
Simple and rapid detection of bacteria using a nuclease-responsive DNA probe†
Kyung Jin
Lee‡
abc,
Wang Sik
Lee‡
bc,
Ahreum
Hwang
bd,
Jeong
Moon
b,
Taejoon
Kang
abc,
Kyoungsook
Park
 *a and
Jinyoung
Jeong
*abc
*a and
Jinyoung
Jeong
*abc
aBioNano Health Guard Research Center, 125 Gwahak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: jyjeong@kribb.re.kr; marsp@kribb.re.kr
bHazards Monitoring Bionano Research Center, Korea Research Institute of Bioscience and Biotechnology, 125 Gwahak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea
cDepartment of Nanobiotechnology, KRIBB School of Biotechnology, University of Science and Technology, Daejeon 34113, Republic of Korea
dDepartment of Chemistry, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea
First published on 24th November 2017
Abstract
We demonstrate simple and rapid bacterial detection using a nuclease-responsive DNA probe. The probe consisting of a fluorescent dye and a quencher at the 5′ and 3′ termini, respectively, was designed to be cleaved by nucleases such as endonucleases, exonucleases, and DNases, which are released from bacteria using an optimized lysis buffer. The fluorescence signal of the cleaved DNA probe correlates with the number of Gram-negative Escherichia coli and Gram-positive Staphylococcus aureus, and the detection limit was 103 CFU for E. coli and 104 CFU for S. aureus. Moreover, this method is specific for live bacteria and takes just one minute to get the signal including sample collection. These features make the present bacterial detection method a powerful on-site bacterial contamination assay which is simple, rapid, and quantitative.
Introduction
Bacterial detection is important for improving hygiene and the quality of life. Environmental surfaces that we frequently touch such as cell-phones, drawer handles and tables can be easily contaminated with microorganisms, including opportunistic pathogens.1,2 Furthermore, contamination at food processing plants and various places in the public sector, such as hospitals and schools, threatens our health and causes serious economic loss via infection with healthcare-associated pathogens.3–5 Thus, rapid and sensitive detection of microbial contamination is necessary to ensure safety and prevent bacterial infections.6–11For bacterial detection, traditional culture-based methods are commonly used, but they take several days and require specialized equipment and species-specific protocols.12,13 Polymerase chain reaction (PCR)-based methods are another conventional system with high sensitivity and specificity. However, PCR has drawbacks including a long detection time and the need for specialized equipment.6,14,15 Recently, an adenosine triphosphate (ATP)-based bioluminescence technique has been commercially used for rapid monitoring of environmental bacterial contamination.16 ATP is a common biological energy source that is present in various microorganisms at approximately 0.47 fg per cell. ATP in bacterial cells can be catalyzed by luciferase, producing oxyluciferase, adenosine monophosphate (AMP), and bioluminescence.17 Therefore, the bioluminescent signal is proportional to the ATP concentration and indicates the amount of bacteria in the sample.18–20 ATP systems have been employed to measure surface cleanliness including the presence of organic debris and microbial contamination.21 However, the degree of contamination is often overestimated because ATP-based assays have difficulty in distinguishing live and dead bacteria.22,23 Moreover, non-bacterial ATP and extracellular ATP of organic debris could induce false-positive signals.
Herein, we developed a novel bacterial sensing method by using a nuclease-responsive DNA probe. In general, bacteria make various nucleases to defend against the damage caused by foreign DNA, UV, and oxidative stress.24 Based on this fact, we proposed to quantify the degree of microbial contamination by measuring the activity of nucleases released from microbial cells. In order to measure the activity of nucleases, we adopted the fluorescence resonance energy transfer (FRET) phenomenon of an oligonucleotide containing a fluorophore and a quencher. We developed a well-designed DNA probe which can be cleaved by nucleases efficiently and optimized the lysis buffer carefully. Therefore, the present nuclease-based bacterial assay can detect Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) quantitatively in one minute. Furthermore, this method can distinguish live and dead bacteria, preventing false-positive signals unlike the ATP-based assay. For the real-world application of the developed method, several environmental samples were collected and tested. We successfully detected bacteria from environmental samples and the results agreed well with the commercial ATP-based assay. We anticipate that the newly developed simple and rapid bacterial method will practically be used for on-site bacterial contamination assays soon.
Experimental
Materials
E. coli K12 (KCTC 1116) and S. aureus (KCTC 1621, ATCC 25923) were obtained from the Korean Collection for Type Cultures (KCTC, Korea). All of the colony-purified strains were minimally passaged and stored at −80 °C in Luria–Bertani (LB) broth (BD Diagnostics, USA) with 15% glycerol (Sigma-Aldrich, USA) prior to use in this study. Mung bean nuclease, lambda exonuclease, and DNase I were obtained from New England Biolabs Inc. (NEB, USA). A protease inhibitor cocktail was purchased from Roche Diagnostics (Switzerland). Triton X-100 and 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS) were purchased from Thermo Fisher Scientific (USA). Lysozyme, lysostaphin, ammonium persulfate (APS), and N,N,N′,N′-tetramethylethylenediamine (TEMED) were purchased from Sigma-Aldrich (USA). The chemicals used were of analytical grade and used without further purification. All of the water used was Milli-Q ultrapure grade (EMD Millipore, USA).Preparation of the nuclease-responsive DNA probe
The DNA probe used in this study was designed to encode an optimal sequence for a variety of nuclease activities by combining single-strand, mismatched strand, and double-strand DNA into one probe. The probe was labeled with a fluorescent dye (FAM) at the 5′-end and a quencher (BHQ-1) at the 3′-end. Dual-labeled oligonucleotide probes were obtained from GenoTech Corp. (Korea). Oligonucleotide probes were dissolved in distilled water at a concentration of 100 μM. The complementary oligonucleotides at the same molar concentration were mixed in 50 μL annealing buffer (50 mM NaCl, 10 mM Tris-HCl pH 7.9, 10 mM MgCl2, 100 μg mL−1 bovine serum albumin (BSA)). The final concentration of the DNA probe was 2 μM. After denaturation at 95 °C for 10 min, the probe was hybridized at 45 °C for 70 min. Then, a fluorophore and a quencher on the double-strand probe were in close proximity, and the fluorescence intensity was minimized. The annealed probe was stored at 4 °C or frozen.Bacterial culture, lysis, and detection using the nuclease-responsive DNA probe
E. coli and S. aureus were cultured in LB broth at 37 °C for 14–16 h in a shaking incubator. The respective bacterial densities were measured from the optical density at 600 nm (Beckman Coulter DU-730, USA) to be 108 CFU mL−1, which is in the logarithmic growth phase of bacteria. Bacterial samples (108 CFU mL−1) were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The LB supernatant was discarded, and the bacteria were suspended in 1× phosphate buffered saline (PBS, pH 7.4) to prepare bacterial sample solutions. The solutions were serially diluted with 1× PBS ranging from 104 CFU mL−1 to 1010 CFU mL−1. Ten microliters of each solution were treated in 90 μL of bacterial lysis buffer at room temperature (RT) for 20 s. The lysed bacteria were diluted with reaction buffer (50 mM Tris-HCl pH 8.0, 5 mM CaCl2) 10 times to protect the DNA probe and enhance the nuclease activity. Prior to measuring the samples, 10 μL of 2 μM nuclease-responsive DNA probe was mixed with 100 μL of the reaction sample and each sample was loaded in triplicate into a 96-well black microwell plate (Greiner bio-one, Germany). The fluorescence intensity of each bacterial sample ranging from 101 to 107 CFU per assay was measured using a multimode microplate reader (Infinite® M200 PRO, Tecan Trading AG, Switzerland). The wavelength of excitation was 470 nm and emission was 520 nm. All of the data were obtained and analyzed by using i-control™ software (Tecan Trading AG, Switzerland).
000 rpm for 10 min. The LB supernatant was discarded, and the bacteria were suspended in 1× phosphate buffered saline (PBS, pH 7.4) to prepare bacterial sample solutions. The solutions were serially diluted with 1× PBS ranging from 104 CFU mL−1 to 1010 CFU mL−1. Ten microliters of each solution were treated in 90 μL of bacterial lysis buffer at room temperature (RT) for 20 s. The lysed bacteria were diluted with reaction buffer (50 mM Tris-HCl pH 8.0, 5 mM CaCl2) 10 times to protect the DNA probe and enhance the nuclease activity. Prior to measuring the samples, 10 μL of 2 μM nuclease-responsive DNA probe was mixed with 100 μL of the reaction sample and each sample was loaded in triplicate into a 96-well black microwell plate (Greiner bio-one, Germany). The fluorescence intensity of each bacterial sample ranging from 101 to 107 CFU per assay was measured using a multimode microplate reader (Infinite® M200 PRO, Tecan Trading AG, Switzerland). The wavelength of excitation was 470 nm and emission was 520 nm. All of the data were obtained and analyzed by using i-control™ software (Tecan Trading AG, Switzerland).
To prepare dead bacterial samples, each of the bacterial cell pellets (108 CFU) was resuspended in 100 μL of PBS for heat treatment or in 100 μL of 70% ethanol (EtOH). Resuspended bacteria in PBS were treated by heating at 100 °C for 30 min in a heating block. Bacterial cells in 70% EtOH were incubated at RT for 2 h. Ten microliters of dead samples were treated with 90 μL of bacterial lysis buffer at RT for 20 s. And then, the lysates were diluted ten-fold in reaction buffer. One hundred microliters of reaction samples (106 CFU rxn−1) were mixed with 10 μL of 2 μM nuclease-responsive DNA probe. The fluorescence intensity was measured using a multimode microplate reader.
Activity test of the nuclease-responsive DNA probe with various nucleases
To confirm the activity of the nuclease-responsive DNA probe with various nucleases, the probe was treated with endonuclease (Mung bean nuclease), exonuclease (Lambda exonuclease), and DNase (DNase I). Mung bean nuclease is a single-strand specific endonuclease that degrades single-stranded extensions from the ends of DNA. Lambda exonuclease degrades double-stranded DNA. DNase I is an endonuclease that nonspecifically cleaves single- and double-stranded DNA. To measure the probe sensitivity for the endonuclease and exonuclease, 50 μL of 2 μM probe was incubated with 10 units of endonuclease or exonuclease in 450 μL working buffer. To provide optimal conditions, each enzyme was supplied with its working buffer. The working buffer for Mung bean nuclease consists of 30 mM NaCl, 50 mM NaOAc, and 1 mM ZnSO4 (pH 5). Lambda exonuclease reaction buffer contains 67 mM glycine-KOH, 2.5 mM MgCl2, and 50 μg mL−1 BSA (pH 9.4), and DNase I reaction buffer contains 10 mM Tris-HCl, 2.5 mM MgCl2, and 0.5 mM CaCl2 (pH 7.6). After 5 min of incubation at RT, an increased fluorescence intensity was observed. For DNase I, the same amount of probe was mixed with 2 units of DNase I and measured directly at RT without incubation. The fluorescence signal was also measured using a multimode microplate reader.Standard plate counting method
To measure the number of surviving bacterial cells, a standard plate counting method was used. After treatment with heating or 70% EtOH, the sample was serially diluted 10-fold with 1× PBS until the bacteria were dilute enough to count accurately when spread on a plate. For colony counts, 0.1 mL aliquots of the diluted sample were plated onto LB agar and incubated at 37 °C. After incubation for 24 h, the colonies on the plate were counted and the result was expressed in CFU rxn−1.Confirmation of nuclease-responsive DNA probe cleavage using polyacrylamide gel
To visualize the cleavage of the DNA probe by nucleases and bacteria, a gel-based degradation assay was performed. To prepare a 15% polyacrylamide gel, 20 mL of 15% gel solution was mixed with 10 mL of 30% acrylamide (29:1), 2 mL of 10× Tris-borate buffer (TBE, 0.89 M Tris base, 0.89 M boric acid, 20 mM EDTA pH 8.0), 800 μL of 10% APS and 20 μL of TEMED. The gel solution was poured into an assembled plate. After polymerization, the gel was removed from the gel caster and inserted into a gel box with 1× TBE running buffer. For each degradation assay sample, 15 μL of 2 μM probe was mixed with 15 μL of the bacterial lysate (106 CFU rxn−1) at RT for 15 min. In the case of the nuclease activity assay, 15 μL of 2 μM probe was mixed with 10 units of Mung bean endonuclease or Lambda exonuclease in 15 μL of each working buffer at RT for 20 min. For DNase I, the same probe was incubated with 0.4 units of DNase I in 15 μL of DNase I working buffer at RT for 5 min. Assay samples were added to 5 μL of 6× DNA gel-loading dye (60 mM Tris HCl with pH 8.0, 6 mM EDTA with pH 8.0, 60% glycerol, 0.25% bromophenol blue), incubated for 6 min at 95 °C, transferred to ice for 5 min, briefly centrifuged and kept on ice until loading. Then, 35 μL of the total samples were loaded in each well. A DNA ladder (Bioneer, Korea) with molecular weight markers ranging from 10 to 100 bp in 10 bp increments was used. The gel was run at 100 V for approximately 1 h. A fluorescence image of the gel was visualized on a chemiluminescence imaging system (WSE-6200H LuminoGraph II, ATTO Corp., Japan) by detecting the emitted fluorescence signal at 515 nm. After staining the gel for approximately 5 min with GelRed nucleic acid staining solution (Komabiotech, Korea), an image of the DNA fragments was obtained from a gel documentation system (AE-9000 E-graph, ATTO Corp., Japan) by exposure to UV light.
Bacterial detection using the nuclease-responsive DNA probe from environmental samples
To determine the potential of this system for use in environmental cleaning and monitoring, an environmental surface that was easily contaminated by bacteria was directly swabbed, and the number of bacteria on the surface was measured via fluorescence intensity. A sitting mat, desk, and bathroom doorknob that were frequently touched by many people were swabbed using FLOQ swabs (Copan Diagnostics Inc., USA). The swab was put into a mixture of 45 μL lysis buffer and 450 μL of reaction buffer. After the swab was removed, 100 μL of the sample solution was mixed with 10 μL of 2 μM DNA probe. The fluorescence signal was measured with a multimode microplate reader. The fluorescence signal increased proportionally to the contamination level of the samples.Bacterial detection using the ATP-based bioluminescence method
To compare the DNA probe method with a commercial method for bacterial contamination, the ATP-based bioluminescence method was performed on the same surfaces, including the sitting mat, desk, and bathroom doorknob. The bacterial measurements using the ATP-based system were performed by following the commercial manual (Luminester PD-30, Kikkoman Biochemifa Company, Japan). The bacterial sample was dropped on a swab of LucipacPen. The swab stick was returned to the holder, and the handle was pushed down to pass it through the extraction buffer. Then, to thoroughly dissolve the powdered reagent, the swab stick was shaken well. By inserting the LucipacPen into the Lumitester PD-30, the cleanliness was measured. Thus, the luminescence intensity of samples with different bacterial concentrations was obtained. Dead bacterial samples were also assessed for luminescence in the same way.Results and discussion
Fig. 1 illustrates the detection scheme for microorganisms such as E. coli and S. aureus using a synthetic double-strand DNA probe with a fluorophore (FAM dye) label at the 5′ end and a quencher (BHQ-1) label at the 3′ end. The fluorophore and quencher are in close proximity, and the fluorescence intensity is minimized due to FRET. The DNA probe was designed to target microbial nucleases, including single-strand and double-strand exonucleases and double-strand endonucleases. In the presence of these nucleases, which are released from the bacteria after lysis, the DNA probe is cleaved, and the fluorophore is separated from the quencher. The fluorescence signal of the probe increases with increasing bacterial concentrations, allowing quantification of the bacteria.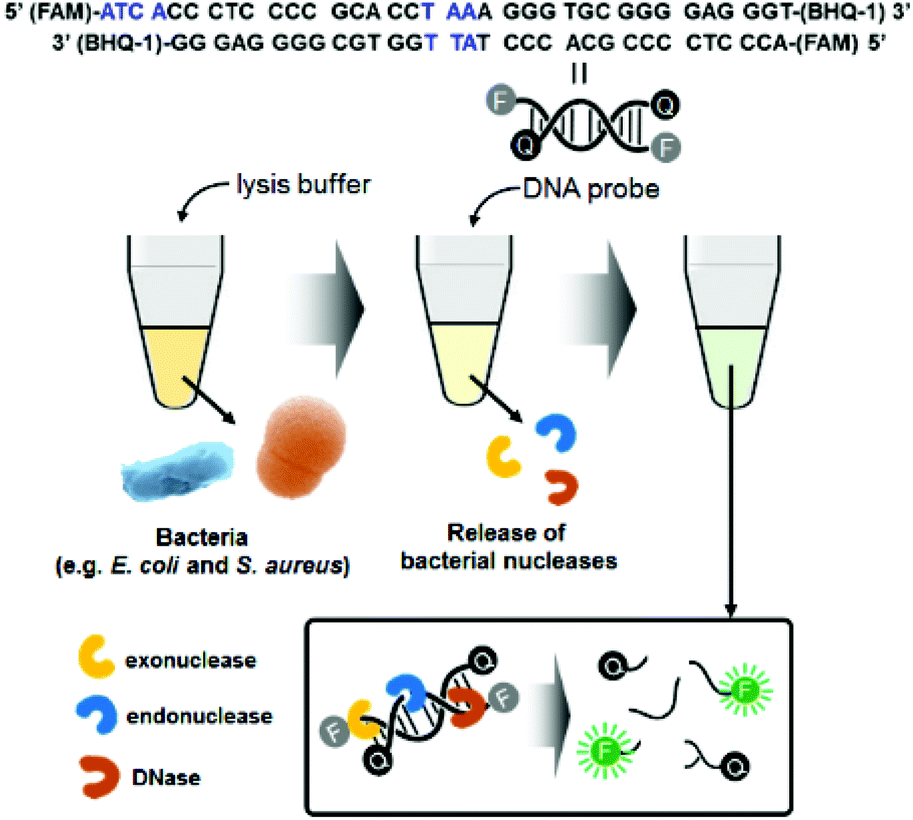 | ||
| Fig. 1 Schematic illustration of bacterial detection using the nuclease-responsive DNA probe. Blue marks in the DNA probe sequence indicate the target points of exodonuclease and endonuclease. | ||
For this method, we prepared an optimized lysis buffer containing CHAPS (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate) and Triton X-100 (t-octylphenoxypolyethoxyethanol). Both CHAPS and Triton X-100 are mild detergents, minimizing damage to bacterial proteins. In particular, CHAPS is a zwitterionic detergent that is incorporated into the membrane surface and does not penetrate deeply into the bilayer. Furthermore, Triton X-100 is a non-ionic detergent with a strong membrane-perturbing effect, attributed to its bulky polar moiety, which causes bacterial lysis in a short amount of time.25,26 Moreover, to lyse both Gram-negative and Gram-positive bacteria at once, lysozyme and lysostaphin were included in the lysis buffer. Lysostaphin is widely known as an antimicrobial enzyme that cleaves the crosslinking pentaglycine bridges in the cell wall of staphylococci.27 Thus, the combination of two detergents and lysogenic enzymes in the lysis buffer increases the bacterial lysis efficiency. To confirm that the cells were thoroughly lysed, standard plate counting methods were used. The lysate was spread on a LB agar plate and incubated at 37 °C for 16 h. The number of colonies was dramatically reduced after lysis for 20 s; thus, the bacteria were thoroughly lysed in the lysis buffer (Fig. S1†). The SEM images in Fig. S2† also support the notion that bacterial lysis was efficient in the lysis buffer. Both E. coli and S. aureus were clearly observed with their characteristic shapes before lysis. However, only bacterial cell debris was visible after lysis, indicating that the bacteria were damaged and dead.
Prior to the detection of bacteria, we measured the fluorescence of the DNA probe with various nucleases including endonuclease, exonuclease, and DNase I with each working solution using a multimode microplate reader in wavelength scanning mode. Fig. 2a shows that the fluorescence of the DNA probe increased after treatment of these nucleases. In particular, the fluorescence of the probe cleaved with DNase I exhibits a very high signal, although the concentration of the enzyme used was one fifth lower than the other enzymes because the fluorescence signal at the same concentration was saturated. It is assumed that this probe is comprised of mainly double-strand DNA which DNase I can cleave and a short length of mismatch and a blunt or stick end group on which endonuclease and exonuclease may work. The fragmentation of the DNA probe by nuclease was confirmed by gel-based degradation analysis. The images of the DNA gel were obtained using gel documentation under UV exposure for obtaining the image of the DNA ladder and using a chemiluminescence imaging system under 485 nm excitation for the fluorescence image of the fragmented probe by nucleases. As seen in Fig. 2c, the band of the probe with DNase I in lane 3 was clearly seen near 10 bp. The band of the probe with Mung bean endonuclease was also seen in lane 2 very weakly. As was revealed by the fluorescence spectra of the probe with the same nucleases, the gel data of the DNA probe cleaved by nucleases consistently support the fact that the probe is mainly cleaved by DNase I and partially by endonuclease and exonuclease.
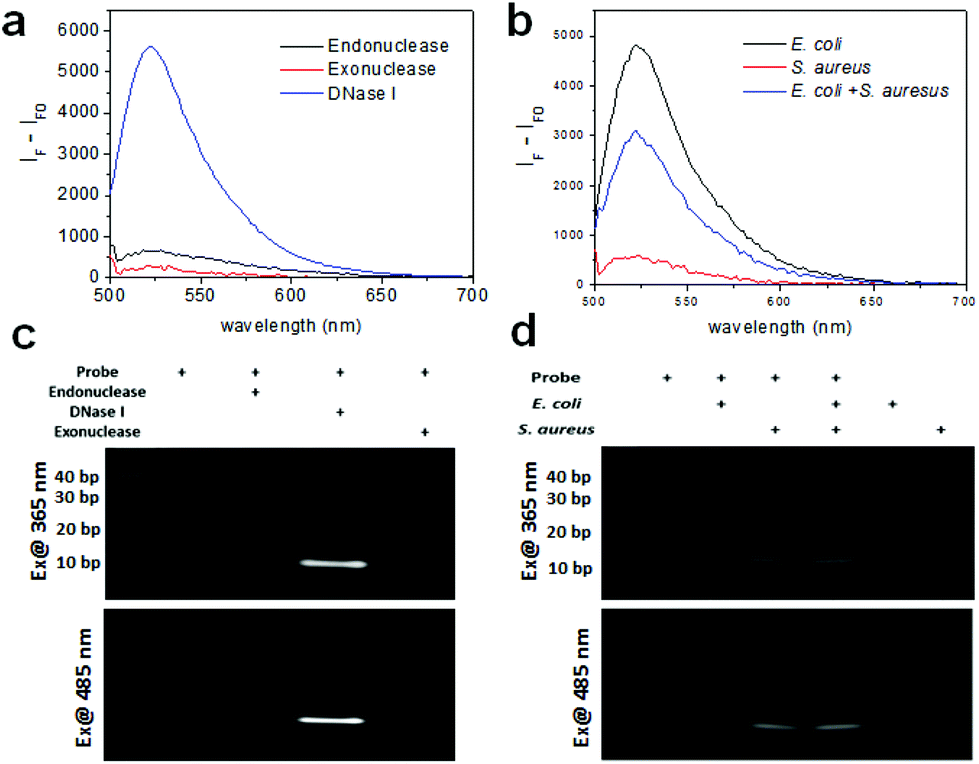 | ||
| Fig. 2 (a) Fluorescence spectra of the nuclease-responsive DNA probe with exonuclease, endonuclease, and DNase I. (b) Fluorescence spectra of the DNA probe with bacteria (E. coli with 106 CFU rxn−1, S. aureus with 106 CFU rxn−1, and the mixture of E. coli and S. aureus with 5 × 105 CFU rxn−1 for each). (c) DNA gel images of the DNA probes cleaved by various nucleases, and (d) DNA gel images of the DNA probes cleaved by bacteria under UV and 485 nm excitation. | ||
We also tested the enzyme activity of these nucleases using the DNA probe either in the working buffer or the reaction buffer prepared in this work to confirm the activities of nucleases under these conditions. We observed that the activity of DNase I was similar both in working buffer and reaction buffer. Otherwise, the signals of Mung bean endonuclease and lambda exonuclease in each reaction buffer were half or less than those in working buffer (data not shown). It indicates that the probe can work with various nucleases in the presence of their working solution, but may not work with some nucleases in the presence of reaction buffer.
As a proof-of-concept, the fluorescence of the DNA probe was measured in the presence of bacteria (i.e., 106 CFU rxn−1 of E. coli and S. aureus) using a multimode microplate reader in wavelength scanning mode. In Fig. 2b, the fluorescence of the probe with E. coli and S. aureus showed a significant signal at 520 nm, indicating that the probe was cleaved by nucleases from the bacteria. Although the fluorescence signal observed was different for E. coli and S. aureus at the same concentration, assuming that the nucleases from S. aureus might have different activities from those from E. coli, the probe appears to work for both Gram-negative and Gram-positive bacteria. Moreover, a mixture containing half the concentration of bacteria exhibited the predicted intensity, indicating that the detectable fluorescence correlates with the bacterial concentration. The cleavage of the probe by bacteria was also confirmed by using gel experiment. In Fig. 2d, the DNA gel image shows the DNA fragments cleaved by E. coli and S. aureus in lanes 2 and 3, respectively. The DNA cleaved by E. coli was approximately 20 base pairs, whereas the DNA cleaved by S. aureus was closer to 10 base pairs. Furthermore, the DNA bands cleaved by the mixture of bacteria were in the same location for each species in lane 4. These results indicate that E. coli and S. aureus express different nucleases as it is known that different bacterial strains express different types of restriction endonucleases to defend against viruses.28 The DNA gel image under 485 nm excitation also clearly showed that the fluorophores from the cleaved DNA migrated to the same position under UV illumination. Moreover, in the lane containing the E. coli and S. aureus mixture, two bands located at 10 bps and 20 bps were shown, indicating that the probe was cleaved by each of the bacteria. Meanwhile, the lanes without the probe or with the probe only (36-mer) as controls did not show fluorescence under UV or 485 nm excitation, indicating that the fluorophore (FAM) was completely quenched by BHQ-1.
To determine the limit of bacterial detection, the concentration-dependent fluorescence signal was measured from 0 to 107 CFU rxn−1 for each bacterium. Fig. 3a shows that the fluorescence signal gradually increases from 102 to 104 CFU rxn−1, and the response to E. coli dramatically increases at 107 CFU rxn−1. For S. aureus, although the fluorescence signal was relatively lower compared to that of E. coli, the cell number-dependent fluorescence signal gradually increased up to 107 CFU rxn−1 (Fig. 3b). Based on these data, the detection limit of this method is 103 CFU for E. coli and 104 CFU for S. aureus. To compare this detection limit with other methods, we performed the cell number-dependent measurement using ATP-based bioluminescence (Fig. S3†). Although the bioluminescence signal was seen in a low cell number (103–104 CFU), it was clearly detectable at 105 CFU for both E. coli and S. aureus and the signal dramatically increased 4-fold at 107 CFU bacteria. In fact, FRET-based sensing methods are widely used for the detection of bacteria and bacterial enzymes.29–33 However, some challenges such as development of an efficient DNA probe and reduction of the reaction time still remain for simple and rapid bacterial detection. This result indicates that the nuclease-based method is a quantitative measurement for practical bacterial detection with sensitive detection limit compared to the ATP-based method.
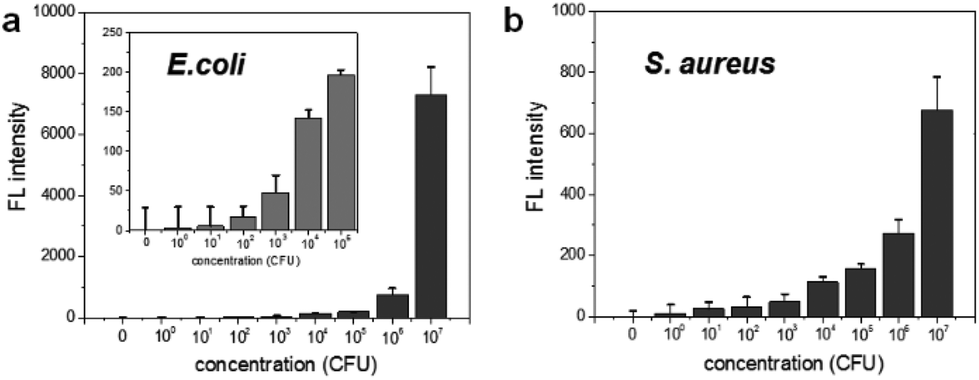 | ||
| Fig. 3 Bacterial concentration dependent fluorescence signal of nuclease-responsive DNA probe cleavage in the presence of (a) E. coli and (b) S. aureus. The concentration range is from 0–107 CFU rxn−1. | ||
To evaluate the specificity for the detection of live bacteria, dry heating and a chemical disinfectant were applied in this study as simple and economical sterilization methods. Ethanol as a well-known chemical disinfectant is widely used in hospitals and food processing. To prepare dead bacterial samples, 106 CFU of E. coli and S. aureus were inactivated by heating at 100 °C for 30 min or treating with 70% EtOH for 2 h. The dead bacteria were added in lysis buffer and mixed with the DNA probe. After reaction with the DNA probe, the fluorescence intensity was measured. As shown in Fig. 4a, the fluorescence signal from live E. coli was significantly higher than the signal observed after heating or 70% EtOH treatment. For S. aureus, the fluorescence was relatively high in live cells while the signal from the cells treated with 70% EtOH was almost 10 times lower (Fig. 4b). Interestingly, the fluorescence intensity in heat-treated S. aureus was slightly higher than that of the 70% ETOH-treated one, assuming that heat resistant nucleases remaining intact in dead bacteria might react with the DNA probe.34 In the same manner, we also investigated a live bacteria-selective detection using the ATP-based bioluminescence method and there was no difference between live and dead bacteria for both E. coli and S. aureus. Moreover, the signal of heat-treated bacteria was more than 10 times higher than live bacteria (Fig. S4†). It is assumed that since ATP is relatively stable compared to other enzymes such as nucleases under the sterilization methods, dead bacteria are still detectable by the ATP-based method as we confirmed that bacteria after heating or treatment with 70% EtOH were dead using the standard plate counting method (Fig. S5†). Based on this result, the nuclease-based method is advantageous for live bacteria-selective detection.
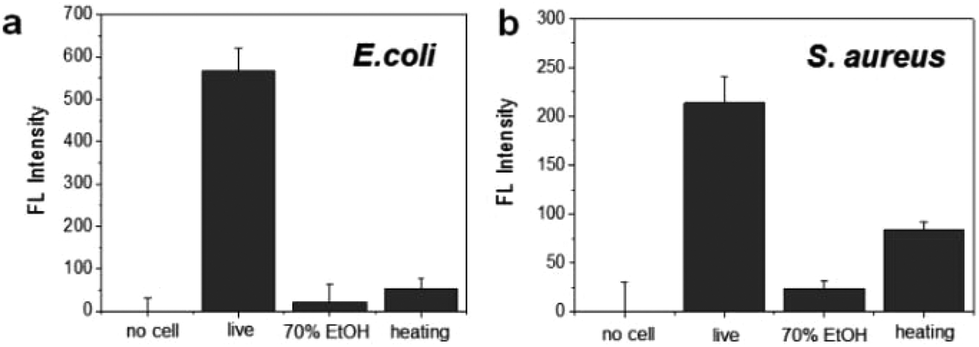 | ||
| Fig. 4 Selective detection of live and dead bacteria using the nuclease-responsive DNA probe in the presence of (a) E. coli and (b) S. aureus. The number of bacteria was 1 × 106 CFU rxn−1 for each. | ||
Finally, we investigated the detection of bacteria from environmental samples such as doorknobs, tables, desks, and hands and compared these results with the commercial ATP-based bioluminescence method. As seen in Fig. 5, the signal pattern of bacterial detection between the two methods was similar in each location, indicating that this system is comparable to a conventional ATP-based method. Moreover, the measurement time using this method was about one minute from sample collecting to obtaining the signal due to the rapid nuclease activity with the help of an optimized lysis buffer. This feature will make this method a practical assay for on-site environmental bacterial detection. There are various bacterial detection methods including PCR, immunology-based, and ATP-based methods and we compared these methods with the nuclease-based method studied in this work (Table S1). Combining the detection limit, time for assay, and selectivity, the nuclease-based method can be an alternative bacterial detection method.
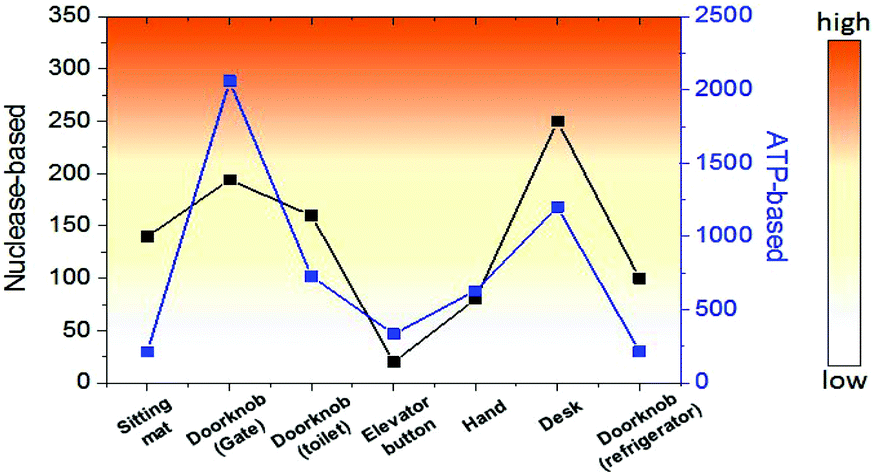 | ||
| Fig. 5 Comparison of bacterial detection in various environments between nuclease-based and ATP-based methods. | ||
Conclusions
In this work, a nuclease-based bacterial contamination detection method was developed using a synthetic double-strand DNA probe labelled with a fluorescent dye and a quencher at the 5′ and 3′ termini respectively and an optimized lysis buffer. In this method, both Gram-negative and Gram-positive bacteria were detected with the detection limit at 103–104 CFU. Moreover, it is selective for live bacteria, which is an advantage over the ATP-based method. This method is simple to operate, does not require the use of complex equipment or reagents, and can determine the degree of microbial contamination. Therefore, this method could be practical for environmental bacterial analysis and monitoring public health.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Global Frontier Project (H-GUARD_2013 M3A6B2078950, H-GUARD_2014 M3A6B2060489) through the Center for BioNano Health-Guard funded by the Ministry of Science and ICT (MSIT) of Korea, Innopolis Foundation (A2017-01-DD-002), and KRIBB Initiative Research Program.Notes and references
- J. M. Boyce, J. Hosp. Infect., 2007, 65, 50–54 CrossRef PubMed.
- N. Kajigaya, Y. Hirose, S. Koike, T. Fujita, N. Yokota, S. Hata, M. Ikenaga, N. Kobayashi and T. Takahashi, BMC Res. Notes, 2015, 8, 352 CrossRef PubMed.
- K. S. Park, C.-H. Huang, K. Lee, Y.-E. Yoo, C. M. Castro, R. Weissleder and H. Lee, Sci. Adv., 2016, 2, e1600300 Search PubMed.
- S. S. Magill, J. R. Edwards, W. Bamberg, Z. G. Beldavs, G. Dumyati, M. A. Kainer, R. Lynfield, M. Maloney, L. McAllister-Hollod, J. Nadle, S. M. Ray, D. L. Thompson, L. E. Wilson and S. K. Fridkin, N. Engl. J. Med., 2014, 370, 1198–1208 CrossRef CAS PubMed.
- A. Marchetti and R. Rossiter, J. Med. Econ., 2013, 16, 1399–1404 CrossRef PubMed.
- J. W.-F. Law, N.-S. Ab Mutalib, K.-G. Chan and L.-H. Lee, Front. Microbiol., 2013, 3, 98–101 Search PubMed.
- S. P. Olivier, B. M. Jayarao and R. A. Almeida, Foodborne Pathog. Dis., 2005, 2, 115–137 CrossRef PubMed.
- R. L. Scharff, J. Food Prot., 2012, 75, 123–131 CrossRef PubMed.
- A. K. Deisingh and M. Thompson, J. Appl. Microbiol., 2004, 96, 419–429 CrossRef CAS PubMed.
- Y. Sun, C. Zhao, Z. Yan, J. Ren and X. Qu, Chem. Commun., 2016, 52, 1–4 RSC.
- Z. Wang, Z. Chen, N. Gao, J. Ren and X. Qu, Small, 2015, 11, 4970–4975 CrossRef CAS PubMed.
- P. K. Mandal, A. K. Biswas, K. Choi and U. K. Pal, Am. J. Food Technol., 2011, 6, 87–102 CrossRef.
- X. Zhao, L. R. Hilliard, S. J. Mechery, Y. Wang, R. P. Bagwe, S. Jin and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15027–15032 CrossRef CAS PubMed.
- A. P. Kilungo, N. Carlton-Carew and L. S. Powers, J. Biosens. Bioelectron., 2013, S12, 002 Search PubMed.
- N. Lee, K. Y. Kwon, S. K. Oh, H.-J. Chang, H. S. Chun and S.-W. Choi, Foodborne Pathog. Dis., 2014, 11, 574–580 CrossRef CAS PubMed.
- G. Shama and D. J. Malik, Int. J. Hyg. Environ. Health, 2013, 216, 115–125 CrossRef CAS PubMed.
- J. Luo, X. Liu, Q. Tian, W. Yue, J. Zeng, G. Chen and X. Cai, Anal. Biochem., 2009, 394, 1–6 CrossRef CAS PubMed.
- D. Champiat, N. Matas, B. Monfort and H. Fraass, Luminescence, 2001, 16, 193–198 CrossRef CAS PubMed.
- D. E. Turner, E. K. Daugherity, C. Altier and K. J. Maurer, J. Am. Assoc. Lab. Anim. Sci., 2010, 49, 190–195 CAS.
- D. J. Squirrell, R. L. Price and M. J. Murphy, Anal. Chim. Acta, 2002, 457, 109–114 CrossRef CAS.
- C. A. Davidson, C. J. Griffith, A. C. Peters and L. M. Fielding, Luminescence, 1999, 14, 33–38 CrossRef CAS PubMed.
- B. Bottari, M. Santarelli and E. Neviani, Trends Food Sci. Technol., 2015, 44, 36–48 CrossRef CAS.
- K. Venkateswaran, N. Hattori, M. T. La Duc and R. Kern, J. Microbiol. Methods, 2003, 52, 367–377 CrossRef CAS PubMed.
- T. Nishino and K. Morikawa, Oncogene, 2002, 21, 9022–9032 CrossRef CAS PubMed.
- M. Nazari, M. Kurdi and H. Heerklotz, Biophys. J., 2012, 102, 498–506 CrossRef CAS PubMed.
- P. M. Rodi, B. Gianello, M. C. Corregido and A. M. Gennaro, Biochim. Biophys. Acta, Biomembr., 2013, 1838, 859–866 CrossRef PubMed.
- S. N. Kulikov and Y. A. Shumkova, Bull. Exp. Biol. Med., 2014, 157, 243–245 CrossRef CAS PubMed.
- R. C. Dubey, Advanced Biotechnology, S. Chand & Company P Ltd, New Delhi-55, 1st edn, 2014, pp. 245–257 Search PubMed.
- J. Cao, C. Feng, Y. Liu, S. Wang and F. Liu, Biosens. Bioelectron., 2014, 57, 133–138 CrossRef CAS PubMed.
- V. V. Didenko, BioTechniques, 2001, 31, 1106–1116 CAS , 1118, 1120–1121.
- B. Jin, S. Wang, M. Lin, Y. Jin, S. Zhang, X. Cui, Y. Gong, A. Li, F. Xu and T. J. Lu, Biosens. Bioelectron., 2017, 90, 525–533 CrossRef CAS PubMed.
- J. J. Li, R. Geyer and W. Tan, Nucleic Acids Res., 2000, 28, E52 CrossRef CAS PubMed.
- S. Sato and S. Takenaka, Sensors, 2014, 14, 12437–12450 CrossRef CAS PubMed.
- W. R. Chesbro and K. Auborn, Appl. Microbiol., 1967, 15, 1150–1159 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01384a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |