
Rational design of a molecularly imprinted polymer for dinotefuran: theoretical and experimental studies aimed at the development of an efficient adsorbent for microextraction by packed sorbent†
Camilla Fonseca
Silva
,
Keyller Bastos
Borges
 * and
Clebio Soares
do Nascimento
Jr.
*
* and
Clebio Soares
do Nascimento
Jr.
*
Departamento de Ciências Naturais, Universidade Federal de São João del-Rei, Campus Dom Bosco, Praça Dom Helvécio 74, Fábricas, 36301-160, São João del-Rei, Minas Gerais, Brazil. E-mail: keyller@ufsj.edu.br
First published on 20th October 2017
Abstract
In this work, we studied theoretically the formation process of a molecularly imprinted polymer (MIP) for dinotefuran (DNF), testing distinct functional monomers (FM) in various solvents through density functional theory calculations. The results revealed that the best conditions for MIP synthesis were established with methacrylic acid (MAA) as FM in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 stoichiometry and with chloroform as the solvent. This protocol showed the most favourable stabilization energies for the pre-polymerization complexes. Furthermore, the formation of the FM/template complex is enthalpy driven and the occurrence of hydrogen bonds between the DNF and MAA plays a major role in the complex stability. To confirm the theoretical results, MIP was experimentally synthesized considering the best conditions found at the molecular level and characterized by scanning electron microscopy and thermogravimetric analysis. After that, the synthesized material was efficiently employed in microextraction by packed sorbent combined with high-performance liquid chromatography in a preliminary study of the recovery of DNF from water and artificial saliva samples.
:4 stoichiometry and with chloroform as the solvent. This protocol showed the most favourable stabilization energies for the pre-polymerization complexes. Furthermore, the formation of the FM/template complex is enthalpy driven and the occurrence of hydrogen bonds between the DNF and MAA plays a major role in the complex stability. To confirm the theoretical results, MIP was experimentally synthesized considering the best conditions found at the molecular level and characterized by scanning electron microscopy and thermogravimetric analysis. After that, the synthesized material was efficiently employed in microextraction by packed sorbent combined with high-performance liquid chromatography in a preliminary study of the recovery of DNF from water and artificial saliva samples.
1. Introduction
Molecular imprinting technology has been considered an important synthetic approach to the development of molecular recognition that can mimic natural recognition entities, such as biological receptors.1 Molecularly imprinted polymers (MIP) have gained remarkable attention in analytical analysis due to their use in separation/purification processes. Recently, MIP have been found to be a good replacement for biological macromolecules in various areas of research, including practical applications.2,3The synthesis of MIP involves the formation of a complex between a template or target molecule and the functional monomers (FM) in an appropriate solvent. Immediately after the polymerization step, the template is withdrawn and cavities are left in the polymeric matrix, which are complementary in both functionality and size arrangement to those of the template molecules.4,5 Thus, MIP can recognize in a selective way the template molecule from structurally related compounds. This kind of macromolecular material can be used in sample preparation and pre-determined selectivity. Furthermore, it has been applied in chromatography and capillary electrophoresis as a separating agent,6,7 in the synthesis of molecularly imprinted nanoparticles,8,9 as chemical sensors10–12 and in controlled drug release.13–15
Although the synthesis of MIP is relatively easy, the optimization of MIP demands the selection of the best FM, which should present the most effective interactions with the template to achieve high selectivity and rebinding capacity, and the best suitable polymerization solvent for each selected template, considering various reagents.16 Experimentally, a standard imprinting protocol synthesis is time consuming and tedious. Routinely, the selection of the best imprinting conditions has been mainly made in an empirical way based on a trial-and-error method and chemical intuition. In this sense, to improve the properties of MIP, computer-aided studies have proved an effective and useful tool in the search for optimal imprinting conditions, which can save substantial labour and laboratory resources.17 The use of theoretical calculations in MIP modelling has allowed the suitable preparation of high affinity polymers through a systematic rational design procedure.18
Recently, some publications reporting the applicability of density functional theory (DFT) methods for use in chromatographic,19 electrophoretic,20,21 and enantioselective methods have been found in the literature. In addition, the use of quantum mechanics methods to rationalize MIP design for application in the sample preparation process has increased considerably since the end of the last decade.22–32 DFT allows the accurate study of novel molecules of theoretical interest to be carried out at a lower computational cost as compared with pure post-Hartree–Fock ab initio methods. In a very recent work, our group has investigated MIP formation for tramadol, an analgesic drug. Distinct FM were used for the evaluation of the interaction process of tramadol, in distinct solvents employing DFT calculations at the B3LYP/6-311G(d,p) level. We could establish that the best MIP synthesis conditions were obtained with acrylic acid (AA) as FM in a 1:3 mole ratio and using chloroform as the solvent.32
Dinotefuran (DNF) (Fig. S1†), a nitroguanidine neonicotinoid insecticide usually found as a racemic mixture, was chosen as the template molecule. It has been used to control a wide range of pests, such as whiteflies, leafhoppers, aphids, mealy bugs, stink bugs, leaf miners, ants, cockroaches, fleas, flies, crickets and gnats.33 Thus, because it is a relatively new, widely marketed and low-cost molecule, monitoring it in environmental, food or biological fluid samples is useful as a strategy to guarantee food quality and consumer safety, as well as preservation of waterways, soils and biodiversity.
In this context, the main goals of this work were to (i) select the best-suited FM using DFT calculations of structural and energetic parameters, (ii) find the most stable mole ratio between FM and the template molecule, (iii) find the most suitable solvent for the MIP synthesis, (iv) prepare and characterize a non-imprinted polymer (NIP) and MIP by thermogravimetric analysis (TGA) and scanning electron microscopy (SEM), (v) optimize the enantioselective analysis of DNF enantiomers and (vi) evaluate the potential of MIP in a preliminary study of the recovery and precision of DNF enantiomers from water and artificial saliva samples employing microextraction by packed sorbent (MEPS) as the sample preparation technique. It is worth emphasizing that to our knowledge, this is the first work involving theoretical and experimental studies (rational synthesis) of MIP for the DNF molecule.
2. Experimental
2.1. Materials and reagents
For the synthesis of MIP, the following reagents were employed: DNF Pestanal® (analytical standard, template), methacrylic acid (MAA) inhibited with 250 mg L−1 hydroquinone (as the FM), chloroform (porogenic solvent), and ethylene glycol dimethacrylate (EGDMA) inhibited with 100 mg L−1 monomethyl ether hydroquinone (cross-linking agent) were obtained from Sigma–Aldrich (Steinheim, Germany) and 4,4′-azo-bis-(4-cyanopentaenoic) (radical initiator) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).HPLC grade hexane, methanol and ethanol were obtained from J.T. Baker (Mexico City, MX, Mexico). Diethylamine was obtained from Sigma–Aldrich (Steinheim, Germany). The components for artificial saliva, i.e. NaHCO3 (0.5208 g), K2HPO4 (0.1045 g), NaCl (0.8770 g), KCl (0.4770 g), CaCl2·2H2O (0.4410 g), were obtained from Sigma–Aldrich (Steinheim, Germany) and transferred to a 1 L volumetric flask, and sonicated and the volume was filled with ultrapure water to the line. All other chemicals were of analytical grade with the highest purity available. Water was distilled and purified using a Millipore Milli-Q Plus system (Bedford, MA, USA).
2.2. Preparation of stock and standard solutions
DNF stock solution was prepared in methanol at a concentration of 1 mg mL−1, and stored at −20 °C in the absence of light. Standard solutions were prepared daily using serial dilutions of this stock solution in water or artificial saliva to obtain concentrations of 20 ng mL−1 and 100 ng mL−1, which were also stored at −20 °C in the absence of light.2.3. Instrumentation
:ethanol:methanol (83:10:7, v/v/v) plus 0.5% diethylamine at a flow rate of 1.2 mL min−1 and detected at 270 nm. All chromatographic procedures were conducted at 25 ± 3 °C and the injection volume was 10 μL for standards and samples. An Agilent Open LAB Chromatography Data System® was used to control the HPLC system and for data acquisition.
2.4. Theoretical methodology
The thermodynamic properties such as enthalpy (ΔH), Gibbs free energy (ΔG) and the entropic contribution (TΔS) regarding the formation process of FM/template complexes were calculated using eqn (1)–(3) as follows:
| ΔH = Hcomplex − [HDNF + nEFM], | (1) |
| ΔG = Gcomplex − [GDNF + nGFM], | (2) |
| TΔS = TScomplex − [TSDNF + nTSFM], | (3) |
:1, 1:2, 1:3 and 1:4 stoichiometries.
Posteriorly, the solvent effect was considered to predict the stability of the FM/template complexes using the integral equation formalism of the polarizable continuum model.36 The solvents chosen for testing were water, chloroform, acetonitrile, methanol, acetone and dimethyl sulfoxide. All theoretical calculations were carried out using the Gaussian 2009 quantum mechanics package.37
2.5. Experimental methodology
:acetic acid solution (9:1, v/v) to remove the DNF and then with methanol to remove the residual acetic acid. After washing, the material was dried at approximately 65 °C for 48 h. Finally, the material obtained was sieved using a 100-mesh sieve. The NIP was synthesized under the same MIP conditions, but without the addition of the template molecule.
3. Results and discussion
3.1. Theoretical studies
:1, 1:2, 1:3 and 1:4 stoichiometries.
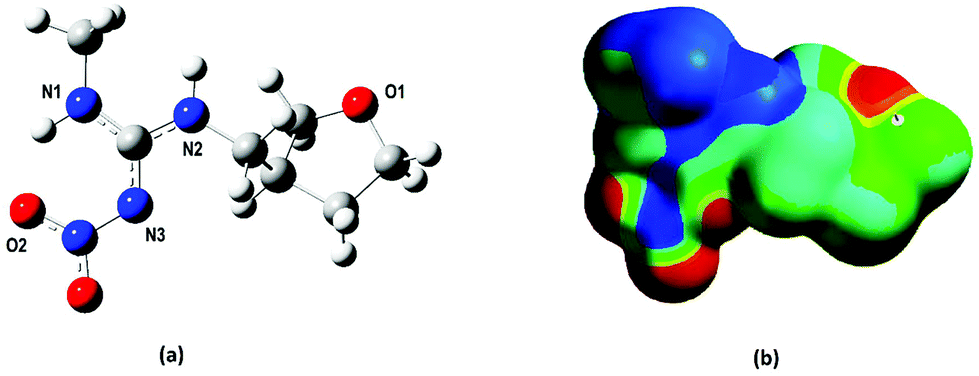 | ||
| Fig. 1 (a) DFT-optimized geometry for the dinotefuran template molecule and (b) electrostatic potential map highlighting the three most susceptible regions (1, 2 and 3) to interact via hydrogen bonds with the FMs (regions in red). | ||
Our first attempt was to evaluate the interaction of every FM (AA, MAA, APV and TFMAA) with the furanosyl oxygen (O1) leading to 1:1 mole ratio complexes. The DFT-optimized geometries for the 1:1 FM–template complexes are shown in Fig. S2.† The establishment of two hydrogen bonds in each of the complexes can be noticed: one between the hydrogen of the FM's hydroxyl group and the DNF furanosyl oxygen (O⋯H), the other between the FM's carboxyl oxygen and the DNF hydrogen located in the N2 group (N⋯H).
The calculated B3LYP thermodynamic properties, ΔH, ΔG and TΔS in the gas phase for the 1:1 FM–template complexes, are given in Table 1. From the results, it is possible to notice that the DNF-(AA)1, DNF-(APV)1 and DNF-(TFMAA)1 complexes exhibited very small energy differences, around 1.0 kcal mol−1 (ΔH and ΔG). On the other hand, the best complex, which means the most stable one, is achieved with MAA functional monomer. The DNF-(MAA)1 complex is about 3.0 kcal mol−1 more favourable than the other 1:1 species, both for ΔH and ΔG, as can be observed in Table 1.
:1, 1:2, 1:3 and 1:4 molar ratios. Values are given in kcal mol−1
| Complexes | ΔH | ΔG | TΔS | H bonds |
|---|---|---|---|---|
| In the last column is given the number of hydrogen bonds established per complex. | ||||
| DNF-(AA)1 | −16.5 | −4.8 | −11.7 | 2 |
| DNF-(MAA)1 | −19.6 | −8.3 | 11.3 | 2 |
| DNF-(APV)1 | −16.5 | −4.8 | −11.7 | 2 |
| DNF-(TFMAA)1 | −17.8 | −5.1 | −12.7 | 2 |
| DNF-(AA)2 | −28.7 | −6.4 | −22.3 | 4 |
| DNF-(MAA)2 | −35.5 | −13.7 | −22.3 | 4 |
| DNF-(APV)2 | −28.9 | −6.4 | −22.5 | 4 |
| DNF-(TFMAA)2 | −30.6 | −6.7 | −23.9 | 4 |
| DNF-(AA)3 | −40.7 | −9.5 | −31.2 | 5 |
| DNF-(MAA)3 | −50.5 | −19.6 | −30.9 | 5 |
| DNF-(APV)3 | −40.5 | −10.6 | −29.9 | 5 |
| DNF-(TFMAA)3 | −41.1 | −11.0 | −30.1 | 5 |
| DNF-(AA)4 | −54.3 | −12.5 | −41.8 | 6 |
| DNF-(MAA)4 | −67.4 | −25.0 | −42.4 | 6 |
| DNF-(APV)4 | −53.9 | −12.2 | −41.7 | 6 |
| DNF-(TFMAA)4 | −55.2 | −13.3 | −41.9 | 6 |
Then, a second FM was added to the 1:1 optimized species at the nitro group (NO2), resulting in the formation of 1:2 FM/template complexes. Observing Table 1, it is clearly seen that the same behaviour was found as for the 1:1 complexes, i.e. the DNF-(AA)2, DNF-(APV)2 and DNF-(TFMAA)2 possess almost degenerate energies and the DNF-(MAA)2 remains the most stable 1:2 complex (about 6.0 kcal mol−1 more favourable than the other complexes). For all 1:2 species, four hydrogen bonds can be found between the template and FM, as shown in Fig. S3.† The two new hydrogen bonds are formed as follows: one of them between the hydrogen of the FM's hydroxyl group and the oxygen (O2) located in the DNF nitro group (O⋯H), and the other between the FM's carboxyl oxygen and the hydrogen located in the N2 group of DNF (N⋯H).
Following this, a third FM was added to the 1:2 species at the N1 amine group of DNF (see Fig. 1), resulting in the formation of 1:3 FM/template complexes. In Table 1, it is easy to notice again the same systematic behaviour as that found for the 1:1 and 1:2 complex energies. The DNF-(MAA)3 continues to be the most stable complex (about 10.0 kcal mol−1 more favourable than the other 1:3 species). For all 1:3 species, five hydrogen bonds are formed between the template and FM, as can be observed in Fig. S4.† The newest hydrogen bond is formed between the hydrogen of the FM's hydroxyl group and the N1 amine nitrogen of DNF (N⋯H).
Finally, a fourth FM was added to the 1:3 optimized species at the N2 amine group of DNF (see Fig. 1), resulting in the formation of 1:4 complexes. The same systematic behaviour as that found for the 1:1, 1:2 and 1:3 complexes can be observed in Table 1. Again, the DNF-(MAA)4 continues to be the most stable complex (about 13.0 kcal mol−1 more favourable than the other complexes). In 1:4 complexes, six hydrogen bonds are formed between the template and FM, as can be observed in Fig. 2. The newest hydrogen bond is detected between the hydrogen of the FM's hydroxyl group and the N3 amine nitrogen of DNF (N⋯H).
 | ||
| Fig. 2 B3LYP/6-311G(d,p)-optimized structures for the 1:4 FM/template complexes: (a) DNF-(AA)4, (b) DNF-(MAA)4, (c) (DNF-APV)4, and (d) DNF-(TFMAA)4. The hydrogen bonds are drawn in dotted lines to facilitate visualization. | ||
It can be seen from Table 1 that the complexes formed with a 1:4 stoichiometry are the most stable in comparison with the mole ratios 1:1, 1:2 and 1:3. Besides, the hydrogen bonds found between the DNF and FM molecules play a crucial role in the FM/template complex formation. Still in Table 1, one can notice that the entropic contribution [TΔS] is almost constant for all complexes, regardless of the differing mole ratios. Thus, we can state that the formation of the complexes is not governed by entropy. Instead, it is a spontaneous and enthalpy-driven process. In this sense, the DFT methods offer a very computationally feasible way to include electron correlation effects in quantum chemical calculations, which is important to describe intermolecular interactions.22
From these results, we selected MAA as the best FM for the MIP synthesis of DNF, based on a systematic structural and energetic analysis. Besides, we could also determine the ideal stoichiometry, with 1:4 being found as the best mole ratio for synthesis purposes.
| Solvents | DNF-(MAA)4 complex | |
|---|---|---|
| ΔH | ΔG | |
| Water | −70.7 | −16.5 |
| Acetone | −78.5 | −21.8 |
| Dimethyl sulfoxide | −72.2 | −17.7 |
| Chloroform | −86.9 | −30.3 |
| Acetonitrile | −73.5 | −18.5 |
| Methanol | −76.6 | −19.7 |
As can be seen in Table 2 the lowest ΔH (−86.9 kcal mol−1) and ΔG (−30.3 kcal mol−1) values found for DNF-(MAA)4 are in the presence of chloroform as the solvent. The interaction between FM and the target molecule is accomplished through hydrogen bonds; thus a non-polar solvent with a low dielectric constant, such as chloroform, can be considered a more adequate medium for the interactions. Furthermore, higher dielectric constant solvents, such as acetonitrile, may be used; however, the MIP obtained will possess a lower ability in rebinding to the template molecule. For protic solvents, such as water and methanol, they will release hydrogen ions into the medium, which will compete in the formation of hydrogen bonds with those that can be formed between FM and the template molecule. Thus, it can provide a compromised functionality polymer. We concluded that chloroform should be the best solvent for MIP synthesis.
3.2. Experimental studies
:ethanol:methanol (85:5:10, v/v/v) as the mobile phase at a flow rate of 1.0 mL min−1 and detection at 270 nm. In this work, we evaluated the influence of the percentage composition of the mobile phase employing the Chiralpak® IA column (100 mm × 4.6 mm, 3 μm). Fig. S5(A)† presents the initial tests for the chromatographic separation of the DNF enantiomers employing hexane:ethanol:methanol (85:5:10, v/v/v) at a flow rate of 1.0 mL min−1 as described in the literature.38 Although the chiral selectors of the ChromegaChiral CCA (a polysaccharide-coated chiral stationary phase) and Chiralpak® IA (a polysaccharide-immobilized chiral stationary phase) columns were the same, both formed by 3,5-dimethylphenyl carbamate amylose, the chromatographic behaviours were different. A baseline separation was not observed. Fig. S5(B)† shows the chromatogram employing 80% hexane, 10% ethanol, 10% methanol plus 0.1% diethylamine at a flow rate of 12 mL min−1. As can be seen, this condition did not show a good separation. Fig. S5(C)† refers to the chromatogram with 83% hexane, 11% ethanol, 6% methanol plus 0.25% diethylamine and a flow rate of 1.2 mL min−1. A better separation was observed between the enantiomers, although they appear with posterior tails. Finally, Fig. 3 show the best conditions for the enantioseparation of DNF, which was obtained with hexane:ethanol:methanol (83:10:7, v/v/v) plus 0.5% diethylamine and a flow rate of 1.2 mL min−1. Under these conditions, (+)-(S)-DNF eluted at 4.19 min and (−)-(R)-DNF eluted at 5.03 min, presented a resolution of 1.9, more than 2000 theoretical plates and asymmetry around 1.1.
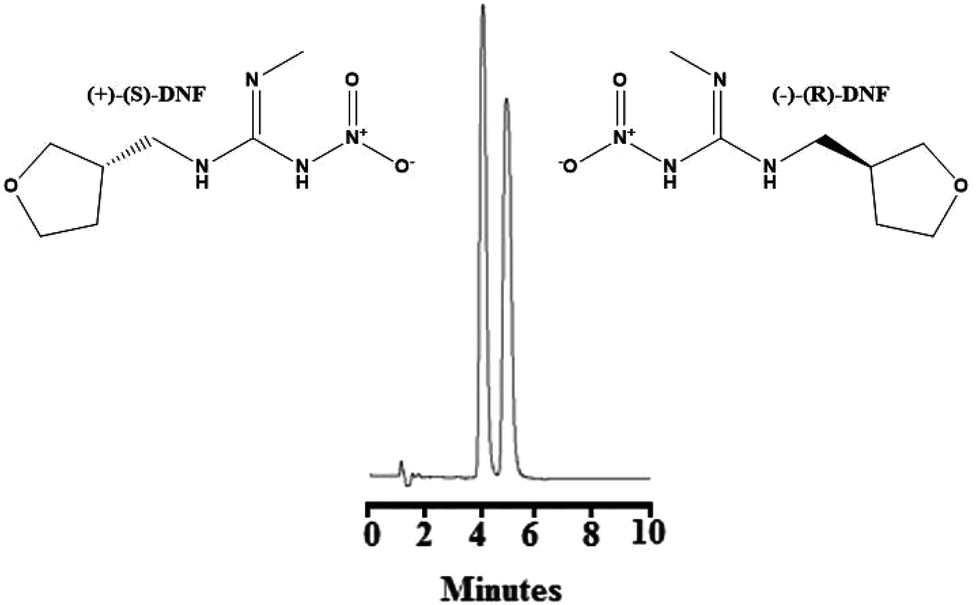 | ||
| Fig. 3 Chromatogram referring to optimized chromatographic conditions for the enantioseparation of dinotefuran. Conditions: Chiralpak® IA (100 mm × 4.6 mm, 3 μm), hexane:ethanol:methanol (83:10:7, v/v/v) plus 0.5% diethylamine, 10 μL volume of injection, flow rate 1.2 mL min−1, and detection at 270 nm. (+)-(S)-DNF eluted at 4.19 min and (−)-(R)-DNF eluted at 5.03 min. | ||
The synthesis of NIP and MIP was conducted by bulk polymerization, in which the polymers were synthesized first and ground into particles for their application in the sample preparation techniques, such as solid-phase extraction, MEPS and others. In fact, the size of the particles could influence the extraction efficiency and selectivity of the MIP. Low-porosity polymers with a small surface area have a low molecular recognition capacity, basically due to the slow diffusion of the analytes towards the selective sites located in the micropores.39–44 Therefore, the morphological structures of NIP and MIP were investigated by SEM. Fig. S6(A and B)† shows SEM micrographs under 180× for NIP and 200× for MIP. As expected, the heating-induced bulk polymerization method carried out in a homogeneous system containing the monomer, the template molecule, the solvent, the cross-linking reagent and the radical initiator in the absence of oxygen resulted in the formation of relatively large particles of irregular shape and non-uniform size.42–44 However, it is known that these irregularities do not limit the application of this material as an efficient adsorbent.
Fig. S6(C and D)† shows the curves obtained by TGA, performed in a temperature range of 25 to 600 °C for the NIP and MIP. Our research group has been working on the synthesis of MIP using MAA as FM,39–44 in which it is possible to determine the presence of three main thermal events. The first one (between approximately 25 and 80 °C) presents a small loss of mass due to the evaporation of residual water. The second event (between approximately 250 and 450 °C) indicates the beginning of the decomposition process of the materials showing a rapid loss of mass. The third (between approximately 450 and 600 °C) corresponds to a small loss of mass due to the possible formation and evaporation of some volatile compounds. Therefore, it has been observed that the decomposition of the materials occurs at relatively high temperatures, about 300 °C, thus demonstrating good thermal stability.
:methanol (95:5, v/v) mixture. To start the optimization process, some conditions were set: 5 mg of MIP, 200 μL of a water:methanol (95:5, v/v) mixture, 200 μL of spiked sample at 100 ng mL−1 of DNF (water or artificial saliva), 200 μL of washing solvent, 200 μL of elution solvent, sample pH equal to 5.5 (water or saliva) and without the addition of salt. After each extraction, the eluent was dried under nitrogen flow and resuspended in 50 μL of ethanol prior to HPLC injection.
3.2.3.1 Effect of washing solvent. In this parameter, the best washing solvent is the one that extracts a small amount of analyte, but removes interference from the matrix. For this study, water, ethanol and methanol were evaluated. Methanol extracted 3.32 ± 0.13 and 3.27 ± 0.38%, ethanol extracted 2.59 ± 0.31 and 3.07 ± 0.42% and water 2.64 ± 0.16 and 2.34 ± 0.19% of (+)-(S)-DNF and (−)-(R)-DNF, respectively (Fig. 4A). Water was more appropriate because it could remove interference from the matrix and extracted a very small amount of DNF enantiomers.
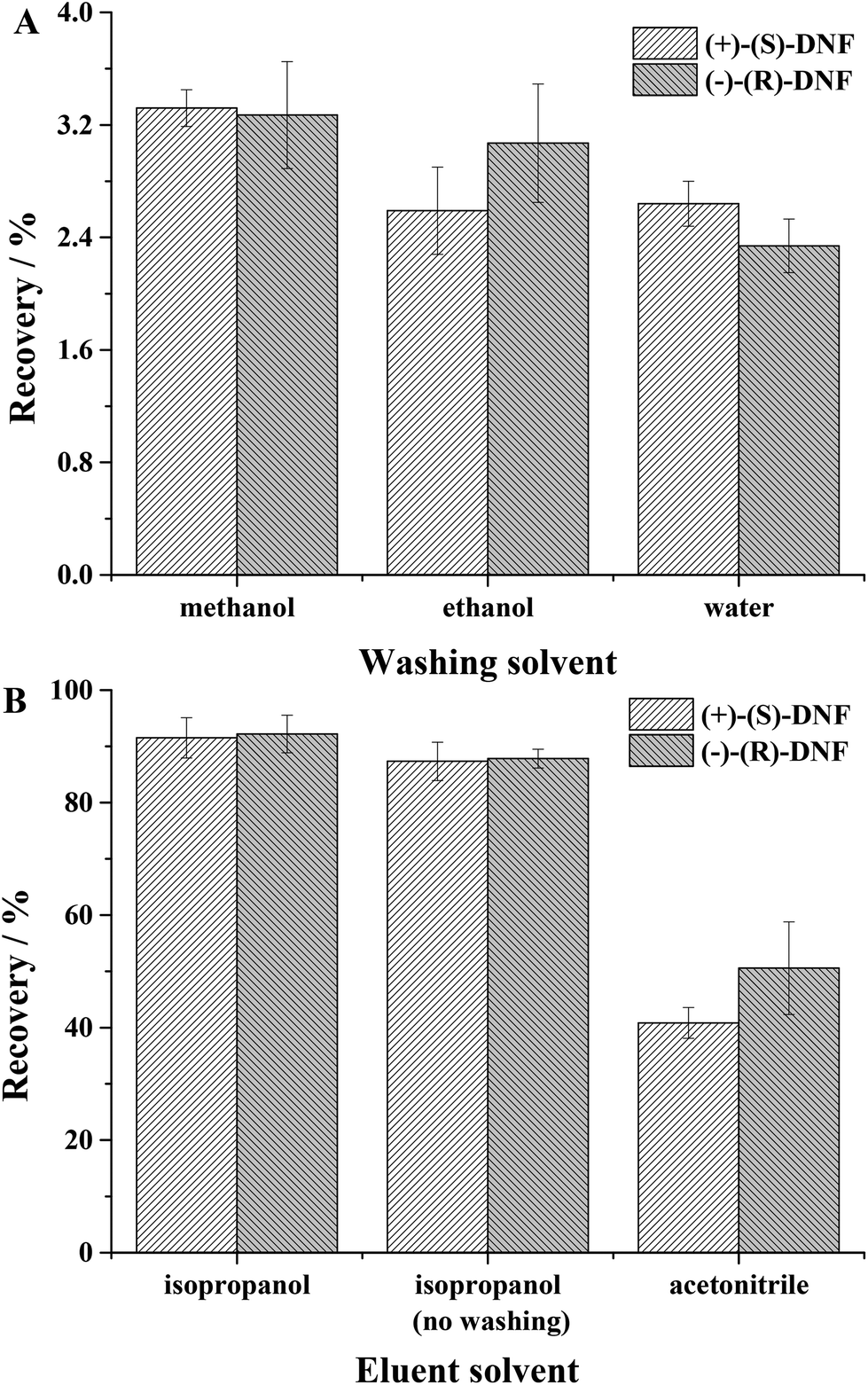 | ||
| Fig. 4 (A) Effect of washing solvent: water, ethanol and methanol; (B) effect of elution solvent: isopropanol, isopropanol without the previous washing step and acetonitrile. | ||
3.2.3.2. Effect of elution solvent. This step used isopropanol, isopropanol without a previous washing step and acetonitrile (Fig. 4B). Although the recoveries were similar for the solvents isopropanol and isopropanol without the previous washing step, we opted for the first due to the removal of interference from the matrix. After only two initial tests, washing solvent and elution solvent, the recoveries/standard deviation for (+)-(S)-DNF and (−)-(R)-DNF reached 91.51 ± 3.58% and 92.21 ± 3.34%, respectively. The recovery values for water and artificial saliva (90.43 ± 4.76% and 91.09 ± 4.58%) did not present a significant difference. Our synthesized material, used as an adsorbent in MEPS, was superior to other sorbents described in the literature,45 in which the recovery of DNF ranged from 15.6 ± 1.4 to 57.2 ± 3.1%. In addition, the MIP synthesized by rational design and non-rational design presented recoveries ranging from 89.03 to 92.68 and 54.87 to 59.56, respectively. Table 3 presents the recoveries and precision using rational and non-rational design for the MIP syntheses. Finally, the technique after two simple experiments proved to be effective for the extraction of DNF enantiomers from water and artificial saliva, using small volumes of solvents and a small mass of MIP with recoveries around 100%.
| Analytes | Concentration (ng mL−1) | Rational designa | Non-rational designb | ||
|---|---|---|---|---|---|
| % Recovery (mean ± SD) (n = 6) | Precision RSD% (n = 6) | % Recovery (mean ± SD) (n = 6) | Precision RSD% (n = 6) | ||
| a Conditions for MIP synthesis using rational design: 1 mmol of DNF, 4 mmol of MAA, 10 mL of chloroform, 20 mmol of EGDMA and 0.3 mmol of 4,4′-azo-bis-(4-cyano pentaenoic). b Conditions for MIP synthesis using non-rational design: 1 mmol of DNF, 2 mmol of AA, 10 mL of acetonitrile, 20 mmol of EGDMA and 0.3 mmol of 4,4′-azo-bis-(4-cyano pentaenoic). | |||||
| (+)-(S)-DNF | 20 | 89.03 | 3.91 | 54.87 | 1.85 |
| 100 | 89.87 | 4.64 | 56.44 | 5.08 | |
| (−)-(R)-DNF | 20 | 92.68 | 3.63 | 58.65 | 8.70 |
| 100 | 91.90 | 4.03 | 59.56 | 7.94 | |
4. Concluding remarks
In the present paper, we report for the first time theoretical and experimental studies for the interaction process of FM with DNF aimed at obtaining the best MIP synthesis conditions. This is a summary of the main conclusions drawn from our results: (i) the best FM/template mole ratio was 1:4 based on structural and energetic data, (ii) the best FM was found to be MAA, (iii) chloroform, a non-polar solvent with a low dielectric constant, was predicted to be the best solvent, (iv) the hydrogen bonds formed between the DNF and FM play a major role in the complex stability, (v) the complex formation process is enthalpy driven because the entropic contribution is almost constant for each DNF-(FM)n complex regardless of the different stoichiometries, (vi) an enantioselective HPLC method was developed and improved compared with previous work, (vii) MIP for DNF was rationally synthesized and characterized by TGA and SEM, being an excellent material for the adsorption process, (viii) MEPS proved to be an efficient and simple technique for sample preparation, (ix) this strategy can be applied to other systems to economize time and cost, improve the efficiency of extraction, reduce matrix effects and prolong the lifetime of the instruments.
Abbreviations
| MIP | Molecularly imprinted polymer |
| NIP | Non-imprinted polymer |
| DNF | Dinotefuran |
| FM | Functional monomers |
| DFT | Density functional theory |
| MAA | Methacrylic acid |
| AA | Acrylic acid |
| APV | p-Vinyl benzoic acid |
| TFMAA | 2-(Trifluoromethyl)acrylic acid |
| EGDMA | Ethylene glycol dimethacrylate |
| TGA | Thermogravimetric analysis |
| SEM | Scanning electron microscopy |
| MEPS | Microextraction by packed sorbent |
Conflicts of interest
All authors declare no conflict of interest, particularly no financial and personal relationships with other people or organizations that could inappropriately influence this work.Acknowledgements
The authors would like to thank the Brazilian agencies CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais) for financial support. This study is also part of a project involving the Rede Mineira de Química (RQ-MG) supported by FAPEMIG (project REDE-113/10; project CEX – RED-0010-14).References
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- C. H. Lu, Y. Zhang, S. F. Tang, Z. B. Fang, H. H. Yang, X. Chen and G. N. Chen, Biosens. Bioelectron., 2012, 31, 439–444 CrossRef CAS PubMed.
- J. L. Urraca, C. S. A. Aureliano, E. Schillinger, H. Esselmann, J. Wiltfang and B. Sellergren, J. Am. Chem. Soc., 2011, 133, 9220–9223 CrossRef CAS PubMed.
- M. Tabandeh, S. Ghassimipour, H. Aqababa, M. Tabatabaei and M. Hasheminejad, J. Chromatogr., B: Biomed. Appl., 2012, 898, 24–31 CrossRef CAS PubMed.
- W. Wan, M. Biyikal, R. Wagner, B. Sellergren and K. Rurack, Angew. Chem., Int. Ed., 2013, 52, 7023–7027 CrossRef CAS PubMed.
- C. Kulsing, R. Knob, M. Macka, P. Junor, R. I. Boysen and M. T. W. Hearn, J. Chromatogr. A, 2014, 1354, 85–91 CrossRef CAS PubMed.
- H. Y. Zong, X. Liu, Z. S. Liu and Y. P. Huang, Electrophoresis, 2015, 36, 818–824 CrossRef CAS PubMed.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed.
- A. Poma, A. P. F. Turner and S. A. Piletsky, Trends Biotechnol., 2010, 28, 629–637 CrossRef CAS PubMed.
- N. Karimian, M. Vagin, M. H. A. Zavar, M. Chamsaz, A. P. F. Tuerner and A. Tiwari, Biosens. Bioelectron., 2013, 50, 492–498 CrossRef CAS PubMed.
- M. P. Tiwari and B. B. Prasad, J. Chromatogr. A, 2014, 1337, 22–31 CrossRef CAS PubMed.
- J. Zhou, N. Gan, T. Li, F. Hu, X. Li, L. Wang and L. Zheng, Biosens. Bioelectron., 2014, 54, 199–206 CrossRef CAS PubMed.
- N. Mirzaei, S. A. H. Najafabadi, M. Abdouss, S. Azodi-Deilami, E. Asadi, M. R. M. Hosseini and M. Piramoon, J. Appl. Polym. Sci., 2013, 128, 1557–1562 Search PubMed.
- E. V. Piletska, B. H. Abd, A. S. Krakowiak, A. Parmar, D. L. Pink, K. S. Wall, L. Wharton, E. Moczko, M. J. Whitcombe, K. Karim and S. A. Piletsky, Analyst, 2015, 140, 3113–3120 RSC.
- F. Puoci, S. Hampel, O. I. Parisi, A. Hassan, G. Cirillo and N. Picci, J. Appl. Polym. Sci., 2013, 130, 829–834 CrossRef CAS.
- I. A. Nicholls, K. Adbo, H. S. Andersson, P. O. Andersson, J. Ankarloo, J. H. Dahlstrom, P. Jolela, J. G. Karlsson, L. Olofsson, J. Rosengren, S. Shoravi, J. Svenson and S. Wikman, Anal. Chim. Acta, 2001, 435, 9–18 CrossRef CAS.
- S. Subrahmanyan and S. A. Piletsky, Computational Design of Molecularly Imprinted Polymers, Springer Science, LCC, 2009 Search PubMed.
- I. A. Nicholls, H. S. Andersson, C. Charlton, H. Henschel, B. C. G. Karlsson, J. G. Karlsson, J. O'Mahony, A. M. Rosengren, K. J. Rosengren and S. Wikman, Biosens. Bioelectron., 2009, 25, 543–552 CrossRef CAS PubMed.
- A. Carotti, F. Ianni, S. Sabatini, A. Di Michele, R. Sardellaa, G. W. Kaatz, W. Lindner, V. Cecchetti and B. Natalini, J. Pharm. Biomed. Anal., 2016, 129, 182–189 CrossRef CAS PubMed.
- C. S. Nascimento Jr., L. Guimaraes, J. F. Lopes and K. B. Borges, Analyst, 2014, 139, 3901–3910 RSC.
- M. C. Fonseca, R. C. S. da Silva, C. S. Nascimento Jr. and K. B. Borges, Electrophoresis, 2017, 38, 1860–1868 CrossRef PubMed.
- T. Cowen, K. Karim and S. Piletsky, Anal. Chim. Acta, 2016, 936, 62–74 CrossRef CAS PubMed.
- L. A. de Barros, L. A. Pereira, R. Custódio and S. Rath, J. Braz. Chem. Soc., 2014, 25, 619–628 Search PubMed.
- F. Ahmadi, E. Yawari and M. Nikbakht, J. Chromatogr. A, 2014, 1338, 9–16 CrossRef CAS PubMed.
- M. Torkashvand, M. B. Gholivand and F. Taherkhani, Mater. Sci. Eng., 2015, 55, 209–217 CrossRef CAS PubMed.
- N. Karimian, M. B. Gholivand and F. Taherkhani, J. Electroanal. Chem., 2015, 740, 45–52 CrossRef CAS.
- L. S. Fernandes, P. Homem-de-Mello, E. C. Lima and K. M. Honório, Eur. Polym. J., 2015, 71, 364–371 CrossRef CAS.
- K. K. Tadi, R. V. Motghare and V. Ganesh, RSC Adv., 2015, 5, 99115–99124 RSC.
- K. M. Muzyka, J. Nano-Electron. Phys., 2015, 7, 5 Search PubMed.
- A. Nezhadali, S. Senobari and M. Mojarrab, Talanta, 2016, 146, 525–532 CrossRef CAS PubMed.
- B. B. Prasad, R. Singh and A. Kumar, Carbon, 2016, 102, 86–96 CrossRef CAS.
- M. C. Fonseca, C. S. Nascimento Jr. and K. B. Borges, Chem. Phys. Lett., 2016, 645, 174–179 CrossRef CAS.
- D. Goulson, J. Appl. Ecol., 2013, 50, 977–987 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- V. Barone, M. Cossi and J. A. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillom, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 2009, revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- X. Chen, F. Dong, X. Liu, J. Xu, J. Li, Y. Li, Y. Wang and Y. Zheng, J. Sep. Sci., 2012, 30, 200–205 CrossRef PubMed.
- H. L. Oliveira, S. S. Anacleto, A. T. M. da Silva, A. C. Pereira, W. S. Borges, E. C. Figueiredo and K. B. Borges, J. Chromatogr., B: Biomed. Appl., 2016, 1033, 27–39 CrossRef PubMed.
- R. C. S. Silva, V. Mano, A. C. Pereira, E. C. Figueiredo and K. B. Borges, Anal. Methods, 2016, 8, 4075–4085 RSC.
- M. M. C. Borges, H. L. Oliveira and K. B. Borges, Electrophoresis, 2017, 38, 2150–2159 CrossRef PubMed.
- A. T. M. da Silva, H. L. de Oliveira, C. F. Silva, M. C. Fonseca, T. F. D. Pereira, C. S. Nascimento Jr., E. C. Figueiredo and K. B. Borges, J. Chromatogr., B: Biomed. Appl., 2017, 1061–1062C, 399–410 CrossRef PubMed.
- C. N. Boscari, G. R. Mazzuia, C. Wisniewski, K. B. Borges and E. C. Figueiredo, Electrophoresis, 2017, 38, 1083–1090 CrossRef CAS PubMed.
- T. P. Rao, K. Ramakrishnan and S. Daniel, Anal. Chim. Acta, 2006, 578, 105–116 CrossRef CAS PubMed.
- J. Zhang, Y. Wei, H. Li, E. Y. Zeng and J. You, Talanta, 2017, 170, 392–398 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01324h |
| This journal is © The Royal Society of Chemistry 2018 |