
Towards the electrochemical diagnostic of influenza virus: development of a graphene–Au hybrid nanocomposite modified influenza virus biosensor based on neuraminidase activity†
Ülkü
Anik
 *a,
Yudum
Tepeli
a,
Maher
Sayhi
bc,
Jihene
Nsiri
b and
Mohamed Fethi
Diouani
*b
*a,
Yudum
Tepeli
a,
Maher
Sayhi
bc,
Jihene
Nsiri
b and
Mohamed Fethi
Diouani
*b
aMugla Sitki Kocman University, Faculty of Science, Chemistry Department, Kotekli/Mugla, Turkey. E-mail: ulkuanik@mu.edu.tr
bLaboratory of Epidemiology and Veterinary Microbiology (LEMV), Institut Pasteur de Tunis, University of Tunis El Manar, LR11IPT03, Tunis-Belvédère 1002, Tunisia. E-mail: fethi.diouani@pasteur.rns.tn
cFaculty of Sciences of Tunis, University of Tunis El Manar, Tunisia
First published on 26th October 2017
Abstract
An effective electrochemical influenza A biosensor based on a graphene–gold (Au) hybrid nanocomposite modified Au-screen printed electrode has been developed. The working principle of the developed biosensor relies on the measurement of neuraminidase (N) activity. After the optimization of experimental parameters like the effect of bovine serum albumin addition and immobilization times of fetuin A and PNA lectin, the analytical characteristics of the influenza A biosensor were investigated. As a result, a linear range between 10−8 U mL−1 and 10−1 U mL−1 was found with a relative standard deviation value of 3.23% (for 10−5 U mL−1 of N, n:3) and a limit of detection value of 10−8 U mL−1 N. The developed biosensor was applied for real influenza virus A (H9N2) detection and very successful results were obtained.
Introduction
The influenza virus A can be described as a negative stranded RNA virus which belongs to the Orthomyxoviridae family. The virus contains two surface glycoproteins namely hemagglutinin (H) and neuraminidase (N). The classification of the virus subtype can be made according to the antigenic properties of these 18 H (1–18) and 11 N (1–11) glycoproteins.1–14 Since the influenza virus is very mutagenic, it can easily change the antigenic portions of H and N proteins and as a result a very serious antigenic drift has occurred.15 Seasonal influenza viruses can easily be affected by these antigenic drift mutations which cause millions of serious infections and approximately 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths every year.15,16 Also, sometimes two or more influenza A viruses of different origin infect the same cell and as a result, new strains or subtypes emerge because of the occurrence of genetic reassortment.1 As an example, the reassortment between human and avian virus strains was the reason for the influenza A pandemics in 1957 (H2N2) and in 1968 (H3N2) while in 2009 the (H1N1) pandemic virus was found to be a reassortment containing gene segments from human, avian and swine influenza viruses.2,4 Besides these, highly mortal avian viruses like H5N1 and H7N9 have been found to transmit from birds to humans.4,17 Because of these, some researchers define the virus as causing “continuously emerging influenza disease” which means it still continues to be a threat for animals and human beings.15
000 deaths every year.15,16 Also, sometimes two or more influenza A viruses of different origin infect the same cell and as a result, new strains or subtypes emerge because of the occurrence of genetic reassortment.1 As an example, the reassortment between human and avian virus strains was the reason for the influenza A pandemics in 1957 (H2N2) and in 1968 (H3N2) while in 2009 the (H1N1) pandemic virus was found to be a reassortment containing gene segments from human, avian and swine influenza viruses.2,4 Besides these, highly mortal avian viruses like H5N1 and H7N9 have been found to transmit from birds to humans.4,17 Because of these, some researchers define the virus as causing “continuously emerging influenza disease” which means it still continues to be a threat for animals and human beings.15
Although rapid detection of influenza virus is mandatory, present diagnostic techniques cannot compete with the mutagenic behavior of the virus. For example, viral culture, which has been accepted as a golden standard method, needs two or three days to retrieve the results which is a somewhat long time for the diagnosis of the influenza virus.18–20 On the other hand, quantitative polymerase chain reaction (qPCR), which is chosen frequently by health personnel, can be described as sensitive but relatively slow (about 2 h) method for influenza detection. qPCR is also expensive and can be performed only by specialists.20–22 Apart from these methods, various immune based rapid tests have been fabricated recently to compensate the need for the fast diagnosis of the virus. From this point of view, these tests manage to show the results in 15 to 30 min. However, they lack the sensitivity and the precision.20,21,23,24
Electrochemical techniques are accepted as practical techniques for many applications.25,26 The combination of electrochemical techniques with biosensor systems results in electrochemical biosensors which provide not only practicality but also selectivity and sensitivity.27,28 For these reasons, electrochemical biosensors can be accepted as good candidates to be used in point of care systems.29 Recently electrochemical biosensors were combined with nanomaterials and as a result, more sensitive and accurate results were obtained.30–32 Graphene is a kind of two-dimensional nanomaterial and provides properties like higher surface area and good conductivity which are very important for electrochemical biosensors.33–35 Apart from graphene, recently graphene–metallic nanocomposites were produced and used in these systems. These nanocomposites provide more sensitive and selective results when combined with the biosensor systems.36–43
Considering the influenza biosensors, it can be stated that, until now almost only H based systems have been developed.7–10,44–48 Lately our group managed to develop a N based electrochemical influenza biosensor.14 In that work, as preliminary data, only the electrochemical impedance spectroscopy (EIS) diagrams of the fabrication of a biosensor were presented.14 For that earlier work, a glassy carbon paste electrode was used as the main electrode. First fetuin A which is a kind of glycoprotein, was attached onto the electrode. Then, N was immobilized onto fetuin A. Fetuin A includes terminal 12–14 sialic acid residues per molecule and N cleaves fetuin A from these sialic acid ends. Lastly, peanut agglutinin (PNA) lectin was immobilized onto the electrode surface to monitor this cleavage.14
In this work, as a continuation of our previous paper, we improve our system by using a gold screen-printed electrode (AuSPE) together with a graphene–Au hybrid nanocomposite. Also the experimental parameters of the developed system were optimized and the analytical characteristics were examined. Lastly, the developed biosensor was applied for real influenza A virus (H9N2) detection.
Experimental
Chemicals
Graphite powder, NaNO3, chloroauric acid (HAuCl4·H2O), K3[Fe(CN)6], K4[Fe(CN)6]·3H2O, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), fetuin A, bovine serum albumin (BSA), N, PNA lectin, MES monohydrate and ethylene glycol (EG) were purchased from Sigma-Aldrich. H2SO4, H2O2, KMnO4, KH2PO4, NaOH, NaCl and KCl were obtained from Merck. Double distilled water was used for the preparation of all solutions. All chemicals were of analytical grade and were used without needing further purification.Instrumentation
Electrochemical measurements were carried out by using μ-AUTOLAB Type III with the FRA 2 module electrochemical measurement system from Metrohm B.V. that is controlled by NOVA 1.10 software. AuSPE was used as a working electrode and was purchased from Dropsens. SEM and EDS measurements were performed using a JSM-7600 F FEG-SEM at 15.0 kV. During the preparation procedure of a graphene–Au nanocomposite, a Thermo Electron Corporation pH-meter, an IKA ® C-MAG HS7 hotplate, a Bandelin Sonorex sonicator and a NUVE vacuum oven were used. For the incubation of biological materials at 37 °C an incubator shaker was used. ELLA measurements were carried out using an automatic plate reader with a 490 nm filter.Synthesis procedure of graphene–Au nanocomposite
In order to synthesize a graphene–Au nanocomposite, graphene oxide (GO) was used as the starting material. Therefore, first GO was synthesized from graphite by using a modified Hummers–Offeman method.38,49 For this purpose, 1 g of graphite powder was added into 23 mL of 98% H2SO4 solution and stirred at room temperature for 24 h. Then, 100 mg of NaNO3 was added into the mixture and stirred for another 30 min. After the mixture was cooled to 5 °C by using an ice-bath, 3 mg of KMnO4 was added to the medium and then the stirred mixture was heated up to 40 °C. 46 mL of double distilled water was added into the above mixture for a period of 25 min. Finally, 140 mL of double distilled water and 10 mL of 30% H2O2 were added into the mixture to stop the reaction. The unexploited graphite in the resulting mixture was removed by centrifugation. Then the mixture was dried in a desiccator at room temperature.Additionally, for the graphene–Au nanocomposite synthesis step, a 10 mg portion of GO powder which was synthesized by using the modified Hummers–Offeman method was dispersed in 10 mL of water by sonication for 1 h to form a stable GO colloid.50,51 Then, 20 mL of EG solution and 0.5 mL of 0.01 M HAuCl4·H2O were added to the GO colloid and stirred for 30 min. After that, the mixture was heated at 100 °C for 6 h by applying magnetic stirring. Subsequently, the graphene–Au nanocomposites were separated from the EG solution via centrifugation and washed with deionized water five times. The resulting product was dried in a vacuum oven at 60 °C for 12 h.37 Finally, the synthesized graphene–Au nanocompsite was dispersed to 10 mg mL−1 in double distilled water by ultrasonication and stored at 4 °C until it was used.38,40,41,52
Preparation of developed electrochemical influenza A biosensor
AuSPE was used as the supporting electrode for the preparation of the developed electrochemical influenza A biosensor. First, AuSPE was modified with a graphene–Au hybrid nanocomposite. For this purpose, 6 μL of graphene–Au nanocomposite dispersion (10 mg mL−1, in double distilled water) was dropped onto the surface of AuSPE and dried at room temperature for 1 h. Then, 10 μL of a 50 mM EDC/NHS mixture (in 50 mM MES pH: 5.5 solution) was dropped onto the electrode surface and left for 1 h. Subsequently, the electrode surface was rinsed with MES and phosphate buffered saline (PBS) with a pH value of 7.4 (137 mM NaCl, 10 mM KH2PO4, and 2.7 mM KCl). After that, 10 μL of 250 μg/250 μL of fetuin A in 0.1 M PBS was immobilized onto the electrode surface and left for 30 min. The electrode surface was then washed with PBS and then 10 μL of 1% BSA (in 0.1 PBS) solution was dropped onto the electrode surface for performing BSA effect experiments. Other than that, BSA was not used during the fabrication of the influenza A virus biosensor. After that, the electrode was rinsed with PBS and 10 μL of different concentrations of N (in 0.1 M pH: 7.4 PBS) was dropped onto the electrode surface and kept for 18 h at 37 °C under stirring. After the electrode surface was washed with PBS, 10 μL of 50 μg mL−1 PNA lectin (in 0.1 M pH: 7.4 PBS) solution was dropped onto the developed electrochemical biosensor for 1 h at +4 °C. PNA lectin here specifically binds to the galactose molecules that appear after N cleaves the sialic acids from the fetuin A molecule. In this way, the N activity of the developed influenza A biosensor was determined electrochemically (Scheme 1). The characterization of the developed electrochemical influenza A biosensor and the determination of N activity studies were carried out by using the EIS method.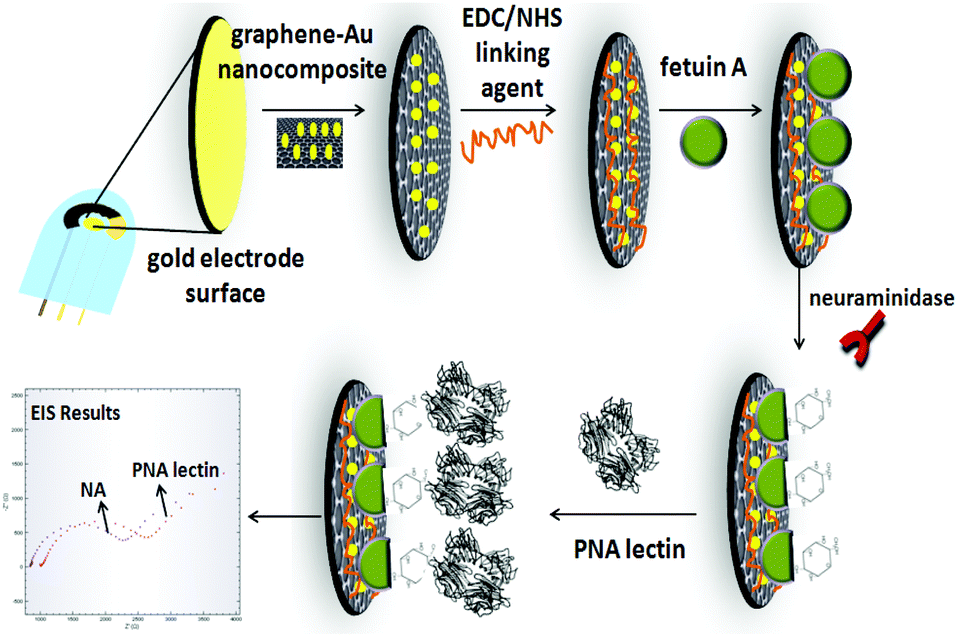 | ||
| Scheme 1 Schematic diagram depicting the steps of the developed electrochemical influenza A biosensor. | ||
Influenza type A virus replication in embryonated eggs
H9N2 influenza type A virus (A/Equi/1/Prague 56) was propagated in 11-day old specific-pathogen free (SPF) chicken embryonated eggs via the allantoic route. The eggs were incubated at 37 °C for three days and the allantoic fluid was collected, and clarified by centrifugation at 3000g for 15 min. The viral titer, in the collected allontoic fluids, was monitored by performing a hemagglutination test.ELLA measurements
An Enzyme-Linked Lectin Assay (ELLA) was performed to measure influenza A virus N activity as described by Couzens, L. et al.3 A fetuin-coated plate was prepared by dispensing 100 μl per well of 25 μg ml−1 fetuin working solution into each well. The plate was covered and placed at +4 °C for 24 hours. Then, the plate was washed 3 times with 200 μl per well PBS wash buffer for 3 minutes. 100 μl sample diluent buffer was added in column 12 as a negative control and 50 μl serial dilution antigen (influenza virus, N and non-infected allantoic fluid) was transferred from the dilution plate to each well in columns 1–11 of the fetuin-coated plate containing an equal volume of the sample diluent buffer. The plate was incubated in a humidified incubator with 5% CO2 for 18 hours at 37 °C, and then washed 6 times as described before. 100 μl per well PNA-peroxidase was dispensed into each well and incubated at room temperature for 2 hours. After this step, 3 time wash was performed before the addition of 100 μl per well freshly prepared substrate solution for 10 min in the dark at room temperature. The color reaction was stopped by the addition of 100 μl per well stop solution. The plates were read at 490 nm using a 96-well automatic plate reader.Results and discussion
Characterization of synthesized graphene–Au hybrid nanocomposite
Fig. 1 shows the SEM images and EDS results of the graphene–Au nanocomposite. It can be seen from Fig. 1 that AuNps are located on the graphene sheets as white dots (average diameter of 250 nm). EDS results show that the atomic dispersion percentages of the graphene–Au nanocomposite are 83.73%, 14.97%, and 1.06% and the mass percentages of these elements are 69.29%, 14.66% and 14.32% for C, O and Au, respectively. | ||
| Fig. 1 (A), (B) SEM images of a graphene–Au nanocomposite and (C) EDS results of the graphene–Au nanocomposite. | ||
Electrochemical characterization of developed electrochemical influenza A biosensor
The fabrication of the developed influenza A biosensor was monitored via EIS in the presence of 10 mM K3[Fe(CN)6]/K4[Fe(CN)6]. From the Nyquist plots of EIS (Fig. 2), it can be seen that bare AuSPE has a small semi-circle domain because of the high conductivity of the Au surface (curve a). After the electrode modification with the graphene–Au hybrid nanocomposite, the semi-circle domain increases a little bit (curve b). After that, when the electrode surface was covered with fetuin A, the resistance of the electrode surface increased since the active surface area was blocked (curve c). Then, N was immobilized on to fetuin A from the sialic acid sides. Therefore, the electrode surface was covered with N and the charge transfer ability of the electrode surface was reduced resulting in the increment in the semicircle diameter (curve d). Lastly, PNA lectin, which shows specificity to galactose molecules that appear after the cleavage of fetuin A with N, was used. When the PNA lectin binds onto the galactose molecules, the electron transfer gets even tougher and the semi-circle diameter also increases due to the resistance increase on the electrode surface (curve e). | ||
| Fig. 2 Nyquist plots of the developed influenza A viral biosensor. a. Plain AuSPE, b. AuSPE/graphene-AuNp, c. AuSPE/graphene-AuNp/fetuin A, d. AuSPE/graphene-AuNp/fetuin A/N, and e. AuSPE/graphene-AuNp/fetuin A/N/PNA lectin. The EIS procedure was set to measure the electron transfer resistance in the frequency range of 0.1 Hz–10 kHz at a potential of 0.1 V and 10 mM K3[Fe(CN)6]/K4[Fe(CN)6] (in pH: 7.4, 0.1 M PBS) was used as a redox probe. | ||
Optimization of experimental parameters
The experimental parameters consisting of the effect of BSA addition on the developed system, the immobilization time of fetuin A, and the immobilization time of PNA lectin were investigated by EIS. Since the developed system is considered to be applied for human samples like throat swabs in future, physiological human pH and body temperature were chosen as the working conditions so that all the experiments are carried out at pH 7.4 and the incubation of N was done at 37 °C.The effect of BSA on the developed system
As is well known, BSA is generally used for the blockage of any unspecific interaction on the electrode surface, after the immobilization of the analyte. However, even for this step, there is a need to wait for a period for the completion of the attachment of BSA. In order to observe this effect, the developed influenza A biosensor was fabricated in the presence of BSA, in the absence of BSA and with the simultaneous immobilization of fetuin A. All these steps were monitored by EIS under the previously mentioned working conditions. The obtained EIS diagrams (ESI, Fig. S1†) demonstrate that for our system there is no necessity for BSA addition which means that, there is no unspecific binding during the fabrication of the developed influenza A biosensor. Also by eliminating the addition of BSA, the preparation procedure of the biosensor is shortened and as a result, the practicality of the fabrication procedure is increased. In conclusion, no BSA attachment onto the electrode surface was done for the future experiments.Optimization of fetuin A immobilization time
Fetuin A immobilization time experiments were carried out for 15 min, 30 min, 45 min, 60 min and 90 min incubation times and the results were monitored by using EIS. The obtained EIS diagrams are presented in the ESI, Fig. S2† in addition to the Excel plots. It can be seen from Fig. S2† that the best result was obtained at 30 min. Incubation times less than 30 min resulted in a lower difference in electrode resistances, demonstrating that more time is needed probably for the exchange reactions between fetuin A amino groups and NHS to be completed. On the other hand, incubation times more than 30 min resulted in a decrease in the resistance difference that might be attributed to the separation of the non-specific bonding from the electrode surface. As a result, further experiments were conducted by using 30 min as the optimum incubation time for fetuin A.The effect of PNA lectin immobilization time
PNA lectin immobilization time experiments were carried out for 30 min, 45 min, 60 min and 90 min immobilization times by using EIS. The obtained EIS diagrams are presented in the ESI, Fig. S3† and the Excel plots are also demonstrated in Fig. S3†. It can be seen from Fig. S3† that the best result was obtained at 60 min. Immobilization times less than 60 min resulted in a lower difference in electrode resistances demonstrating that more time is needed probably for the specific binding reactions between PNA lectin and galactose molecules to be completed. Immobilization times that lasted longer than 60 min resulted in a decrease in the resistance difference that might be attributed to the falling out of the PNA-galactose structure from the electrode surface.11Analytical characteristics
After the optimization of the experimental parameters, the analytical characteristics were examined. For obtaining a calibration graph, the specific interactions between PNA lectin and galactose molecules that occur after the cleavage of N from fetuin A were monitored. When the concentration of N is increased, since more fetuin A will undergo cleavage, more galactose molecules will appear and as a result, more PNA lectin will link to the galactose ends. All these changes were monitored via the resistance values that were obtained from EIS measurements (Fig. 3). As can be seen from Fig. 3, the linear range and limit of detection (LOD) values which are based on the N concentration are obtained between 10−8 U mL−1 and 10−1 U mL−1 with the equations of y = 2746x + 31.69 (R2 = 0.99) and 1 × 10−8 U mL−1 (1.27 ng mL−1) respectively. The relative standard deviation (R.S.D) value was calculated for 10−5 U mL−1 N (n = 3) and found to be 3.23%. The obtained analytical characteristic values of the developed electrochemical biosensor were compared with the influenza A virus enzyme-linked immunosorbent (ELISA) assay results in the literature. These ELISA assays relied on the N activity or antibody–antigen interactions. From these ELISA assays LOD values such as 0.7 ng mL−1 antigen,53 33 ng mL−1 antigen,53 0.5 ng mL−1 protein54 and 102–103 Tissue Culture Infectious Dose 50% (TCID50)55 can be obtained. From these results it is clear that the developed sensor is sensitive enough. On the other hand considering its rapidity and practicality, it can be said that the developed electrochemical biosensor could definitely offer better performance.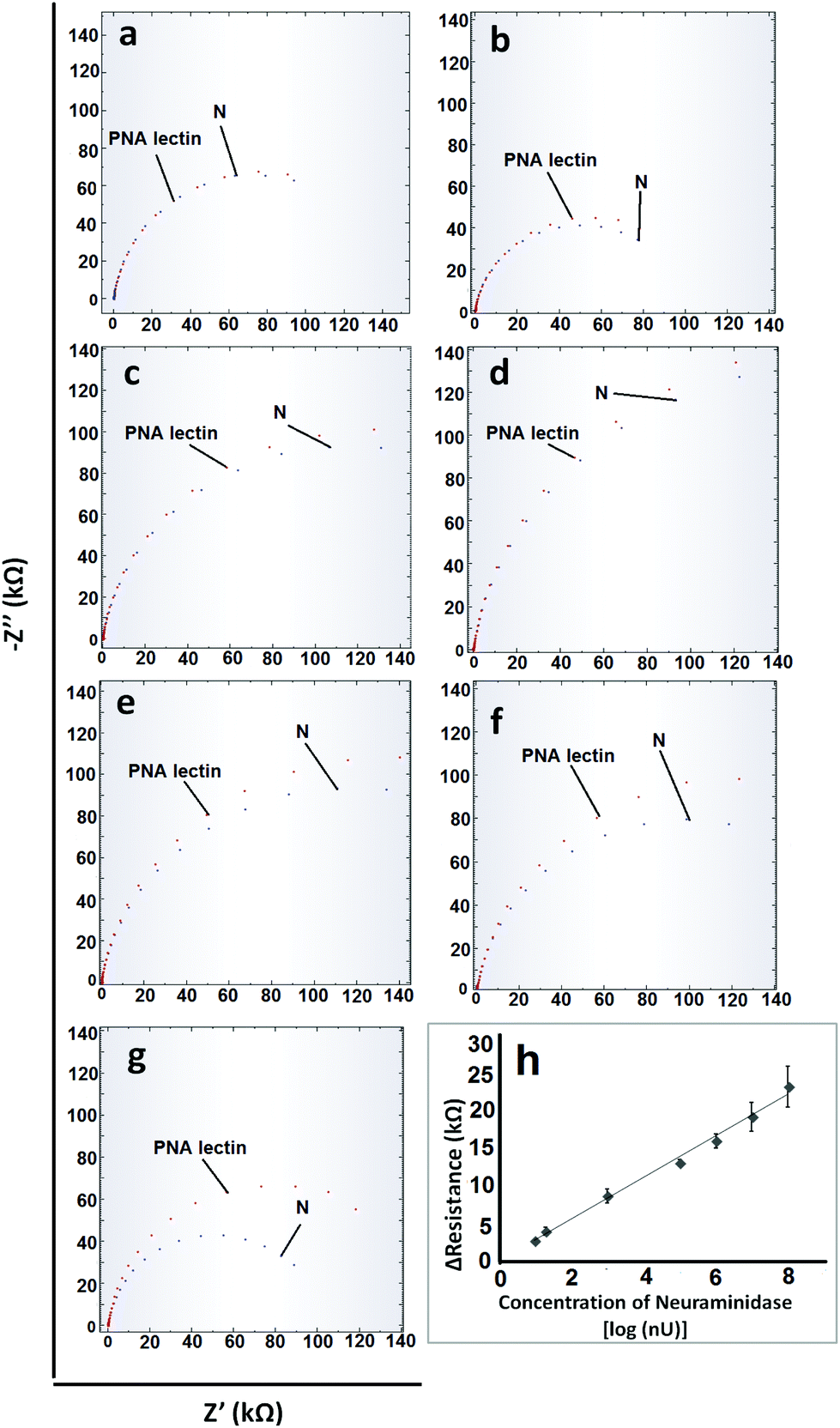 | ||
| Fig. 3 Nyquist plots of different concentrations of N: (a) 10−8 U mL−1, (b) 2 × 10−8 U mL−1, (c) 10−6 U mL−1, (d) 10−5 U mL−1, (e) 10−3 U mL−1, (f) 10−2 U mL−1 and (g) 10−1 U mL−1, which were used for a calibration graph and (h) the calibration graph of the developed electrochemical influenza virus A biosensor. | ||
Sample application and control studies
The control study was also carried out by using an uninfected egg sample (allantoic fluid). It can be seen from Fig. 4 that the developed electrochemical influenza biosensor detects influenza A virus selectively.
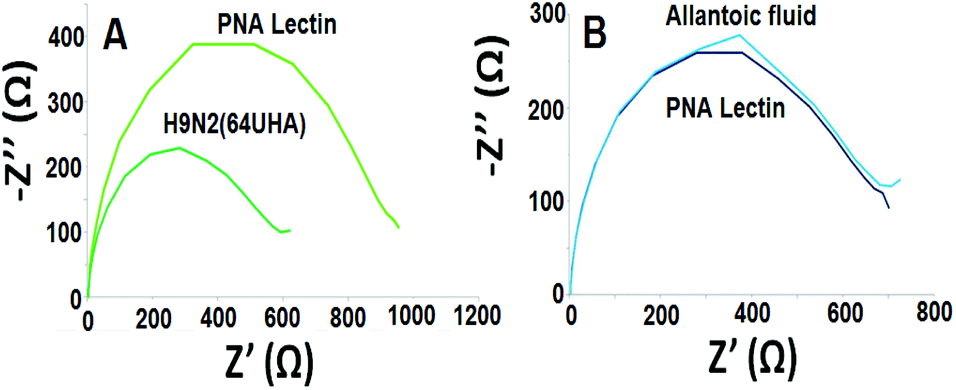 | ||
| Fig. 4 Nyquist plots for (A) 64 UHA H9N2 influenza virus, and (B) the control group by using the developed electrochemical influenza virus A biosensor. | ||
Conclusions
Another novel and effective electrochemical influenza A biosensor based on graphene–Au modified Au-SPE has been developed. This work is the continuation of our previous work in which the working principle relies on the measurement of N activity.14 From the LOD values, it is clear that with the developed electrochemical influenza A biosensor more sensitive results were obtained compared to those with the ELLA assay. We believe that, the wide linear range and sensitive results from the real influenza A sample (H9N2) analysis increase the potential of usage of our system as a point of care tool for the diagnosis of influenza virus A in the future.56Conflicts of interest
There are no conflicts to declare.Acknowledgements
The grants from The Technical and Scientific Council of Turkey (TUBITAK) Project no: 114Z654 and Tunisian Ministry of Higher Education and Scientific Research (MHESRT): Projet Tuniso-Turque 2015–2017 are gratefully acknowledged.References
- Y. Wu, Y. Wu, B. Tefsen, Y. Shi and G. F. Gao, Trends Microbiol., 2014, 22, 183–191 CrossRef CAS PubMed.
- N. Sriwilaijaroen and Y. Suzuki, Proc. Japan Acad. Ser., B Phys Biol Sci., 2012, 88, 226–249 CrossRef CAS.
- L. Couzens, J. Gao, K. Westgeest, M. Sandbulte, V. Lugovtsev, R. Fouchier and M. J. Eichelberger, Virol. Methods, 2014, 210, 7–14 CrossRef CAS PubMed.
- D. R. Peaper and M. L. Landry, Clin. Lab. Med., 2014, 34, 365–385 CrossRef PubMed.
- M. K. Abraham, J. Perkins, G. M. Vilke and C. J. Coyne, J. Emerg. Med., 2016, 50, 536–542 CrossRef PubMed.
- C. M. Mair, K. Ludwig, A. Herrmann and C. Sieben, Biochim. Biophys. Acta, 2014, 1838, 1153–1168 CrossRef CAS PubMed.
- J. H. Han, D. Lee, C. H. C. Chew, T. Kim and J. J. Pak, Sens. Actuators, B, 2016, 228, 36–42 CrossRef CAS.
- U. Jarocka, R. Sawicka, A. Gora-Sochacka, A. Sirko, W. Zagorski-Ostoja, J. Radecki and H. Radecka, Biosens. Bioelectron., 2014, 55, 301–306 CrossRef CAS PubMed.
- S. K. Arya, P. Kongsuphol, C. C. Wong, L. J. Polla and M. K. Park, Sens. Actuators, B, 2014, 194, 127–133 CrossRef CAS.
- U. Jarocka, R. Sawicka, A. Gora-Sochacka, A. Sirko, W. Dehaen, J. Radecki and H. Radecka, Sens. Actuators, B, 2016, 228, 25–30 CrossRef CAS.
- L. J. Mitnaul, M. N. Matrosovich, M. R. Castrucci, A. B. Tuzikov, N. V. Bovin, D. Kobasa and Y. Kawaoka, J. Virol., 2000, 74, 6015–6020 CrossRef CAS PubMed.
- M. N. Matrosovich, T. Y. Matrosovich, T. Gray, N. A. Roberts and H. D. Klenk, J. Virol., 2004, 78, 12665–12667 CrossRef CAS PubMed.
- J. C. Dortmans, J. Dekkers, I. N. Wickramasinghe, M. H. Verheije, P. J. Rottier, F. J. Van Kuppeveld, E. De Vries and C. A. De Haan, Sci. Rep., 2013, 3, 3058 CrossRef CAS PubMed.
- U. Anik, Y. Tepeli and M. F. Diouani, Anal. Chem., 2016, 88, 6151–6153 CrossRef CAS PubMed.
- J. K. Park and J. K. Taubenberger, ACS Infect. Dis., 2016, 2, 5–7 CrossRef CAS PubMed.
- WHO. Influenza (seasonal), http://www.who.int/mediacentre/factsheets/fs211/en/ (accessed March 23, 2017).
- H. Yu, B. J. Cowling, L. Feng, E. H. Lau, Q. Liao, T. K. Tsang, Z. Peng, P. Wu, F. Liu, V. J. Fang, H. Zhang, M. Li, L. Zeng, Z. Xu, Z. Li, H. Luo, Q. Li, Z. Feng, B. Cao, W. Yang, J. T. Wu, Y. Wang and G. M. Leung, Lancet, 2013, 382, 138–145 CrossRef.
- L. Krejcova, D. Hynek, V. Adam, J. Hubalek and R. Kizek, Int. J. Electrochem. Sci., 2012, 7, 10779–10801 CAS.
- J. Bell, A. Bonner, D. M. Cohen, R. Birkhahn, R. Yogev, W. Triner, J. Cohen, E. Palavecino and R. Selvarangan, J. Clin. Virol., 2014, 61, 81–86 CrossRef CAS PubMed.
- K. Leirs, P. T. Kumar, D. Decrop, E. Perez-Ruiz, P. Leblebici, B. V. Kelst, G. Compernolle, H. Meeuws, L. V. Wesenbeeck, O. Lagatie, L. Stuyver, A. Gils, J. Lammertyn and D. Spasic, Anal. Chem., 2016, 80, 8450–8458 CrossRef PubMed.
- C. H. Cho, M. K. Woo, J. Y. Kima, S. Cheong, C.-K. Lee, S. A. An, C. S. Lim and W. J. Kim, J. Virol. Methods, 2013, 187, 51–56 CrossRef CAS PubMed.
- L. Chen, Y. Tian, S. Chen and O. Liesenfield, Eur. J. Microbiol. Immunol., 2015, 5, 236–245 CrossRef CAS PubMed.
- B. Hazelton, T. Gray, J. Ho, V. M. Ratnamohan, D. E. Dwyer and J. Kok, Influenza Other Respir. Viruses, 2015, 9, 151–154 CrossRef CAS PubMed.
- D. E. Sutter, S. A. Worthy, D. M. Hensley, A. M. Maranich, D. M. Dolan, G. W. Fischer and L. T. Daum, J. Med. Virol., 2012, 84, 1699–1702 CrossRef CAS PubMed.
- M. Santhiago, M. Strauss, M. P. Pereira, A. S. Chagas and C. C. B. Bufon, ACS Appl. Mater. Interfaces, 2017, 9, 11959–11966 CAS.
- C. Batchelor-McAuley, E. J. F. Dickinson, N. V. Rees, K. E. Toghill and R. G. Compton, Anal. Chem., 2012, 84, 669–684 CrossRef CAS PubMed.
- J. E. Frew and H. A. O. Hill, Anal. Chem., 1987, 59, 933A–944A CrossRef CAS PubMed.
- Y. Xu, L. Liu, Z. Wang and Z. Dai, ACS Appl. Mater. Interfaces, 2016, 8, 18669–18674 CAS.
- U. Anik, Med. Biosens. for POC Appl., in Electrochemical medical biosensors for POC applications, ed. R. J. Narayan, Woodhead Publishing, 2016, ch. 12, vol. 2017, pp. 275–292 Search PubMed.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS PubMed.
- J. Wang, Analyst, 2005, 130, 421–426 RSC.
- Li, X. Su and Y. Lu, Analyst, 2015, 140, 2916–2943 RSC.
- J. N. Tiwari, V. Vij, K. C. Kemp and K. S. Kim, ACS Nano, 2016, 10, 46–80 CrossRef CAS PubMed.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- S. Wu, Q. He, C. Tan, Y. Wang and H. Zhang, Small, 2013, 9, 1160–1172 CrossRef CAS PubMed.
- X. Gan and H. Zhao, Sens. Mater., 2015, 27, 191–215 CAS.
- C. Xu, X. Wang and J. Zhu, J. Phys. Chem. C, 2008, 112, 19841–19845 CAS.
- Y. Tepeli and U. Anik, Electroanalysis, 2016, 28, 3048–3054 CrossRef CAS.
- Z. Zhang, L. Luo, L. Zhu, Y. Ding, D. Deng and Z. Wang, Analyst, 2013, 138, 5365–5370 RSC.
- S. Aslan and U. Anik, Microchim. Acta, 2016, 183, 73–81 CrossRef CAS.
- S. C. Sultan and U. Anik, Talanta, 2014, 129, 523–528 CrossRef CAS PubMed.
- M. Giovanni, H. L. Poh, A. Ambrosi, G. Zhao, Z. Sofer, F. Sanek, B. Khezri, R. D. Webster and M. Pumera, Nanoscale, 2012, 4, 5002–5008 RSC.
- C. Tan, X. Huang and H. Zhang, Mater. Today, 2013, 16, 29–36 CrossRef CAS.
- M. F. Diouani, S. Helali, I. Hafaid, W. M. Hassen, M. A. Snoussi, A. Ghram, N. Jaffrezic-Renault and A. Abdelghani, Mater. Sci. Eng., C, 2008, 28, 580–583 CrossRef CAS.
- T. L. Kamikawa, M. G. Mikolajczyk, M. Kennedy, P. Zhang, W. Wang, D. E. Scott and E. C. Alocilja, Biosens. Bioelectron., 2010, 26, 1346–1352 CrossRef CAS PubMed.
- M. Veerapandian, R. Hunter and S. Neethirajan, Talanta, 2016, 155, 250–257 CrossRef CAS PubMed.
- J. Huang, Z. Xie, Z. Xie, S. Luo, L. Xie, L. Huang, Q. Fan, Y. Zhang, S. Wang and T. Zeng, Anal. Chim. Acta, 2016, 913, 121–127 CrossRef CAS PubMed.
- Z. Wu, C. H. Zhou, J. J. Chen, C. Xiong, Z. Chen, D. W. Pang and Z. L. Zhang, Biosens. Bioelectron., 2015, 68, 586–592 CrossRef CAS PubMed.
- W. S. J. Hummers and R. J. Offeman, Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. Li, M. B. Mgller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- Y. Tepeli and U. Anik, Electrochem. Commun., 2015, 57, 31–34 CrossRef CAS.
- W. Honquan, I. Sultana, L. K. Couzens and S. Mindaye, J. Virol. Methods, 2017, 244, 23–28 CrossRef PubMed.
- G. Edevag, M. Eriksson and M. Granström, J. Biol. Stand., 1986, 14, 223–230 CrossRef CAS PubMed.
- S. Velumani, Q. Du, B. J. Fenner, M. Prabakaran, L. C. Wee, L. Y. Nuo and J. Kwang, J. Virol. Methods, 2008, 147, 219–225 CrossRef CAS PubMed.
- Y. Tepeli and U. Anik, Sens. Actuators, B, 2018, 254, 377–384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01537b |
| This journal is © The Royal Society of Chemistry 2018 |