
Revisiting monosaccharide analysis – quantitation of a comprehensive set of monosaccharides using dynamic multiple reaction monitoring†
Gege
Xu
 a,
Matthew J.
Amicucci
a,
Zhi
Cheng
a,
Ace G.
Galermo
a and
Carlito B.
Lebrilla
*abc
a,
Matthew J.
Amicucci
a,
Zhi
Cheng
a,
Ace G.
Galermo
a and
Carlito B.
Lebrilla
*abc
aDepartment of Chemistry, University of California, Davis, CA 95616, USA. E-mail: cblebrilla@ucdavis.edu
bDepartment of Biochemistry and Molecular Medicine, University of California, Davis, CA 95616, USA
cFoods for Health Institute, University of California, Davis, CA 95616, USA
First published on 23rd November 2017
Abstract
A rapid method for the quantitation of sixteen neutral and acidic monosaccharides, from both animal and plant sources was developed using ultra-high performance liquid chromatography triple quadrupole mass spectrometry (UHPLC/QqQ-MS) in dynamic multiple reaction monitoring (dMRM) mode. Monosaccharides including three pentoses (ribose, xylose, arabinose), two deoxyhexoses (rhamnose, fucose), five hexoses (fructose, mannose, allose, glucose, galactose), two hexuronic acids (glucuronic acid, galacturonic acid), and two N-acetyl-hexosamines (GlcNAc, GalNAc), were derivatized with 1-phenyl-3-methyl-5-pyrazolone (PMP), while underivatized sialic acids, Neu5Ac and Neu5Gc, were simultaneously analyzed with a 10-minute run. With the optimized UHPLC conditions, baseline separations of the isomers were achieved. The sensitivity and calibration ranges of this method were determined. The limits of detection were between femtomoles and attomoles with linear ranges spanning four to six orders of magnitude and coefficients of variation (CVs) ≤7.2%. Spiking experiments performed on a pooled fecal sample demonstrated the high accuracy of this method even when applied to samples with complicated matrices. The validated method was applied to fecal samples from an infant transitioning from breast milk to weaning foods. Major milk monosaccharides including galactose, fucose, glucose, GlcNAc, and Neu5Ac were found to be the most abundant components in the feces of milk-fed infants. PMP-derivatives of nine other monosaccharides including apiose, lyxose, altrose, talose, gulose, glucosamine, galactosamine, mannosamine, and N-acetylmannosamine (ManNAc) were also tested and could be added to the quantitation method depending on the need. The speed and sensitivity of the method makes it readily adaptable to rapid throughput analysis of monosaccharides in biological samples.
Introduction
Carbohydrates including monosaccharides, oligosaccharides, polysaccharides, and the glycan moieties of glycoconjugates, make up one of the most abundant groups of compounds in nature and are found in all living organisms. Monosaccharides including glucose, galactose, and others play key roles in nearly every biological process. Oligosaccharides and polysaccharides play a large host of additional roles by providing structural components in tissues, as antigens, and as post-translational modifications of lipids and proteins. For example, free oligosaccharides in human milk are known to promote the growth of beneficial bacteria, thus establishing protective gut microbiota for the infants.1–3 Some plant pectic polysaccharides have immune system modulating activities and can, therefore, inhibit tumor growth and metastasis by stimulating host immunity.4,5 Glycans presented on proteins play vital roles in protein folding, trafficking, function, and recognition.6–8To determine the numerous roles of carbohydrates in biological systems, methods that can elucidate their monosaccharide compositions are necessary. Methods are currently available for monosaccharide analysis, however, fast, sensitive, and accurate methods for absolute monosaccharide quantitation particularly in studies involving large biological sample sets are still needed. Monosaccharides often differ by a single stereocenter, but these minor structural variations can have large biological consequences. To identify structural isomers, efficient separation techniques are often essential for the analysis of monosaccharides. Conventional methods for monosaccharide quantitation involve gas chromatography (GC) separation in combination with mass spectrometry (MS)9,10 or flame ionization detection (FID).11–13 Carbohydrates lack volatility and require multiple derivatization steps to make suitable for GC, however, they still require long elution times to achieve isomer separation. High pH anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD),14–16 and capillary electrophoresis (CE) or high performance liquid chromatography (HPLC) separation with UV detection are increasingly more common techniques for separating and quantifying monosaccharides.17–19 To improve the efficiency of chromatographic separation and sensitivity of UV detection, derivatization of monosaccharides is necessary. Among the various labels available, 1-phenyl-3-methyl-5-pyrazolone (PMP) developed by Honda et al.20 has been widely used because it can quantitatively react with carbohydrates under mild conditions. During the derivatization reaction, two PMP molecules are added to the reducing end of each saccharide by Michael addition. The hydrophobic PMP molecules can facilitate the separation of monosaccharides by reversed phase liquid chromatography employing C18 as the stationary phase.21,22 However, the specificity of UV detection is relatively low because many other molecules can also absort UV light. Quantitation by UV often greatly depends on effective chromatographic separation of all analytes from possible impurities, resulting in long analysis time.
LC-MS methods have been developed and have brought remarkable advances in terms of sensitivity and specificity to the general analysis of carbohydrates.23,24 Rühmann et al.25 coupled UV and electrospray ionization with ion trap detection and achieved faster analysis of saccharide-PMP derivatives. However, the sensitivity was not greatly improved. Compared with other MS-based methods, multiple reaction monitoring (MRM) on a triple quadrupole instrument is a more selective and sensitive technique for the targeted quantitation of analytes in complex biological samples because it involves mass filtering of both parent and product ions. MRM has been applied to the absolute quantitation of glycoproteins,26,27 metabolites,28,29 and oligosaccharides.30,31 For monosaccharides, Hammad et al. used an aminopropyl column to separate the acetate adducts of reducing monosaccharides with subsequent detection using MRM.32,33 Due to the high similarity of monosaccharide structures, isomers such as glucose and galactose were not well separated. Recently, Han et al.34 developed a MRM-based method to quantify neutral mono- and disaccharides derivatized by 3-nitrophenylhydrazine employing 13C-labeled internal standards in negative ion mode. Isomeric compounds such as xylose and arabinose were also not well separated and subsequently co-eluted. In addition, the amino and acidic sugars, that are widely present in functional carbohydrates, were not included.
In this study, we have developed a comprehensive method for quantitation of both neutral and acidic monosaccharides using ultra-high performance liquid chromatography triple quadrupole mass spectrometry (UHPLC/QqQ-MS) in dynamic multiple reaction monitoring (dMRM) mode. The current method achieved simultaneous quantitation of fourteen PMP-derivatized monosaccharides including fructose, and two sialic acids with label-free detection. With the optimized UHPLC conditions, chromatographic separation of all isomer groups could be achieved within 7.5 minutes of a 10-minute total run time. This method was validated and applied to the quantitation of free monosaccharides in infant fecal samples. The method is thus far the most sensitive, while also providing rapid monosaccharide quantitation covering common monosaccharides found in both plants and animals. Furthermore, it can be readily adapted for the analysis of various less common monosaccharides such as gulose, lyxose and talose. Its speed, high sensitivity, and wide linear range make this method ideal for large clinical sample sets.
Experimental section
Samples and materials
D-Glucose, D-galactose, D-mannose, D-allose, D-fructose, N-acetyl-D-glucosamine (GlcNAc), N-acetyl-D-galactosamine (GalNAc), D-arabinose, D-xylose, D-ribose, L-rhamnose, L-fucose, D-glucuronic acid (GlcA), D-galacturonic acid (GalA), N-acetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc), D-apiose, D-talose, D-altrose, L-gulose, D-glucosamine hydrochloride (GlcN), D-galactosamine hydrochloride (GalN), D-mannosamine (ManN), N-acetyl-D-mannosamine (ManNAc), ammonium acetate, PMP, ammonia (28.0–30.0%), and chloroform were purchased from Sigma-Aldrich (St Louis, MO). D-Lyxose was purchased from TCI America (Portland, OR). Acetonitrile (LC-MS grade) was purchased from Honeywell (Muskegon, MI). Five fecal samples from the same infant were collected after the infant was fed with different diets including breast milk only (sample 1 and sample 3), breast milk with water (sample 2), breast milk with squash (sample 4), and squash only (sample 5). All five samples were collected within a two-week period at six months postpartum. Informed consent was obtained from the subjects (infants’ parent) based on the study protocol approved by the University of California, Davis Institutional Review Board. The UCD IRB Administration is tasked to follow federal regulations to protect the rights and welfare of human subjects.Extraction of free monosaccharides from fecal samples
The five fecal samples were weighed, diluted 10-fold (10 μL mg−1) with nanopure water, and homogenized in a shaker overnight at 4 °C. After centrifugation at 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 minutes, the supernatants were transferred and stored at −20 °C. A pool was prepared by mixing 100 μL of supernatant from each sample and was used for recovery experiments. Samples and the pool were further diluted by 5-fold and centrifuged at 21000g for 30 minutes before derivatization and injection for LC-MS analysis.
000g for 30 minutes, the supernatants were transferred and stored at −20 °C. A pool was prepared by mixing 100 μL of supernatant from each sample and was used for recovery experiments. Samples and the pool were further diluted by 5-fold and centrifuged at 21000g for 30 minutes before derivatization and injection for LC-MS analysis.
Derivatization of monosaccharides using PMP
A mixed standard solution containing 1 mg mL−1 of 14 monosaccharides was prepared and serially diluted to concentrations of 0.1 ng mL−1 to 500 μg mL−1. Neu5Ac and Neu5Gc standard solutions were prepared separately and not derivatized. Both the standard mixtures and extracted monosaccharides from fecal samples were subjected to derivatization using PMP. The derivatization conditions optimized by Wu et al.24 were used with some modifications. Briefly, 50 μL of monosaccharide standards or fecal samples were mixed with 200 μL ammonia solution (28.0–30.0%) in water and 200 μL 0.2 M PMP solution in methanol. The mixtures were allowed to react at 70 °C for 30 minutes then dried by vacuum centrifugation. The dried samples were reconstituted in 500 μL water and washed 2 times with 500 μL chloroform. Next, 20 μL of the aqueous layer from each fecal sample and standard solution was mixed with 20 μL of underivatized sample or sialic acid standards, respectively. A 1 μL aliquot of the mixture was directly injected to UHPLC/MRM-MS for complete monosaccharide analysis.UHPLC/MRM-MS analysis
Detection and quantitation of derivatized monosaccharides and underivatized Neu5Ac and Neu5Gc were performed using an Agilent 6490 triple quadrupole mass spectrometer equipped with an Agilent 1290 infinity UHPLC system and an Agilent ZORBAX Eclipse Plus C18 column (2.1 mm × 100 mm i.d., 1.8 μm particle size). For UHPLC separation, the aqueous mobile phase A was 25 mM ammonium acetate in 5% acetonitrile in water (v/v) with the pH adjusted to 8.2 using ammonia. The organic mobile phase B was 95% acetonitrile in water (v/v). Separation of PMP-labeled monosaccharides was performed using an optimized 10-minute binary gradient of 0.0–7.0 min, 12–15% B; 7.1–8.5 min, 99% B; 8.6–10.0 min, 12% B. The flow rate was set at 0.5 mL min−1 and the column temperature was set at 35 °C.Electrospray ionization (ESI) was used as the ionization source and was operated in positive ion mode. The MS parameters were as follows: the drying gas temperature and sheath gas temperature were set at 290 °C and 300 °C, respectively. Drying gas flow rate and sheath gas flow rate were set at 11 L min−1 and 12 L min−1, respectively. Nebulizer pressure was set at 30 psi. The capillary voltage and fragmentor voltage were set at 1800 V and 280 V, respectively. The amplitudes of RF voltage for high-pressure and low-pressure ion funnels were 150 V and 60 V, respectively. For scanning product ions, the mass range was set at m/z 50–600 for PMP-labeled monosaccharides and m/z 50–350 for free Neu5Ac and Neu5Gc.
Results and discussion
Optimization of PMP concentration
The concentration of PMP is critical to ensure completion of the derivatization reaction for different types of monosaccharides in the samples and minimization of ion suppression caused by the excess reagent. To determine the optimal concentration of PMP, a series of derivatization reactions for a mixture of twelve monosaccharide standards (100 μg mL−1 each) were carried out using varying molarities of PMP from 20 mM to 500 mM. The yields varied between different monosaccharides and were very low for monosaccharides such as GlcNAc and glucose when only 20 mM PMP was used (Fig. S1†). When PMP concentration increased from 20 mM to 50 mM, the yields of labeled monosaccharides increased dramatically and reached a maximum with PMP concentration of 100 mM and 200 mM. When the concentration was increased further, however, slight decreases were observed for some monosaccharides possibly due to ion suppression from the excess amount of PMP. Therefore, 200 mM was used for the complete derivatization of all the monosaccharides in both standards and fecal samples.MRM transitions for quantitation
MRM transitions of the fourteen derivatized monosaccharides and two unlabeled sialic acids, Neu5Ac and Neu5Gc, were obtained by scanning their respective fragment ions using product ion mode of the QqQ. After derivatization, each monosaccharide is labeled with two PMP molecules. The protonated precursor ion of each derivatized monosaccharide corresponding to the initial monosaccharide mass plus the mass of two PMP residues ([M + 330 + H]+) was subjected to collisional induced dissociation (CID). Two major fragment ions from the MS/MS spectra were used as product ions for MRM transitions. As shown in Fig. 1, m/z 175.0 corresponding to [PMP + H]+ was the most abundant product ion for the derivatized monosaccharides. Therefore, m/z 175.0 was used as the quantifying ion transition for the PMP-derivatized monosaccharides to achieve the best sensitivity. The second most abundant fragment ion was related to the cleavage of the C2–C3 bond on the monosaccharide, and one of the C1–PMP bonds. The resulting ions of m/z 216.1 for GalNAc and GlcNAc and m/z 217.1 for all the other derivatized monosaccharides were used as qualifying ions to confirm the presence of target analytes. Protonated molecular ions [M + H]+ of unlabeled Neu5Ac and Neu5Gc with a m/z 310.3 and 326.3 were used as their precursor ions, and the most abundant fragment ions of m/z 274.2 and 290.2, corresponding to loss of two water molecules, were used as quantifiers. The qualifying ion chosen for these two compounds was m/z 167.1, which results from a cross-ring cleavage. The MRM transitions and retention times of the sixteen monosaccharides are shown in Table 1.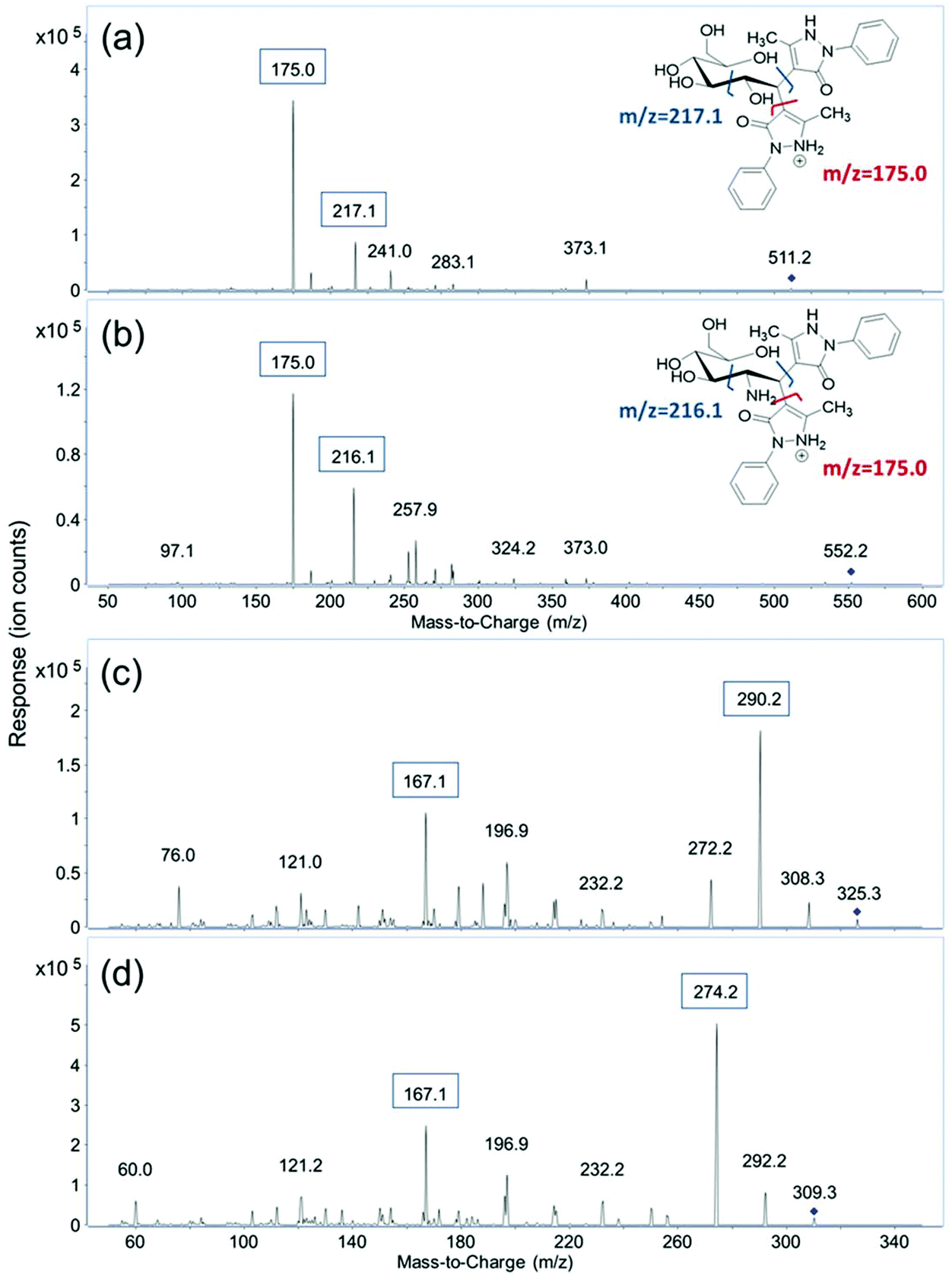 | ||
| Fig. 1 MS/MS spectra of (a) PMP-labeled glucose; (b) PMP-labeled GlcNAc; (c) label-free NeuAc and (d) label-free NeuGc. | ||
| Compound | Mass | MRM transitions | Collision energy (eV) | Retention time (min) | Calibration curve | R 2 | Linear range (μg mL−1) | LOD (fmol) | CV (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Precursor ion | Product ions | |||||||||
| D-Ribose | 150.1 | 481.2 |
![[1 with combining low line]](https://www.rsc.org/images/entities/b_char_0031_0332.gif) ![[7 with combining low line]](https://www.rsc.org/images/entities/b_char_0037_0332.gif) ![[5 with combining low line]](https://www.rsc.org/images/entities/b_char_0035_0332.gif) ![[. with combining low line]](https://www.rsc.org/images/entities/b_char_002e_0332.gif) ![[0 with combining low line]](https://www.rsc.org/images/entities/b_char_0030_0332.gif) , 217.1 , 217.1 |
25 | 3.46 |
y = 35459x − 656 |
0.996 | 0.0005–200 | 0.067 | 4.1 |
| D-Xylose | 150.1 | 481.2 |
, 217.1 |
25 | 6.36 |
y = 66315x − 32445 |
0.997 | 0.002–200 | 0.67 | 4.5 |
| D-Arabinose | 150.1 | 481.2 |
, 217.1 |
25 | 6.68 |
y = 38321x + 17246 |
0.997 | 0.001–500 | 0.067 | 2.2 |
| L-Rhamnose | 164.2 | 495.2 |
, 217.1 |
25 | 3.82 |
y = 34309x − 2893 |
0.997 | 0.001–300 | 0.061 | 4.6 |
| L-Fucose | 164.2 | 495.2 |
, 217.1 |
25 | 7.25 |
y = 52865x − 65834 |
0.997 | 0.001–200 | 0.30 | 3.1 |
| D-Fructose | 180.2 | 511.2 |
, 217.1 |
20 | 0.73 | y = 1007x − 2134 | 0.997 | 0.01–300 | 5.6 | 7.2 |
| D-Mannose | 180.2 | 511.2 |
, 217.1 |
25 | 2.94 |
y = 18559x − 48930 |
0.998 | 0.001–500 | 0.56 | 3.4 |
| D-Allose | 180.2 | 511.2 |
, 217.1 |
25 | 3.48 |
y = 20597x − 9591 |
0.995 | 0.005–200 | 0.56 | 2.9 |
| D-Glucose | 180.2 | 511.2 |
, 217.1 |
25 | 5.70 |
y = 24914x − 167109 |
0.997 | 0.001–500 | 0.056 | 1.8 |
| D-Galactose | 180.2 | 511.2 |
, 217.1 |
25 | 6.31 |
y = 22034x − 58374 |
0.998 | 0.002–200 | 0.56 | 4.6 |
| D-Glucuronic acid | 194.2 | 525.2 |
, 217.1 |
25 | 3.52 |
y = 13553x − 29466 |
0.997 | 0.01–200 | 2.6 | 3.0 |
| D-Galacturonic acid | 194.2 | 525.2 |
, 217.1 |
25 | 4.10 |
y = 15292x − 125100 |
0.996 | 0.01–500 | 2.6 | 4.8 |
| GlcNAc | 221.2 | 552.2 |
, 216.1 |
30 | 5.85 |
y = 6136x − 27902 |
0.995 | 0.01–300 | 4.5 | 6.0 |
| GalNAc | 221.2 | 552.2 |
, 216.1 |
30 | 6.32 |
y = 10264x − 18018 |
0.999 | 0.01–200 | 4.5 | 5.2 |
| Neu5Ac | 309.3 | 310.3 |
![[2 with combining low line]](https://www.rsc.org/images/entities/b_char_0032_0332.gif) ![[4 with combining low line]](https://www.rsc.org/images/entities/b_char_0034_0332.gif) , 167.1 , 167.1 |
5 | 0.48 |
y = 18845x + 9125 |
0.998 | 0.01–50.0 | 16 | 1.9 |
| Neu5Gc | 325.3 | 326.3 |
![[9 with combining low line]](https://www.rsc.org/images/entities/b_char_0039_0332.gif) , 167.1 , 167.1 |
5 | 0.49 |
y = 12393x + 5318 |
0.998 | 0.01–50.0 | 15 | 1.0 |
Optimization of collision energies
Collision energies were optimized from 10 to 40 eV for the PMP-labeled monosaccharides, and from 1 to 40 eV for free Neu5Ac and Neu5Gc to obtain the best signal for the quantifying ions. As shown in Fig. S2,† responses of most derivatized monosaccharides first increased with increasing collision energy because higher energy caused more efficient fragmentation of the precursor ions. While the signal dramatically decreased when the collision energy was set greater than 30 eV due to the larger fractions of cross-ring cleavages produced under high-energy collision conditions. It was found that 25 eV yielded the highest responses for most of the PMP-derivatized monosaccharides, with 30 eV for the derivatized amino sugars including GlcNAc and GalNAc, and 20 eV for fructose. For unlabeled Neu5Ac and Neu5Gc, significantly lower collision energy was required for the quantifying product ions, derived mainly from water loss, while relatively higher energy was used for the qualifiers to facilitate cross-ring cleavage. For both Neu5Ac and Neu5Gc, 5 eV was the optimized collision energies for the quantifying product ions.UHPLC separation and dynamic MRM
A UHPLC C18 column was used to elute and separate the fourteen PMP-derivatized monosaccharides as well as the two unlabeled sialic acids in a rapid manner. As discussed by Li et al., buffer system and pH of the mobile phase has a significant effect on the separation of PMP-derivatized monosaccharides.18 As shown in Fig. S3a,† when only water and acetonitrile were used in unbuffered mobile phase A and B, a number of isomers such as glucuronic acid and galacturonic acid were barely separated. After mobile phase A was buffered with 25 mM ammonium acetate at pH 7, complete separations of the isomers were observed except for the pentoses, where only ribose was separated and the other two isomers (xylose and arabinose) coeluted as a single peak (Fig. S3b†). In this method, therefore, the pH of mobile phase A was optimized and a pH of 8.2 was found to provide the best separation efficiency (Fig. S3c†). Fig. 2 shows the dynamic MRM chromatograms of a standard mixture, where the sixteen monosaccharides are annotated. Peaks labeled in the same color represent isomeric monosaccharides. Solid peaks correspond to signals of the quantifying transitions, and dashed peaks correspond to the qualifying transitions. As expected, unlabeled Neu5Ac and Neu5Gc were not retained on C18 column and immediately eluted in 0.5 min. Fructose, the most common ketone monosaccharide, eluted much earlier than its aldehyde isomers at 0.7 min, indicating that the PMP derivative of fructose is much less hydrophobic compared to other labeled monosaccharides. Compounds that coelute such as ribose, allose, and glucuronic acid have different masses and were readily distinguished by MS. With the solvents and gradient optimized in this method, baseline separation of corresponding isomeric structures including two deoxyhexoses (rhamnose and fucose), five hexoses (fructose, mannose, allose, glucose, and galactose), two hexuronic acids (GlcA and GalA), and two N-acetyl-hexosamines (GlcNAc and GalNAc) was readily achieved within a 7.5-minute period during a total run time of 10 minutes (Fig. 2). Compared to other monosaccharide quantitation methods,23,24,35 the separation time in this method is greatly shortened with more compounds included.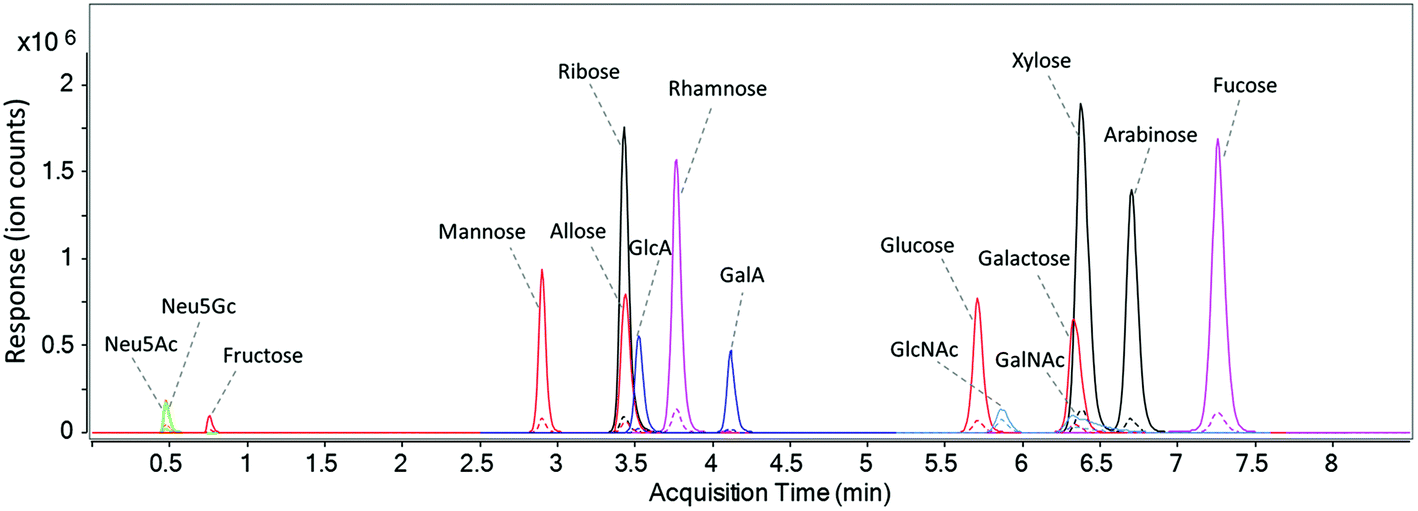 | ||
| Fig. 2 The dynamic MRM chromatogram of fourteen PMP-labeled monosaccharides and native sialic acid standards (10 ng injected for each). | ||
Linearity and sensitivity
Table 1 lists the standard calibration curves, linear regression coefficients (R2), linear ranges, and limits of detection (LODs, S/N ≥ 3) for the derivatized monosaccharides along with unlabeled Neu5Ac and Neu5Gc. In general, structural isomers had similar ionization efficiencies, thus had similar response factors (the slopes of the standard calibration curves), linear ranges, and limits of detection. Among the monosaccharides, pentoses including ribose, xylose, arabinose, deoxyhexoses including rhamnose, fucose, and hexoses including mannose, allose, glucose, galactose had the highest responses. Their linear ranges spanned 5 to 6 orders of magnitude with correlation coefficients higher than 0.995 and the limits of detection were at sub-femtomole or attomole level, which should be sufficient to include the concentration ranges in various biological samples. Ketones are known to have lower reaction efficiency with PMP compared to aldehydes.36Table 1 shows that fructose has a 20-fold lower response factor than other hexoses, resulting in a relatively lower detectability at 5.6 fmol. The limits of detection for hexuronic acids (GlcA and GalA), N-acetylhexosamines (GlcNAc and GalNAc) and unlabeled sialic acids (Neu5Ac and Neu5Gc) were also at the femtomole level due to their lower ionization efficiencies compared to the derivatized compounds. The linear ranges spanned 4 to 6 orders of magnitude for the derivatized monosaccharides and over 3 orders of magnitude for the sialic acids. The coefficients of variance were lower than 5% for most compounds and 5.2%–7.2% for the others including GalNAc, GlcNAc, and fructose based on four replicates. Compared to other recently published quantitation methods for monosaccharides including GC-FID, CE-UV, HPAEC-PAD, and LC-MS using an ion trap instrument (Table S1†),37–40 the current method employing UHPLC-QqQ with dMRM has proven to be the fastest and most sensitive, while covering a wide range of monosaccharides. Owing to the efficient separation, minimal interference, and targeted detection, the LODs for most PMP-derivatized monosaccharides were improved by >1000-fold to 0.056–5.6 femtomoles compared to ≥9 or 0.7 picomoles obtained by UV or ion trap MS, respectively. In addition, the linear calibration ranges for the monosaccharides in this method were two to three orders of magnitude greater than those from other methods, while the reproducibility (CV%) was comparable.Method validation
Monosaccharide analysis is often necessary for samples containing complex biological matrices. To evaluate the effect of matrix on our method, standard addition experiments were conducted on a pooled fecal sample. Fecal samples contain monosaccharides as well as large amounts of lipids, proteins metabolites, and bacterial cells. To evaluate the recovery rates of monosaccharide extraction and derivatization, a mixture of standards was spiked into a pooled fecal sample extract. The spiked-in amounts corresponded to 100% and 300% of the measured monosaccharide concentrations in the pool. After derivatization with PMP and cleanup using chloroform extraction, the monosaccharide concentrations in the original and spiked samples were both measured and the recovery rates were calculated. Table 2 lists the concentrations of monosaccharides in the pooled fecal sample and their measured recoveries and coefficients of variance. Thirteen out of sixteen monosaccharides were quantified in the pooled fecal sample while allose, galacturonic acid, and Neu5Gc were not detected. Therefore, spiking experiments were not performed for these three compounds. With 100% spike, the recoveries of PMP-derivatized monosaccharides ranged from 88.9% for GlcNAc to 115.2% for fructose with relative standard deviations lower than 6.9% (n = 4) except for xylose, arabinose, and rhamnose, which had higher variations due to their low concentrations. With 300% spike, better recoveries ranging from 92.9% for arabinose to 112.8% for glucose were obtained with all of the relative standard deviations lower than 7.0% (n = 4). For Neu5Ac, the recovery was approximately 80% possibly due to the minor ion suppression from coeluting compounds in the sample. Nevertheless, in both experiments, recovery rates of most monosaccharides were within 100 ± 5% placing them within acceptable ranges.| Monosaccharides | Content in sample (μg g−1) | Recovery of spiked-in standards | |||
|---|---|---|---|---|---|
| 100% spiking | CV% | 300% spiking | CV% | ||
| o-Ribose | 161 | 97.6% | 2.7 | 96.0% | 1.0 |
| o-Xylose | 6.2 | 110.0% | 9.5 | 106.5% | 3.5 |
| o-Arabinose | 5.0 | 94.6% | 19.7 | 92.9% | 3.7 |
| L-Rhamnose | 7.8 | 102.0% | 10.8 | 103.5% | 2.1 |
| L-Fucose | 3626 | 102.6% | 1.2 | 100.9% | 1.7 |
| D-Fructose | 162 | 115.2% | 6.9 | 105.9% | 7.0 |
| o-Mrumose | 23.2 | 101.4% | 6.7 | 99.2% | 1.3 |
| o-Allose | ND | — | — | — | — |
| o-Glucose | 2258 | 107.8% | 1.2 | 112.8% | 1.2 |
| o-Galactose | 4913 | 103.2% | 3.0 | 97.4% | 2.6 |
| o-Glucuronic acid | 30.4 | 102.8% | 4.1 | 101.8% | 1.2 |
| o-Galacturonic acid | ND | — | — | — | — |
| GlcNAc | 3029 | 88.9% | 1.4 | 95.5% | 2.0 |
| GalNAc | 181 | 102.0% | 0.8 | 98.2% | 2.4 |
| Neu5Ac | 1137 | 80.5% | 1.3 | 80.4% | 1.8 |
| Neu5Gc | ND | — | —— | — | — |
Quantitation of free monosaccharides in fecal samples
With the validation completed, we used this method to measure free monosaccharide concentrations in fecal samples from an infant transitioning from breast milk to weaning foods. Infant feces were collected after milk only, milk with water, milk with weaning food (squash), and weaning food only. The absolute and relative concentrations of free monosaccharides in each sample are listed in Table 3. In all five samples, thirteen out of sixteen monosaccharides were detected while allose, galacturonic acid, and Neu5Gc were below their limits of detection. In sample 1 and sample 3, where the infant was fed with only breast milk, the total concentrations of free monosaccharides were 22.2 mg g−1 and 21.5 mg g−1, respectively, corresponding to approximately 2.2% of the fecal content. This number decreased to 12.9 mg g−1 (1.29%) in sample 2 where the infant was fed with breast milk and water and 17.4 mg g−1 (1.74%) in sample 4 where the infant was fed with breast milk and squash. In sample 5, where the infant was fed with only squash, it further decreased to 3.0 mg g−1 (0.30%). Among the thirteen monosaccharides detected, galactose, fucose, glucose, GlcNAc, and Neu5Ac were the most abundant and these five monosaccharides accounted for 96.3% to 98.0% of the free monosaccharides in sample 1, 2, 3, and 4 where breast milk is the dominant component of the diet. Not coincidently, these are the major monosaccharide building blocks of milk components including lactose, human milk oligosaccharides, glycoproteins, and glycolipids.3 In sample 5, although they are still the most abundant compounds, the concentrations of these five monosaccharides decreased to 48.8–791 μg g−1, and their total relative abundance decreased to 84.9%. In contrast, the concentrations of ribose and GalNAc in sample 5 were 184 μg g−1 and 215 μg g−1, respectively, and were comparable to those in other samples. The relative abundances of ribose and GalNAc increased from about 1% to 6.13% and 7.17% in sample 5. Trace amounts of mannose, glucuronic acid, and plant monosaccharides including xylose, arabinose, rhamnose, and fructose were also detected in all five samples. The six compounds accounted for less than 2% of the total free monosaccharides in the fecal samples and may be present in the mother's milk through her diet. These results are consistent with the indications that carbohydrates from breast milk, which are mainly lactose, oligosaccharides, and N-/O-glycans, were better utilized in the infant gut than those from plants, which are mainly composed of polysaccharides, when transitioning from breastfeeding to weaning.41| Monosaccharides | Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Conc. (μg g−1) | %Abund. | Conc. (μg g−1) | %Abund. | Conc. (μg g−1) | %Abund. | Conc. (μg g−1) | %Abund. | Conc. (μg g−1) | %Abund. | |
| D-Ribose | 242 ± 11 | 1.09 | 103.8 ± 2.0 | 0.80 | 140 ± 4 | 0.65 | 137.1 ± 3.4 | 0.79 | 184 ± 8 | 6.13 |
| D-Xylose | 5.9 ± 0.1 | 0.03 | 4.7 ± 0.1 | 0.04 | 7.5 ± 0.2 | 0.03 | 10.9 ± 1.1 | 0.062 | 4.3 ± 60 | 0.14 |
| D-Arabinose | 8.3 ± 0.1 | 0.04 | 3.4 ± 0.6 | 0.03 | 6.1 ± 0.2 | 0.03 | 4.9 ± 0.7 | 0.028 | 1.8 ± 0.1 | 0.06 |
| L-Rhamnose | 6.8 ± 0.1 | 0.0 | 6.5 ± 0.3 | 0.05 | 8.3 ± 0.1 | 0.04 | 14.6 ± 1.4 | 0.08 | 5.4 ± 0.3 | 0.18 |
| L-Fucose | 4893 ± 209 | 22.06 | 4246 ± 60 | 32.82 | 5344 ± 91 | 24.81 | 3649 ± 135 | 20.95 | 518 ± 11 | 17.27 |
| D-Fructose | 99.3 ± 12.9 | 0.45 | 69.7 ± 16 | 0.54 | 108 ± 5 | 0.50 | 177 ± 66 | 1.02 | 23.1 ± 4.2 | 0.77 |
| D-Mannose | 26.9 ± 0.1 | 0.12 | 10.7 ± 0.1 | 0.08 | 32.4 ± 0.9 | 0.15 | 41.6± 1.0 | 0.24 | 6.7 ± 0.2 | 0.22 |
| D-Allose | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| D-Glucose | 4310 ± 73 | 19.43 | 2439 ± 89 | 18.85 | 2533 ± 59 | 11.76 | 1969 ± 60 | 11.30 | 407 ± 14 | 13.57 |
| D-Galactose | 7841 ± 246 | 35.36 | 4307 ± 44 | 33.29 | 6674 ± 72 | 30.99 | 5693 ± 123 | 32.69 | 778 ± 14 | 25.94 |
| D-Glucuronic acid | 43.4 ± 0.7 | 0.20 | 13.0 ± 0.2 | 0.10 | 49.1 ± 1.2 | 0.23 | 38.1 ± 0.2 | 0.22 | 15.9 ± 0.5 | 0.53 |
| D-Galacturonic acid | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| GlcNAc | 3683 ± 170 | 16.61 | 1270 ± 24 | 9.82 | 5048 ± 166 | 23.44 | 4402 ± 205 | 25.27 | 791 ± 58 | 26.38 |
| GalNAc | 251 ± 12 | 1.13 | 49.9 ± 1.2 | 0.38 | 133 ± 5 | 0.62 | 221 ± 8 | 1.27 | 215 ± 6 | 7.17 |
| Neu5Ac | 766 ± 6 | 3.46 | 412 ± 11 | 3.18 | 1455 ± 16 | 6.76 | 1058 ± 29 | 6.07 | 48.8 ± 0.9 | 1.63 |
| Neu5Gc | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Total | 22177 |
100 | 12936 |
100 | 21538 |
100 | 17416 |
100 | 2999 | 100 |
Analysis of other monosaccharides
The sixteen monosaccharides discussed above are the most common building blocks of carbohydrates in nature. To demonstrate the applicability of the MRM method for other monosaccharides, the PMP-derivatives of nine more common and uncommon monosaccharides including four amino sugars (glucosamine, galactosamine, mannosamine, ManNAc), two pentoses (apiose, lyxose) and three hexoses (altrose, talose, gulose) were also analyzed. The transitions and collision energies optimized for neutral and amino sugars above were utilized and shown in Table S2.† Using the same gradient at pH 8.2, talose, apiose, GalN and ManNAc were well resolved from their corresponding isomers. Whereas coelution was observed for ribose and lyxose, as well as mannose and gulose. When switching to an optimized gradient at pH 7.0, better separation was achieved between the above isomers. However, the two important plant pentoses, xylose and arabinose, could not be resolved. The coelution of altrose with galactose could also interfere with the quantitation (Fig. S4†). Therefore, the optimized UHPLC conditions at pH 8.2 were used in this study for analyzing the more common monosaccharides. It should be noted that gulose, talose, and altrose, tested here, occur very rarely in nature, and the quantitation of these sugars were not extensively tested in this work. For the specific analysis of these rare monosaccharides, further optimization of separation conditions is required. Other stationary phase supports such as phenyl-based reversed-phase supports could be tested for this purpose.Conclusions
The simultaneous quantitation of sixteen monosaccharides using dynamic MRM represents the most sensitive and comprehensive method for monosaccharide analysis thus far. This method provides high sensitivity with accurate measurement of many common monosaccharides including fructose, which is typically analyzed separately, and other neutral, amino and anionic monosaccharides. The monosaccharides were derivatized with PMP, with the exception of two sialic acids, while employing the speed of UHPLC/MRM-MS. UHPLC buffer and gradient were optimized to achieve isomeric separation within a 10-minute run. With the optimal derivatization and fragmentation conditions, the limits of detection for certain monosaccharides reached attomole level compared to picomole level obtained by previous methods, and the linear ranges spanned up to six orders of magnitude. Recovery experiments on fecal samples demonstrated the accuracy of this method for samples with complicated matrices. The analysis of nine other common and rare monosaccharides showed the applicability of this MRM method to monosaccharides from different sources with modification of UHPLC conditions. The method is readily applicable to various biological samples. The speed and accuracy of the method provides a tool for large sample size studies to determine the relationship of dietary carbohydrates and human health.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Research reported in this publication was supported in part by National Institute of General Medical Sciences under the award numbers R01GM049077 and RO1AT007079.References
- D. Barile and R. A. Rastall, Curr. Opin. Biotechnol., 2013, 24, 214–219 CrossRef CAS PubMed.
- S. Musilova, V. Rada, E. Vlkova and V. Bunesova, Benefic. Microbes, 2014, 5, 273–283 CrossRef CAS PubMed.
- J. T. Smilowitz, C. B. Lebrilla, D. A. Mills, J. B. German and S. L. Freeman, Annu. Rev. Nutr., 2014, 34, 143–169 CrossRef CAS PubMed.
- S. B. Han, C. W. Lee, M. R. Kang, Y. D. Yoon, J. S. Kang, K. H. Lee, W. K. Yoon, K. H. Lee, S. K. Park and H. M. Kim, Cancer Lett., 2006, 243, 264–273 CrossRef CAS PubMed.
- K. W. Yu, H. Kiyohara, T. Matsumoto, H. C. Yang and H. Yamada, Carbohydr. Polym., 2001, 46, 125–134 CrossRef CAS.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- M. J. Kailemia, D. Park and C. B. Lebrilla, Anal. Bioanal. Chem., 2017, 409, 395–410 CrossRef CAS PubMed.
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed.
- T. Murata, S. Takahashi, T. Takeda and S. Onishi, Abstr. Pap. Am. Chem. Soc., 1979, 115–115 Search PubMed.
- P. M. Medeiros and B. R. T. Simoneit, J. Chromatogr. A, 2007, 1141, 271–278 CrossRef CAS PubMed.
- J. S. Sawardeker, J. H. Sloneker and A. Jeanes, Anal. Chem., 1965, 37, 1602–1604 CrossRef CAS.
- C. Hoebler, J. L. Barry, A. David and J. Delortlaval, J. Agric. Food Chem., 1989, 37, 360–367 CrossRef CAS.
- J. Ma, L. Adler, G. Srzednicki and J. Arcot, Food Chem., 2017, 220, 100–107 CrossRef CAS PubMed.
- H. Yokota, K. Mori, H. Yamaguchi, H. Kaniwa and N. Saisho, J. Pharm. Biomed. Anal., 1999, 21, 767–774 CrossRef CAS PubMed.
- C. C. Y. Ip, V. Manam, R. Hepler and J. P. Hennessey, Anal. Biochem., 1992, 201, 343–349 CrossRef CAS PubMed.
- Z. Q. Zhang, N. M. Khan, K. M. Nunez, E. K. Chess and C. M. Szabo, Anal. Chem., 2012, 84, 4104–4110 CrossRef CAS PubMed.
- Y. Lv, X. B. Yang, Y. Zhao, Y. Ruan, Y. Yang and Z. Z. Wang, Food Chem., 2009, 112, 742–746 CrossRef CAS.
- H. L. Li, C. N. Long, J. Zhou, J. Liu, X. B. Wu and M. N. Long, Biotechnol. Lett., 2013, 35, 1405–1409 CrossRef CAS PubMed.
- E. Grill, C. Huber, P. Oefner, A. Vorndran and G. Bonn, Electrophoresis, 1993, 14, 1004–1010 CrossRef CAS PubMed.
- S. Honda, E. Akao, S. Suzuki, M. Okuda, K. Kakehi and J. Nakamura, Anal. Biochem., 1989, 180, 351–357 CrossRef CAS PubMed.
- J. A. Saba, X. D. Shen, J. C. Jamieson and H. Perreault, J. Mass Spectrom., 2001, 36, 563–574 CrossRef CAS PubMed.
- D. J. Strydom, J. Chromatogr. A, 1994, 678, 17–23 CrossRef CAS.
- P. Zhang, Z. F. Wang, M. M. Xie, W. L. Nie and L. J. Huang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2010, 878, 1135–1144 CrossRef CAS PubMed.
- X. D. Wu, W. Jiang, J. J. Lu, Y. Yu and B. Wu, Food Chem., 2014, 145, 976–983 CrossRef CAS PubMed.
- B. Rühmann, J. Schmid and V. Sieber, J. Chromatogr. A, 2014, 1350, 44–50 CrossRef PubMed.
- J. C. Huang, M. J. Kailemia, E. Goonatilleke, E. A. Parker, Q. T. Hong, R. Sabia, J. T. Smilowitz, J. B. German and C. B. Lebrilla, Anal. Bioanal. Chem., 2017, 409, 589–606 CrossRef CAS PubMed.
- E. W. Song, S. Pyreddy and Y. Mechref, Rapid Commun. Mass Spectrom., 2012, 26, 1941–1954 CrossRef CAS PubMed.
- A. R. S. Ross, S. J. Ambrose, A. J. Cutler, J. A. Feurtado, A. R. Kermode, K. Nelson, R. Zhou and S. R. Abrams, Anal. Biochem., 2004, 329, 324–333 CrossRef CAS PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593–599 CrossRef CAS PubMed.
- Q. T. Hong, L. R. Ruhaak, S. M. Totten, J. T. Smilowitz, J. B. German and C. B. Lebrilla, Anal. Chem., 2014, 86, 2640–2647 CrossRef CAS PubMed.
- G. Xu, J. C. C. Davis, E. Goonatilleke, J. T. Smilowitz, J. B. German and C. B. Lebrilla, J. Nutr., 2017, 147, 117–124 CrossRef CAS PubMed.
- L. A. Hammad, M. M. Saleh, M. V. Novotny and Y. Mechref, J. Am. Soc. Mass Spectrom., 2009, 20, 1224–1234 CrossRef CAS PubMed.
- L. A. Hammad, D. Z. Derryberry, Y. R. Jmeian and Y. Mechref, Rapid Commun. Mass Spectrom., 2010, 24, 1565–1574 CrossRef CAS PubMed.
- J. Han, K. Lin, C. Sequria, J. C. Yang and C. H. Borchers, Electrophoresis, 2016, 37, 1851–1860 CrossRef CAS PubMed.
- Y. G. Chua, S. H. Chan, B. C. Bloodworth, S. F. Y. Li and L. P. Leong, J. Agric. Food Chem., 2015, 63, 279–289 CrossRef CAS PubMed.
- C. X. Wu, L. Xia, L. J. Liu, F. L. Qu, Z. W. Sun, X. N. Zhao and J. M. You, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2016, 1011, 14–23 CrossRef CAS PubMed.
- X. Wang, L. Zhang, J. Wu, W. Xu, X. Wang and X. Lu, Food Chem., 2017, 220, 198–207 CrossRef CAS PubMed.
- Y. Hu, T. Wang, X. Yang and Y. Zhao, Carbohydr. Polym., 2014, 102, 481–488 CrossRef CAS PubMed.
- Z. Zhang, N. M. Khan, K. M. Nunez, E. K. Chess and C. M. Szabo, Anal. Chem., 2012, 84, 4104–4110 CrossRef CAS PubMed.
- B. Ruhmann, J. Schmid and V. Sieber, J. Chromatogr. A, 2014, 1350, 44–50 CrossRef CAS PubMed.
- J. E. Koenig, A. Spor, N. Scalfone, A. D. Fricker, J. Stombaugh, R. Knight, L. T. Angenent and R. E. Ley, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4578–4585 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01530e |
| This journal is © The Royal Society of Chemistry 2018 |