
On-chip conductometric detection of short DNA sequences via electro-hydrodynamic aggregation†
B.
Venzac
 *abc,
M. L.
Diakité
abc,
D.
Herthnek
d,
I.
Cissé
e,
U.
Bockelmann
e,
S.
Descroix
abc,
L.
Malaquin
abcf and
J.-L.
Viovy
*abc
*abc,
M. L.
Diakité
abc,
D.
Herthnek
d,
I.
Cissé
e,
U.
Bockelmann
e,
S.
Descroix
abc,
L.
Malaquin
abcf and
J.-L.
Viovy
*abc
aLaboratoire Physico Chimie Curie, Institut Curie, PSL Research University, CNRS UMR168, 75005, Paris, France. E-mail: bastienvenzac@gmail.com; jean-louis.viovy@curie.fr
bSorbonne Universités, UPMC Univ Paris 06, 75005, Paris, France
cInstitut Pierre-Gilles de Gennes, 75005, Paris, France
dScience for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Solna, Sweden
eLaboratoire Nanobiophysique, École Supérieure de Physique et de Chemie Industrielles de la Ville de Paris, Centre National de la Recherche Scientifique (CNRS), Unité Mixte de Recherche (UMR) Gulliver 7083, 75005 Paris, France
fUniv. de Toulouse, LAAS, F-31400 Toulouse, France
First published on 24th November 2017
Abstract
Fluorescence measurement is the main technology for post-amplification DNA detection in automated systems. Direct electrical reading of DNA concentration in solution could be an interesting alternative to go toward more miniaturized or less expensive devices, in particular in the pathogen detection field. Here we present the detection of short bacterial biomarkers with a direct impedancemetric measurement, within solutions of amplified and elongated DNA sequences in a microchannel. This technology relies on the electrohydrodynamic instability occurring in solutions of long charged macromolecules in a strong electric field. This instability specifically induces the aggregation of long DNAs and triggers conductivity variations that can be monitored by on-contact conductometry. An innovative isothermal amplification and elongation strategy was developed, combining SDA and HRCA reactions, in order to yield long DNAs suitable to be detected by the above principle, from a dilute initial DNA target. In contrast with previous label-free detection methods, this new strategy is very robust to matrix effects, thanks to the unique molecular weight dependence of the instability, coupled with this specific DNA amplification strategy. We demonstrate the detection of a 1 pM gene sequence specific to Staphylococcus aureus, in a portable system.
Introduction
In the field of clinical diagnostics, detection of pathogenic biomarkers through lab-based techniques such as qPCR1 or ELISA2 has enabled quicker and more precise diagnosis as compared to classical microorganism cultures. Automated systems developed allow for a decrease in analysis time and a reduction of human-based errors, such as the GeneXpert System from Cepheid, the Razor-EX from Biofire Defense or the Cobas Liat system from Roche. In term of cost, these systems have been shown to be good competitors to classical tests3 and are able to treat raw samples, extract bacterial or viral DNA, and to amplify and detect low amounts of DNA via qPCR. These systems offer a good sensitivity and excellent specificity (90% and 99% respectively for tuberculosis detection with the GeneXpert).4 But they still rely on optical detection of fluorescence, require expensive sensors, light sources and a robust and bulky configuration to maintain a good alignment of the optical elements. Cheaper, simpler and portable systems were developed in academia using smartphone camera and low-cost optics5–7 for point-of-care diagnosis in low-resources settings, but they could not compete in terms of performances for precise applications.Detection of amplified DNA with a direct electrical reading appears as an intriguing alternative to optics. Electrodes are easy to integrate into miniaturized cartridges and microfluidic systems. Moreover, techniques to fabricate electrodes exist for several substrates: silicon, glass, plastic,8 paper,9 fabrics…10 thus enabling an easy integration and widespreading of the technology. However, direct conductometric detection is very sensitive to the solution's content in small ions, making it mostly unsuitable to direct DNA detection in raw sample. Several strategies to overcome this limitation were proposed: nanopore sensing of large amplicons,11,12 electrochemical detection of captured DNAs,13 metallization of DNA bridges between two electrodes.14,15
Our group previously proposed a new concept for the conductometric detection of long DNA molecules in a microfluidic format,16 using an electrohydrodynamic aggregation of DNA17,18 (ElectroHydroDynamic Aggregation, EHDA). When subjected to a strong AC electric field (>100 V cm−1, 10 Hz) in a confined space, a homogeneous solution of concentrated DNA molecules longer than 5 kbp is destabilized, and highly dense regions of DNA called aggregates are created. The conductivity of these aggregates differs from that of the solution and this variation could be monitored using planar electrodes. This technique is however limited by the mechanism of electrohydrodynamic aggregation that occurs only for DNA longer than 5 kbp.17 This gives rise to two major limitations. First, most of the known genomic biomarkers are smaller than 1 kbp, so their PCR amplicons cannot be detected directly by aggregation. Second, an amplification step is required to reach a number of long DNA molecules (typically around 1 nM in Tris-EDTA buffer)19 that is sufficient to induce electrohydrodynamic aggregation and lead to a measurable variation of conductivity. For instance, long-range PCR20 was successfully applied to the amplification of fragments above 5 kbp that were further detected through electrohydrodynamic aggregation.16 The use of such amplification method, strictly limited to long DNAs, was however a strong limitation towards a wide application of this detection principle.
Detecting fragments with the usual size of genomic biomarkers, typically from one to few hundred base pairs, with EHDA, would thus require both amplification and elongation of DNA. To our knowledge, no amplification technique allows the production of long DNAs (over 5 kbp) with an amplification factor above 106 in a single step. Loop-mediated Isothermal Amplification (LAMP)21 creates longer products than the target, without reaching these lengths. Rolling-Circle Amplification (RCA) can elongate targets until 100 kbp,22,23 and several exponential upgrades have been developed: Hyperbranched RCA (HRCA),23,24 Primer Generation-RCA (PG-RCA)25 and Circle-to-Circle Amplification (C2CA).26 The two first ones mainly produce DNA smaller than 5 kbp, while C2CA requires several steps and sometimes the use of magnetic particles.
In this paper, we developed a 3-steps elongation and amplification reaction combining a first amplification step with a Strand Displacement Amplification (SDA)27–29 and an elongation step with a HRCA. This protocol provides a simple and straightforward method for both the elongation and amplification of DNA oligonucleotides, starting from 1 pM of 165 nt-long targets and producing more than 100 nM of products longer than 50 kbp. It only required addition of reagents, without washing or purification steps, and 3 temperature plateaus (see Fig. 1a). Amplification products prepared by this new protocol were successfully detected via an improved, fully portable EHDA detection system, with no limitation in the size of the recognition sequence, thus opening the route to a vast range of point-of-care applications.
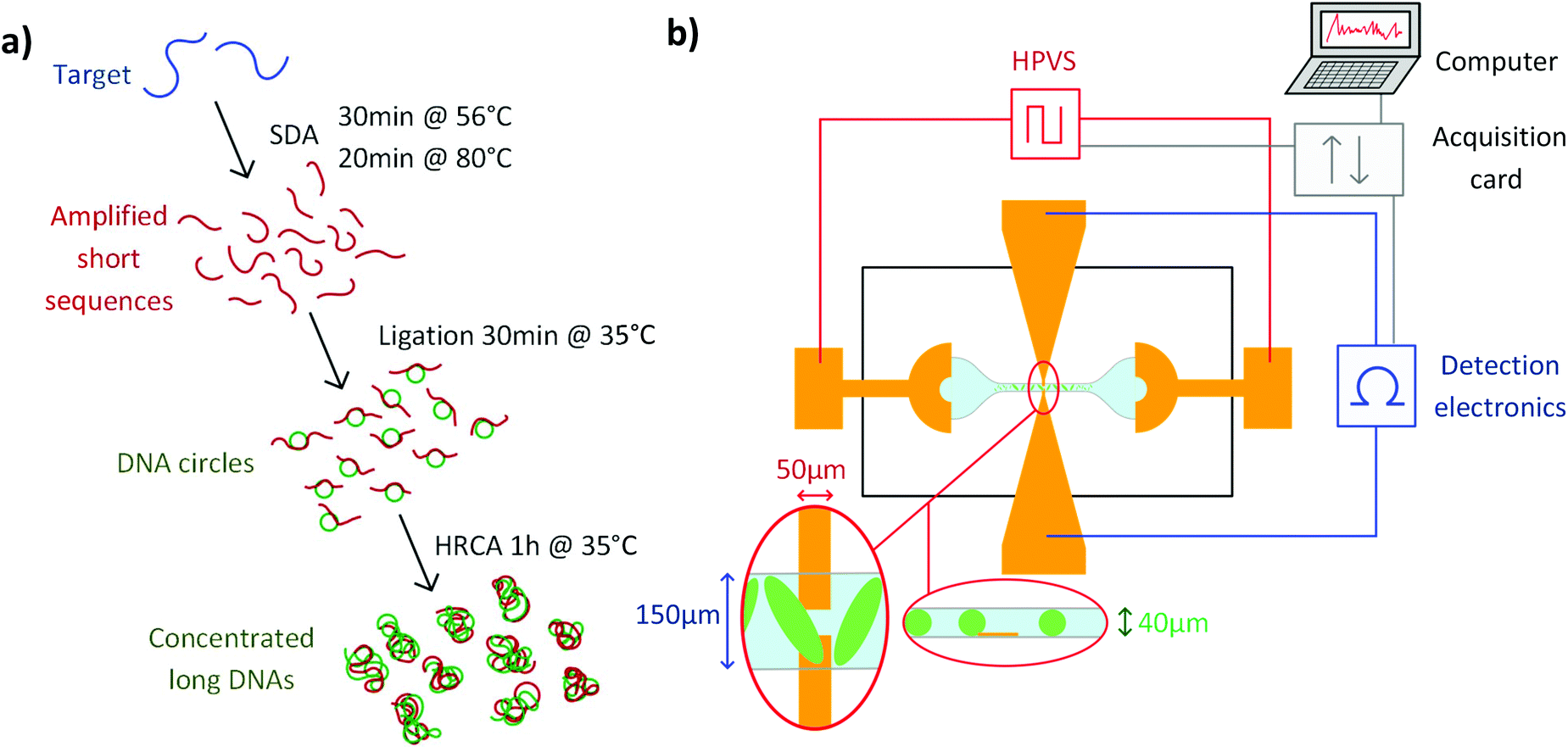 | ||
| Fig. 1 (a) Simplified protocol for the amplification and elongation reaction. (b) Schematics of the detection system, including the microchip, the detection and excitation electronics, the computer and acquisition card. Close views of the inter-electrode space from top and from the side are represented in the red ellipses. | ||
Materials and methods
Microfabrication
The microfluidic device was composed of a PDMS chip mounted on a glass substrate patterned with gold electrodes (see Fig. 1b). Microfluidic channel were fabricated by PDMS casting. Sylgard 184 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base/curing agent ratio) was poured and cured on a brass mould created by micro-milling (Minitech Machinery). The milled structures were composed of a long channel (3 mm × 150 μm × 40 μm) connected to two 2.2 mm-wide circular chambers using a progressive channel enlargement, to avoid strong electric field gradients. 500 μm-deep, 1.8 mm-wide holes were milled inside the two chambers and transferred as pillars in the PDMS replica (see ESI Fig. S1†). These pillars were used as guides to align 2 mm-puncher in the centre of the circular chambers, producing two 2 mm-wide reservoirs in the cured PDMS with high reproducibility.
:1 base/curing agent ratio) was poured and cured on a brass mould created by micro-milling (Minitech Machinery). The milled structures were composed of a long channel (3 mm × 150 μm × 40 μm) connected to two 2.2 mm-wide circular chambers using a progressive channel enlargement, to avoid strong electric field gradients. 500 μm-deep, 1.8 mm-wide holes were milled inside the two chambers and transferred as pillars in the PDMS replica (see ESI Fig. S1†). These pillars were used as guides to align 2 mm-puncher in the centre of the circular chambers, producing two 2 mm-wide reservoirs in the cured PDMS with high reproducibility.
Electrodes were created by deposition of chromium (adhesion layer) and gold on a circular, 2-inches glass slide. A first photolithography step was used to create the electrodes patterns in a 2 μm-thick S1813 positive resist (Microposit). The electrode design integrated two electrodes pairs: excitation electrodes were composed of a crescent shape of 3 mm of diameter linked to a bonding pad. The detection electrodes, facing each other in the middle of the channel, had a width of 50 μm and a gap of 50 μm. 10 nm of chromium and 100 nm of gold were deposited by evaporation (Auto 500 Edwards). The two electrode pairs were revealed by lift-off after an ultrasonic bath with acetone. The PDMS part and the patterned glass slide were bonded together by exposition to oxygen plasma and alignment of the excitation electrodes at the bottom of the reservoirs and the detection electrodes at the middle of the channel. Devices were stored for 48 h at 70 °C prior to experiments in order to restore the hydrophobicity of the PDMS surface. Electric wires were then bonded to the connecting pads.
Electronics
Our detection scheme relied on contact conductivity measurement, meaning that no dielectric layer was inserted between the solution and the detection electrodes. This configuration is more sensitive than contactless detection, and does not require electronics working at higher frequencies.30 It is, however, more delicate to implement. In particular, any coupling between the high electric field used for aggregates generation and the detection electronics could lead to electrolysis, bubble formation and electronic parasites. For that, we used a balanced and isolated electronic module for the conductometric measurement, as previously described.16 In short, a resistor bridge was used to monitor the voltage variation between the two detection electrodes when the solution conductivity changed, under an imposed current (31.25 kHz, 220 nA). This tension was then amplified and recorded via a NI-USB 6380X National Instrument acquisition card. Low-cost ISO124P isolation amplifiers were located between the electronic module and the National Instruments card, to avoid transfer of the common mode voltage between the different elements of the set-up. Balanced circuit was designed to eliminate electromagnetic interferences. Finally, the detection module was supplied with 6 batteries (9 V) to avoid ground loops.The AC, symmetric high electric field was generated by a dedicated high voltage power supply (HVPS). A PIC microcontroller was used to generate a digital square signal with 52% duty cycle, 10 ms rise and fall times and 100 ms period. After conversion to an analogue signal, a PA343 amplifier increased the voltage to ±170 V centred on 0 V. A symmetric signal was produced by inverting the signal for the second excitation electrode. The excitation module was supplied by two 12 V-lithium batteries and the voltage parameters could be changed from dedicated software by USB link, and stored into an internal memory. The schematics of the excitation module can be found in the ESI.†
Signal acquisition
The tension measured and amplified by the detection module was acquired via a National Instrument acquisition card with a sample rate of 312.5 kHz and cut into samples of 100 ms, triggered by a digital signal synchronized with the period of the excitation field from the HVPS. To remove the artefacts produced by the high electric field inversion, only 45 ms of constant voltage in each period were kept. Demodulation at 10 Hz was then performed to obtain the envelope modulation (Fig. S3†).Wavelet treatment
Baseline drifts and electronic noise above 10 Hz are usual in conductometric measurement and have to be eliminated after acquisition. The aggregation process is random and does not lead to periodic events. Information about the physically significant conductivity changes due to the passage of aggregates were extracted from the raw signal with a Matlab discrete wavelet transform, as described previously.16 In short, wavelet transform was achieved by convoluting the conductometric signal with patterns called wavelets, with temporal extensions and shapes tuned to roughly mimic the shape and duration of the peaks analysed. From the coefficients obtained by convolution, a signal centred on 0 V, containing only the aggregate contributions could be reconstructed (Fig. S3†). To characterise the amplitude and the number of the EHDA-related peaks, a quantification parameter Q equal to the mean of the absolute value of the reconstructed signal is used. The detail of the wavelet treatment can be found in ESI.†Experimental procedure
The microfluidic chip was first connected to the excitation and detection modules. The samples used were raw HRCA products or diluted 48 kbp Lambda-DNA (from New England Biolabs) in a 1× Tris-EDTA buffer. The 8 μl sample was injected into the microchannel and reservoirs with a micropipette. The sample level was equilibrated in the two reservoirs with a syringe to avoid hydrostatic flows. Excitation and detection modules were controlled via a Labview program. The high electric field was generated during 40 s: at this point, aggregates reached a quasi-static regime. The detection signal was then recorded and demodulated during 100 s. 3 measurements separated by at least 2 minutes were performed for each experiment. As a facultative control during experiment and to check the apparition of aggregates, live fluorescent monitoring was achieved with the FITC channel of a Zeiss Axiovert 135TV epifluorescent microscope and a Nikon DS-Q1Mc camera, with a final concentration of SyBr Green DNA dye of 5X, added before injection in the microsystem.SDA–HRCA
All the enzymes and reagents were purchased from New England Biolabs and the oligonucleotides were from Integrated DNA Technologies. Oligonucleotide sequences are given in ESI.† Human genomic DNA was purchased from Sigma Aldrich.SDA protocols and oligonucleotide sequences were adapted from Pr. Yager's papers.29,31 The SDA mix was composed of 1× Cutsmart buffer, 1 mM of dNTPs, 50 nM of Bump Primer 1, 50 nM of Bump Primer 2, 250 nM of Extension Primer 1, 500 nM of Extension Primer 2. Ldh1 gene target was added to the mix before the addition of 800 mU μl−1 of Bst 2.0 Warmstart Polymerase and 200 mU μl−1 of Nt·BbvCl nicking enzyme. The complete mix was incubated at 56 °C during 30 min in a thermocycler (Biometra), followed by a heat inactivation step at 80 °C during 20 min.
For the ligation step, 2.5 μl of raw product were then mixed with the HRCA components to obtain a 10 μl HRCA mix. This solution was composed of 0.75× T4 ligase buffer (final concentration), 1 mM of dNTP, 1.25 μg μl−1 of BSA, 100 nM of RCA Padlock probe and 25 mU μl−1 of T4 ligase. The mix was incubated at 37 °C during 30 minutes.
HRCA was then performed by adding 0.5 μl of HRCA Primer 1, 0.5 μl of HRCA Primer 2 (final concentrations 1 μM) and 5 U of Phi29 polymerase to the previous mix. Polymerisation was realised during 1 h at 35 °C, followed by a facultative heat inactivation at 65 °C during 2 minutes.
Results and discussion
Long DNA aggregation via electrohydrodynamic instabilities
Electrohydrodynamic aggregation of long poly-electrolytes has been demonstrated in several conditions: square glass capillaries,17 Hele-Shaw geometry18 and rectangular glass–PDMS hybrid microchannel.16 The main parameters which favour the aggregation are well known.19 Aggregation is increased when: the DNA size and concentration increase, the electric field intensity increases, the electric field frequency decreases, the buffer conductivity decreases. The maximum aggregate width is similar to the channel height. We first checked optically the behaviour of long λ-DNA in a 1× TE buffer inside our microsystem under a high electric field of 680 V cm−1, square, 10 Hz and 52% of duty cycle.As seen on Fig. 2a–e, aggregates were visible for concentration above 300 pM. Aggregates volume increased as the concentration increased. At the highest concentrations, aggregates were elongated and tilted with regards to the field direction at an angle of roughly 45°, as predicted by an analytical model of Isambert et al.18 The 52% duty cycle of the square high electric field led to a net movement of the aggregates from the right to the left of the channel, used to transform the spatial distribution of aggregates into a temporal evolution of the detected conductometric signal.
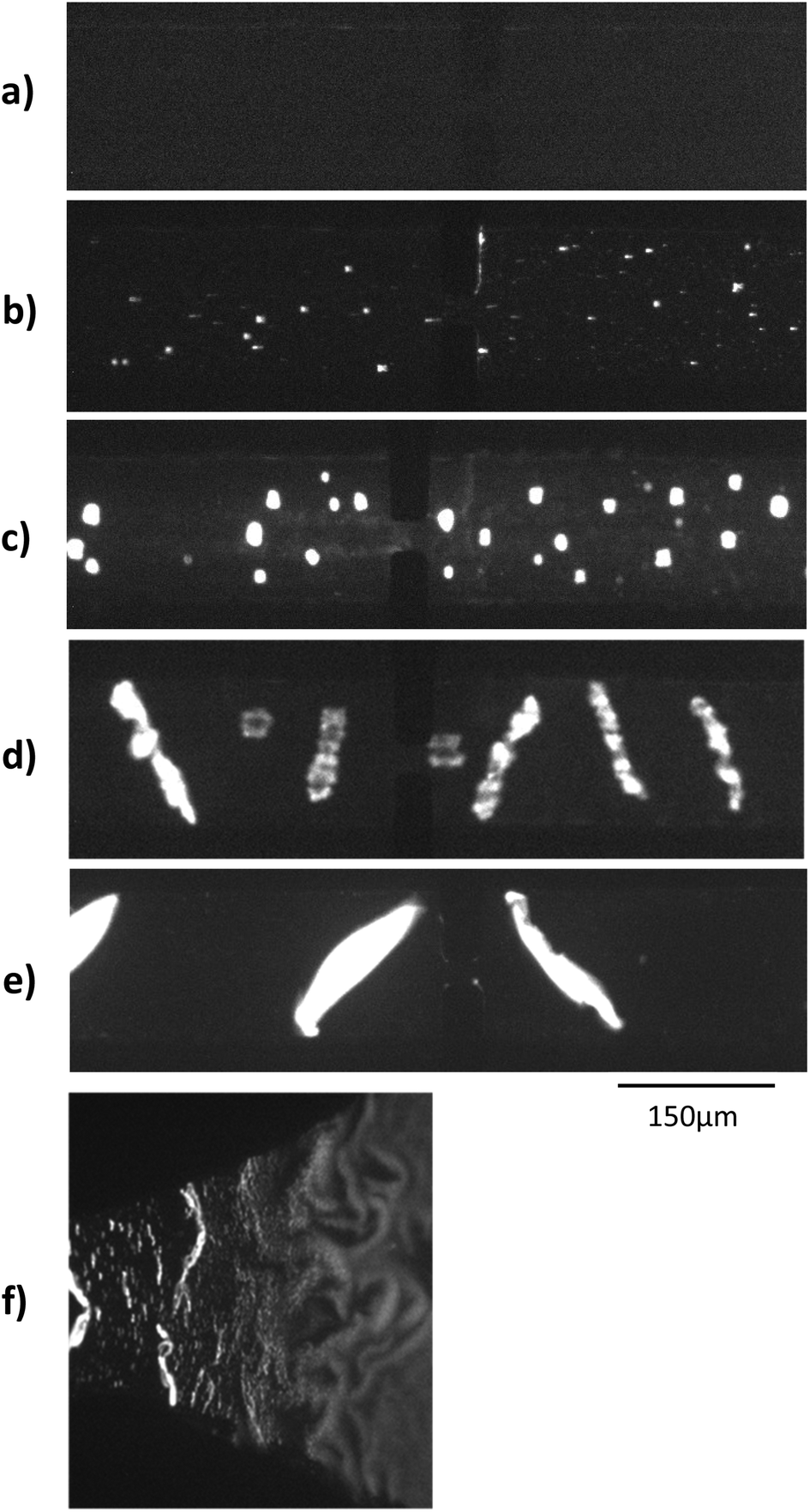 | ||
| Fig. 2 Fluorescent microscopic observation of Lambda-DNA aggregation, for different initial concentration: (a) 0 nM (b) 30 pM (c) 300 pM (d) 1.5 nM (e) 2.7 nM. Panel (f) corresponds to the transition between the reservoir and the channel for a Lambda-DNA concentration of 1.5 nM. | ||
In a first design similar to that in Diakité et al. paper,16 longitudinal electric field was generated by platinum wires plunged into the reservoirs. This configuration led to solution level changes and hydrostatic flow when the wires were immerged after the channel filling. Also, the microchip consisted of a long channel directly connected to reservoirs, with a combined 2D narrowing and 3D step at the channel entrance. This induced a strong electric field gradient, which tended to stimulate the formation of aggregates and their trapping at the channel entrance. The microchip structure was thus modified to increase the control over the aggregate formation and movement. Gold planar electrodes were patterned at the bottom of the reservoirs in order to generate a reproducible high electric field and to eliminate the hydrostatic flow produced by the previous platinum wires (see Fig. 1b). With the gentle transition between the 150 μm-wide channel and the 2 mm-wide reservoir, DNAs started to aggregate only in the slowly narrowing transition (Fig. 2f).
The new HVPS also offers a better control of electric field characteristics, as compared to the previous analogue one, while being more miniaturized (20 cm × 10 cm), battery-supplied and computer-free.
Conductometric measurement
Long, concentrated DNA formed optically detectable aggregates under high electric field in our microsystem. Direct-electrical detection of the conductivity change produced by the DNA accumulation could offer a label-free, easily miniaturizable technique.We chose to detect these conductivity variations with a contact-mode measurement, using planar electrodes directly in contact with the solution. A balanced, isolated detection module allowed decoupling the low-field detection module from the high electric field. The quick changes of polarity of the strong electric field, however, still produced a residual noise via capacitive or inductive coupling. These parasites were successfully removed by implementing a new acquisition procedure. One example of the signal acquired during aggregation of λ-DNA (1.5 nM) in 1× TE-buffer, after treatment and demodulation, is visible in Fig. 3, with a blank control. As usual in conductometric measurements, the blank control was subjected to various perturbations, like slow baseline shift (pH changes), electric noise above 10 Hz and quick variations of the signal, attributed to bubble movements in the reservoirs. The passage of aggregates above the detection electrodes led to voltage peaks with amplitudes around 1 mV and durations around 2 seconds. The time interval between the passages of two adjacent aggregates was not fixed and varied along the course of the measurement, as well as the aggregate size. To isolate the conductivity peaks due to DNA aggregation, a wavelet treatment was developed. A reconstructed signal was obtained without the slow baseline variation and the noise around 10 Hz. To obtain a quantification of the number and amplitude of these conductivity peaks, a quantification parameter Q was calculated as the average of the mean value of the reconstructed signal, on the intervals without artefacts. On Fig. 4, this quantification was performed on measurements of the conductivity during aggregation of the Lambda-DNA solutions presented on the Fig. 2. λ-DNA concentrations above 1.5 nM in TE 1× buffer were successfully detected.
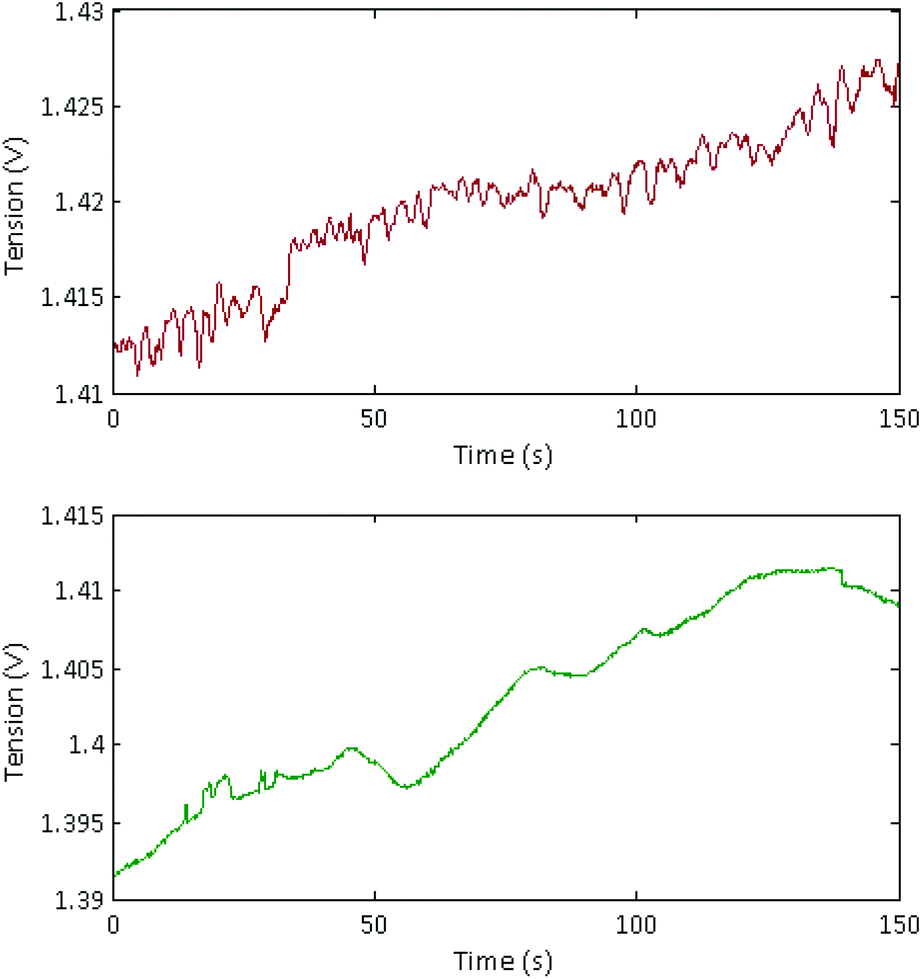 | ||
| Fig. 3 Signal acquired and demodulated during the conductometric detection protocol. Top: Conductometric signal from an aggregated Lambda-DNA solution (1.5 nM) in 1× TE buffer. Bottom: Blank control with 1× TE buffer. | ||
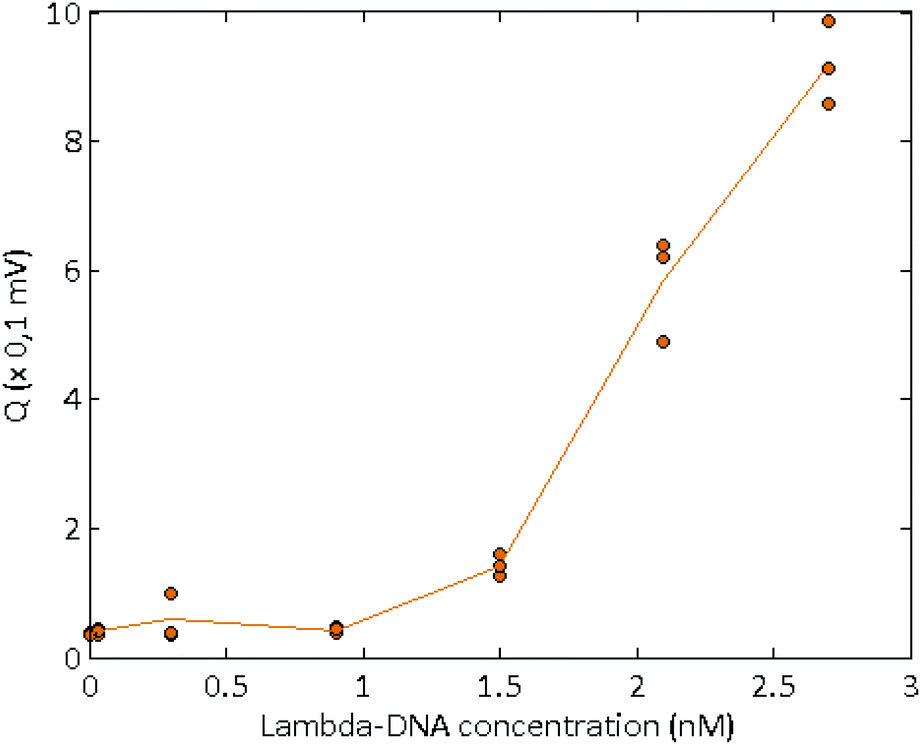 | ||
| Fig. 4 Quantification parameters obtained for the conductometric detection of several solutions of Lambda-DNA, at various concentrations in 1× TE buffer. | ||
Detection of short sequences amplified by SDA–HRCA
In order to detect the presence in solution of short biomarkers, we developed a new elongation and amplification scheme combining SDA and HRCA. SDA is an isothermal amplification using a polymerase with a strand-displacement activity, and a nicking enzyme.27–29 The mechanism is illustrated in the ESI Fig. S4.† The sequence to be amplified hybridizes with an extension primer partly complementary to the target (Fig. S4a†). This primer also carries a recognition sequence for a nicking enzyme on the second part. Both the target and the extension primer are extended by the polymerase (Fig. S4b†). The nicking enzyme is then able to recognise the double-stranded nicking site and to cut only the newly extended primer (Fig. S4c†). The nicked strand acts as a priming site. The polymerase can thus elongate the cut strand from the 3′ side of the nick, and remove the ssDNA located between the nick and the 5′ end of the template (Fig. S4d†). This nicking/polymerizing cycle is repeated and leads to linear accumulation of ssDNA. These ssDNAs hybridize to primers (Fig. S4e†), and are elongated to create new dsDNA templates for the nicking/polymerizing cycle, leading to exponential amplification (Fig. S4f–h†). To obtain a single-stranded and delimited target, an initiation step is necessary. Extension primers containing the specific sequence for the nicking enzyme in their non-template specific part were elongated after hybridization with the separated template. Bumper primers bind the template behind the primer, and the polymerase removes the elongated primer while replicating the substrate from the bumper. The same process occurs again with this removed ssDNA as template, leading to a dsDNA flanked with nicking-specific sequences at both ends, ready for the amplification.Here, we use a 165 nt-target corresponding to the NO˙-inducible L-lactate dehydrogenase (ldh1) gene, specific to Staphylococcus aureus. In Fig. 5, the amplification products were visualized on an agarose gel (parameters on ESI†), for several initial concentrations of target. One can observe a background and parasite amplifications for target concentrations below 10 pM (initial concentration of the sample before dilution in the reaction mix), which is a well-known drawback of SDA,27 which nevertheless does not limit the use of this technique for sensitive amplification of bacterial traces when coupled with a detection method which specifically detect the amplicon sequence.29,32 The target amplicons were visible above a target initial concentration of 1 pM. The asymmetry in extension primer concentrations allowed the formation of an excess of ssDNA products at the end of the reaction.
 | ||
| Fig. 5 Agarose gel of SDA products, for different ldh1 target concentrations, stained with SyBr Gold. | ||
In order to detect these with EHDA, SDA fragments have to be elongated above 5 kbp. For that, we first selected the RCA method, which is known to yield large multimers of a single padlock probe, in the presence of target. During RCA, two adjacent sequences in the SDA single-stranded amplicon are recognized by a RCA Padlock probe33 (ESI Fig. S5a†). The probe is then circularized by a ligase only if the probe is completely complementary to the target34 (Fig. S5b†). This step is highly specific and circularization does not occur with the parasite products of the SDA. During a second step, a polymerase with a strand-displacement activity and a primer complementary to probe skeleton sequences is added (Fig. S5c†). The complementary primer binds to the padlock probe and is elongated by a polymerase with a strand-displacement activity, which rolls around the DNA circle while extending a concatemeric ssDNA that can attain 100 kbp.22,23 To monitor the elongation, the RCA products were introduced into the microdevice and tested for aggregation with both optical recording and conductometric measurements.
In a first series of SDA–RCA experiments, which did not involve an inactivation step at the end of the SDA, false positive results were obtained. We attributed it to the simultaneous presence of the T4 ligase and the Bst 2.0 polymerase, favouring non-templated circularization of the padlock probe, and added an inactivation step after the SDA to hinder the polymerase activity in the subsequent amplification. This procedure eliminated the false positives (see Fig. 6a).
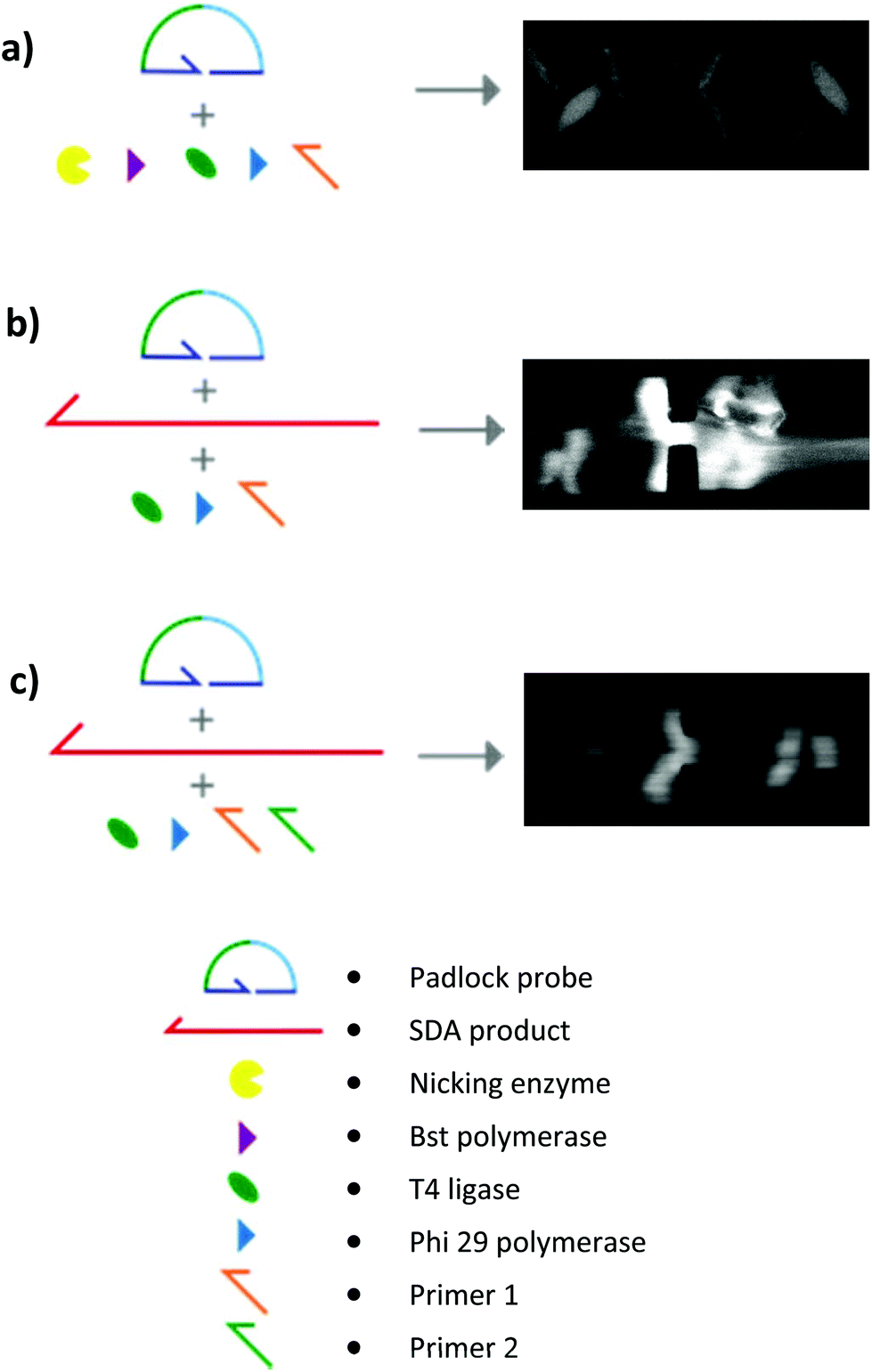 | ||
| Fig. 6 Several conditions for the elongation step. (A) RCA elongation without ldh1 target and without inactivation step in the SDA. The nicking enzyme and the Bst polymerase were still active during the RCA, leading to false positive, as shown on the fluorescent DNA aggregate during the conductometric detection. (b) RCA elongation with ldh1 target and an inactivation step in the SDA. Presence of highly dense, fluorescent DNA network in the solution. (c) HRCA elongation with ldh1 target and an inactivation step in the SDA. Presence of fluorescent DNA aggregation by EHDA. | ||
By microscopy, however, we could observe that the RCA products were inhomogeneously distributed in the solution, with regions comprising a highly dense DNA network and regions with no visible traces of DNA (Fig. 6b). This in turn affected the reproducibility and robustness of conductometric detection. This drawback was overcome by replacing simple RCA by Hyperbranched RCA. In HRCA, a second primer allows the generation of dsDNA branches, which are then displaced, multiplying the total number of molecules in the system and increasing the molecular mass distribution width. The production of dsDNA branches in HRCA improved the homogeneity of the solution and the aggregation of the HRCA products in the microdevice as compared to the RCA ones (Fig. 6c). Results of optical recording and conductometric measurements are presented in Fig. 7. The negative control, with no ldh1 target for the SDA step, did not show any visible aggregates, even if a large amount of parasite DNA was present in the SDA products. This is a strong advantage of coupling SDA with HRCA (or RCA), which has a strong specificity regarding the target sequence. Visible aggregates appeared for [ldh1] = 100 fM and aggregates were detectable by conductometry for ldh1 concentration above 1 pM. This result is in accordance with the amplification results of the SDA shown on Fig. 5, and the EHDA detection was even more sensitive than gel electrophoresis.
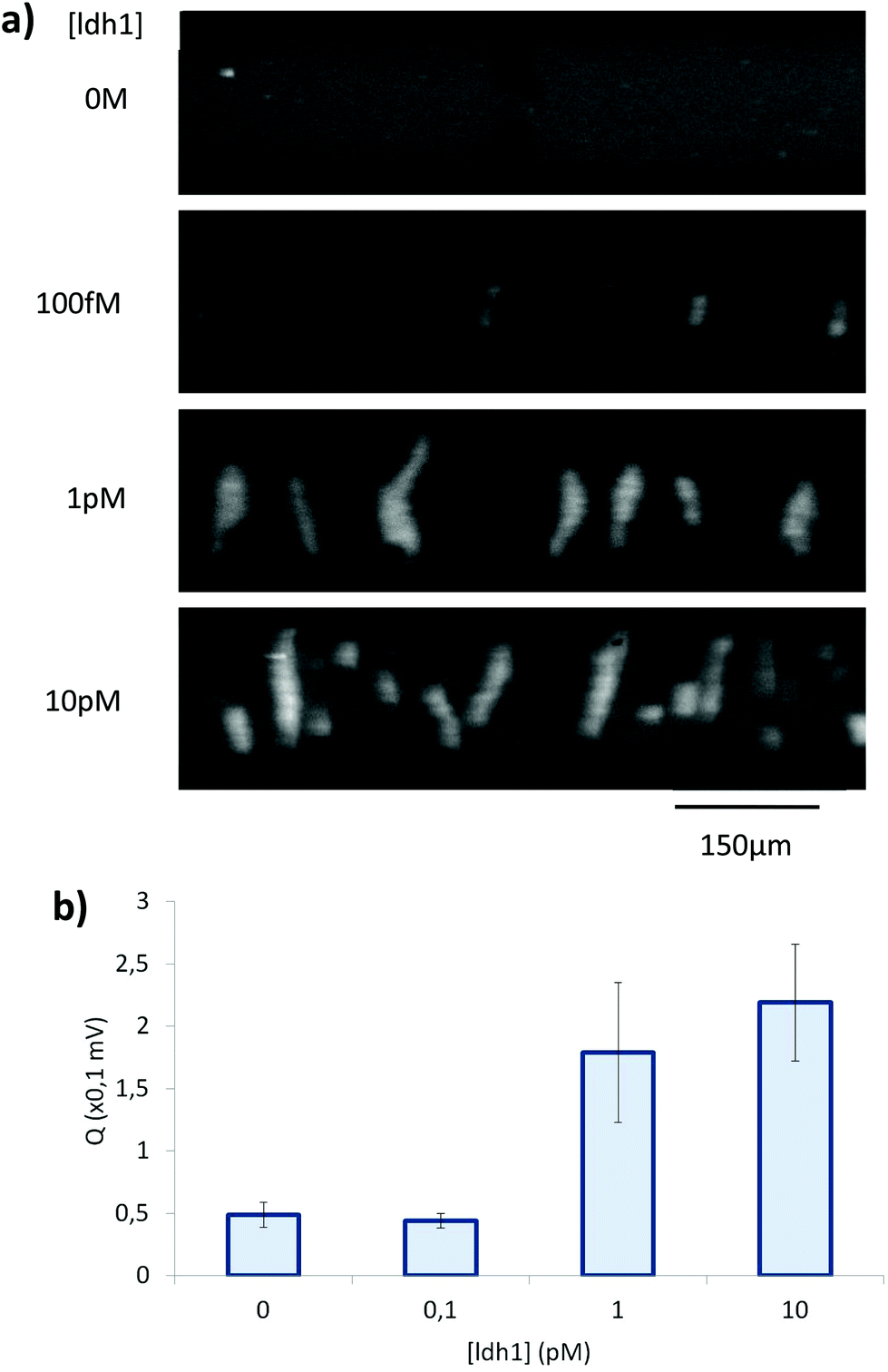 | ||
| Fig. 7 (a) Pictures taken during fluorescent microscopic observation of SDA–HRCA product aggregation, for different initial concentration of target. (b) Conductometric detection of SDA–HRCA product aggregation for 3 experiments per initial concentration of target. | ||
Optimisation of the buffer conductivity was critical, as both aggregation efficiency and detection sensitivity decreases when the buffer conductivity increases. In our system, the final conductivity of the optimised buffer used for HRCA and detection is around 500 mS m−1 (versus of around 3.5 mS m−1 for 1× TE buffer). This conductivity difference leads to a large difference between the limit of detection observed for λ-DNA in TE buffer (1.5 nM) and the final concentration of detected HRCA products, which we estimate around 100 nM (unpublished data).
Specificity of the detection method
The ligation step present in HRCA is known to be highly specific, with the possibility to discriminate a single-nucleotide mutation for an optimized protocol.23 Moreover, the EHDA detection method only detects long DNA, and is thus insensitive to contamination by short DNA either produced by the SDA background reaction or present in the original sample. To confirm this property, we first replaced the ldh1 oligonucleotide by another sequence with a similar length corresponding to the mecA gene, also present in the methycillin-resistant Staphylococcus aureus. We then performed the detection of the ldh1 gene in a highly concentrated human genomic DNA background (200 ng, corresponding to approximately the DNA content of 30000 cells).
Without ldh1 target in the solution, the presence of 10 pM of the mecA sequence or 200 ng of human genomic DNA does not trigger the formation of DNA aggregates, leading to conductometric signals similar to the negative control of the previous experiment (Fig. 8). The simultaneous presence of the ldh1 target and the highly concentrated human DNA background did not compromise the amplification of the target and DNA aggregates were successfully detected. Nevertheless, the obtained signal was lower than the positive control without human genomic DNA, and the aggregate were smaller, as proved by the optical control. This could be explained by a larger amount of parasite reactions during the SDA step, which decreased the amplification yield of the bacterial target gene.
 | ||
| Fig. 8 Conductometric detection of SDA–HRCA product aggregation for specificity testing. When present, MecA and ldh1 genes concentrations were 10 pM. Human genomic DNA quantity was 200 ng. | ||
Conclusions and perspectives
We reported here the detection of small (around 160 nt) DNA sequences from Staphylococcus aureus by conductometry, using a new combination of DNA aggregation by electrohydrodynamic instabilities (EHDA), conductometric measurements, and an original amplification and elongation reaction, combining two isothermal amplifications: SDA and HRCA. We succeeded to detect 1 pM of target sequences after 2h20 of reaction. The sensitivity we reached in this first proof-of-concept is below that of state-of-the-art detection of amplification products via fluorescence reporters. One should remember, however, that this conventional technology benefited from now twenty years of improvements and major industrial involvement, and that its sensitivity in the early stages was even lower that achieved here. One can already envision several tracks to improve the sensitivity of our technology, which we believe to be limited, for instance, by the performance of the SDA step. A coupling between HRCA and another isothermal amplifications like helicase-dependent isothermal amplification (HDA),35 recombinase polymerase amplification (RPA)36 or LAMP could in a next future improve the method sensitivity to detect pertinent level of bacterial DNA.As shown in this study, fluorescence could also be a way to detect the presence and the shape of aggregates of amplification products. This solution could be easily implemented with miniaturized LED and sensors and should be able to further decrease the limit of detection of the system, as the aggregates were visible in the present study, at 100 fM without being detected by conductometry. This was achieved without particular efforts in optimizing the optics or image processing, so a gain by two orders of magnitude, on top of what could be gained by further progress on the amplification protocol, should be rather easy to reach.
Ultimately, however, we believe that the main potential use of EHDA detection does not lie in the search of ultimate sensitivity, but in the development of low-cost, portable point-of-use system.
In the current system, the conductometric detection is performed with a portable prototype, comprising two electronic modules working on battery, a laptop for the acquisition and control of the detection electronic, and a miniaturized microfluidic chip. The only manual operation in the detection protocol is the filling of the microfluidic channel by pipetting. No pump or pressure control are required. No expensive components are used for the electronics and the detection is label-free and did not need any labelled oligonucleotides or capture molecules as antibodies. In contrast with conventional conductometric detection, EHDA-based conductometry can accommodate background ions concentrations much higher than the average DNA concentration in the solution, and is robust to small variations in this concentration. It is also independent of the presence in the solution of even a large excess of DNA fragments below 1 kbp. SDA–HRCA allows the formation of long DNA molecules (>5 kbp), and is specific regarding the target sequence. Overall, the scheme is quite insensitive to variations or contaminations in the matrix and background electrolyte. Finally, the detection is volume-based, so the technology should also be less sensitive to non-specific adsorption of contaminant molecules, than surface-based techniques such as electrochemistry of FET-based label-free detections. This combination of advantages is to our knowledge a strong novelty as compared to all previous label-free DNA detection, and makes it particularly suitable for the development of simple, “sample-to-result” protocols.
The combined amplification and elongation reaction only needed addition of reagents over the course of this 3 step strategy, and 3 different temperature plateaus (56 °C, 80 °C and 35 °C). Using a thermostable ligase and the Bst2.0 as the HRCA polymerase, this reaction could perhaps become fully isothermal. This improvement would lead to an easier integration of the DNA amplification and elongation steps into a microfluidic format.
As the ligation step could recognize short sequences (20 bp) inside a larger DNA strand, this SDA–HRCA strategy would be particularly suited for nested and multiplexed amplifications,37,38 with the SDA targeting a common and preserved gene over different bacteria species, and the HRCA focusing on specific sequences.
Regarding size and power consumption, finally, through industrialization, both the field generation and the detection can be further miniaturized considerably, to be finally adaptable to the power source of a smartphone, and the size of a USB key.
Conflicts of interest
Some authors are coinventors of patent WO2014006561 to CNRS and Institut Curie.Acknowledgements
We thank Robert Breton for assistance in the electronic development, P. Silberzan and A. Buguin for access to the clean room of the Curie Institute and Jérôme Champ for biological support. Our gratitude also goes to Mats Nilsson (Science for Life Laboratory, Stockholm University) for fruitful discussion and help on the DNA amplification scheme. This work has received the support of Institut Pierre-Gilles de Gennes (équipement d'excellence, “Investissements d'avenir”, program ANR-10-EQPX-34). This work was supported by a Direction Générale de l'Armement (DGA) PhD fellowship to BV and by the ERC Advanced Grant CellO (FP7-IDEAS-ERC-321107).References
- C. Heid, J. Stevens, K. Livak and P. Williams, Real time quantitative PCR, Genome Res., 1996, 6, 986–994 CrossRef CAS PubMed.
- E. Engvall and P. Perlmann, Enzyme-linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G, Immunochemistry, 1971, 8, 871–874 CrossRef CAS PubMed.
- H. W. Choi, K. Miele, D. Dowdy and M. Shah, Cost-effectiveness of Xpert® MTB/RIF for diagnosing pulmonary tuberculosis in the United States, Int. J. Tuberc. Lung Dis., 2013, 17, 1328–1335 CrossRef CAS PubMed.
- C. C. Boehme, et al., Feasibility, diagnostic accuracy, and effectiveness of decentralised use of the Xpert MTB/RIF test for diagnosis of tuberculosis and multidrug resistance: A multicentre implementation study, Lancet, 2011, 377, 1495–1505 CrossRef.
- M. D. Borysiak, K. W. Kimura and J. D. Posner, NAIL: Nucleic Acid detection using Isotachophoresis and Loop-mediated isothermal amplification, Lab Chip, 2015, 15, 1697–1707 RSC.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Diagnostics for the developing world: microfluidic paper-based analytical devices, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- J. S. Cybulski, J. Clements and M. Prakash, Foldscope: Origami-Based Paper Microscope, PLoS One, 2014, 9, e98781 Search PubMed.
- S. Wünscher, et al., Chip-on-foil devices for DNA analysis based on inkjet-printed silver electrodes, Lab Chip, 2014, 14, 392–401 RSC.
- N. Kurra and G. U. Kulkarni, Pencil-on-paper: electronic devices, Lab Chip, 2013, 13, 2866 RSC.
- D. Dendukuri, T. Choudhary and G. P. Rajamanickam, Woven Electrochemical Fabric-based Test Sensors (WEFTS): A new class of multiplexed electrochemical sensors, Lab Chip, 2015, 15, 2064–2072 RSC.
- L. J. Steinbock, G. Stober and U. F. Keyser, Sensing DNA-coatings of microparticles using micropipettes, Biosens. Bioelectron., 2009, 24, 2423–2427 CrossRef CAS PubMed.
- M. Kühnemund and M. Nilsson, Digital quantification of rolling circle amplified single DNA molecules in a resistive pulse sensing nanopore, Biosens. Bioelectron., 2015, 67, 11–17 CrossRef PubMed.
- B. Lam, Z. Fang, E. H. Sargent and S. O. Kelley, Polymerase Chain Reaction-Free, Sample-to-Answer Bacterial Detection in 30 Minutes with Integrated Cell Lysis, Anal. Chem., 2012, 84, 21–25 CrossRef CAS PubMed.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, DNA-templated assembly and electrode attachment of a conducting silver wire, Nature, 1998, 391, 775–778 CrossRef CAS PubMed.
- C. Russell, et al., Gold Nanowire Based Electrical DNA Detection Using Rolling Circle Amplification, ACS Nano, 2014, 8, 1147–1153 CrossRef CAS PubMed.
- M. L. Y. Diakité, et al., A low-cost, label-free DNA detection method in lab-on-chip format based on electrohydrodynamic instabilities, with application to long-range PCR, Lab Chip, 2012, 12, 4738 RSC.
- L. Mitnik, C. Heller, J. Prost and J. L. Viovy, Segregation in DNA solutions induced by electric fields, Science, 1995, 267, 219–222 CrossRef CAS PubMed.
- H. Isambert, A. Ajdari, J.-L. Viovy and J. Prost, Electrohydrodynamic patterns in macroion dispersions under a strong electric field, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 5688–5704 CrossRef CAS.
- S. Magnúsdóttir, H. Isambert, C. Heller and J. Viovy, Electrohydrodynamically induced aggregation during constant and pulsed field capillary electrophoresis of DNA, Biopolymers, 1999, 49, 385–401 CrossRef.
- W. M. Barnes, PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2216–2220 CrossRef CAS.
- T. Notomi, et al., Loop-mediated isothermal amplification of DNA, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- J. Banér, M. Nilsson, M. Mendel-Hartvig and U. Landegren, Signal amplification of padlock probes by rolling circle replication, Nucleic Acids Res., 1998, 26, 5073–5078 CrossRef.
- P. M. Lizardi, et al., Mutation detection and single-molecule counting using isothermal rolling-circle amplification, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- D. Yong Zhang, M. Brandwein, T. Chun Hung Hsuih and H. Li, Amplification of target-specific, ligation-dependent circular probe, Gene, 1998, 211, 277–285 CrossRef.
- T. Murakami, J. Sumaoka and M. Komiyama, Sensitive isothermal detection of nucleic-acid sequence by primer generation-rolling circle amplification, Nucleic Acids Res., 2009, 37, e19–e19 CrossRef PubMed.
- F. Dahl, et al., Circle-to-circle amplification for precise and sensitive DNA analysis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4548–4553 CrossRef CAS PubMed.
- G. T. Walker, et al., Strand displacement amplification–an isothermal, in vitro DNA amplification technique, Nucleic Acids Res., 1992, 20, 1691–1696 CrossRef CAS PubMed.
- A. Joneja and X. Huang, Linear nicking endonuclease-mediated strand DNA Amplification, Anal. Biochem., 2011, 414, 58–69 CrossRef CAS PubMed.
- B. J. Toley, et al., Isothermal strand displacement amplification (iSDA): a rapid and sensitive method of nucleic acid amplification for point-of-care diagnosis, Analyst, 2015, 140, 7540–7549 RSC.
- R. M. Guijt, C. J. Evenhuis, M. Macka and P. R. Haddad, Conductivity detection for conventional and miniaturised capillary electrophoresis systems, Electrophoresis, 2004, 25, 4032–4057 CrossRef CAS PubMed.
- L. K. Lafleur, et al., A rapid, instrument-free, sample-to-result nucleic acid amplification test, Lab Chip, 2016, 16, 3777–3787 RSC.
- C. A. Spargo, et al., Detection of M. tuberculosis DNA using thermophilic strand displacement amplification, Mol. Cell. Probes, 1996, 10, 247–256 CrossRef CAS PubMed.
- M. Nilsson, et al., Padlock probes: circularizing oligonucleotides for localized DNA detection, Science, 1994, 265, 2085–2088 CAS.
- D. Wu and R. Wallace, Specificity of the nick-closing activity of bacteriophage T4 DNA ligase, Gene, 1989, 76, 245–254 CrossRef CAS PubMed.
- M. Vincent, Y. Xu and H. Kong, Helicase-dependent isothermal DNA amplification, EMBO Rep., 2004, 5, 795–800 CrossRef CAS PubMed.
- M. Euler, et al., Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus, J. Clin. Virol., 2012, 54, 308–312 CrossRef CAS PubMed.
- N. Boon, et al., Evaluation of nested PCR-DGGE (denaturing gradient gel electrophoresis) with group-speci′c 16S rRNA primers for the analysis of bacterial communities from di¡erent wastewater treatment plants, Archaea, 2002, 39, 101–112 CAS.
- G. Czilwik, et al., Rapid and fully automated bacterial pathogen detection on a centrifugal-microfluidic LabDisk using highly sensitive nested PCR with integrated sample preparation, Lab Chip, 2015, 15, 3749–3759 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an00798a |
| This journal is © The Royal Society of Chemistry 2018 |