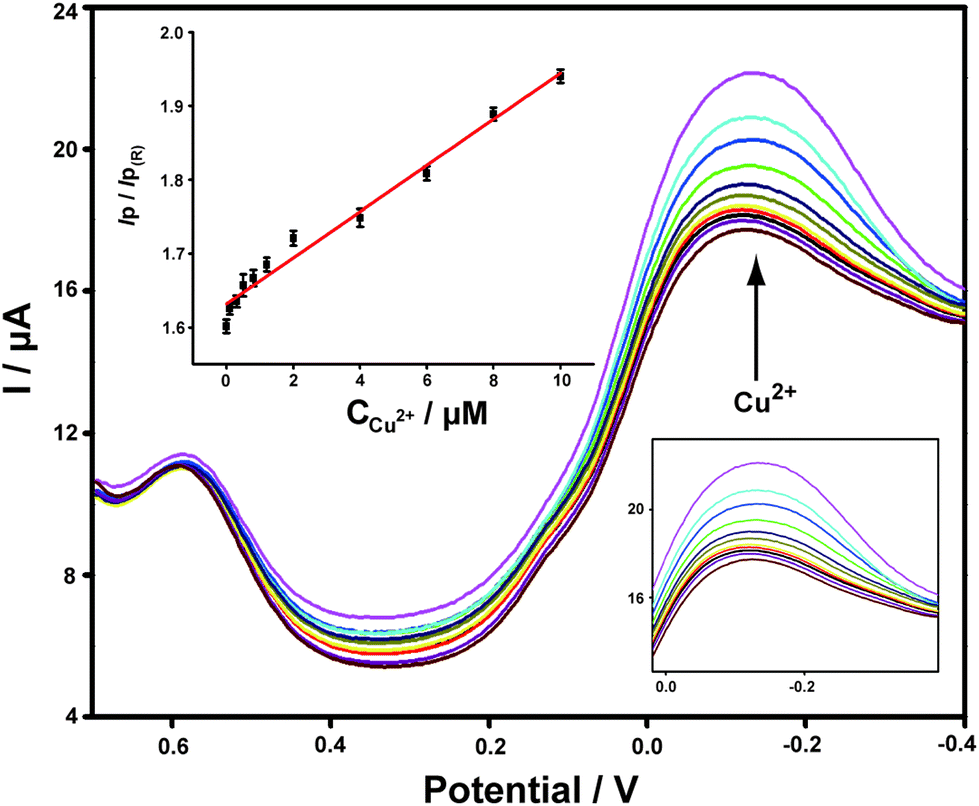 | ||
| Fig. 3 DPV curves of the ABTS-PDDA/CNTs-NKB-GC toward successive additions of Cu2+ at the concentrations of 0, 0.1, 0.3, 0.5, 0.8, 1.2, 2.0, 4.0, 6.0, 8.0, and 10.0 μM. The inset is the corresponding linear plot of the IP/IP(R) ratios versus the absolute concentrations of Cu2+. | ||
Ratiometric determination of Aβ1–42 by ABTS-PDDA/CNTs-NKB-GC
Aβ has always been considered as the direct pathogenic factor for AD by way of oligomerization and fibrillization to form neutric plaques. Several research groups have reported the binding of Cu2+ to Aβ peptides.53,54 On the basis of the abovementioned Cu2+ determination, it was therefore expected that Cu2+ acted as the complexing agent for Aβ, after which, the current responses of Cu2+ on the ABTS-PDDA/CNTs-NKB-GC would basically decrease in the presence of Aβ due to the release of Cu2+ from the [CuII(NKB)2] complex, hence developing a novel methodology for Aβ monitoring. The DPV responses obtained at the ABTS-PDDA/CNTs-NKB electrode in 6.0 μM Cu2+ solution before (curve a) and after additions of 1.8 and 3.8 μg mL−1 Aβ1–42 (curves b and c) are shown in Fig. 4A. Upon the first addition of Aβ1–42, the reduction current at −0.12 V increased whereas the signal at 0.58 V decreased. This phenomenon was normally caused by the perturbation of the detection electrolyte by the variation in pH as the Aβ standard solution was prepared in NaOH. However, in the second test, the reduction peak located at −0.12 V decreased, while that observed at 0.58 V remained constant. The decline in the current was attributed to the release of Cu2+ from the [CuII(NKB)2] complex when bound to Aβ, probably due to the reason that Aβ has a stronger coordination capability to Cu2+ than NKB, and thus Cu2+ was replaced from the [CuII(NKB)2] complex with the addition of Aβ. In order to verify that the signal change was reliably induced by Aβ, and not by other factors, two control experiments were carried out thereafter. Firstly, in the absence of Cu2+, the continuous additions of 1.8 and 3.8 μg mL−1 Aβ1–42 to the blank PBS didn't cause any visible change in the reduction currents at both −0.12 V and 0.58 V (Fig. 4B). Meanwhile, as suggested from Fig. 4C, although the initial addition of NaOH solution, a solvent for solubilizing Aβ, to 6.0 μM Cu2+-contained PBS buffer induced the same result as that of Aβ1–42, no further change was observed when NaOH was introduced into the electrolyte for the second time. These results clearly demonstrated that this single ratiometric sensor provided a sensitive and accurate platform for determination of Cu2+ and Aβ1–42 in one electrochemical system.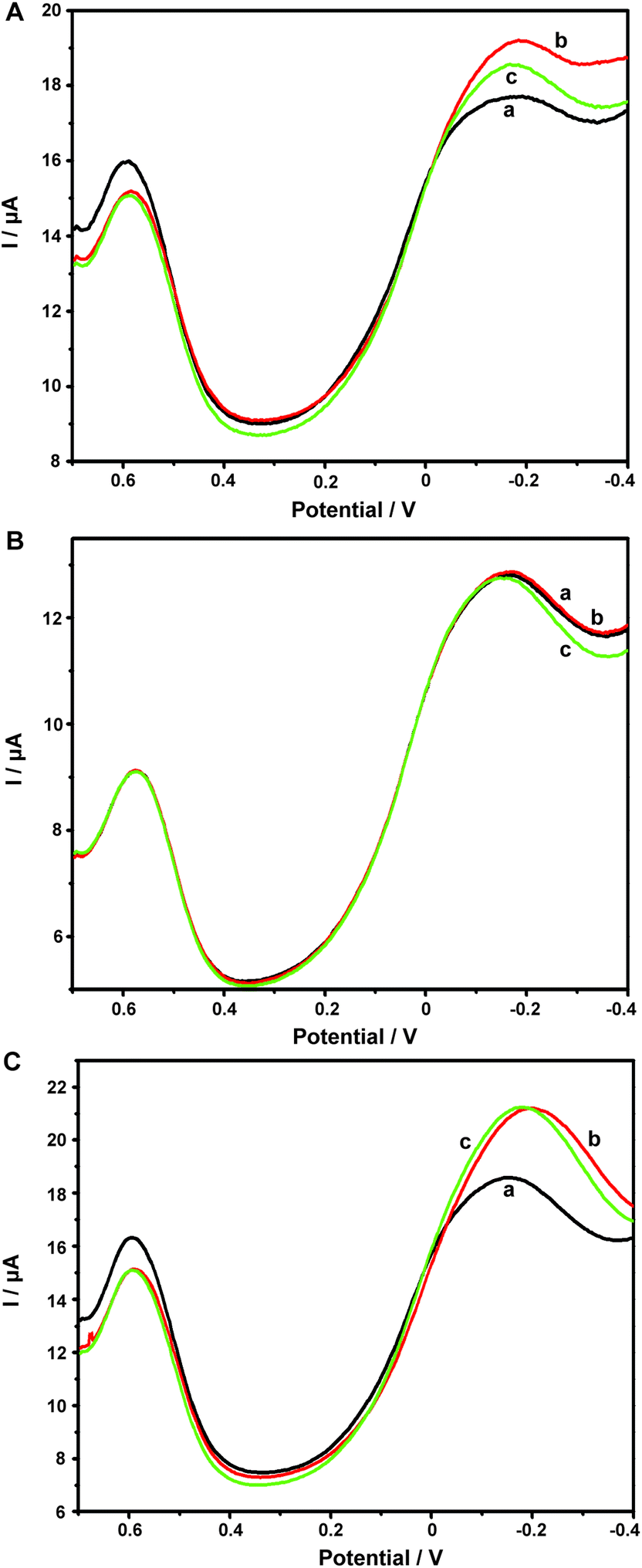 | ||
| Fig. 4 DPV curves obtained on ABTS-PDDA/CNTs-NKB-GC toward 1.8 (red curve, b) and 3.8 μg mL−1 Aβ1–42 (green curve, c) in the presence (A) and absence (B) of 6.0 μM Cu2+ PBS buffer. (C) The DPV responses of 10 μL NaOH additions (b and c) to 6.0 μM Cu2+ PBS buffer. | ||
It has been widely discussed that Cu2+ was capable of inhibiting or accelerating the aggregation of Aβ peptide by binding to Aβ and forming the Cu2+–Aβ complex.23 Therefore, it was quite interesting whether this binding was concentration-dependent or not. Herein, we examined the interaction between Cu2+ and Aβ with three concentrations of Cu2+ by an electrochemical technique. As shown in Fig. 5C, when the experiment was performed in 5.70 μM Cu2+ solution, the peak current obtained at −0.12 V gradually and linearly decreases on additions of increasing concentrations of Aβ1–42 (from 0.1 to 3.8 μg mL−1). More interestingly, we found that the linear current changes of Aβ1–42 on the ABTS-PDDA/CNTs-NKB-GC could only be acquired in a fixed concentration of Cu2+. For example, in Fig. 5B, when a lower concentration of Cu2+ (0.95 μM) was contained in PBS, the lowest detectable concentration of Aβ1–42 was down to 0.01 μg mL−1, one order of magnitude lower than that in a 5.70 μM Cu2+ solution. Likewise, when the concentration of Cu2+ continued to decrease down to 0.38 μM, the linear range of Aβ1–42 started at a much lower concentration (1 ng mL−1) in comparison with that in 0.95 and 5.70 μM Cu2+ (Fig. 5A). We also tested the response of Aβ1–42 in the Cu2+ solution with a much lower concentration than 0.38 μM. Unfortunately, no similar tendency was observed as those depicted above (data not shown). The corresponding linear regression equations and linear correlation coefficients of Aβ1–42 on the ABTS-PDDA/CNTs-NKB biosensor at different concentrations of Cu2+ are shown in the insets of Fig. 5.
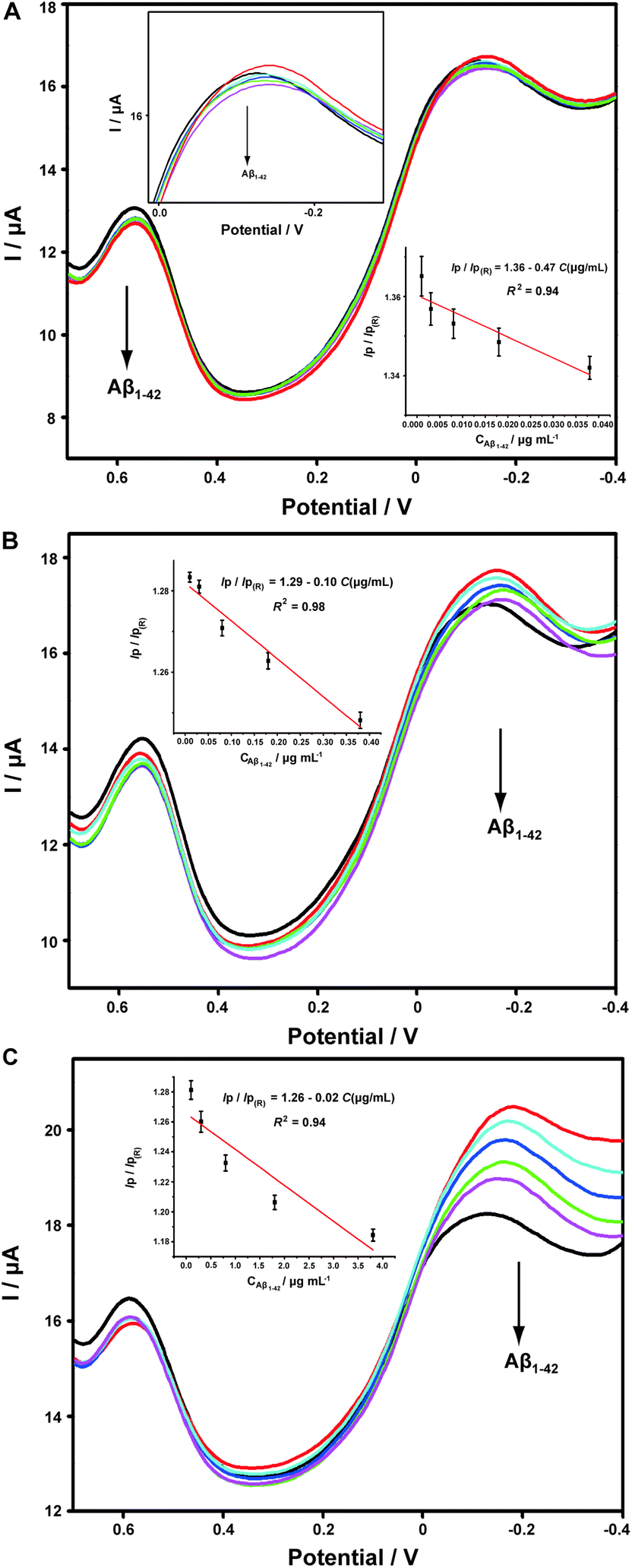 | ||
| Fig. 5 DPV curves of the ABTS-PDDA/CNTs-NKB-GC toward successive additions of Aβ1–42 in 0.38 (A), 0.95 (B) and 5.70 μM (C) Cu2+-contained PBS buffer. The inspected concentrations of Aβ1–42 was 0.001, 0.003, 0.008, 0.018, and 0.038 μg mL−1 for 0.38 μM Cu2+; 0.01, 0.03, 0.08, 0.18, and 0.38 μg mL−1 for 0.95 μM Cu2+; 0.1, 0.3, 0.8, 1.8, and 3.8 μg mL−1 for 5.70 μM Cu2+. The insets are the corresponding linear plots of the IP/IP(R) ratios versus the absolute concentrations of Aβ1–42. | ||
It has been reported that Cu bound Aβ could function as a peroxidase and possesses peroxidase activity.55 Therefore, to verify the above DPV results, we measured the peroxidase activity of the Cu2+–Aβ complex with three concentrations for the catalytic oxidation of ABTS in the presence of H2O2 by UV-vis. As shown in Fig. S5,† in the range of 600–800 nm, two strong absorption peaks located at around 620 and 700 nm were observed, which were ascribed to the catalytic oxidation of ABTS by the formed Cu–Aβ complex in the presence of H2O2. Additionally, by lowering the incubated concentration of Cu2+ and Aβ1–42 in sequence, the peak intensities at 620 and 700 nm gradually decreased, especially the peak at 700 nm, indicating that the concentrations of Cu2+ and Aβ played key roles in the catalytic capability of the Cu2+–Aβ complex for the oxidation of ABTS by H2O2, which were consistent with the electrochemical results.
Selectivity, reproducibility and stability investigations
The selectivity of the developed electrochemical system for Cu2+ and Aβ determination was evaluated in two ways: one was by individually monitoring and comparing the electrochemical signals of Cu2+ and interferences in PBS, and the other was by adding interferences to the Cu2+-contained PBS solution and comparing the signals with that of single Cu2+. Metal ions (Ca2+, Fe3+, Mg2+, Na+, Cd2+, Co2+, Mn2+, Ni2+, Pb2+, Zn2+, and Cu+), amino acids (Cys, Phe, Glu, Met, Lys, Gly, Arg, Ser, Leu, Val, Thr, and His) and other common endogenous substances, including ascorbic acid (AA), dopamine (DA), uric acid (UA), lactic acid (Lac) and glucose (Glu) were chosen as the potential interferences. As shown in Fig. S4,† compared with Cu2+, additions of the tested interferences to PBS buffer didn't cause significant changes in the current intensity. Likewise, the IP/IP(R) values were almost unperturbed in the simultaneous presence of these species and Cu2+ relative to that of single Cu2+. More importantly, we also examined the potential interferences from the aggregated Aβ. The results shown in Fig. S6C† clearly illustrate that the developed electrochemical method was suitable only for the monitoring of monomeric forms of Aβ, rather than oligomeric or other forms. All these pieces of evidence indicated the high selectivity of the fabricated ABTS-PDDA/CNTs-NKB-GC for Cu2+ and Aβ biosensing against metal ions, amino acids and endogenous species coexisting in the biological system.In addition, the relative standard deviations (RSDs) were evaluated to be 4.9% and 7.1% for three different electrodes in parallel for Cu2+ and Aβ1–42, respectively, indicating the acceptable reproducibility for the dual determination of two analytes. The biosensor also showed reasonable stabilities for the analytes, in which 91.5% and 87.4% of the current responses for Cu2+ and Aβ1–42 were retained after the modified electrode was stored at 4 °C for one week.
Estimation of Cu2+ and Aβ1–42 variations in the plasma and hippocampus of normal and AD rats
Finally, we applied the developed ABTS-PDDA/CNTs-NKB-GC to the estimation of Cu2+ and Aβ1–42 variations in the plasma and hippocampus of normal and AD rats. Fig. 6 presents the DPV curves obtained on the ABTS-PDDA/CNTs-NKB-GC for Cu2+ in the plasma from normal and AD rat brains. As can be seen, the electrochemical responses of Cu2+ showed significant differences between normal and AD groups. A clear increase in the concentration of Cu2+ was witnessed in AD rat plasma compared with that in normal rats, which were in good agreement with previously reported results, as well as our findings.47,56 Based on our post-calibration, the concentration of Cu2+ was calculated to be 1.11 ± 0.19 μM in normal rat plasma and this value increased to 9.20 ± 1.15 μM in AD rat plasma (n = 3). For Aβ1–42 determination, as for the coexistence of many different isoforms of Aβ in the biological system including Aβ1–42, the method of real sample detection was different from Cu2+, in which various amounts of Aβ1–42 were firstly spiked with the hippocampus homogenate and then added to Cu2+-contained PBS buffer. As listed in Table S1,† the recoveries of these measurements were in the range of 91.7%–110% under optimal conditions, indicating that this method was reliable and practical.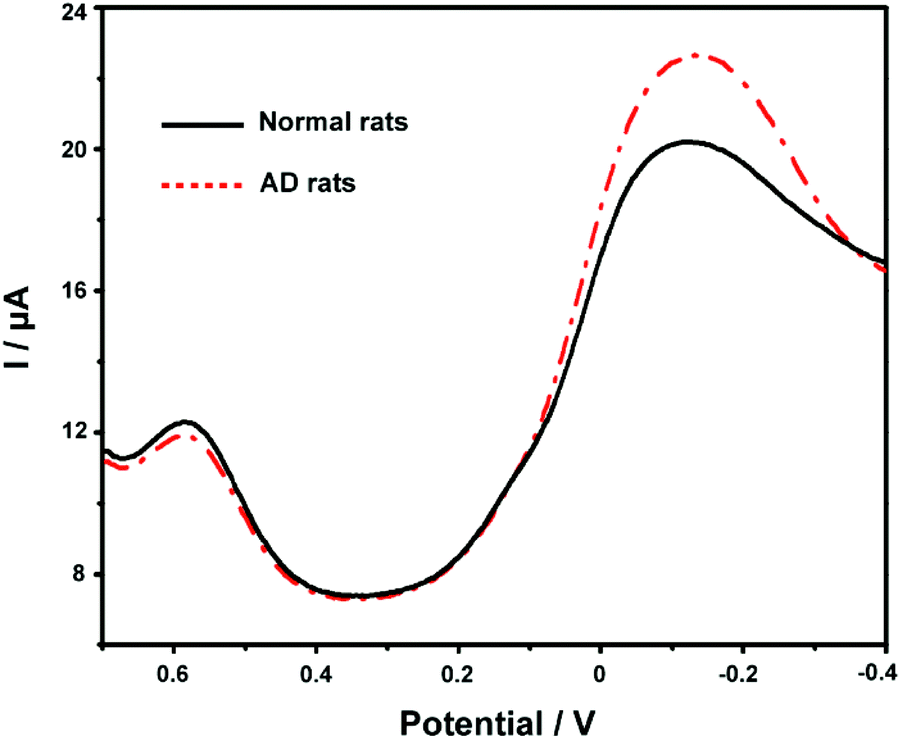 | ||
| Fig. 6 DPV responses of the ABTS-PDDA/CNTs-NKB-GC on Cu2+ in the plasma of normal and AD rats. | ||
Conclusions
In summary, with the demand for revealing the roles of Aβ peptide and metal ions in the treatment of AD, we presented herein a ratiometric electrochemical detection system to sensitively probe Cu2+ and Aβ co-variations, both of which were associated with the development of AD. Due to the stronger complexation of Cu2+ with Aβ, Cu2+ was released from the as-formed [CuII(NKB)2] complex once Aβ was added. Therefore, besides Cu2+, Aβ could also be easily monitored by the same ABTS-PDDA/CNTs-NKB biosensor immediately after Cu2+ determination. The theoretically simple, low technical and instrumental demands as well as the high sensitivity and selectivity made this methodology suitable and reliable enough for the real sample detection. Our results showed that the present approach provided us a crucial route for simultaneously monitoring two important biological species, which were closely linked with each other, in one single analytical system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate financial support from the National Natural Science Foundation of China (no. 21675137 and 21205102), the Natural Science Foundation of Jiangsu Province (no. BK20161170), China Postdoctoral Science Special Foundation (no. 2016T90504) and China Postdoctoral Science Foundation funded projects (no. 2015M580471), the Jiangsu Planned Project for Postdoctoral Research Funds (no. 1501087B), the Program for Distinguished Talents of Six Domains in Jiangsu Province (no. 2016-SWXY-060), the Jiangsu “333” project of cultivation of high-level talents, the Qing Lan Project of Jiangsu Province and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- I. Choi and L. P. Lee, ACS Nano, 2013, 7, 6268–6277 CrossRef CAS PubMed.
- M. Meier, J. Kennedy-Darling, S. H. Choi, E. M. Norstrom, S. S. Sisodia and R. F. Ismagilov, Angew. Chem., Int. Ed., 2009, 48, 1487–1489 CrossRef CAS PubMed.
- S. Chimon and Y. Ishii, J. Am. Chem. Soc., 2005, 127, 13472–13473 CrossRef CAS PubMed.
- A. A. Reinke, P. M. U. Ung, J. J. Quintero, H. A. Carlson and J. E. Gestwicki, J. Am. Chem. Soc., 2010, 132, 17655–17657 CrossRef CAS PubMed.
- S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S. H. Jeong, A. Ramamoorthy and N. A. Kotov, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef CAS PubMed.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- A. T. Petkova, R. D. Leapman, Z. H. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed.
- N. Xia, L. Liu, M. G. Harrington, J. Wang and F. Zhou, Anal. Chem., 2010, 82, 10151–10157 CrossRef CAS PubMed.
- T. E. Golde, C. B. Eckman and S. G. Younkin, Biochim. Biophys. Acta, 2000, 1502, 172–187 CrossRef CAS.
- L. A. Munishkina and A. L. Fink, Biochim. Biophys. Acta, 2007, 1768, 1862–1885 CrossRef CAS PubMed.
- R. Picou, J. P. Moses, A. D. Wellman, I. Kheterpal and S. D. Gilman, Analyst, 2010, 135, 1631–1635 RSC.
- Y. Y. Yu, X. Y. Sun, D. Q. Tang, C. L. Li, L. Zhang, D. X. Nie, X. X. Yin and G. Y. Shi, Biosens. Bioelectron., 2015, 68, 115–121 CrossRef CAS PubMed.
- Y. Y. Yu, L. Zhang, C. L. Li, X. Y. Sun, D. Q. Tang and G. Y. Shi, Angew. Chem., Int. Ed., 2014, 53, 12832–12835 CrossRef CAS PubMed.
- Y. Y. Yu, L. Zhang, X. Y. Sun, C. L. Li, Y. Qiu, H. P. Sun, D. Q. Tang, Y. W. Liu and X. X. Yin, Chem. Commun., 2015, 51, 8880–8883 RSC.
- Y. L. Zhou, J. Wang, L. T. Liu, R. R. Wang, X. H. Lai and M. T. Xu, ACS Chem. Neurosci., 2013, 4, 535–539 CrossRef CAS PubMed.
- J. H. Viles, Coord. Chem. Rev., 2012, 256, 2271–2284 CrossRef CAS.
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef CAS PubMed.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. S. Kim, X. D. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665–676 CrossRef CAS PubMed.
- R. A. Cherny, J. T. Legg, C. A. McLean, D. P. Fairlie, X. D. Huang, C. S. Atwood, K. Beyreuther, R. E. Tanzi, C. L. Masters and A. I. Bush, J. Biol. Chem., 1999, 274, 23223–23228 CrossRef CAS PubMed.
- L. Guilloreau, S. Guilloreau, A. Sournia-Saquet, H. Mazarguil and P. Faller, ChemBioChem, 2007, 8, 1317–1325 CrossRef CAS PubMed.
- C. C. Curtain, F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, K. Beyreuther, C. J. Barrow, C. L. Masters, A. I. Bush and K. J. Barnham, J. Biol. Chem., 2001, 276, 20466–20473 CrossRef CAS PubMed.
- D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545–15552 CrossRef CAS PubMed.
- A. Tiiman, P. Palumaa and V. Tougu, Neurochem. Int., 2013, 62, 367–378 CrossRef CAS PubMed.
- J. Zou and N. Sugimoto, Protein Pept. Lett., 1999, 6, 373–378 CAS.
- J. Zou and N. Sugimoto, J. Chem. Soc., Perkin Trans. 2, 2000, 10, 2135–2140 RSC.
- Y. P. Luo, Y. Tian and Q. Rui, Chem. Commun., 2009, 21, 3014–3016 RSC.
- M. Xu, R. h. Wang and Y. b. Li, Analyst, 2002, 9, 990–991 Search PubMed.
- X. M. Zhuang, H. H. Wang, T. He and L. X. Chen, Microchim Acta, 2016, 183, 3177–3182 CrossRef CAS.
- X. M. Zhuang, D. L. Wang, Y. Q. Lin, L. F. Yang, P. Yu, W. Jiang and L. Q. Mao, Anal. Chem., 2012, 84, 1900–1906 CrossRef CAS PubMed.
- Q. Wang, Y. Song, Y. Q. Chai, G. Q. Pan, T. Li, Y. L. Yuan and R. Yuan, Biosens. Bioelectron., 2014, 60, 118–123 CrossRef CAS PubMed.
- P. Jing, W. J. Xu, H. Y. Yi, Y. M. Wu, L. J. Bai and R. Yuan, Analyst, 2014, 139, 1756–1761 RSC.
- N. Zhou, J. H. Li, H. Chen, C. Y. Liao, Z. P. Chen and L. X. Chen, Analyst, 2013, 138, 1091–1097 RSC.
- S. M. Oja, M. Wood and B. Zhang, Anal. Chem., 2013, 85, 473–486 CrossRef CAS PubMed.
- L. T. Qu and L. M. Dai, J. Am. Chem. Soc., 2005, 127, 10806–10807 CrossRef CAS PubMed.
- Y. T. Kim, K. Ohshima, K. Higashimine, T. Uruga, M. Takata, H. Suematsu and T. Mitani, Angew. Chem., Int. Ed., 2006, 45, 407–411 CrossRef CAS PubMed.
- L. Cao, F. Scheiba, C. Roth, F. Schweiger, C. Cremers, U. Stimming, H. Fuess, L. Q. Chen, W. T. Zhu and X. P. Qiu, Angew. Chem., Int. Ed., 2006, 45, 5315–5319 CrossRef CAS PubMed.
- B. H. Wu, D. Hu, Y. J. Kuang, B. Liu, X. H. Zhang and J. H. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS PubMed.
- J. Wang and M. Musameh, Analyst, 2004, 129, 1–2 RSC.
- K. Karnicka, K. Miecznikowski, B. Kowalewska, M. Skunik, M. Opallo, J. Rogalski, W. Schuhmann and P. J. Kulesza, Anal. Chem., 2008, 80, 7643–7648 CrossRef CAS PubMed.
- D. Wang, Z. C. Li and L. W. Chen, J. Am. Chem. Soc., 2006, 128, 15078–15079 CrossRef CAS PubMed.
- L. Tao, G. J. Chen, G. Mantovani, S. York and D. M. Haddleton, Chem. Commun., 2006, 47, 4949–4951 RSC.
- S. Y. Wang, X. Wang and S. P. Jiang, Langmuir, 2008, 24, 10505–10512 CrossRef CAS PubMed.
- S. Wang, S. P. Jiang and X. Wang, Nanotechnology, 2008, 19, 265601 CrossRef PubMed.
- I. V. Anoshkin, I. I. Nefedova, D. V. Lioubtchenko, I. S. Nefedov and A. V. Räisänen, Carbon, 2017, 116, 547–552 CrossRef CAS.
- H. Thakur, N. Kaur, D. Sareen and N. Prabhakar, Talanta, 2017, 171, 115–123 CrossRef CAS PubMed.
- Y. Y. Yu, C. Yu, T. X. Yin, S. S. Ou, X. Y. Sun, X. R. Wen, L. Zhang, D. Q. Tang and X. X. Yin, Biosens. Bioelectron., 2017, 87, 278–284 CrossRef CAS PubMed.
- Z. j. Lin, F. Q. Luo, T. Q. Dong, L. Y. Zheng, Y. X. Wang, Y. W. Chi and G. N. Chen, Analyst, 2012, 137, 2394–2399 RSC.
- H. Xu, J. Yan, X. She, L. Xu, J. Xia, Y. Xu, Y. Song, L. Huang and H. Li, Nanoscale, 2014, 6, 1406–1415 RSC.
- L. E. Guo, J. F. Zhang, X. Y. Liu, L. M. Zhang, H. L. Zhang, J. H. Chen, X. G. Xie, Y. Zhou, K. Luo and J. Yoon, Anal. Chem., 2015, 87, 1196–1201 CrossRef CAS PubMed.
- L. Zhang, Y. Han, F. Zhao, G. Shi and Y. Tian, Anal. Chem., 2015, 87, 2931–2936 CrossRef CAS PubMed.
- A. Zhu, Q. Qu, X. Shao, B. Kong and Y. Tian, Angew. Chem., Int. Ed., 2012, 51, 7185–7189 CrossRef CAS PubMed.
- C. S. Atwood, R. C. Scarpa, X. D. Huang, R. D. Moir, W. D. Jones, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Neurochem., 2000, 75, 1219–1233 CrossRef CAS PubMed.
- C. D. Syme, R. C. Nadal, S. E. J. Rigby and J. H. Viles, J. Biol. Chem., 2004, 279, 18169–18177 CrossRef CAS PubMed.
- D. Pramanik and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 81–87 CrossRef CAS PubMed.
- Y. P. Luo, L. M. Zhang, W. Liu, Y. Y. Yu and Y. Tian, Angew. Chem., Int. Ed., 2015, 54, 14053–14056 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS characterization of the nanocomposite, electrochemical experiments for the electrode, UV-vis spectra, and real sample detection. See DOI: 10.1039/c7an01683b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |