
Sample preparation for polar metabolites in bioanalysis
Nicolas
Drouin
 ,
Serge
Rudaz
and
Julie
Schappler
*
,
Serge
Rudaz
and
Julie
Schappler
*
School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Rue Michel-Servet 1, 1211 Geneva 4, Switzerland. E-mail: julie.schappler@unige.ch; Fax: +41 22 379 68 08; Tel: +41 22 379 60 49
First published on 27th November 2017
Abstract
Sample preparation is a primary step of any bioanalytical workflow, especially in metabolomics analysis where maximum information has to be obtained without spoiling the analytical instrument. Because of their biological implication, highly polar metabolites, such as amino acids, nucleobases, and catecholamines seem to attract growing interest in the field of comprehensive metabolomics analysis although their extraction from the matrix remains a real challenge. In this paper, we discuss about the actual practice and issues of hydrophilic metabolites' extraction, including new solutions and perspectives to improve their phase transfer from a complex biological sample to a clean extract prior to analysis.
1. Introduction
Metabolomics is a scientific discipline, which consists in studying the complex chemical fingerprint of a biological system to describe its status. Focus is made toward any chemical species with a molecular weight lower than 1000–1500 Da, produced, consumed, or present in the biological system, also called metabolome. Metabolites are a direct consequence of gene expression, metabolism, dietary habits, and environmental exposure.1,2 Endogenous and exogenous metabolites are involved in organism homeostasis, as well as pathological or toxic processes. Comprehensive or untargeted metabolomics involves the whole metabolites’ compartment and is complicated due to the large variety of physicochemical properties the metabolites possess. This complex procedure involves the implementation of a large set of analytical methods to cover the diversity of the metabolome in terms of biochemical and physicochemical properties (volatility, solubility, lipophilicity, pKa, etc.). Based on lipophilicity, the metabolome can be classified into three main categories (Fig. 1): (i) lipophilic metabolites with partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) values from 5 to 10, such as fatty acids and triglycerides, (ii) moderately lipophilic metabolites with logP from 0 to 5, such as drug metabolites and steroids, and (iii) hydrophilic metabolites with logP below 0, such as amino acids, nucleobases, and nucleotides.
P) values from 5 to 10, such as fatty acids and triglycerides, (ii) moderately lipophilic metabolites with logP from 0 to 5, such as drug metabolites and steroids, and (iii) hydrophilic metabolites with logP below 0, such as amino acids, nucleobases, and nucleotides.
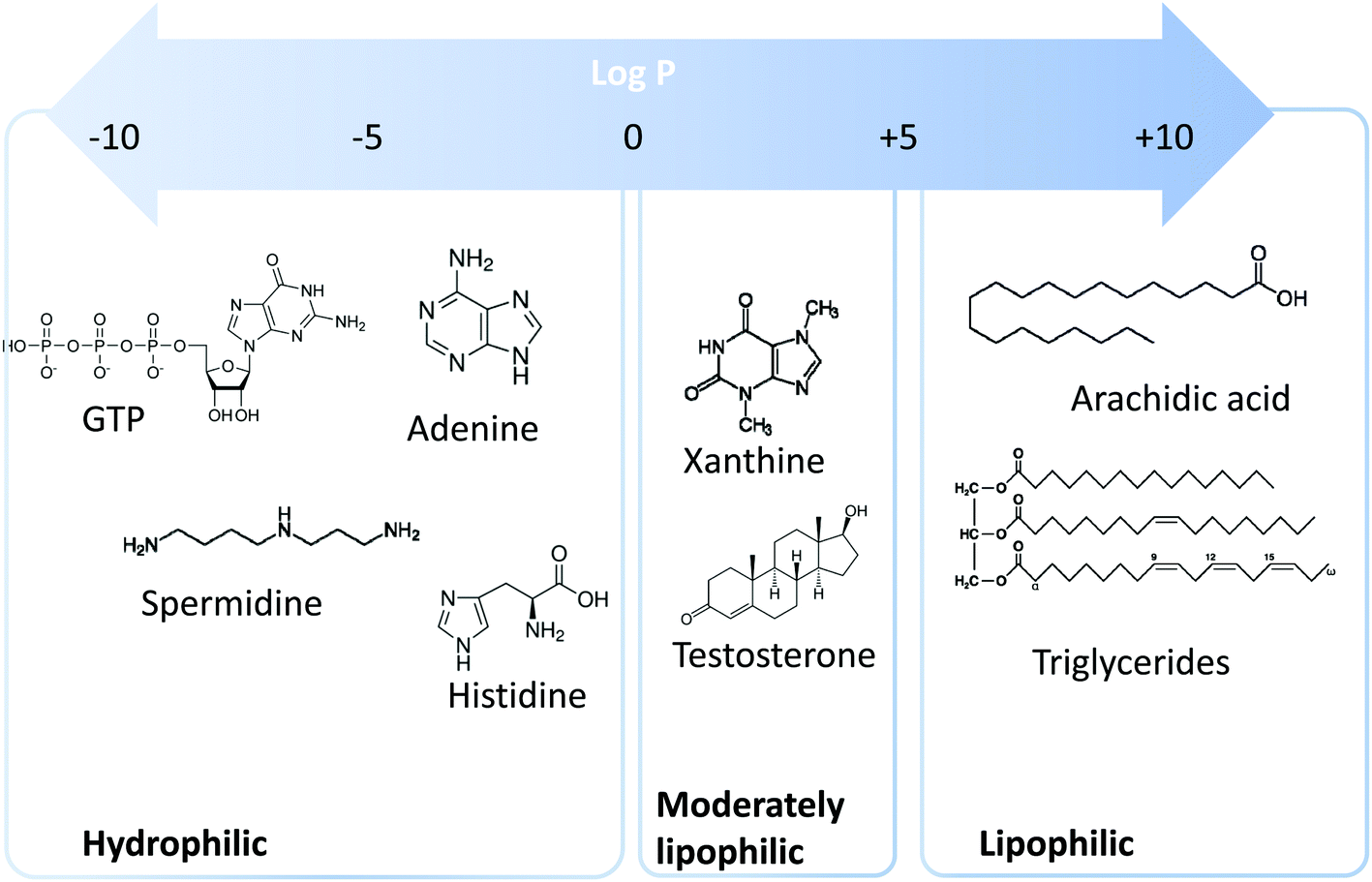 | ||
| Fig. 1 Lipophilicity range of metabolites encountered in metabolomics. | ||
A typical metabolomics workflow consists of several steps, including the sampling, sample preparation, data acquisition, and finally data treatment. Data acquisition is mainly performed using a separation method along with mass spectrometry (MS) such as LC-MS/MS in RPLC or HILIC mode, GC-MS/MS, and CE-MS/MS, especially for targeted analysis.3 Sample preparation includes all procedures and operations applied to the sample prior to its analysis. This step is essential when MS-based techniques are used. It is implemented to (i) stabilize the sample, (ii) remove the sample contaminants, (iii) enable sample enrichment, (iv) improve the analysis selectivity, and/or (v) avoid fouling of the mass spectrometer. Sample preparation approaches can be divided into two groups, namely sample pretreatment and sample extraction methods (Fig. 2).
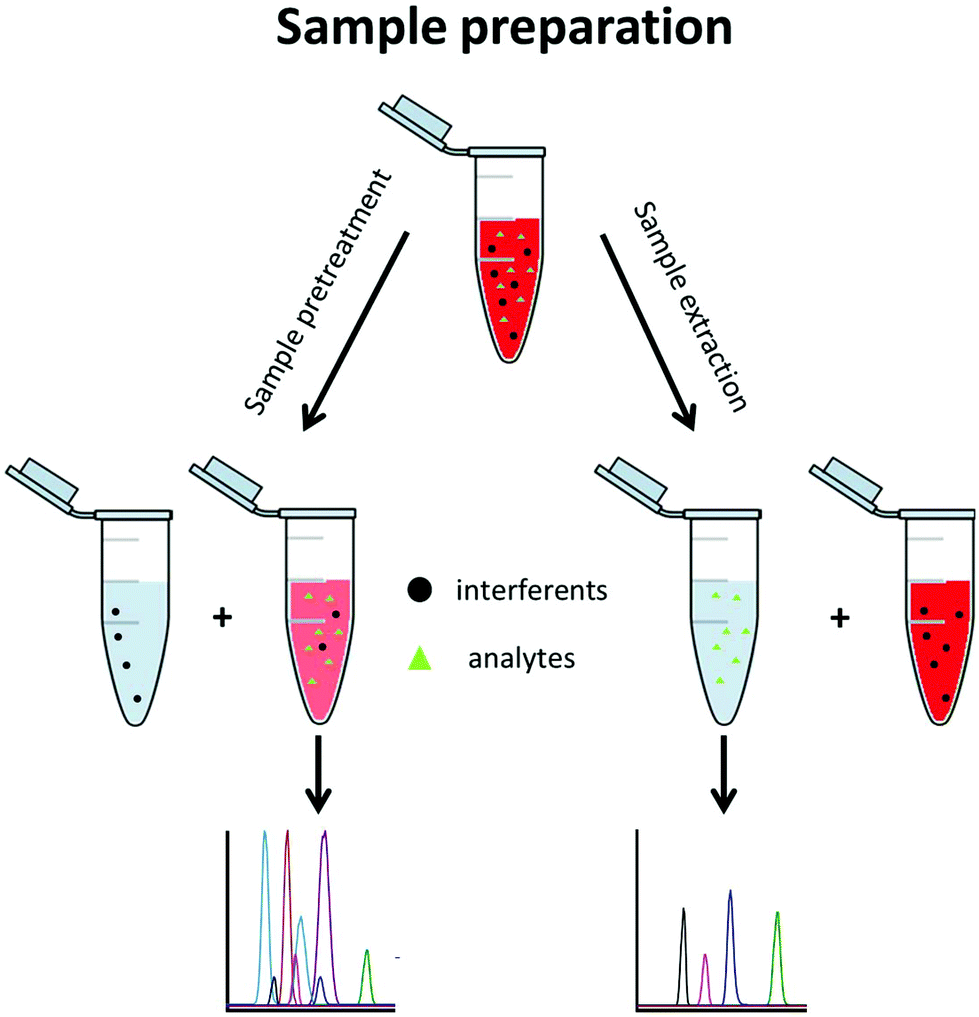 | ||
| Fig. 2 The two main approaches of sample preparation. | ||
Sample pretreatment consists of removing or decreasing the concentration of interferents from the original sample to obtain the analyte(s) with a low amount of contaminants. This approach is fast, generic for a large variety of matrices, and enables a large metabolite coverage, so it is favoured for untargeted metabolomics. With this approach, interferents are often not completely discarded and are still present in the pretreated aqueous sample where the polar metabolites are also found. This generally decreases the repeatability of the method and its sensitivity due to a matrix effect.4,5 Hydrophilic metabolites are also diluted in the process so the sensitivity of the analytical technique can be an issue further. Dilute-and-shoot6,7 and protein precipitation8,9 belong to this group and are mostly used for urine and blood-based matrices such as plasma and serum.
With sample extraction, metabolites of interest are extracted from the original sample matrix and transferred to another interferent-free phase. This approach can be very selective, so it is primarily used in targeted metabolomics, where preselected metabolites are searched for. Sample extraction methods allow a higher purification level than sample pretreatment, reducing the matrix effect and important analytical issues such as carry over. Solid phase extraction (SPE) belongs to this group and is the most commonly used technique, regardless of the application. Although SPE and liquid–liquid extraction (LLE) are also commonly used in untargeted metabolomics, they are not applied to extract the hydrophilic metabolites but rather to eliminate the lipophilic interferents. From a fundamental point of view, SPE and LLE in this case are used as sample pretreatment approaches since the hydrophilic metabolites remain in the sample phase.
Nowadays, both untargeted and targeted metabolomics analyses often need enhanced sensitivity and repeatability to identify and/or quantify new biomarkers. Improvements can be made in sample preparation by enriching the metabolites or by reducing the matrix effect. Sample extraction is required in this context. Lipophilic metabolites can be easily extracted with efficient phase transfers, from the original aqueous sample to an immiscible organic phase, due to the favorable partition coefficient (logP) of these metabolites. Hydrophilic compounds, including ionized analytes, are however not straightforwardly extracted due to their negative logP values, and a fortiori negative distribution coefficient (logD) value, which is unfavorable for such phase transfer. In this review, the focus is made on the extraction of hydrophilic metabolites, and it includes the current achievements, issues, and the future perspectives for hydrophilic or ionized metabolite extraction.
2. Current practice and issues
In most metabolomics studies, sample pretreatment, typically protein precipitation, is often performed.5,8 When high selectivity towards polar metabolites or improved sample clean-up is required, LLE is performed with water immiscible organic solvents such as dichloromethane or hexane (addition of a co-solvent can be performed to slightly modify the selectivity) to remove most of the lipophilic interferents and retain the hydrophilic metabolites in the original aqueous phase.10 SPE is also used to remove proteins and sphingolipids11,12 and is generally followed by conventional LLE.13 These multi-step approaches feature the advantage of compartmentalizing the sample: proteins, lipophilic metabolites, and hydrophilic metabolites are all recovered in different fractions, which can be separately analyzed and detected with appropriate analytical techniques.14 In this case, the analysis of hydrophilic metabolites is only improved by the better extraction of lipophilic interferents. From a fundamental point of view, this approach is not considered selective for hydrophilic metabolites, since the latter are not transferred to another phase and due to the presence of a large amount of salts and other contaminants remaining in the same aqueous matrix. Without efficient sample preparation, these interferents may in LC (i) induce injector carry over, (ii) saturate the stationary phases from unwanted proteins or other endogenous compounds, and (iii) cause high back pressure (and leaking during the run), which all may lead to peak broadening, a shift in retention times, and/or instrument fouling. It is even worth noting that when MS detection is used, it may result in (i) the enhancement or suppression of the analyte signal (i.e., the matrix effect), (ii) the degradation of the method performance, and hence (iii) a decrease in the metabolites that can be accurately quantified.15If higher selectivity or sensitivity is needed for ionized or ionizable metabolites, ion exchange SPE (IE-SPE) is also used. IE-SPE sorbents are made of cationic or anionic charges, permanent or not, to retain anionic compounds in the case of anion exchange or cationic compounds with cation exchange. IE-SPE is adapted for the extraction of ionic compounds such as amino acids,16 dopamine,17 nucleotides and their analogs,18 and purines,19 and has already been used a few times for large metabolomics studies.20 Elution is performed either by switching the ionic charge (of the compounds or the support) with a buffer at an appropriate pH or by exchanging the analytes with a high salt concentration solution. The drawbacks of IE-SPE are (i) the limited loading capacity of the supports18,21 and (ii) the use of salts or buffers for the elution, which can be deleterious for the further analytical method, particularly with MS detection.
3. Future trends
One major drawback of LLE is the time needed to obtain the phase equilibrium (e.g. several hours in the case of polar compounds for low recovery) and the large volumes of toxic organic solvents required. Based on the same principle as LLE, dispersive liquid–liquid microextraction (DLLME) is an interesting alternative (Fig. 3). Due to the formation of a microemulsion between the sample and the extraction solvent, the contact surface is drastically enhanced and the equilibrium is reached very quickly. Enrichment can be very important, up to 100-fold due to the very low volume of the extraction solvent compared to that of the sample and can even counterbalance the low recovery obtained with most of the polar metabolites.22,23 The use of a low amount of an organic solvent is also an advantage in terms of toxicity and solvent consumption. Several studies have demonstrated the applicability of DLLME for very polar metabolites, such as neurotransmitters24 or folate derivatives.25 Research is still needed to find other organic solvents, which can improve the extraction recovery of hydrophilic compounds.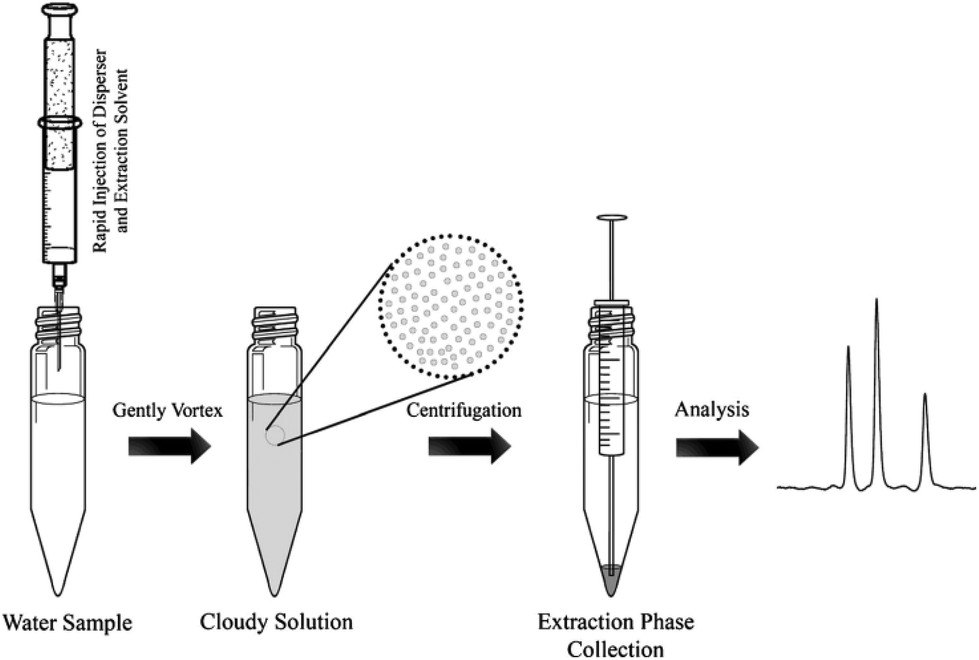 | ||
| Fig. 3 Dispersive liquid–liquid microextraction procedure workflow,16 with the permission of Springer. | ||
Solid phase microextraction (SPME) is another sample preparation method adapted to hydrophilic metabolites. This sample preparation technique consists of a thin polymeric coating on a support where analytes are adsorbed onto according to their partition coefficient. The extraction is maximum when the equilibrium is reached, although pre-equilibrium extraction is also possible and sometimes necessary. Considering the thickness and the mass of the coating, SPME cannot be considered an exhaustive extraction method but a sampling approach selective to the compounds of interest.26 A large variety of supports exist, from fiber, stir-bars, needles or syringes to small blades for the 96 well-plate format,27–29 leading to an excellent versatility of SPME in terms of sample volume. Moreover, new biocompatible sorbents are now used for direct in vivo sampling of living systems.30,31 Around ten coatings are commercially available now. If they prove their usefulness in inter-laboratory studies on various matrices and applications, they will be mainly suitable for lipophilic compounds. New coatings are thus in development for hydrophilic compounds and should be available in the near future.32–34
Electromembrane extraction (EME) is a recent approach for the extraction of ionized compounds. This sample preparation technique is based on the application of an electrical field between two compartments, the sample and the acceptor compartments, separated by a polymeric membrane impregnated by an organic solvent, known as the supported liquid membrane (SLM) (Fig. 4).35 When a voltage is applied between the two compartments, ionic compounds migrate under the electrical field through the SLM and are concentrated into the acceptor compartment.36 EME is a one-step sample preparation procedure and features the recovery of the analytes in an aqueous phase, directly compatible with the further analytical device. Interferents such as proteins, salts or phospholipids do not cross the SLM and generally a low matrix effect such as ionization alteration is observed.37,38 Because EME can extract either cations or anions, it can be an effective fractionation method according to the charge of the metabolite, in the case of very complex matrices. Nowadays, EME is largely used for the extraction of moderately lipophilic ionized compounds such as drugs and drug metabolites,39,40 inorganic ions,41,42 from a large variety of matrices such as plasma and waste water. As for other extraction techniques, the extraction of hydrophilic endogenous compounds remains the principal challenge for EME. The modification of the SLM with carriers43 or the application of new organic solvents44 appears as a promising solution to enhance the mass transfer of charged hydrophilic metabolites. Although no large metabolomics studies using EME have been reported yet, a few studies have already described the implementation of EME for the extraction of peptides,45,46 amino acids,47 and catecholamines.43
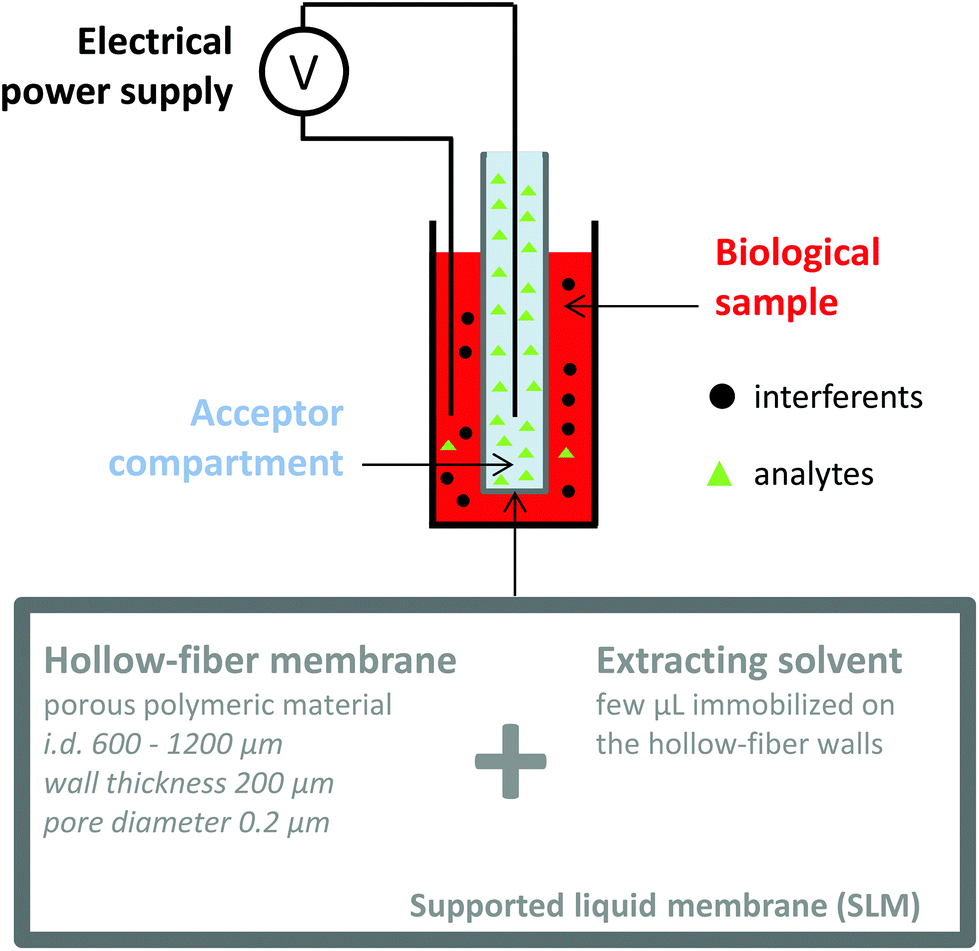 | ||
| Fig. 4 Schematic principle of an electromembrane extraction device. | ||
High throughput is nowadays another important aspect of sample preparation in metabolomics. A large batch of samples has to be prepared every day with low variability. The 96 well-plate format is generally used according to the practical considerations such as volume samples, injector modules, or robotic liquid handlers.48 Until now, only SPME is commercially available in a 96 well-plate format compatible with the automatic workflow.27 No commercial device exists for DLLME or EME yet, although efforts are currently made in EME to develop such a parallel approach.47,49
4. Concluding remarks
Because hydrophilic metabolites, such as amino acids, nucleobases, catecholamines, play important roles in biological systems, there is a need for their analysis in complex biological matrices. The sample extraction of these metabolites is still challenging and most of the time, only pretreatment approaches are applied to the sample, with the issue of the matrix effect. Today, other strategies exist and allow the phase transfer of hydrophilic metabolites from the matrix phase to a clean interference-free phase.SPME can be used as an automated sampling method adapted for the first screening of a large set of metabolites according to the coating. SPME offers numerous supports and sorbents, which are reusable and biocompatible, allowing even in vivo sampling or high throughput extraction. Because it is commercially available, SPME offers excellent repeatability and robustness, although no commercial sorbent dedicated to hydrophilic compounds are available yet. By increasing the contact surface of the extracting solvent with the sample compared to classical LLE, DLLME allows decreasing the extraction time from a few hours in LLE to a few seconds, while improving the enrichment obtained for hydrophilic compounds, although solvents suitable for these analytes are still lacking. Automation has not been reported as well, circumventing the widespread use of DLLME. EME also appears to be a promising approach for hydrophilic metabolites. The electrical field forces the extraction through a supported liquid membrane, enabling very good extraction performance in terms of recovery and enrichment, while reducing the extraction time and the matrix effect. In the near future, new organic solvents and 96 well-plate devices are expected to further expand the use of EME.
List of abbreviations
| DLLME | Dispersive liquid–liquid microextraction |
| EME | Electromembrane extraction |
| IE-SPE | Ion exchange solid phase extraction |
| LLE | Liquid–liquid extraction |
| SPE | Solid-phase extraction |
| SPME | Solid-phase microextraction |
| SLE | Supported liquid membrane |
Conflicts of interest
There are no conflicts to declare.References
- Y. Pico, Advanced sample preparation techniques for MS analysis, John Wiley & Sons, Inc., 2015 Search PubMed.
- C. S. Clendinen, M. E. Monge and F. M. Fernandez, Analyst, 2017, 142, 3101–3117 RSC.
- O. Begou, H. G. Gika, I. D. Wilson and G. Theodoridis, Analyst, 2017, 142, 3079–3100 RSC.
- R. Romero-Gonzalez, F. J. Liebanas, R. Lopez-Ruiz and A. G. Frenich, J. AOAC Int., 2016, 99, 1395–1402 CrossRef CAS PubMed.
- E. Rico, O. Gonzalez, M. E. Blanco and R. M. Alonso, Anal. Bioanal. Chem., 2014, 406, 7641–7652 CrossRef CAS PubMed.
- Z. Cao, E. Kaleta and P. Wang, J. Anal. Toxicol., 2015, 39, 335–346 CrossRef CAS PubMed.
- I. Rodin, A. Braun, A. Stavrianidi, T. Baygildiev, O. Shpigun, D. Oreshkin and I. Rybalchenko, J. Anal. Toxicol., 2015, 39, 69–74 CrossRef CAS PubMed.
- J. Chen, W. Hou, B. Han, G. Liu, J. Gong, Y. Li, D. Zhong, Q. Liao and Z. Xie, Anal. Bioanal. Chem., 2016, 408, 2527–2542 CrossRef CAS PubMed.
- Y. Zeng, L. Luo, W. Hou, B. Lu, J. Gong, J. Chen, X. Zhang, B. Han, Z. Xie and Q. Liao, J. Sep. Sci., 2017, 40, 3221–3230 CrossRef CAS PubMed.
- A. Armirotti, A. Basit, N. Realini, C. Caltagirone, P. Bossu, G. Spalletta and D. Piomelli, Anal. Biochem., 2014, 455, 48–54 CrossRef CAS PubMed.
- J. Han, Y. Liu, R. Wang, J. Yang, V. Ling and C. H. Borchers, Anal. Chem., 2015, 87, 1127–1136 CrossRef CAS PubMed.
- S. Tulipani, X. Mora-Cubillos, O. Jauregui, R. Llorach, E. Garcia-Fuentes, F. J. Tinahones and C. Andres-Lacueva, Anal. Chem., 2015, 87, 2639–2647 CrossRef CAS PubMed.
- K. Skov, N. Hadrup, J. Smedsgaard and H. Frandsen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 978–979, 83–88 CrossRef CAS PubMed.
- S. Tulipani, X. Mora-Cubillos, O. Jauregui, R. Llorach, E. Garcia-Fuentes, F. J. Tinahones and C. Andres-Lacueva, Anal. Chem., 2015, 87, 2639–2647 CrossRef CAS PubMed.
- P. Panuwet, R. E. Hunter Jr., P. E. D'Souza, X. Chen, S. A. Radford, J. R. Cohen, M. E. Marder, K. Kartavenka, P. B. Ryan and D. B. Barr, Crit. Rev. Anal. Chem., 2016, 46, 93–105 CrossRef CAS PubMed.
- Y. Wang, M. Zhao, M. Wang and C. Zhao, Pharm. Biol., 2016, 54, 2254–2263 CrossRef CAS PubMed.
- D. Zhang, L. Wu, D. S. Chow, V. H. Tam and D. R. Rios, J. Pharm. Biomed. Anal., 2016, 117, 227–231 CrossRef CAS PubMed.
- L. C. Jimmerson, L. R. Bushman, M. L. Ray, P. L. Anderson and J. J. Kiser, Pharm. Res., 2017, 34, 73–83 CrossRef CAS PubMed.
- Y. Xu, J. Liu, H. Zhang, M. Jiang, L. Cao, M. Zhang, W. Sun, S. Ruan and P. Hu, Talanta, 2016, 151, 172–178 CrossRef CAS PubMed.
- L. Pont, F. Benavente, J. Jaumot, R. Tauler, J. Alberch, S. Gines, J. Barbosa and V. Sanz-Nebot, Electrophoresis, 2016, 37, 795–808 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard, A. Gjelstad and T. G. Halvorsen, in Bioanalysis of Pharmaceuticals, John Wiley & Sons, Ltd, 2015, pp. 73–122, DOI:10.1002/9781118716830.ch6.
- O. Zuloaga, M. Olivares, P. Navarro, A. Vallejo and A. Prieto, Bioanalysis, 2015, 7, 2211–2225 CrossRef CAS PubMed.
- M. Saraji and M. K. Boroujeni, Anal. Bioanal. Chem., 2014, 406, 2027–2066 CrossRef CAS PubMed.
- L. Konieczna, A. Roszkowska, M. Niedzwiecki and T. Baczek, J. Chromatogr. A, 2016, 1431, 111–121 CrossRef CAS PubMed.
- Y. Nojavan, M. Kamankesh, F. Shahraz, M. Hashemi and A. Mohammadi, Talanta, 2015, 137, 31–37 CrossRef CAS PubMed.
- E. Boyaci, A. Rodriguez-Lafuente, K. Gorynski, F. Mirnaghi, E. A. Souza-Silva, D. Hein and J. Pawliszyn, Anal. Chim. Acta, 2015, 873, 14–30 CrossRef CAS PubMed.
- F. Mousavi, B. Bojko, V. Bessonneau and J. Pawliszyn, J. Proteome Res., 2016, 15, 963–975 CrossRef CAS PubMed.
- H. Kataoka, Bioanalysis, 2015, 7, 2135–2144 CrossRef CAS PubMed.
- H. Piri-Moghadam, M. N. Alam and J. Pawliszyn, Anal. Chim. Acta, 2017, 984, 42–65 CrossRef CAS PubMed.
- J. J. Poole, J. J. Grandy, M. Yu, E. Boyaci, G. A. Gomez-Rios, N. Reyes-Garces, B. Bojko, H. V. Heide and J. Pawliszyn, Anal. Chem., 2017, 89, 8021–8026 CrossRef CAS PubMed.
- V. Bessonneau, J. Ings, M. McMaster, R. Smith, L. Bragg, M. Servos and J. Pawliszyn, Sci. Rep., 2017, 7, 44038 CrossRef PubMed.
- E. Gionfriddo, E. Boyaci and J. Pawliszyn, Anal. Chem., 2017, 89, 4046–4054 CrossRef CAS PubMed.
- E. Cudjoe and J. Pawliszyn, J. Chromatogr. A, 2014, 1341, 1–7 CrossRef CAS PubMed.
- E. A. Souza-Silva, N. Reyes-Garces, G. A. Gomez-Rios, E. Boyaci, B. Bojko and J. Pawliszyn, TrAc, Trends Anal. Chem., 2015, 71, 249–264 CrossRef CAS.
- A. Gjelstad and S. Pedersen-Bjergaard, Electrophoresis, 2014, 35, 2421–2428 CrossRef CAS PubMed.
- A. Gjelstad, S. Pedersen-Bjergaard and K. F. Seip, Bioanalysis, 2015, 7, 2203–2209 CrossRef CAS PubMed.
- L. E. Eibak, M. P. Parmer, K. E. Rasmussen, S. Pedersen-Bjergaard and A. Gjelstad, Anal. Bioanal. Chem., 2014, 406, 431–440 CrossRef CAS PubMed.
- L. Vardal, A. Gjelstad, C. Huang, E. L. Oiestad and S. Pedersen-Bjergaard, Bioanalysis, 2017, 9, 631–641 CrossRef CAS PubMed.
- C. Huang, K. F. Seip, A. Gjelstad and S. Pedersen-Bjergaard, J. Pharm. Biomed. Anal., 2015, 113, 97–107 CrossRef CAS PubMed.
- T. K. Rye, D. Fuchs, S. Pedersen-Bjergaard and N. J. Petersen, Anal. Chim. Acta, 2017, 983, 121–129 CrossRef CAS PubMed.
- Z. Tahmasebi and S. S. H. Davarani, Talanta, 2016, 161, 640–646 CrossRef CAS PubMed.
- A. Fashi, M. R. Yaftian and A. Zamani, Food Chem., 2017, 221, 714–720 CrossRef CAS PubMed.
- E. Fernandez, L. Vardal, L. Vidal, A. Canals, A. Gjelstad and S. Pedersen-Bjergaard, Anal. Bioanal. Chem., 2017, 409, 4215–4223 CrossRef CAS PubMed.
- C. Huang, K. F. Seip, A. Gjelstad and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2016, 934, 80–87 CrossRef CAS PubMed.
- C. Huang, A. Gjelstad and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2015, 853, 328–334 CrossRef CAS PubMed.
- N. Drouin, S. Rudaz and J. Schappler, Anal. Chem., 2016, 88, 5308–5315 CrossRef CAS PubMed.
- N. Drouin, J. F. Mandscheff, S. Rudaz and J. Schappler, Anal. Chem., 2017, 89, 6346–6350 CrossRef CAS PubMed.
- N. Zheng, H. Jiang and J. Zeng, Bioanalysis, 2014, 6, 2441–2459 CrossRef CAS PubMed.
- L. E. Eibak, K. E. Rasmussen, E. L. Oiestad, S. Pedersen-Bjergaard and A. Gjelstad, Anal. Chim. Acta, 2014, 828, 46–52 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |