
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom protein engineering to artificial enzymes – biological and biomimetic approaches towards sustainable hydrogen production
C.
Esmieu
,
P.
Raleiras
 and
G.
Berggren
*
and
G.
Berggren
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, SE-75120 Uppsala, Sweden. E-mail: Gustav.berggren@kemi.uu.se
First published on 6th February 2018
Abstract
Hydrogen gas is used extensively in industry today and is often put forward as a suitable energy carrier due its high energy density. Currently, the main source of molecular hydrogen is fossil fuels via steam reforming. Consequently, novel production methods are required to improve the sustainability of hydrogen gas for industrial processes, as well as paving the way for its implementation as a future solar fuel. Nature has already developed an elaborate hydrogen economy, where the production and consumption of hydrogen gas is catalysed by hydrogenase enzymes. In this review we summarize efforts on engineering and optimizing these enzymes for biological hydrogen gas production, with an emphasis on their inorganic cofactors. Moreover, we will describe how our understanding of these enzymes has been applied for the preparation of bio-inspired/-mimetic systems for efficient and sustainable hydrogen production.
1. Introduction
Hydrogen gas is an important base chemical employed in e.g. the Haber–Bosch process and oil refining, and >30 million tonnes of H2 is produced every year, primarily via steam reforming.1–3 Moreover, the high energy content of the H–H bond makes H2 highly promising as a future energy carrier. Indeed, the energy density of H2 is orders of magnitude higher than those of batteries.4 As such there would be large environmental benefits in developing more sustainable methods for H2 production, both in order to cover industrial needs as well as furthering the implementation of renewable energy in e.g. our transportation sector. The idea of light-driven proton reduction providing H2 as a solar fuel, utilizing either photosynthetic organisms or molecular catalysts in combination with a suitable photosensitizer, is particularly appealing in this context.In light of a future hydrogen society it should be stressed that evolution has already developed a hydrogen economy, where the interconversion between protons and molecular hydrogen is catalysed with remarkable efficiencies by ancient gas processing enzymes called hydrogenases.5 The exceptional catalytic capacity of the hydrogenases makes them highly relevant for biological hydrogen production and in device applications as an alternative to Pt-based catalysts in e.g. (photo-)electrolysers and fuel cells, as well as serving as inspiration for the development of synthetic catalysts.6–11 The interest in hydrogenases and related molecular models is underscored by the number of recent excellent reviews on these different topics (see e.g.ref. 11–20).
Herein we will cover both biochemical and biomimetic approaches of relevance to H+/H2 interconversion. Considering the activity of the field and the number of relatively recent reviews on hydrogenase related chemistry, we will not strive to cover the entire field. Instead, we will provide a short overview of the hydrogenases and then primarily focus our attention on efforts made at the molecular level aimed at engineering and optimizing these enzymes, with an emphasis on their inorganic cofactors. Moreover, we will showcase how our understanding of hydrogenases can be applied in the design of novel biomimetic/-inspired catalysts and the preparation of semi-synthetic hydrogenases.
2. Hydrogenases
The first report of organisms displaying hydrogenase activity was published in the 1930s.21 Since then these enzymes have been found in all domains of life and, despite the apparent simplicity of this reaction, Nature has designed different highly elaborate metal cofactors to perform this chemistry. Thus, hydrogenases are divided into different classes depending on the metal content of the catalytic cofactor, i.e. [NiFe], [FeFe] and [Fe] hydrogenases (Fig. 1).22,23 These main classes can in turn be further divided into different subclasses. It should be noted that, despite striking similarities in the cofactor structure and function, these three different classes of hydrogenases are not phylogenetically related. In the following section, we will briefly summarize the current knowledge of these highly efficient catalysts and their biologically unique, organometallic cofactors.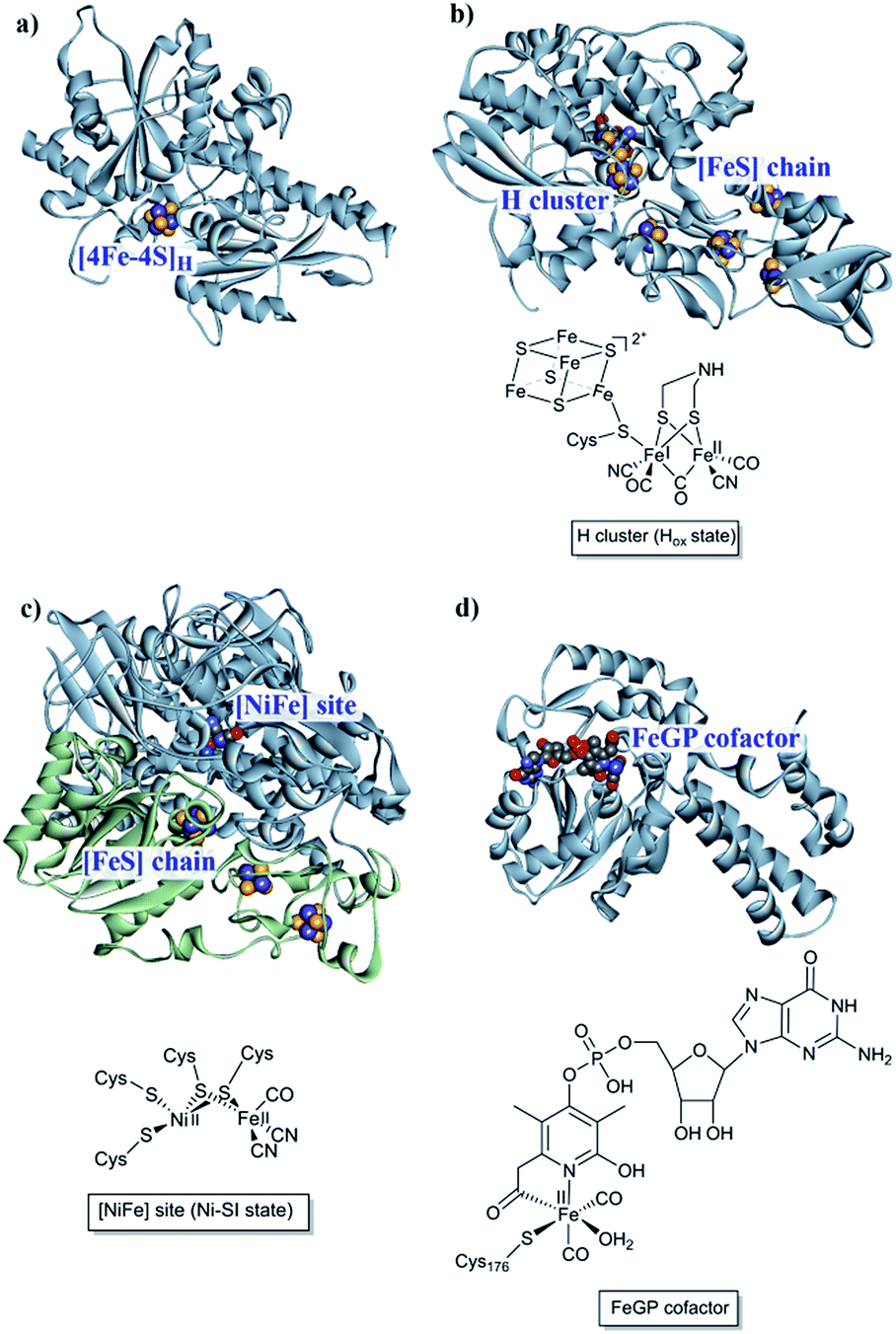 | ||
| Fig. 1 [NiFe], [FeFe] and [Fe] hydrogenases. (a) The “apo” form of Chlamydomonas reinhardtii HydA1 ([FeFe] hydrogenase; PDB: 3LX4);24 (b) Clostridium pasteurianum CpI ([FeFe hydrogenase]; PDB: 4XDC);25 (c) Allochromatium vinosum [NiFe] hydrogenase (PDB: 3MYR);26 (d) Methanocaldococcus jannaschii [Fe] hydrogenase (PDB: 3DAG).27 Protein backbones are shown as solid ribbons and coloured by chain, and metal cofactors are shown as space-filling models and coloured by element (structures drawn using BIOVIA Discovery Studio Visualizer). A schematic representation of the active site of the different subclasses is shown below the respective enzyme structure. Heteroatom colour coding: S: yellow; Fe: indigo; Ni: dark blue; O: red; N: blue; P: orange. | ||
2.1 [NiFe] hydrogenases
[NiFe] hydrogenases form multimeric protein structures, consisting of at least two subunits denoted the large subunit (harbouring the active site) and the small subunit (harbouring a number of iron–sulfur ([FeS]) clusters). These enzymes arguably represent the most studied class of hydrogenases and are often subdivided into different groups based on their molecular phylogeny, but other classification systems are also employed.17,22,23 The reactivity and function of these groups differ quite substantially. Still, they share a dependency on a heterodimeric [NiFe] cofactor, in which the Ni ion is generally coordinated by four cysteine derived thiol ligands. The octahedral Fe ion is bound to the Ni ion by two bridging thiol ligands and further decorated by a CO and two CN− ligands (Fig. 1). The one exception to this reported so far are the so-called [NiFeSe] hydrogenases that form a subgroup within group 1 [NiFe] hydrogenases, in which one of the bridging cysteines is replaced by a selenocysteine residue.13,28,29Most mechanistic studies have been performed on group 1, represented by e.g. Escherichia coli hydrogenase 1 (Hyd-1). The other groups appear to use similar mechanisms, but the specific details may differ between different [NiFe] hydrogenases.30 X-ray crystallography and spectroscopy (primarily FTIR and EPR) in combination with electrochemical methods and DFT calculations have elucidated a number of states of relevance to the catalytic cycle (Fig. 2).12,30–32 In short, purified [NiFe] hydrogenases generally exhibit a mix of oxidised states denoted as the NiA (or un-ready) and NiB (ready) states. Both states feature a [NiIIIFeII] dimer but differ in the nature of the bridging ligand, and both of these states can enter the catalytic cycle upon reduction.33 During catalysis, H2 is heterolytically cleaved upon binding to the [NiIIFeII] cofactor, resulting in the release of a proton to a nearby proton acceptor and the formation of the diamagnetic NiR-state featuring a bridging hydride.32,34 One-electron oxidation of NiR results in the paramagnetic NiC-state [NiIIIFeII] retaining the μ-hydride ligand.35,36 A further one-electron oxidation results in the release of the hydride ligand as a proton and formation of the NiSI-state, a [NiIIFeII] species lacking a bridging ligand. Thus, the Fe ion is believed to remain as FeII throughout the reaction while the Ni ion cycles between NiII and NiIII. Additionally, a [NiIFeII] state denoted as NiL has previously been shown to form upon irradiation of the NiC-state,37,38 and recent spectroscopic studies support the direct involvement of this latter state in the catalytic cycle as an intermediate during the transition between the NiC and the NiSI states.39,40
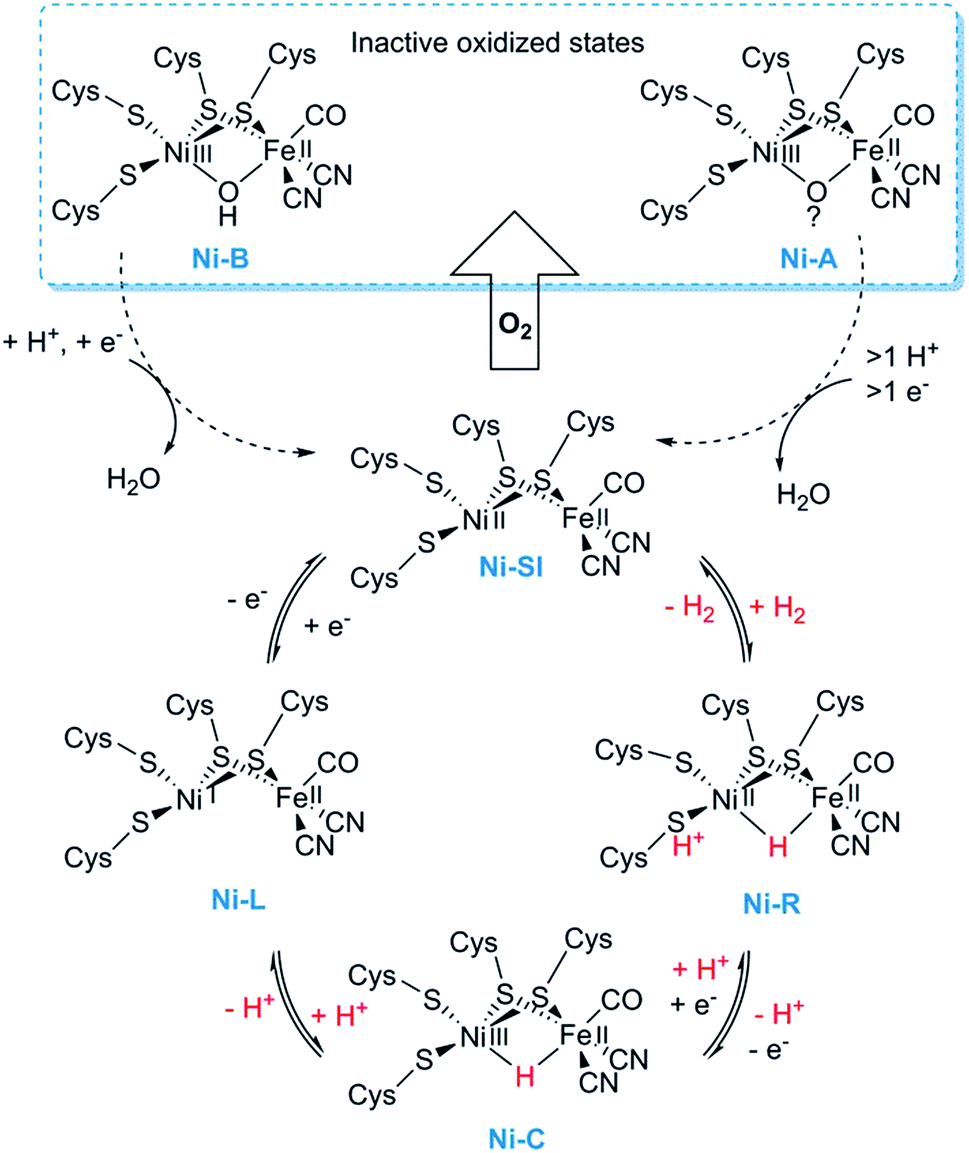 | ||
| Fig. 2 Skeletal representation of the catalytic cycle of the [NiFe] hydrogenase including the NiL-state. | ||
2.2 [FeFe] hydrogenases
In the case of [FeFe] hydrogenases, the reaction is catalysed by a hexanuclear Fe complex, the H-cluster, which is buried in the so-called H-domain. Detailed structural information about [FeFe] hydrogenases is available from X-ray crystallography studies of hydrogenases from Clostridium (C.) pasteurianum (CpI), Desulfovibrio (D.) desulfuricans and Chlamydomonas (Ch.) reinhardtii.24,41–43 In contrast to [NiFe] hydrogenases, the [FeFe] hydrogenases are often monomeric. However, [FeFe] hydrogenases of bacterial origin generally feature additional domains of varying complexity harbouring a series of [FeS] clusters.23,44 The smallest [FeFe] hydrogenases known are the eukaryotic [FeFe] hydrogenases found in green algae that consist of only the H-domain, which features the H-cluster as its only metal site, as exemplified by e.g. HydA1 from Ch. reinhardtii. Still, there are also forms of [FeFe] hydrogenases that feature additional subunits (see for example ref. 44), as exemplified by e.g. the [FeFe] hydrogenases from D. desulfuricans and Thermotoga maritima.43,45–51The H-cluster consists of a canonical cysteine-coordinated [4Fe–4S] cluster ([4Fe–4S]H) coupled to a dinuclear iron complex called the [2Fe] subsite. The [2Fe] subsite features a bridging azadithiolato ligand (adt = [−SCH2NHCH2S−]2−),42,53,54 as well as three CO and two CN− ligands, and is connected to the [4Fe–4S]H-cluster via a bridging cysteine thiol ligand (Fig. 1). The latter represents the only protein derived ligand to the [2Fe] subsite; nevertheless, the proteinous surroundings have a dramatic effect on the reactivity of the cofactor.53,55–59 Many of the details related to the catalytic mechanism are still not fully resolved, but a skeletal mechanism including the recently observed “hydride-state (HHyd)”57,60 is summarized in Fig. 3. It should be noted the mechanism is still intensively studied and alternative protonation behaviour has recently been suggested, including the presence of bridging hydrides in Hred and Hsred, which would argue against their direct involvement in the catalytic cycle.61–63 The paramagnetic Hox-state contains an oxidised cluster and [2Fe] subsite ([4Fe–4S]H2+–[FeIFeII]) with an open coordination site or readily exchangeable ligand on the distal iron. Heterolytic cleavage of H2, facilitated by a bridgehead amine, results in the formation of another paramagnetic species, the HHyd-state, featuring a terminal hydride ligand on the distal Fe ([4Fe–4S]H+–[FeIIFeII–H]). Release of the hydride ligand as a proton with concomitant reduction of the H-cluster generates the Hsred-state ([4Fe–4S]H+–[FeIFeI]). Oxidation of the [4Fe–4S]H-cluster generates the diamagnetic Hred-state ([4Fe–4S]H2+–[FeIFeI]), which is further oxidised back to the Hox-state to close the catalytic cycle.17,57,60
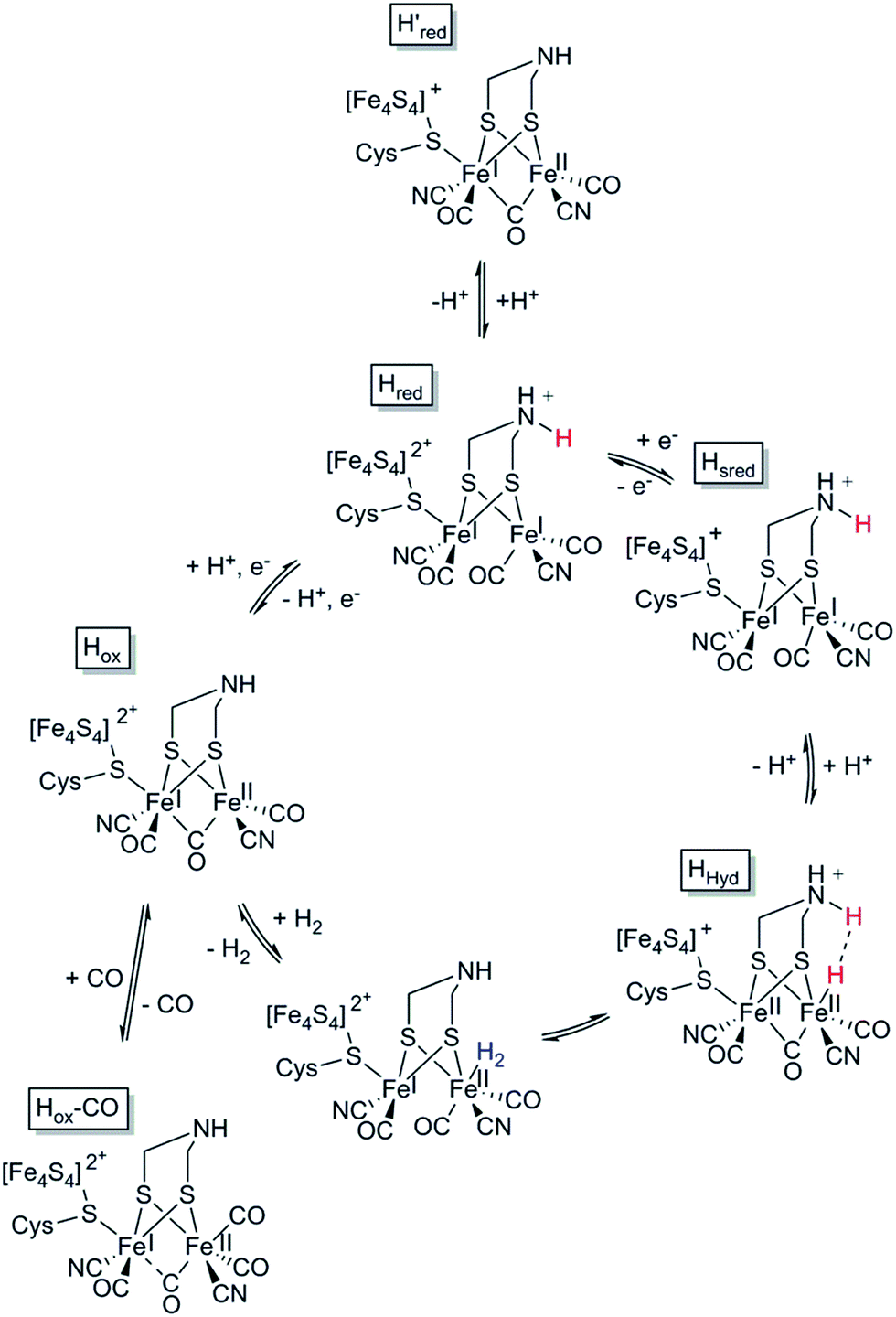 | ||
| Fig. 3 Skeletal representation of the catalytic cycle of the [FeFe] hydrogenase, adapted from Sommer et al.52 | ||
2.3 [Fe] hydrogenases
The third class of hydrogenases, the H2-forming methylene-tetrahydromethanopterin dehydrogenase (Hmd), or [Fe] hydrogenase, is found in methanogenic archaea and was first reported by Thauer and co-workers in 1990.64,65 In comparison to [NiFe] and [FeFe] hydrogenases, the reactivity of Hmd is noticeably different: the enzyme is involved in the reduction of CO2 to CH4. Instead of direct H+/H2 interconversion, Hmd utilizes H2 to catalyse the reversible reduction of N5,N10-methenyl-tetrahydromethanophterin (MPT+) to N5,N10-methylene-tetrahydromethanophterin (HMPT) (Fig. 4).66 The reaction is catalysed by a mononuclear Fe–guanylylpyridinol cofactor (Fe–GP), in which the low-spin Fe ion is further coordinated by two CO ligands and a cysteine-derived thiol ligand (Fig. 1).27,67,68 The Fe ion does not change the oxidation state during catalysis. In the presence of MPT+, the FeII ion instead acts as a Lewis base, enabling the heterolytic cleavage of H2. The substrate acts as a hydride acceptor, while the proton is initially transferred to either the cysteine thiol or the oxygen atom of the pyridinol before it is rapidly exchanged with bulk water.69–71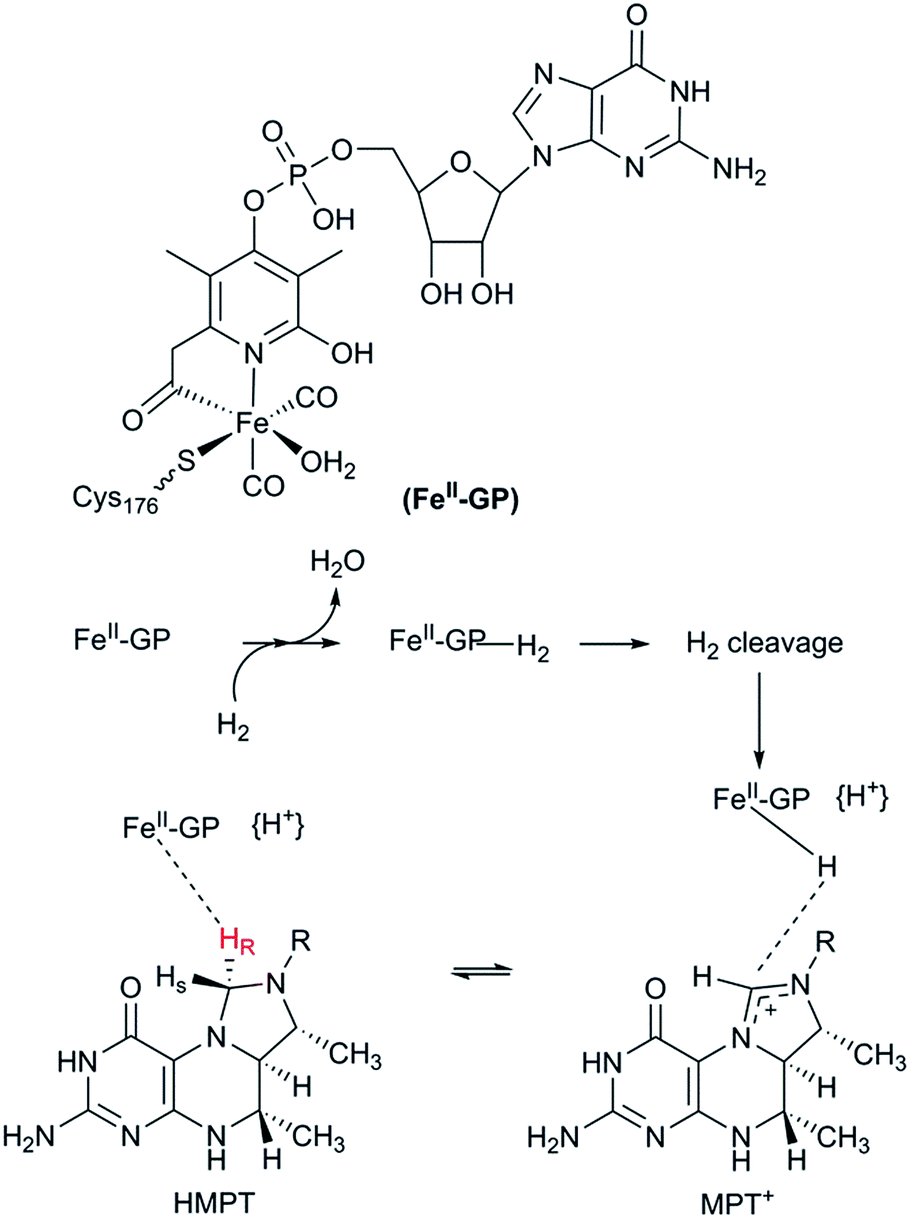 | ||
| Fig. 4 Schematic representation of the catalytic cycle of the [Fe] hydrogenase. | ||
3. Designing robust hydrogenases through protein engineering
Insight into molecular mechanisms of proton activation, gas transfer and electron transfer in hydrogenases has opened the doors to designing robust enzymes for optimal hydrogen production through protein engineering. Two main challenges in engineering hydrogenases are (a) shifting catalytic bias towards hydrogen production and (b) producing enzymes tolerant to dioxygen†(Fig. 5). Other important aspects to consider, but that are outside of the scope of this review, are metabolic engineering to redirect the intracellular electron flow to native hydrogenases and the (over)expression of unmodified hydrogenases for increased hydrogen production.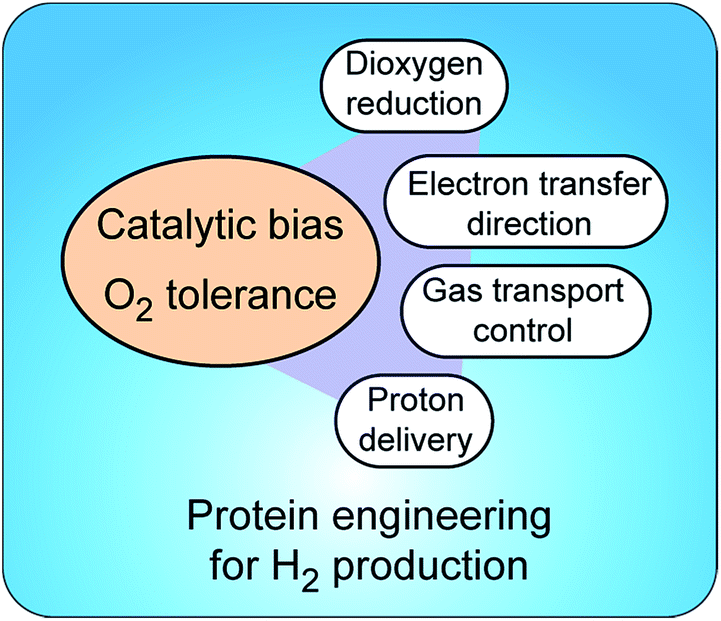 | ||
| Fig. 5 Strategies to control catalytic bias and improve tolerance to O2 in hydrogenases. Protein engineering allows tuning proton and electron delivery and controlling the access of O2 to the active site. | ||
3.1 What is catalytic bias?
Hydrogenases can theoretically perform both H2 oxidation and H2 evolution, but usually one activity predominates significantly over the other, in vivo or in vitro. A straightforward definition of catalytic bias is the ratio between reaction rates for H2 oxidation and H2 production. As pointed out by Abou Hamdan et al., catalytic bias in a given enzyme is classically characterised by parameters related to the equilibrium constant; however, with oxidoreductases, catalytic bias is more often described in terms of the relative redox potentials of the active sites as compared to the potentials of substrate/product pairs.72 Armstrong et al. defined bias in hydrogenases as the ratio between rate constants for the anodic and the cathodic processes, where these constants include both intramolecular electron transfer (ET) and catalytic turnover.12 It is important to point out that this model interprets the behaviour of enzymes containing ET chains immobilized on electrode surfaces under non-limiting substrate conditions and relies on the redox potential of the distal cluster as the determinant factor in ET between the hydrogenase and an electron transfer partner. It should be noted that the direction of the reaction in vivo will be affected by external factors such as cell culture conditions and the availability of intracellular ET partner proteins.In the context of developing H2-producing systems, it is paramount to elucidate the factors modulating catalytic bias to shift it towards H2 production. The [NiFe] hydrogenase from Desulfovibrio fructosovorans‡ is one of the most studied enzymes in the context of understanding catalytic bias. The rate limiting step for H2 production in this hydrogenase was found to be proton transfer and the subsequent release of H2 from the active site, while H2 oxidation is primarily affected by electron transfer (ET) to and from the active site via the three [FeS] cluster relay in the small subunit.72,74
These findings prompt the question: can we change the catalytic bias in a hydrogenase by altering proton and/or electron transport through the enzyme?72 Some answers have recently started to appear in the literature as we gain understanding about proton pathways, gas transport and electron transfer in different types of hydrogenases.
3.2 Tuning electron transfer for the control of catalytic bias
The ET chains in hydrogenases, composed of [FeS] clusters, are a key target for protein engineering to control catalysis. In particular, the effects of non-cysteinyl coordination and the binding pocket properties on the redox and structural properties of [FeS] clusters remain largely unexplored.75,76 It is however not always an easy feat to perform site-directed mutagenesis on residues binding, or in the near vicinity of [FeS] clusters, as mutated proteins may not incorporate iron or be expressed at all.77The ET can presumably be tuned in clostridial [FeFe] hydrogenases by engineering of [FeS] clusters in the ET subunit, using rationales similar to those employed for [NiFe] hydrogenases (see below). For green algal hydrogenases (which do not possess an ET chain), it is the interaction between the catalytic unit and an ET partner protein that becomes more interesting. An example of the latter is Ch. reinhardtii HydA1. Sybirna et al. mutated Arg171, a highly conserved residue involved in the recognition of ferredoxin as an ET partner, to Asp or Trp (Table 1).78 The change in electrostatic charge disrupted the interaction with ferredoxin, as predicted, resulting in lower activity when using this protein as an electron donor. However, when using a methyl viologen assay for H2 evolution, the Arg171Asp mutant presented six times higher Vmax, attributed to an increase in affinity to methyl viologen upon introduction of a positive charge. This study showed that ET was rate-limiting in Ch. reinhardtii HydA1 and suggests that the protein can be engineered to increase the active site accessibility to electron donors.
| Enzyme, organism | Mutation | Main effect | Ref. |
|---|---|---|---|
| HoxG (MBH), [NiFe] R. eutropha | C81A, C81V, C81S, C81T (catalytic subunit) | Higher (variable) O2 tolerance | 98 |
| Periplasmic, [NiFe] D. fructosovorans | V74M, V74C, V74S, V74N (catalytic subunit) | Increased O2 tolerance, decreased H2 oxidation | 33,99,100 |
| HycG (hydrogenase-3), [NiFe] K. oxytoca | G47C, G50C, G113C, G120C, G50C/G120C (ET subunit) | Increased O2 tolerance, unaltered in vivo activity | 107 |
| HydA1, [FeFe] Ch. reinhardtii | R171D (catalytic unit) | Decreased H2 evolution but increased Vmax | 78 |
| HycE (hydrogenase-3), [NiFe] E. coli | Various, random (catalytic unit) | Increased H2 production | 79 |
| Periplasmic, [NiFe] D. fructosovorans | P238C (ET subunit) | Shift in catalytic bias towards H2 production | 84 |
| HynS, [NiFe] A. macleodii | P285C/H230C (ET subunit) | Shift in catalytic bias towards H2 production | 85 |
| HupS, uptake, [NiFe] N. punctiforme | C12P (ET subunit) | Shift in catalytic bias towards H2 production in vivo | 87 |
| HyaA (MBH/hydrogenase-1), [NiFe] E. coli | E73Q (ET subunit) | Shift in catalytic bias towards H2 production | 88 |
Several [NiFe] hydrogenases have been targeted in recent years for the enhancement of ET to/from the active site through protein engineering. A number of crystal structures exist to aid in the design of specific mutations, even if limited to a few groups of hydrogenases. An alternative is found in performing random mutagenesis and screening for enhanced activity. Maeda et al. applied error-prone PCR and saturation mutagenesis on the catalytic subunit, HycE, from hydrogenase-3 in E. coli.79 Seven mutants were found to have up to nine times enhanced H2 evolution, possibly due to strengthening protein–protein interaction between the large and the small subunits of hydrogenase-3, thereby enhancing ET between the [FeS] cluster chain and the catalytic site.
[NiFe] hydrogenases present a highly conserved histidine residue as one of the four ligands of the distal cluster (i.e. the cluster farthest from the active site), a fact that has raised questions on its role in intra- and inter-molecular ET.80 DFT calculations by Petrenko & Stein identified two amino acids as crucial for intermolecular ET, the aforementioned ligand His184 and Ser196, in the D. fructosovorans enzyme.81 Introducing the His184Cys mutation does not significantly change the reorganization energy between the mutant and wild-type enzyme, in contrast to what was suggested by Dementin et al.80 Rather, the all-cysteine coordination affected the electronic coupling to the electrode surface, decreasing the ET rate by several orders of magnitude.81 Thus it seems that the distal cluster in [NiFe] hydrogenases has a crucial role as an entry/exit point for intramolecular ET and in the recognition of an ET partner for intermolecular ET. An interesting exception to the histidine ligation is the cyanobacterial uptake (HupSL) hydrogenases, where a strictly conserved glutamine is found instead of the histidine. Despite this difference, a [4Fe–4S] cluster is assembled in the distal position.82 If this glutamine residue binds the cluster, it might significantly reduce the rate of ET to its electron acceptor.83
In parallel to the work on the distal cluster, efforts have also been invested in the medial and proximal clusters. Almost two decades ago, Rousset et al. reported the conversion of the medial [3Fe–4S] cluster to a [4Fe–4S] metal centre in the [NiFe] hydrogenase from D. fructosovorans.84 This was achieved through a Pro238Cys mutation that inserted a fourth thiolate ligand in a similar position to that found in [NiFeSe] hydrogenases, where a medial [4Fe–4S] cluster occurs naturally. Functionally, the mutant enzyme became a slightly worse H2 oxidiser, whereas the H2 evolution activity increased; intramolecular ET was not deemed to be rate limiting in either wild-type or Pro238Cys hydrogenases. While the enzyme was still a net H2 oxidiser, this work showcased how catalytic bias could be shifted by perturbing the ET chain.
The presence of functional enzymes in photosynthetic organisms would be an important factor for efficient photobiological H2 gas production. In order to address this challenge, it is critical to introduce engineered hydrogenases into such organisms and evaluate their true activity in vivo. An important step was taken by Yonemoto et al. through the heterologous expression of the [NiFe] hydrogenase HynSL from the marine bacterium Alteromonas macleodii in both E. coli and the cyanobacterium Synechococcus elongatus.85 Furthermore, the authors engineered the small subunit HynS through the mutations Pro285Cys (which converts the medial [3Fe–4S] cluster to a [4Fe–4S]) and His230Cys (which alters the coordination sphere of the distal cluster to a full cysteinyl one). It was found that HynSL carrying both mutations showed an effective change in the catalytic bias as measured in lysates of E. coli, observable by an increase in the production of H2. The enzyme was still an overall H2 oxidiser, as uptake activity remained virtually unchanged at levels slightly higher than the production rate. Nevertheless, this modified hydrogenase served as a starting point for further engineering of the electron transfer chain of HynS, through replacement of all twelve coordinating amino acids by [FeS] cluster ligands found in other hydrogenases (Asp, His, Asn and Gln).86 Albeit none of the tested mutants presented a net H2in vivo production, this work further showcased how tweaking the properties of the ET chain may affect the catalytic activity.
Recently, Raleiras et al. demonstrated the in vivo production of H2 in the filamentous cyanobacterium Nostoc (N.) punctiforme ATCC 29133 following engineering of the uptake hydrogenase HupSL.87 The single point mutation Cys12Pro was inserted in the proximal [FeS] cluster, causing its conversion from a [4Fe–4S] to a [3Fe–4S] species; N. punctiforme carrying this modified HupSL presented a net production of H2 (Fig. 6). The authors proposed that particular metabolic conditions, when the reducing power provided by photosynthesis needs an outlet, combined with the structural change in the proximal cluster, facilitate a reverse direction of electron transfer towards the active site, with the concomitant production of H2. Petrenko & Stein have recently suggested that a glutaminate coordination to the distal cluster is probably associated with the possibility to reverse the electron flow in this hydrogenase.83 There is an interesting parallel with the O2 detoxification mechanism of membrane-bound hydrogenases (MBHs), where a reverse electron flow is likely facilitated by the ET chain containing [FeS] clusters with relatively positive redox potentials.
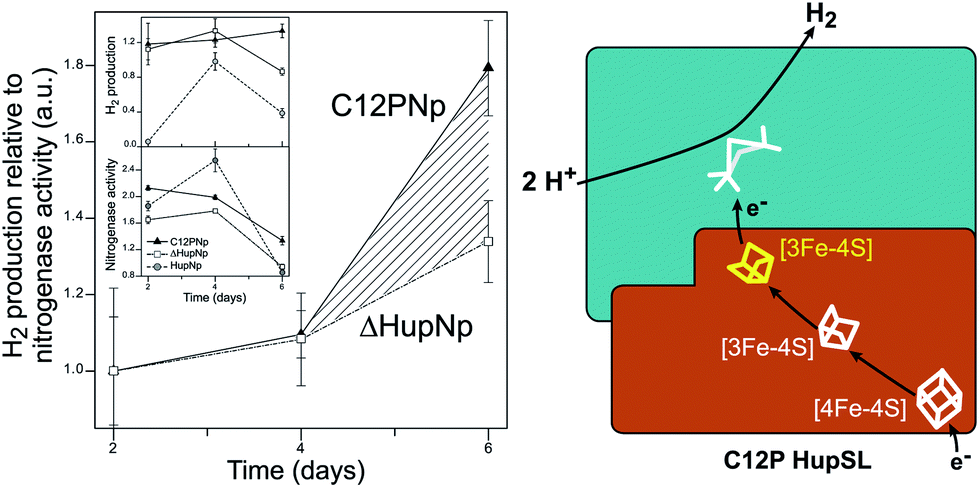 | ||
| Fig. 6 Engineering of the uptake hydrogenase HupSL in Nostoc punctiforme for hydrogen production. Left: N. punctiforme expressing C12P HupS (C12PNp strain) produces H2 in addition to the background production from nitrogenase activity (ΔHupNp strain). Right: the C12P mutation induces a [4Fe–4S] → [3Fe–4S] cluster conversion in the proximal position in HupS, facilitating reverse electron flow towards the active site. The figure was adapted from Raleiras et al.87 with permission from the Royal Society of Chemistry. | ||
The latest effort in a rational tuning of catalytic bias is the attempt to modulate the redox potential of the proximal cluster in the O2-tolerant E. coli hydrogenase-1 by Flanagan et al., through changes in the second coordination sphere.88 In particular, Glu73 was identified as a conserved amino acid in O2-tolerant hydrogenases, in contrast to a glutamine residue present in the homologous position in O2-sensitive enzymes. Introducing the Glu73Gln mutation into hydrogenase-1 did not affect its O2 tolerance but impacted the catalytic bias markedly by doubling the H2 production. The exact cause of this shift in the catalytic bias is so far not fully understood, as there was no significant change in the redox potential of the proximal cluster in the mutant as compared to the wild-type enzyme.
In summary, a number of recent reports support the notion that shifting the relative potentials of the clusters in the ET chain can affect catalytic bias towards H2 production. Further in vivo studies of these modified enzymes in combination with metabolic engineering will be critical to establish the technical feasibility of this approach to biological H2 production.
3.3 The O2 problem: gas channels and O2 reduction
The conversion of sunlight to chemical energy stored as hydrogen through photosynthesis requires conditions where hydrogenases are protected from or tolerant to the presence of O2.15 Fortunately, functional hydrogenases have been found in organisms where exposure to O2 cannot be avoided, which has awakened research into the factors that make a hydrogenase O2-tolerant. [FeFe] hydrogenases have long been considered very sensitive to O2, although a few exceptions exist.89,90 Gas transfer has been studied in [FeFe] hydrogenases, and potential O2 access routes have been identified.91 However, attempts to engineer O2-tolerance in the [FeFe] hydrogenase CpI resulted in a drastic decrease in activity.92 In contrast, [NiFe] hydrogenases are able to reverse O2-mediated inactivation through reduction of the active site, and some [NiFe] hydrogenases actively detoxify O2 during catalysis. For this reason, most efforts have been focused on understanding and improving O2 tolerance in [NiFe] hydrogenases.An early hypothesis concerning the sensitivity to O2 was the selectivity in the access of O2 and H2 to the active site. Gas transport pathways have been identified by structural analysis in several hydrogenases. These pathways are channels within the protein structure, typically lined with hydrophobic amino acids, that connect the active site to the protein surface.93
Compariso n of primary sequences reveals small differences in the immediate vicinity of the active site between O2-sensitive and O2-tolerant [NiFe] hydrogenases. Both H2 and O2 can be transported along the same hydrophobic channels found within the catalytic subunit, as suggested by X-ray crystal structures and molecular dynamics simulations.94,95 Manipulating these channels so that only H2 could selectively move within the protein has been suggested as a way to increase O2 tolerance in [NiFe] hydrogenases. In the O2-tolerant, H2-sensitive [NiFe] hydrogenases from Ralstonia (R.) eutropha§ H16 and Rhodobacter (Rh.) capsulatus, substitution of two particular, bulky amino acid residues (Ile65 and Phe113, Rh. capsulatus numbering) increased sensitivity to O2 by allowing the larger gas molecule to reach the [NiFe] active site.96,97 Ludwig et al. identified Gly80 and Cys81 in the O2-tolerant MBH from R. eutropha H16 as potentially important for the tolerance mechanism.98 The authors noticed, however, that O2 tolerance was likely not due simply to selective gas access to the active site, given that the wild-type enzymes reacted rapidly with O2 without becoming completely inhibited.
Molecular dynamics simulations on the [NiFe] hydrogenase from D. gigas demonstrated that H2 is able to quickly penetrate the catalytic subunit through a number of different channels, but uses preferentially one to reach the active site, possibly using Glu18 and Val67 in a gating mechanism near the active site.93 Dementin et al. and Liebgott et al. mutated the corresponding valine residue in the D. fructosovorans hydrogenase, valine-74 (Fig. 7; Table 1), and found that the mutant enzymes could still competently oxidise H2 in the presence of small concentrations of O2, while the wild-type enzyme is completely inactivated.99,100 The effect seems to be mostly kinetic due to a partial obstruction of O2 access to the active site in the mutated enzyme. An attempt to understand the molecular basis for this higher tolerance to O2 using a larger collection of valine-74 mutants led to the observation that replacing valine with a more hydrophilic amino acid increased the rate of reactivation after O2 exposure.33
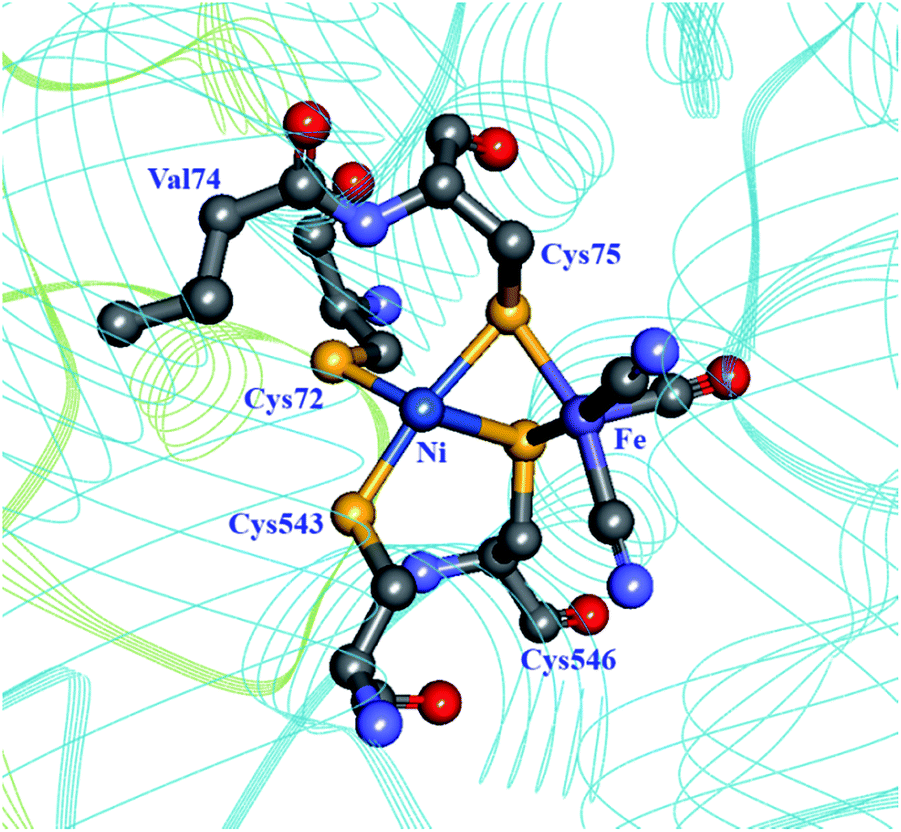 | ||
| Fig. 7 The coordination sphere of the [NiFe] site in the D. fructosovorans hydrogenase (PDB: 1YRQ).101 Through site-directed mutagenesis, valine-74 has been shown to have a role in the gas accessibility to the active site, possibly through a gating action. Blue ribbon: large subunit; green ribbon: small subunit; heteroatom colour coding as in Fig. 1. | ||
An alternative mechanism for O2 tolerance was identified in MBHs from several organisms, which perform H2 oxidation in the presence of low tensions of O2. Enzymes in the oxidised, NiA state typically need long reduction periods to recover to an active state; it follows that if exposure to O2 cannot be avoided, then the enzyme must use part of an available electron pool to reduce O2 to H2O. The proximal [FeS] cluster in MBHs is an unusual [4Fe–3S] cluster ligated by not four (the most common ligation pattern in [FeS] clusters) but rather six cysteines in the small subunit (Fig. 8),102–104 and this cluster has been shown to play an active role in reducing O2 to water as a means to protect the active site from oxidative deactivation.105 While four of the ligating cysteines are homologous to those found in other [NiFe] hydrogenases, the two supernumerary cysteine residues (Cys19 and Cys120 in R. eutropha) were unequivocally shown to be essential for O2 tolerance: replacing these cysteines in the R. eutropha MBH restores the NiA state and abolishes O2 tolerance.103 Lukey et al. pinpointed Cys19 in hydrogenase-1 from E. coli as having the critical role in O2 tolerance as seen by a dramatic decrease in H2 oxidation in the Cys19Gly and Cys19Gly/Cys120Gly mutants.106 All enzyme variants (wild type and Cys19Gly, Cys120Gly, and Cys19Gly/Cys120Gly mutants) present some degree of the NiA state in EPR spectroscopy, but reduction of the [NiFe] site to an active species varies markedly, with the Cys19 mutants requiring much longer reactivation times.
 | ||
| Fig. 8 Structural comparison between proximal [FeS] clusters in O2-tolerant and O2-sensitive [NiFe] hydrogenases. Left: the proximal [4Fe–3S] cluster in the R. eutropha MBH (PDB: 3RGW),103 involved in the direct detoxification of O2. Right: the proximal [4Fe–4S] cluster in the D. fructosovorans hydrogenase (PDB: 1YRQ).108 Cysteine residues directly coordinating the clusters are shown. Heteroatom colour coding as in Fig. 1. | ||
Critically, the [4Fe–3S] proximal cluster undergoes two redox transitions at relatively mild redox potentials (−60 mV and +160 mV vs. SHE in the R. eutropha enzyme),109 necessary for the direct reduction of O2 to H2O.105 Additionally, the other [FeS] clusters in the ET chain also present less reducing than usual redox potentials. This in combination with a structural arrangement allowing fast ET between the distal clusters of two enzymes has been argued to facilitate an efficient electron flow to afford the necessary four electrons for the complete reduction of O2 in a kinetically competent fashion.103,105,109
So far, there are no reports of a successful introduction of O2 tolerance into an O2-sensitive [NiFe] hydrogenase by introducing supernumerary cysteines in homologous positions. Nevertheless, an improvement of pre-existing O2 tolerance of the hydrogenase from Klebsiella oxytoca HP1 was recently reported by Huang et al., upon replacement of several glycine residues by cysteines, including those homologous to Cys19 and Cys120 in the R. eutropha MBH.107 Since no structural or spectroscopic studies have been conducted on these mutants, it is currently not known whether a successful cluster conversion has taken place.
[NiFeSe] hydrogenases are biased towards H2 production and present some degree of tolerance to O2.13 These enzymes do not present the characteristic NiA and NiB inactive states found in O2-exposed [NiFe] hydrogenases. Marques et al. reported recently that substituting the Ni-coordinating selenocysteine in the D. vulgaris Hildenborough [NiFeSe] hydrogenase for cysteine (Sec489Cys) abolished the enzyme's tolerance to O2, effectively turning it into a “regular” [NiFe] hydrogenase.110 The mutation also had a negative effect on the enzyme's ability to incorporate nickel and to produce H2.
So far, attempts to engineer O2-tolerance, primarily via manipulations of gas channels, result invariably in decreased H2 production, but do not affect significantly H2 oxidation. As such, these modified enzymes appear more suitable for exploration in fuel cell applications rather than H2 gas production.
3.4 Proton transport pathways
Proton transport pathways in proteins use either water molecules or amino acid side chains, likely through a Grotthuss mechanism. Understanding how protons move within hydrogenases and how this movement couples with electron transfer is a key step to control reactivity.Three main proton transport pathways have been proposed for [FeFe] hydrogenases, based on computational and structural studies on the enzymes from C. pasteurianum and D. desulfuricans Hildenborough.41,111–113 One pathway seems to dominate, involving proton transport through a tight hydrogen bond network formed by Glu279, Glu282, Cys299, Ser319 (C. pasteurianum numbering) and a water molecule, rather than through a water molecule wire (Fig. 9).111 Interestingly, the hydrogen bond occupancy in this network differs depending on whether the enzyme is producing or oxidising H2,111 which could indicate different preferential pathways depending on the direction of the reaction. The highly conserved Cys299 and its homologue in Ch. reinhardtii HydA1 (Cys169) and C. acetobutylicum HydA (Cys298) cannot be replaced without severely impairing the enzyme activity; the H-cluster is structurally retained in these mutants but mostly locked in an inactive state.56,59,112,114
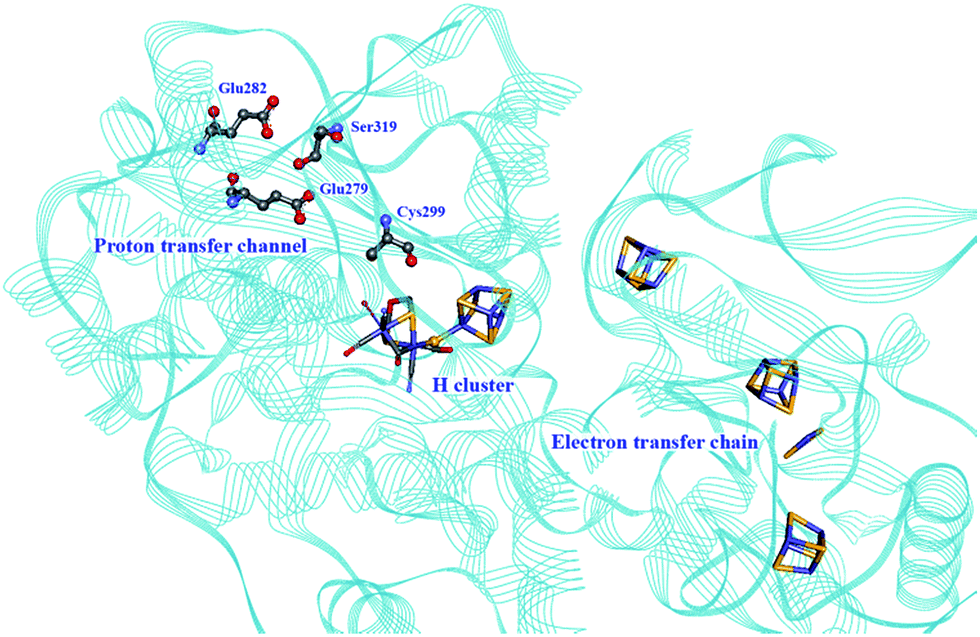 | ||
| Fig. 9 Proton and electron transfer chains in the C. pasteurianum [FeFe] hydrogenase (PDB: 3C8Y).115 Highlighted residues have been identified as part of a hydrogen bond network transporting protons between the protein surface and the active site (H-cluster). Heteroatom colour coding as in Fig. 1. | ||
Mulder et al. found recently that the Cys169Ser mutant slows down catalysis significantly (≈1% residual activity), and this disruption of the H-bonding network allows for accumulation of a terminal hydride on the distal iron of the [2Fe] subsite in the Ch. reinhardtii enzyme.57 A similar species has also been identified by Reijerse et al. after the [2Fe] subsite was replaced with an ether variant (cf. Section 5, semi-synthetic hydrogenases).60
The earliest proposals of proton transport pathways in [NiFe] hydrogenases involved a combined amino acid side chain/structural water molecule wire leading from the active site to a Mg2+-bound water molecule close to the protein surface.101 Alternative pathways were later analysed by Teixeira et al. through Poisson–Boltzmann/Monte Carlo simulations of protonation steps in the enzyme from D. gigas; in particular, a pathway comprising mostly histidine and glutamate residues was found as the most likely one.116 Interestingly, and in contrast to earlier proposals, this pathway includes not only amino acids from the large, catalytic subunit, but also residues from the small subunit in close proximity to the proximal [FeS] cluster. It is still not known if there is proton-coupled electron transfer involving residues around this particular cluster. A parallel study by Fdez. Galván et al. using quantum mechanics/molecular mechanics (QM/MM) on the D. fructosovorans enzyme supported the findings on D. gigas.117 Interestingly, it was found that different pathways have different energy barriers depending on the direction of proton transfer, suggesting that a pathway that is not preferential for one direction may be in use for the reverse reaction. More recently, ultra-high resolution XRD studies of the D. vulgaris Miyazaki F enzyme provided experimental support for the participation of His36 (D. vulgaris numbering) in a well conserved proton transfer network, including residues from both the large and the small subunit.32
No modification has been performed to date to proton channels in hydrogenases that would improve catalysis (in either direction). However, discerning pathways that work towards a particular catalytic bias may help us to design better hydrogenases. It is particularly interesting to notice that in the case of [FeFe] hydrogenases, artificial maturation using variants of the diiron subsite offers new possibilities for controlling proton transfer.16,53
3.5 Summary and outlook
Molecular characterization of diverse types of hydrogenases has provided a portfolio of different solutions used by nature to combine hydrogen production/oxidation and the presence of oxygen. Clearly, there are many starting points to be explored for the engineering of hydrogenases for in vivo and in vitro applications. Results from engineering [FeS] clusters in hydrogenases have been very promising, and it is entirely feasible to take this route for fine tuning intra- and inter-molecular electron transfer and thus controlling catalysis without perturbing the structure of the active site. Further controlling the gas access to the active site may help us to develop more O2-tolerant hydrogenases. It remains to be fully assessed whether high rates of H2 production are compatible with oxygenic conditions, but the tools exist now within our grasp to explore this possibility.4. Synthetic catalysts: from biomolecules to artificial enzymes and bioinspired catalysts
Studies on hydrogenases have repeatedly demonstrated the importance of the protein environment for the catalytic efficiency of their cofactors. This has inspired the biomimetic chemistry community towards the preparation of increasingly elaborate synthetic systems for efficient catalysis.Whereas traditional transition-metal catalysts typically only take advantage of the first coordination sphere to modulate reactivity and selectivity, the preparation of artificial metalloenzymes allows much more elaborate control. This includes the possibility to provide specific microenvironments by controlling the local hydrophobicity/-philicity of the catalyst site, the generation of e.g. specific hydrogen bonding networks and the formation of entatic states via push–pull effects, as well as site isolation and substrate/product specificity, properties that are often referred to as second or outer coordination sphere effects (Fig. 10). Thus, the nature of the metal centres and the first coordination sphere can be modified using standard synthetic techniques. In parallel, the choice of the scaffold environment, ranging from proteins to the presence of a few amino acids or the use of a non-biological matrix, and the mode of ligation between the two partners offer a major opportunity to fine-tune the properties of the assembly. In combination, this provides the chemist with a significantly larger palette for the development of novel catalysts. The artificial enzyme field, born in the 1970s with the pioneering work of Whitesides and co-workers,118 has been constantly growing in the last few decades, as illustrated by recent reviews on e.g. protein design to make efficient artificial enzymes,119–121 and their application for oxygen activation,122 oxidation catalysis123 and asymmetric catalysis.124
 | ||
| Fig. 10 Schematic representation of the insertion of a metallic cofactor in a scaffold highlighting the potential contributions of the outer coordination sphere. | ||
In the context of H2 gas catalysis, the development of artificial hydrogenases is a relatively recent development, which holds great promise for the development of efficient, stable and sustainable catalysts. This section will cover work ranging from repurposing proteins (via metal substitution and metal cofactor replacement) to generate novel artificial enzymes showing hydrogenase activity to selecting purely synthetic systems mimicking specific aspects of enzyme catalysts.
4.1 Re-purposed proteins
The first steps towards the assembly of artificial H2-evolving enzymes relied mainly on the modification of well-known native metalloproteins in order to induce hydrogenase activity by metal centre substitution, co-factor exchange or self-assembly of protein matrices with an active synthetic catalyst. In 1988, Moura et al. described the preparation of nickel-substituted rubredoxins (NiRd), resulting in a mononuclear Ni site coordinated by four cysteine residues.125 This simple metal exchange allowed NiRd to evolve H2 in the presence of reduced methyl viologen, making it the first artificial hydrogenase ever reported. The NiRd (D. desulfuricans ATCC 27774) system has recently been re-investigated, and its electrocatalytic and photocatalytic performances for H2 evolution have been explored.126 Similar to the original study, photochemical or chemical reduction of NiRd allows catalytic H2 evolution in aqueous solution with a moderate turnover frequency (TOF) as compared to native hydrogenases. Protein film voltammetry demonstrated the existence of a proton-coupled electron transfer process within the pH range 3–5. Under these conditions, the overpotential of the NiRd was estimated to be ≈540 mV, and the initial catalytic rate was 20–100 s−1.Myoglobin and other heme-binding proteins have attracted more attention as potential protein scaffolds to assemble artificial hydrogenases, due to their natural ability to incorporate porphyrin-based cofactors in a well-defined environment; in particular in combination with various cobalt based catalysts. Ghirlanda et al. introduced cobaltous protoporphyrin IX (CoP, 1) into an apo-myoglobin, thus replacing the native iron protoporphyrin IX (Fig. 11a).127 Electrochemical investigations of the CoP–myoglobin system in aqueous medium showed significant catalytic waves associated with proton reduction. Interestingly, this catalytic behaviour did not appear to be strongly affected by O2. Conversely, at pH below 6, a loss of activity was observed, attributed to the release of the porphyrin from the heme-binding pocket due to protonation of the ligating His-93. Under photochemical conditions, the CoP–myoglobin was able to produce H2 with a turnover number (TON) ranging from 230 to 520 depending on the pH, significantly superior to free CoP under the same conditions. The influence of the protein environment was probed by the preparation of different mutants, in which histidine residues in vicinity to the metal centre were replaced by alanine. The His97Ala mutant is of particular interest, as it showed a decreased TON of 120 at pH 6.5. This value is close to that observed for free CoP, suggesting decreased cofactor stability for this mutant. In a related approach, CoP was introduced into the well-characterised electron transfer protein cytochrome b562 (Cyt b562).128 This cofactor substitution resulted in a modest catalyst for photo-induced proton reduction. However, mutating Met7, which potentially blocks the catalyst via axial coordination to the Co ion, resulted in a 2.5 fold increase in the TON. The activity of this new artificial enzyme is close to the activity determined for the CoP–myoglobin system assayed under the same conditions. Another cobalt-substituted cytochrome system was prepared by Bren and co-workers (vide infra, Ht c-552, Fig. 14). Under constant potential electrolysis the resulting artificial enzyme is capable of achieving a very high TON (>270![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) operating at an overpotential of 830 mV.129
000) operating at an overpotential of 830 mV.129
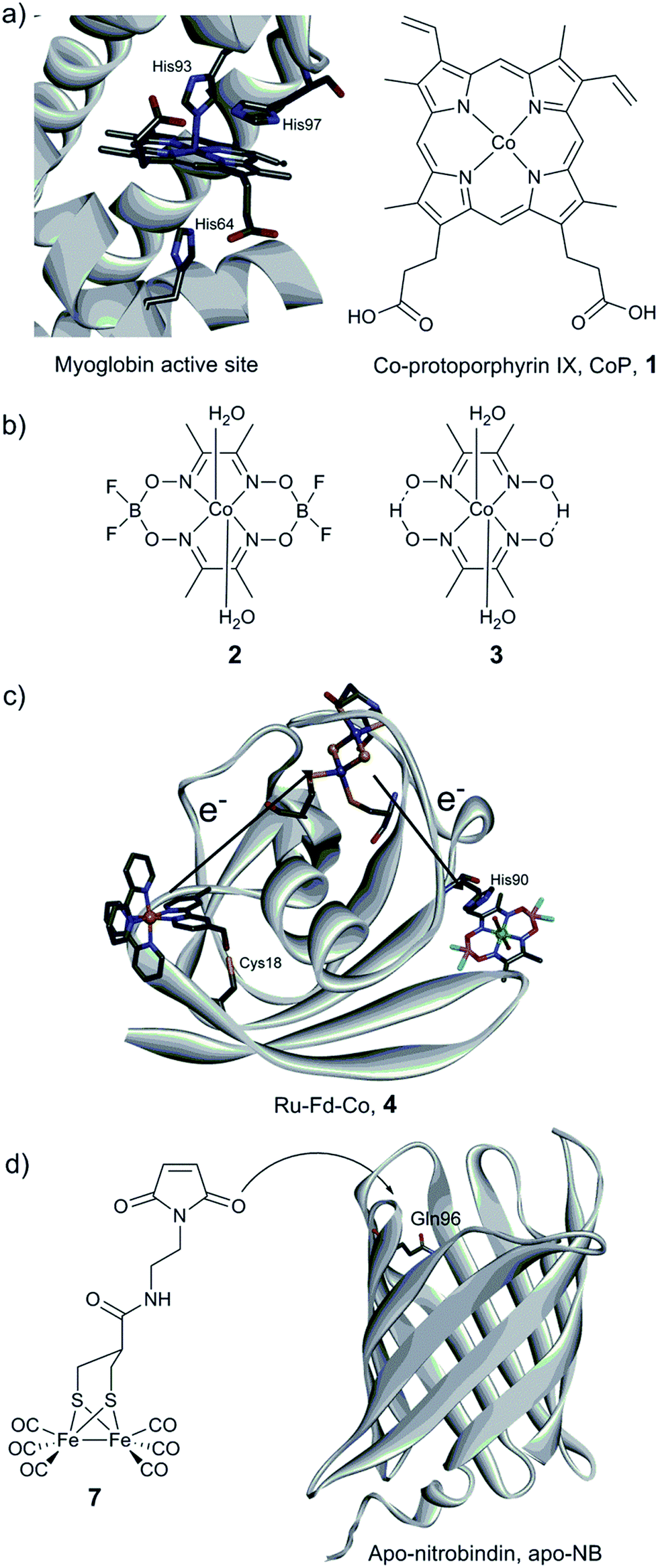 | ||
| Fig. 11 (a) Structure of the active site of myoglobin (PDB ID: 1YOI)135 showing relevant histidine residues and structure of Co-protoporphyrin IX (CoP, 1). (b) Cobalt catalysts (cobaloxime derivatives) inserted into a myoglobin matrix. (c) Scheme of the Ru–Fd–Co, 4 biohybrid, based on the structure of ferredoxin (PDB ID: 1A70) with a potential pathway for electron transfer from the PS to the catalyst. (d) Preparation of the artificial hydrogenase containing a {μ-(SCH2)2CHCOR}Fe2(CO)6 (7) moiety within a Q96C apo-nitrobindin (apoNB) (PDB ID for NB: 2A13).136 | ||
Sperm-whale myoglobin (SwMb) has also been utilized as a host for cobalt based proton reduction catalysts. Artero and co-workers reported on the incorporation of two cobaloxime derivatives, Co(dmgH)2 (2) and Co(dmgBF2)2 (3) (dmgH2 = dimethyl-glyoxime; Fig. 11b), into the cavity of an apo-myoglobin.130 In-depth spectroscopic investigations of these biohybrids clearly indicate cobaloxime insertion in the cavity, most probably through the coordination of His93 to the cobalt centre, as further supported by docking calculations. Cyclic voltammograms recorded at neutral pH have established that a low overpotential (200 mV) is required to evolve H2. Chemical reduction (using Eu(II) complexes at pH 7)131,132 and photocatalytic assays (pH 6) showed the ability of both bio-hybrids to reduce water to hydrogen (TONCR = 0.3–3.2/TONPHOTO = 3–3.8), the catalytic performances being similar to the values reported for the free cobaloxime derivatives under similar conditions. The authors suggested that the Co(III)-hydride generated during the course of the catalytic cycle is quickly deactivated through an intramolecular hydride transfer to the glyoximato ligand, thus leading to the loss of activity.133 In order to overcome this fast deactivation, two analogous heme-binding protein scaffolds providing a similar cavity, but with slightly different binding environments, have also been explored.134 Under optimal conditions, hydrogen evolution was enabled at neutral pH with a 3-fold increase of the TON compared to the isolated cobaloxime.
Cobaloxime catalysts have also been investigated in combination with electron transfer proteins, namely a ferredoxin and an apo-flavodoxin (Fig. 11b and c).137,138 These two proteins have previously been re-engineered to provide them with light-harvesting properties, through the covalent binding of a [Ru(bpy)3] derivative ([Ru(4-CH2Br-4′–CH3–2,2′-bpy)(bpy)2]2+). In both cases, self-assembly of the protein with a cobalt catalyst was achieved (Ru–Fd–Co, 4, Fig. 11c and Ru–apoFld–Co, 5 biohybrids), providing efficient photocatalytic assemblies for proton reduction in aqueous medium. The electron transfer from the photosensitizing unit to the catalytic moieties has been investigated by EPR and ultrafast spectroscopies. When the distance is short enough (10.2 Å), as in the case of flavodoxin, direct ET from the reduced photosensitizer (PS) to the catalyst can occur. However, when the distance between the two partners is increasing (16 Å), as in the case of ferredoxin, an intermediate [2Fe–2S] cofactor is needed to achieve efficient ET.138
A similar biohybrid based on a typical Dubois catalyst [Ni(P2PhN2Ph)2](BF4)2 (6)139 and a previously described Ru–Fld system has been prepared.140 Again, this assembly is able to perform photo-induced proton reduction over a wide pH range (from 3.5 to 12). Notably, a maximum TOF of 410 h−1, associated with a TON of 620, can be reached at pH 6.2. A rather long-lived separated charge state of 20 ms was identified, suggesting a crucial role played by the protein to support and protect relevant catalytic intermediates in an aqueous environment.
Photocatalytic H2 production using biomimetic synthetic diiron carbonyl moieties coupled to a non-hydrogenase protein matrix either by dative anchoring or by covalent linkage has also been investigated. The specific amino acid sequence –CXXC–, found in the pocket of apo-cytochrome c, has been used as an anchoring group for the diiron nonacarbonyl precursor [Fe2(CO)9], thus allowing the formation of a [(μ-S2)Fe2(CO)6] motif mimicking the [2Fe] subsite of [FeFe] hydrogenases.141 Under photocatalytic conditions in aqueous medium (pH 4.7), H2 production was evidenced (up to 80 TON). In contrast, when a smaller heptapeptide having the same –CXXC– sequence was used, H2 production was found to decrease dramatically, providing an example of the importance of catalyst isolation for efficient (photo-)catalysis. Similarly, the photo-induced hydrogen generation of an engineered apo-nitrobindin (apo-NB) containing a [2Fe] subsite model, {μ-(SCH2)2CHCOR}Fe2(CO)6] (7, R = 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethanammonium), covalently linked into its cavity has also been evaluated by Hayashi and co-workers.142 This protein presents a rigid β-barrel structure, where a cysteine residue was introduced (Gln96Cys mutant) into the large internal cavity to allow the coupling with the [2Fe] subsite model via the maleimide moiety (Fig. 11d). Photochemical H2 production experiments employing 7-NB reached a maximum turnover of 130 at pH 4.0. The final TON did not appear to exceed those observed for the [2Fe] mimic alone. However, 7-NB allowed catalysis in pure aqueous media and displayed a higher initial TOF.
The catalytic performance of the different systems described in this subsection is summarised in Table 2. In short, under photocatalytic and other comparable conditions, the activities reported for these re-purposed proteins are in the same range (TONs ranging from tens to hundreds). Thus, the efficiencies of these artificial systems are still far from the efficiencies of the native enzymes, and reports of artificial hydrogenases capable of performing H2 oxidation are still rare. Still, work during the last decade has firmly established the feasibility of preparing artificial enzymes displaying hydrogenase like-activities, i.e. they are capable of producing H2 in aqueous media under reducing conditions. This forms a solid foundation for further optimization and underscores the importance of optimizing both the catalyst and the binding pocket.
| Catalyst centerb | Protein host | Electron supply/conditions | TONc | TOFc (h−1) | Ref. |
|---|---|---|---|---|---|
| a MV = methyl viologen, PS = [Ru(bpy)3]2+, and NaAsc = sodium ascorbate. b In parentheses are the amino acids from the protein host involved in the metal binding. c In parentheses is the duration of the experiment used to determine the corresponding value. d Direct coordination by four Cys from the protein. e nmol per s−1 per mg Rd. f TON is calculated vs. the number of introduced cobalt centers. g PS covalently bound to the protein. h TONs and TOFs are calculated vs. the number of photosensitizers. | |||||
| NiS4d | Rubredoxin (Rd) | Reduced MV/KPi, pH 6.8, 32 °C | — | 594 ± 54e | 125 |
| NiS4d | Rubredoxin (Rd) | 1 mM PS, 0.1 M NaAsc/KPi, pH 6.5, 4 °C | 32 (1 h) | 30 (ini) | 126 |
| CoP, 1 (His93) | Myoglobin | 1 mM PS, 0.1 M NaAsc/KPi, pH 7 | 518 (12 h) | 88 | 127 |
| CoP, 1(His107) | Cyt b562 (Met7Ala mutant) | 1 mM PS, 0.1 M NaAsc/KPi, pH 7 | 310 (8 h) | — | 128 |
| 2 (His93) | SwMb | 20 eq. PS, 0.1 M NaAsc/KPi NaCl (150 mmol L−1), pH 6 | 3.8f (5 min) | — | 130 |
| 3 (His25) | Rat heme oxygenase | 15 eq. [Eu(EGTA) (H2O)]2−/Tris–HCl, pH 7 | 6.2f (5 min) | — | 134 |
| 2 (His90) | S. oleracea ferredoxin | PS derivate,g 0.1 M NaAsc/MES, pH 6.3 | 210h ± 60 (6 h) | 60h (ini) | 137 |
| 2 | S. lividus apo-flavodoxin | PS derivate,g 0.1 M NaAsc/MES, pH 6.3 | 85h ± 35 (6 h) | 30h ± 10 (ini) | 138 |
| 6 | S. lividus apo-flavodoxin | PS derivate,g 0.1 M NaAsc/MES, pH 6.2 | 620 ± 80 (4 h) | 410 ± 30 (ini) | 140 |
| [(μ-S2) Fe2(CO)6] motif (Cys14, Cys17) | Equine heart apo-cyt c | 10 eq. PS, 0.1 M NaAsc/Tris–HCl, pH 4.7 | 80 (2 h) | 126 (ini) | 141 |
| 7 (Cys96) | Apo-NB (Gln96Cys mutant) | PS, 0.1, M NaAsc/Tris–HCl, pH 4.0 | 130 (6 h) | 136 (ini) | 142 |
4.2 Miniaturized enzymes
As an alternative to the use of proteins, small synthetic peptides appear as a promising route towards affordable and efficient artificial hydrogenases, presenting a well-controlled micro-environment for the metallic active sites.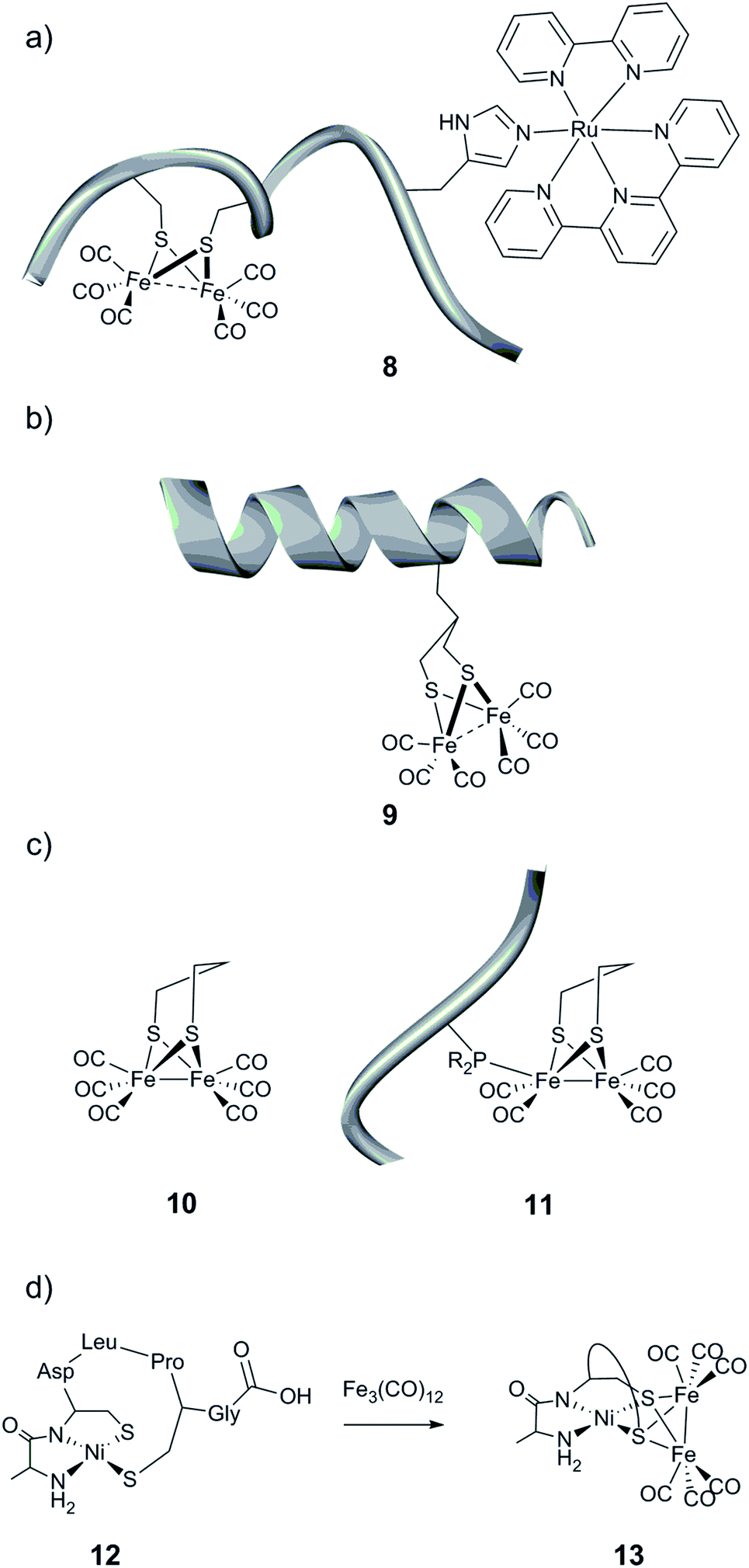 | ||
| Fig. 12 Schematic representation of the (a) cytochrome c-556 fragment containing a [(μ-S2)Fe2(CO)6] moiety and a ruthenium-based photosensitizing unit, (b) helical nonadecapeptide containing a [(μ-S(CH2)3S)Fe2(CO)6] motif, and (c) peptide containing an unnatural amino acid linked to a [(μ-pdt)Fe2(CO)6] (10) complex and (d) formation of a heterobimetallic cluster in a heptapeptide construct. | ||
A second synthetic route to create models even closer to the [FeFe] hydrogenase active site has been explored. Instead of employing cysteine residues present in the peptide sequence to link a diiron carbonyl complex, more engineered peptides relying on the incorporation of an artificial amino acid or post-synthetic modification have been prepared.146,147 To that end, a functionalized propanedithiolate ligand (pdt′ = −S–CH2CHRCH2–S−) was coupled to the amine group of a lysine residue of an octapeptide via amide linkage. Finally, the [μ-(S–CH2CHRCH2–S)Fe2(CO)6] motif was generated by reaction of the [Fe3(CO)12] precursor with the pdt′-containing peptide. While the spectroscopic data indicate the incorporation of the [2Fe] subsite into the peptide, the catalytic properties of the resulting bio-hybrid assembly have not been reported.146 In a related approach, Ghirlanda et al. described the introduction of a non-natural amino acid bearing a 1,3-dithiol moiety into a helical nonadecapeptide, followed by generation of the [μ-(S–CH2CHRCH2–S)Fe2(CO)6] motif within the peptide (Fig. 12b, 9).147 Both photocatalytic and electrocatalytic properties towards proton reduction in water for this miniaturized hydrogenase have been demonstrated.
Phosphines are well-known σ-donor and π-acceptor ligands and have been extensively used in the context of [FeFe] hydrogenase models.11,148 Such a functionality has been introduced into small peptides through two different pathways.149 The resulting phosphine-functionalized peptides were able to react with a [(μ-pdt)Fe2(CO)6] complex (pdt = −S–(CH2)3–S−) (Fig. 12c, 10) via the substitution of a CO ligand (Fig. 12c, 11). This ligand exchange generates an electrocatalyst for H2 production, which however suffers from a large overpotential requirement ranging between 0.75 and 0.9 V in acid-containing acetonitrile.
Among the rare examples of [NiFe]-hydrogenase active site biomimics, Jones et al. investigated the use of a small heptapeptide as a scaffold to coordinate both nickel and iron ions.150 The amino acid sequence used in this work is well known to bind a mononuclear Ni(II) in a square-planar N2S2 environment. After incorporation of the nickel ion into the peptide, the Ni-containing peptide (Fig. 12d, 12) was exposed to a solution of [Fe3(CO)12]. A polynuclear compound, containing two iron and one nickel ions, was then isolated, where the sulfur atoms are the anchoring sites of the iron, forming bridges between the Ni centre and the iron carbonyl fragments [(μ-S2)Fe2(CO)6] (Fig. 12d, 13). The catalytic activity of this hetero-metallic compound has not been reported.
 | ||
| Fig. 13 Schematic representation of mononuclear nickel complexes bearing functionalized PR2NR′2 ligands. | ||
Moreover, the influence of peptide mobility was explored by adding larger and more constrained peptide moieties. To that end, a β-hairpin peptide WIpPRWTGPR-NH2 (WR10, p = D-proline) was attached via an aminophenyl propionic acid (APPA) linker, producing the [Ni(PPh2NAPPA-WR10)2]2+ complex (Fig. 13, 16).159 NMR and CD spectroscopy studies suggest that the incorporation of the decapeptide into the parent complex does not affect the peptide structure, and the peptide construct displays a modest increase in electrocatalytic H2 production as compared to the parent compound ([Ni(PPh2NAPPA)2]2+, Fig. 13). The structure/activity relationship has been investigated further, and the length of the linker (between the phenyl ring and the peptide) as well as the peptide have been varied.160 The octapeptide [Ni(PPh2NAPPA-K8)2]2+ (K8 = WIpKKWTG-NH2, Fig. 13, 17) shows a 2.5-fold current increase compared to the parent compound ([Ni(PPh2NAPPA)2]2+), operating at the same overpotential as the decapeptide (16, 540 mV). The decrease of the linker length from three to one carbon atom (Fig. 13, 18, 19) led to a reduction in the mobility of the peptide but had a negative impact on the catalytic current for both the octa- and the decapeptide.
Bren and co-workers have prepared and studied the H2-evolution properties in water of a series of cobalt-based artificial hydrogenases by constant potential electrolysis (CPE). A small tripeptide model (CoGly–Gly–His, 20, Fig. 14) based on the copper- and nickel-binding (ATCUN) motif found at the N-terminus of albumins was shown to evolve H2, reaching 275 TON after 2.5 h of CPE at near neutral pH with an ∼600 mV overpotential.161
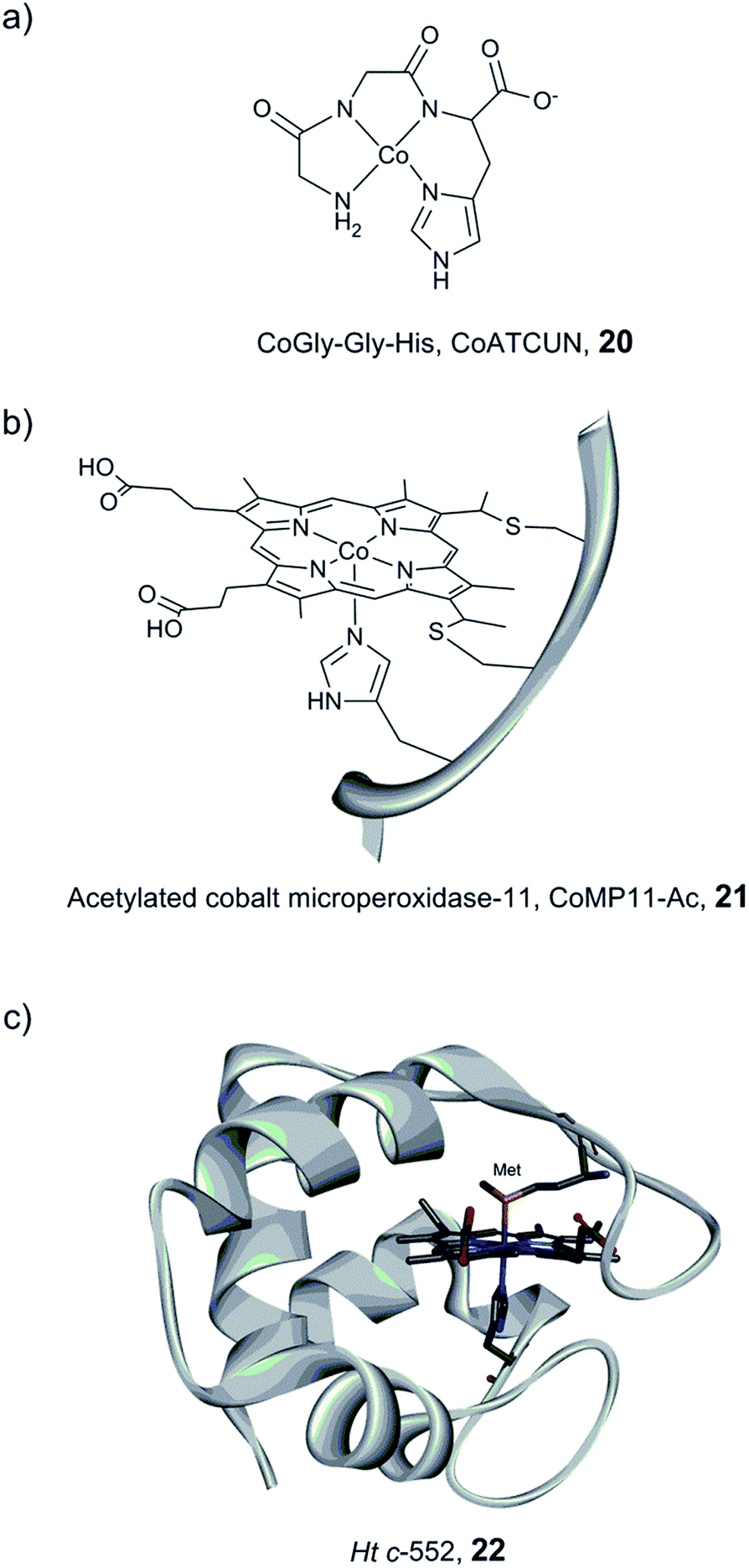 | ||
| Fig. 14 Schematic representation of CoGly–Gly–His (CoATCUN, 20) and CoMP11-Ac, 21, and the structure of cytochrome c-552 (PDB ID: 1YNR)163 (Ht c-552, 22 also showing the native Met-ligand). | ||
A Co–porphyrin based system was prepared via metal substitution of the Fe-ion in the heme-undecapeptide MP11 (CoMP11, 21, Fig. 14). MP11, which is derived from horse cytochrome c, features a porphyrin ligand covalently linked to the peptide fragment and provides an additional axial histidine ligand to the metal centre, leaving an open coordination site. CoMP11, 21, showed efficient H2 production with a TON of over 20000 after 4 h and a surprisingly high tolerance towards O2. However, the maximum current was observed at an overpotential of 852 mV, i.e. significantly higher than that of the ATCUN-based system (20). Furthermore, the electrocatalytic activity started to decline after 15 min (active for a few hours in total), suggesting degradation of the catalyst.162
To overcome the weaknesses encountered with the previous constructs (e.g. peptide flexibility, catalyst exposure to the solvent and fast degradation), a cobalt-substituted cytochrome c-552 system was prepared (Ht-Co, 22, Fig. 14).129 Importantly, the active site was improved for H+ reduction: methionine involved in the coordination of the native heme-Fe ion was removed in order to open up a site for catalysis (Fig. 14). The resulting artificial enzyme is capable of achieving a very high TON (>270000), and with an increased longevity, the catalytic activity showed minimal decline over ∼6 h (active for 24 h in total). Despite this clear improvement, the overpotential stayed nearly unchanged (830 mV). This series of artificial enzyme assemblies nicely exemplify that burying a catalytic site within a protein matrix can enhance longevity, but does not necessarily affect the overpotential.
The catalytic properties of miniaturized hydrogenases discussed in this section are summarized in Table 3. A direct comparison is complicated by the different conditions in which the systems are evaluated. Still, both complexes 16 and 21 nicely highlight the improvements in both the TOF and the TON that can be achieved via manipulations of the microenvironment of molecular catalysts.
| Catalyst center | Peptide host | Electron supply/conditions (overpotential) | TONb | TOFb (s−1) | Ref. |
|---|---|---|---|---|---|
| a EC = electrochemistry, CV = cyclic voltammetry, CPE = constant potential electrolysis, GC = glassy carbon, ini = initial, Ppa = N-Boc-3-(diphenylphosphorothioyl)alanine. b In parentheses is the duration of the experiment used to determine the corresponding value. c Directly coordinated by the His10. | |||||
| [(μ-S2)Fe2(CO)6] | 8 | [Ru(bpy)(tpy)]2+,c 0.1 M NaAsc/Tris–HCl, pH 8.5 | 9 (2 h) | 0.003 (ini) | 145 |
| [(μ-(S–CH2 CHRCH2–S))Fe2(CO)6] | 9 | 15 eq. PS, 0.05 M NaAsc/citrate buffer, pH 4.5 | 84 (2.3 h) | — | 147 |
| [μ-(pdt) Fe2(CO)5] | Val–Ppa–Leu 11 | EC (CV)/GC electrode, CH3CN:H2O (3:1) acid acetic (750 mV) |
— | — | 149 |
| 15 | 15 | EC (CV)/GC electrode, 348 K, 1 atm (N2), H2O, pH 6 | — | 21 ± 5% | 156 |
| [Ni(PPh2NAPPA-WR10)2]2+ | WR10 16 | EC (CV)/GC electrode, CH3CN:H2O (90:10) acidic DMF (540 mV) |
— | 106000 |
159 |
| 20 | Gly–Gly–His 20 | EC (CPE)/Hg pool electrode, MOPS, pH 8, 1 M NaCl (600 mV) | 275 (2.5 h) | 0.031 (2.5 h) | 161 |
| 21 | MP11-Ac 21 | EC (CPE)/Hg pool electrode, KPi, pH 7 (852 mV) | 25000 (4 h) |
6.7 (10 m) | 162 |
| 22 | Ht c-552 (Met61Ala) mutant) 22 | EC (CPE)/Hg pool electrode, KPi, pH 7 (830 mV) | 11000 (6 h) |
— | 129 |
4.3 Artificial active site pockets
Incorporation of [FeFe] hydrogenase active site models into non-biological environments, like polymer matrices, micelles, the hydrophobic cavity of cyclodextrins, or metal–organic frameworks (MOFs), has been developed to realize photocatalytic and electrochemical H2 production in water. These various entities are believed to be capable of mimicking the hydrophobic cavity and the hydrogen bonding sites of the native hydrogenases. In most of the examples reported, incorporation of hydrogenase model complexes into e.g. peptide hydrogels,164 micelles165–167 or the backbone and side chains of polymers168–171 has been shown to keep or enhance the stability and the reactivity of the complex. In the following section we will discuss representative examples.Conversely, when a similar complex was coupled with poly(acrylic acid) (PAA) via an isocyanide ligand [(μ-pdt)Fe2(CO)5(CNR)] (R = diphenylacetylene linker, Fig. 15, 23), the resulting water-soluble polymer catalyst displayed very efficient photocatalytic properties.173 Photocatalytic H2 production in water, in the presence of stabilized CdSe quantum dots (QDs) (MPA-CdSe QDs, MPA = 3-mercaptopropionic acid) as a photosensitizer, resulted in an excellent TON of 27135 after 8 h irradiation. Importantly, this study demonstrated that, aside from solubilizing the diiron mimic, the polymer seems to play a role in protecting CdSe QDs from photo-corrosion and aggregation and ensuring close proximity between the photosensitizer and the catalyst, making the system more efficient. In a similar approach, when polyethylenimine (PEI) is used, the system is photoactive in a broad range of pH, including neutral conditions.174
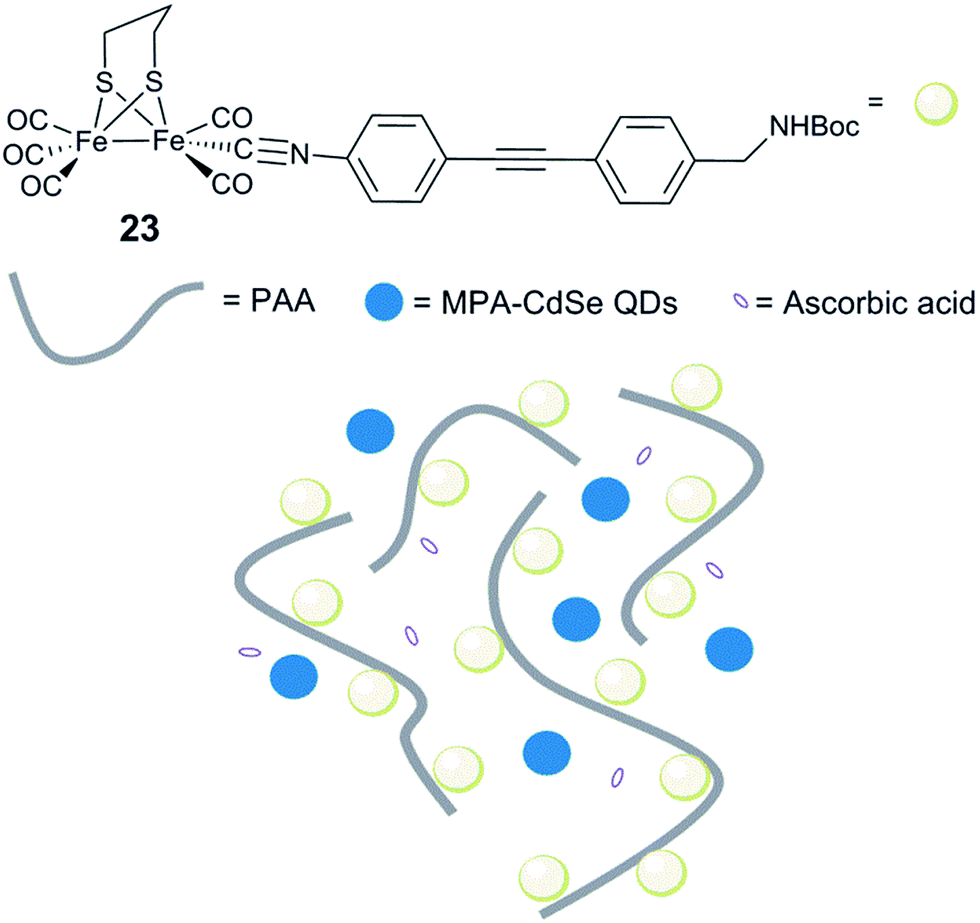 | ||
| Fig. 15 Schematic representation of an artificial system for photocatalytic H2 production, including [(μ-pdt)Fe2(CO)5(CNR)] isocyanide complexes. 23, linked to a water soluble PAA polymer and MPA-CdSe QDs. | ||
In 2013, a system consisting of an iron–thiolate catalyst [(μ-S2)Fe2(CO)6] protected by a dendritic framework achieved a TON of 22000 in 8 h, under photocatalytic conditions in the presence of an Ir-based photosensitiser.175 Interestingly, the TOF and total TON values of the system increased with the size of the dendritic structure, highlighting the effect of site isolation.
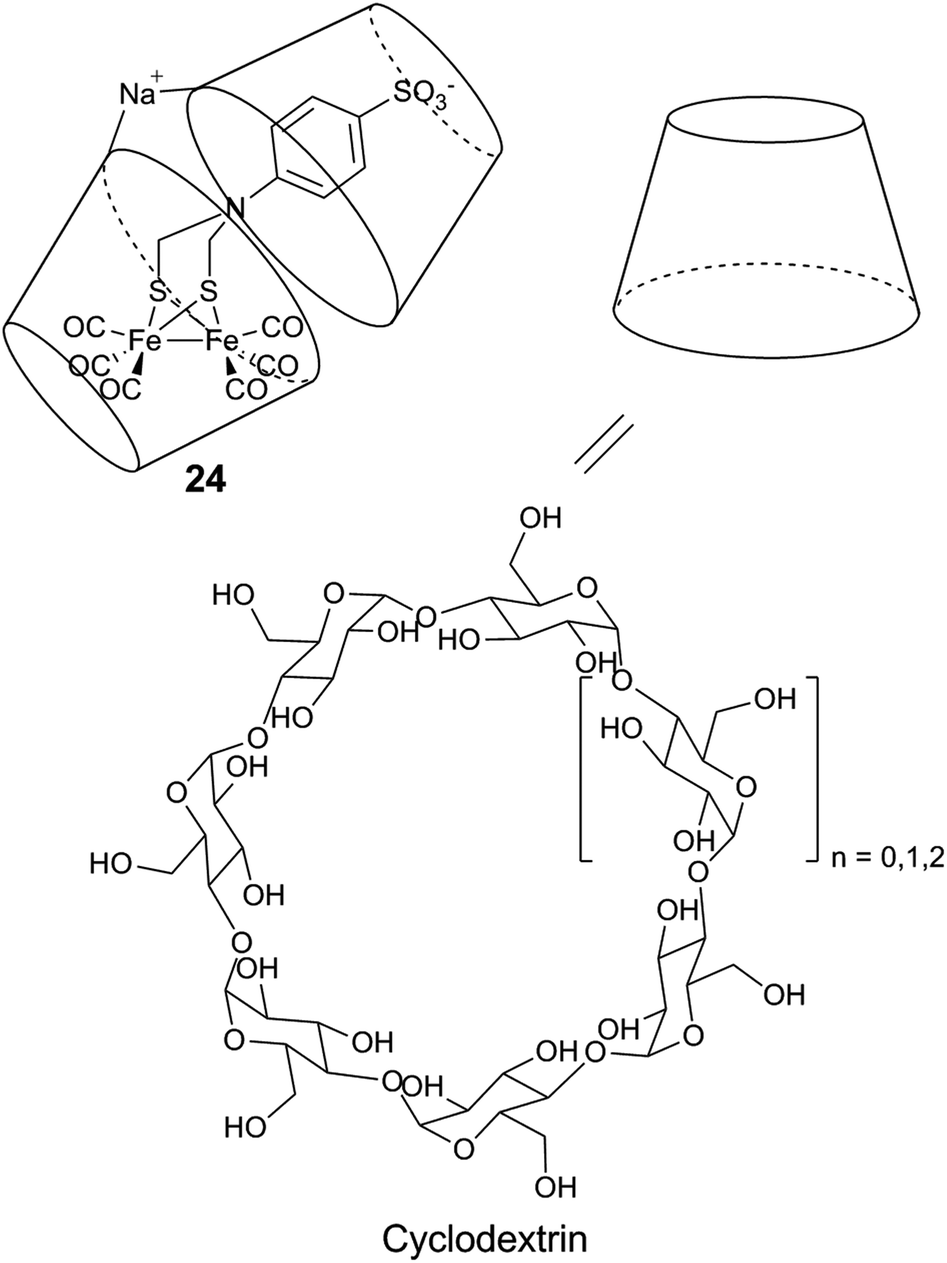 | ||
| Fig. 16 Schematic representation of a diiron mimic encapsulated in a cyclodextrin unit, and the structure of the α (n = 0), β (n = 1), and γ (n = 2) cyclodextrin units. | ||
Similar systems have also been explored under photocatalytic conditions. Introducing a sulfonate-functionalized diiron–thiolate catalyst and a xanthene dye (eosin Y or rose Bengal) into the hydrophobic cavity of cyclodextrin resulted in a substantial increase in the lifetime (3-fold) and TON (9-fold, up to 75) of the resulting photocatalytic system in a basic aqueous solution.178 A more dramatic improvement is observed with chitosan, a polysaccharide which contains primary amines and hydroxyl groups. When protonated, chitosan is able to interact with the [2Fe] subsite model [μ-(SCH2)2NCH2C6H5–Fe2(CO)6] (25) and the PS MPA-CdsTe QDs.179 Photocatalytic activities were demonstrated in a methanol:water 1:3 (v/v) mixture, in the presence of ascorbic acid (H2A). Under optimized conditions, the supramolecular assembly can achieve a TON of up to 52800 and a TOF of 1.40 ± 0.22 s−1, and outperforms the diiron compound without chitosan in terms of both activity and stability.
The highly stable MOF UiO-66(Zr) was used as the host for the [2Fe] subsite mimic [(μ-dcbdt)Fe2 (CO)6] (29, dcbdt = 1,4-dicarboxylbenzene-2,3-dithiolate).185 The molecular catalyst was incorporated into the MOF pores by post-synthetic exchange. The loaded MOF shows an enhanced photo-induced H2 production in the presence of ascorbate and [Ru(bpy)3]2+ compared to the catalyst itself in aqueous media, in terms of both the initial rate and the overall amount of produced hydrogen. Recently, two CO ligands of hexacarbonyl species 29 have been successfully exchanged with phosphine ligands inside the MOF UiO-66(Zr).186 The use of another framework, MIL-101, exhibiting larger pore diameters (29–34 Å), compared to those of UiO-66 (8–11 Å), has been explored. Interestingly, in MIL-101, a larger proportion of the [FeFe] catalysts within the MOF is engaged in photochemical hydrogen production, and the amount of produced hydrogen is proportional to the catalyst loading.187 This result shows the importance of the transport channels through the MOF to generate efficient materials for H2 production.
MOF scaffolds featuring dual functionality have also been reported. Feng et al. described a system in which a [2Fe] subsite mimic was coordinated to the Zn-atom of a porphyrin via the coordination of a pyridine-containing dithiolate bridging ligand in a porphyrin-based MOF (Zr-PF-MOF).188 This system, combining both a photosensitizer and a hydrogen-evolving catalyst, has shown photoactive H2 production in a slightly acidic buffer, in the presence of H2A.
As summarized in Table 4, the encapsulation of [2Fe] subsite mimics generally results in systems with quite remarkable catalytic efficiencies. Unfortunately, in-depth characterisation to explain the enhanced catalytic performances of these embedded catalysts is generally missing. Thus a rationale between the nature of the encapsulation system and the activity is still unclear. This is certainly a challenge that needs to be tackled for future optimisation of the systems. Nevertheless, the work underscores the strong impact of encapsulation on the activity, and this certainly appears as a promising route towards the development of efficient and stable proton reduction catalysts.
| Catalyst center | Encapsulation system | Electron supply/conditions | TONb | TOFb (s−1) | Ref. |
|---|---|---|---|---|---|
| a PAA = poly(acrylic acid), MPA = 3-mercaptopropionic acid, QDs = quantum dots, PEI = polyethylenimine, H2A = ascorbic acid, and SDS = sodium dodecyl sulfate. b In parentheses is the duration of the experiment used to determine the corresponding value. | |||||
| 23 | PAA | MPA-CdSe QDs, 0.1 M H2A/H2O, pH 4 (initial) | 27135 (8 h) |
3.6 (30 m) | 173 |
| 10 | PEI | MPA-CdSe QDs, 0.125 M H2A/pH 6.5 | 10600 (44 h) |
— | 174 |
| [(μ-S2) Fe2(CO)6] motif | Fréchet-type dendron | [Ir(ppy)2(bpy)]+ TEA/acetone/H2O (9:1) |
22000 (8 h) |
2.01 (1 h) | 175 |
| 24 | Cyclodextrin | 1 eq. eosin Y, 10 vol% TEA/H2O, pH 10 | 75 (24 h) | — | 178 |
| 25 | Chitosan | MPA-CdTe QDs, 0.2 M H2A MeOH/H2O (1:3), pH 4.5 (initial) |
52800 (60 h) |
1.4 ± 0.22 | 179 |
| 10 | Polymeric micelles P-NB | 10 eq. PS, 0.045 M H2A/CH3CN/H2O (4:1), pH 4–4.1 |
133 (2 h) | — | 180 |
| 26 | SDS micelle | EC (CPE)/Hg pool electrode, 0.045 M H2A H2O, pH 3 (500 mV) | 52 (1 h) | — | 181 |
| 26 | SDS micelle | 2 eq. eosin Y, 10 vol% Et3N/H2O, pH 10.5 | 117 (4.5 h) | 182 | |
4.4 Summary and outlook
The preparation of artificial hydrogenases based on insights from biological systems is a promising route towards the construction of new water-soluble, O2-tolerant, stable and efficient catalytic systems. As outlined above, several imaginative strategies have been explored, and a range of both biological and synthetic scaffolds have been tested. In short, synthetic and artificial systems still struggle in comparison to enzymes with regard to over-potential requirements and stability. Still, work to date nicely illustrates how site isolation can greatly enhance both photo- and electrocatalytic rates, as well as the stability of otherwise fragile catalysts. Moreover, the capacity to modulate the reactivity by modifying the catalyst microenvironment has been elegantly proven via ligand design of purely synthetic catalysts, as well as through protein scaffold optimization in hybrid systems.In summary this suggests that the incorporation of elaborate molecular catalysts into artificial enzyme pockets should provide a route towards efficient, stable and cheap catalysts for H+/H2 interconversion. Furthermore, such encapsulation should also facilitate the application of these bio-inspired molecular catalysts in technological devices.
5. Semi-synthetic hydrogenases
As discussed above, the H-cluster has long served as an inspiration for synthetic chemists in the development of molecular catalysts for the interconversion of protons and H2.11,189 In parallel, biomimetic model compounds have provided valuable insight into the structural and mechanistic details of the enzyme (see for example ref. 190 and 191). In 2013, it was shown how such synthetic complexes can be introduced into the hydrogenase enzyme itself under in vitro conditions, thus providing a direct link between biomimetic chemistry and biology and allowing the manipulation of the enzyme using synthetic chemistry.53,192 In the following section, hydrogenases prepared this way will be referred to as “semi-synthetic hydrogenases”, reflecting the fact that they consist of a combination of a synthetic cofactor and the native hydrogenase protein scaffold, while differentiating them from the “artificial hydrogenases” discussed above.5.1 Artificial maturation of [FeFe] hydrogenases
The assembly of the H-cluster inside the [FeFe] hydrogenase occurs in at least two discrete steps. The [4Fe–4S] cluster is initially constructed by using house-keeping [FeS] cluster machinery, generating [4Fe–4S]H-HydA.24,193 The enzyme is then activated upon delivery of the [2Fe] subsite, in a process involving a minimum of three HydA specific maturases, denoted as HydE, HydG and HydF.194–198 A precursor of the [2Fe] subsite is assembled on the HydF protein through the activities of HydE and HydG, via a still incompletely characterised process, from where is then transferred to HydA199–202 (for a more in-depth overview of the maturation process see e.g.ref. 16, 17, 203 and 204). The exact structure of the [2Fe] subsite precursor bound to HydF is still unknown. Nevertheless, FTIR spectroscopy indicates that this complex shares structural similarities to the [2Fe] subsite observed in HydA,199,202 and more recently the preparation of semi-synthetic forms of HydF has provided a more detailed picture (see Section 5.4). In 2013, it was shown how a set of [2Fe] subsite mimics, differing in the nature of the bridgehead atom, could be introduced into HydF to generate artificially, or synthetically, loaded forms of the maturase (X-HydF).53 Critically, these semi-synthetic proteins were able to mimic the reactivity of native HydF and transfer their synthetic cargo to [4Fe–4S]-HydA. Shortly thereafter, it was also reported that HydF can be circumvented, and such [2Fe] subsite mimics were directly introduced into HydA (Fig. 17).192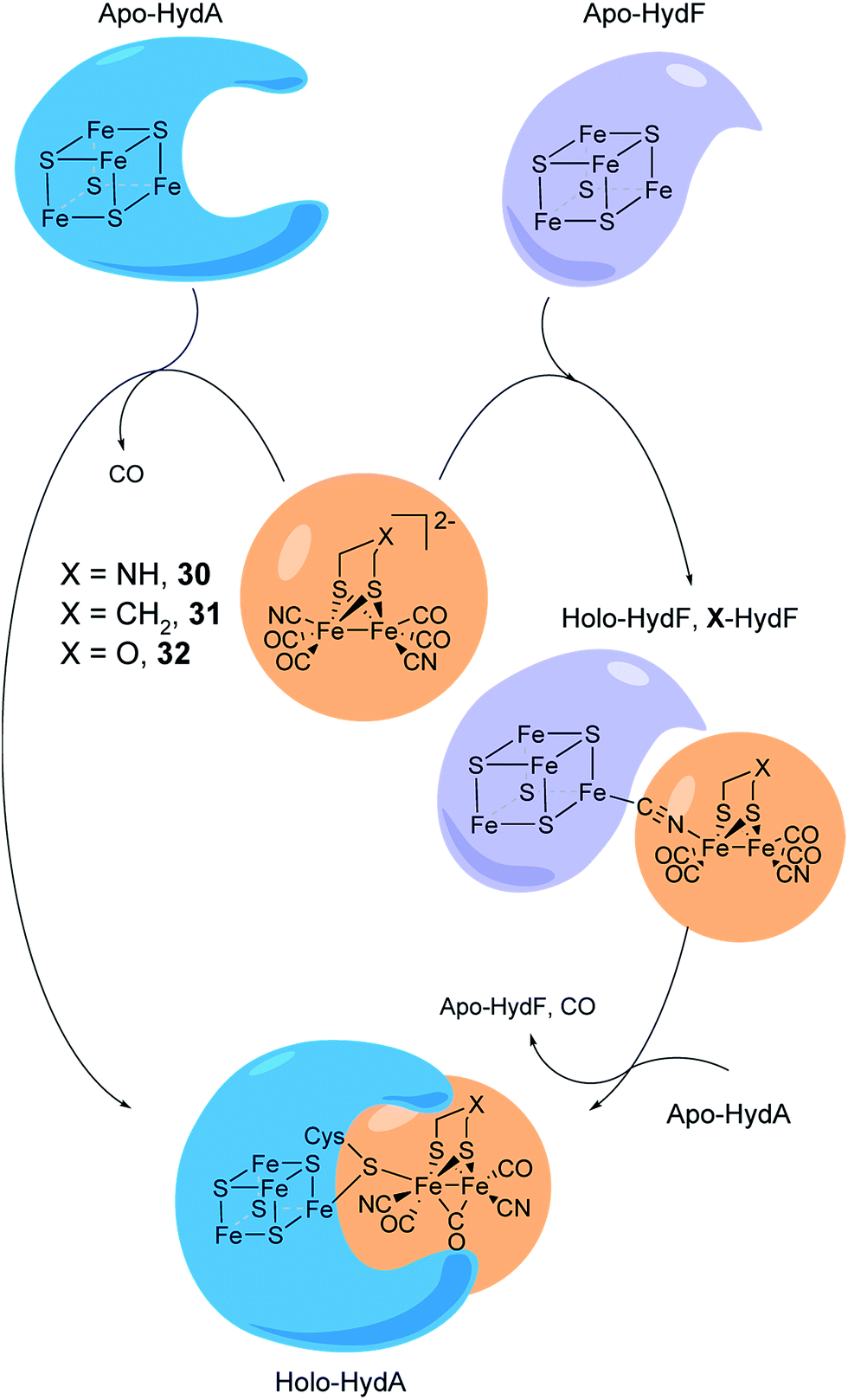 | ||
| Fig. 17 Schematic representation of artificial maturation of the maturase HydF and the hydrogenase enzyme, HydA. | ||
An immediate outcome of this finding was that it unequivocally proved the nature of the bridgehead atom as nitrogen, as suggested by earlier spectroscopic studies.54,190 The introduction of the [(μ-adt)Fe2(CO)4(CN)2]2− complex (adt = (−S–CH2NHCH2–S−)) (30, Fig. 17) resulted in a semi-synthetic enzyme practically indistinguishable from the native enzyme with regard to both spectroscopic and catalytic properties (Fig. 18).53 In the context of synthetic catalyst design it also served to underscore the effect of the protein surroundings and the [4Fe4S]H cluster on the catalytic properties of [2Fe] subsite mimics. Indeed complex 30 does not appear to be catalytic in solution and is highly unstable towards protonation.171,205
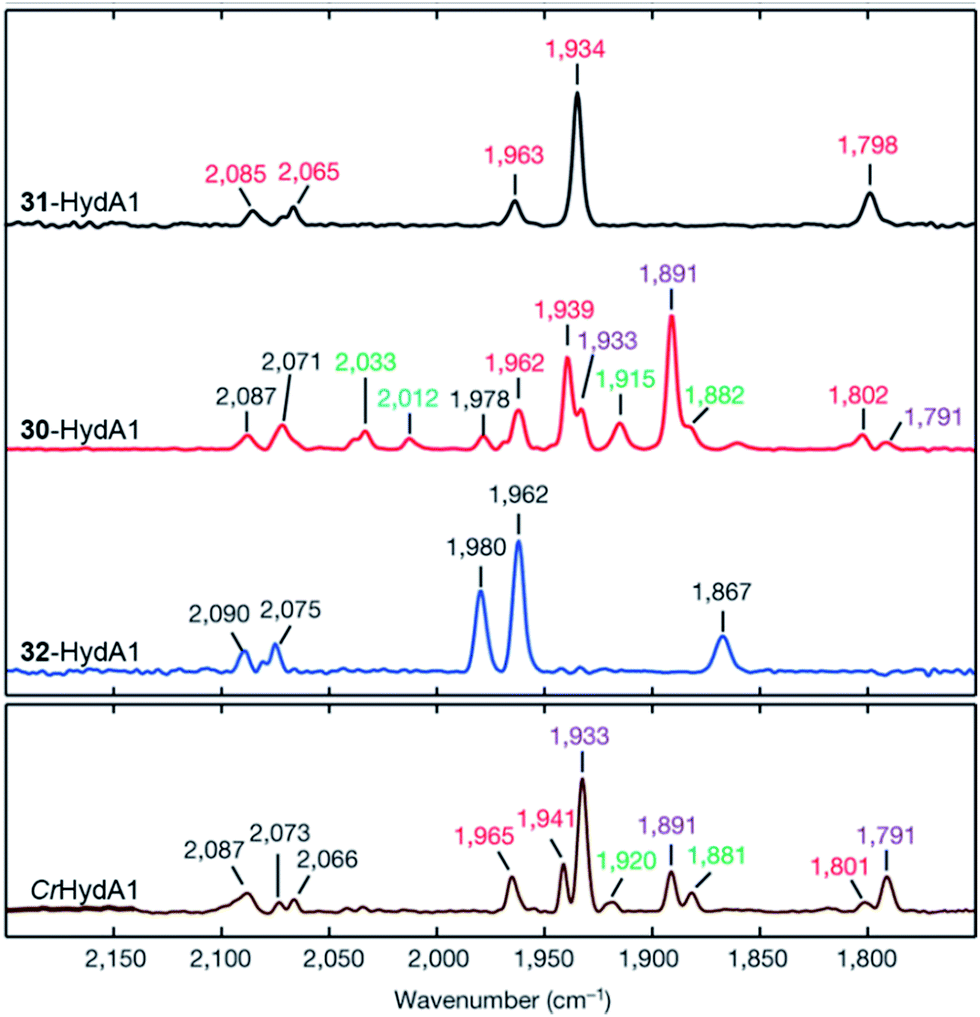 | ||
| Fig. 18 FTIR spectra with normalized intensities of selected forms of semi-synthetic HydA, loaded with synthetic co-factors 30–32 (30–32-HydA1), compared to a form of HydA containing the native co-factor (CrHydA1). The figure was adapted from Berggren et al., Nature, 2013.53 | ||
The newfound capacity to introduce synthetic cofactors into [FeFe] hydrogenases assists spectroscopic studies via the preparations of isotopologues of the H-cluster, and this is nicely exemplified by two extensive spectroscopic characterizations of such systems.206,207 Isotopic labelling of the C and N atoms of the cyanide ligands and bridgehead amine, in combination with HYSCORE and ENDOR spectroscopy, allowed the clear identification of 15N-adt2− ligand HYSCORE features.206 The iron ions found in both [4Fe–4S] clusters, as well as in the [2Fe] subsite, have also been labelled using 57Fe.207 In the latter study, the H-cluster was isolated in the Hox-state and studied by using a number of spectroscopic techniques, including pulsed EPR, Mössbauer and nuclear resonance vibrational spectroscopy (NRVS). It is noteworthy that these initial studies both agree with, and refine findings from, earlier studies using enzymatically maturated forms of the enzyme.54,208,209
The concept of artificial maturation has now been applied to [FeFe] hydrogenases from a wide range of different organisms, including Clostridium pasteurianum (CpI),192Chlamydomonas reinhardtii (CrHydA1),53,192,210Megasphaera elsdenii (MeHydA),192,211 and Desulfovibrio desulfuricans ATCC 7757 (DdHydAB),48 underscoring the general applicability of the technique.
A few years after the initial discovery of the artificial maturation technique, its adaptation for in vivo applications was reported, with the successful activation of CrHydA1 inside living E. coli cells using [2Fe] subsite mimics. Treating anaerobic cell cultures expressing apo-HydA1 with complex 30 resulted in glucose-dependent H2 production.212 More recently, this approach has also enabled the detection of the Hox and Hox–CO states in vivo,213 paving the way for mechanistic studies under physiological conditions. The mechanism of activation under in vivo conditions, including cell entry and enzyme localization, is still unknown, but in vitro assays suggest a high degree of activation of the enzyme. Techniques for the introduction of metal complexes and other small molecules into living cells for the purpose of activating a specific protein are still quite rare.214–219 However, the activation of HydA1 occurred spontaneously in standard E. coli expression strains upon addition of the [2Fe] subsite mimic, suggesting a general applicability of the technique to other expression hosts.
5.2 Reactivity of semi-synthetic hydrogenases
The artificial maturation technique is not restricted to the preparation of “native” hydrogenases. Already in 2013, it was shown that [(μ-pdt)Fe2(CO)4(CN)2]2− (31) and [(μ-odt)Fe2(CO)4(CN)2]2− (odt = (−S–CH2OCH2–S−)) (32) can be incorporated into an apo-enzyme with good yields. However, both 31-HydA and 32-HydA appeared to be catalytically inactive and instead locked the H-cluster in specific oxidation states.53 Since then, a series of studies have been reported involving the incorporation of alternative [2Fe] subsite mimics, and an overview of the complexes assayed for the preparation of semi-synthetic hydrogenases at the time of this review is shown in Fig. 19. The main emphasis of these studies has been on the bridging ligand, including changes to the bridgehead atom and the introduction of increased steric bulk. Moreover, the effect of replacing CN ligands with CO, as well as the possibility to introduce mononuclear Fe complexes, has been studied.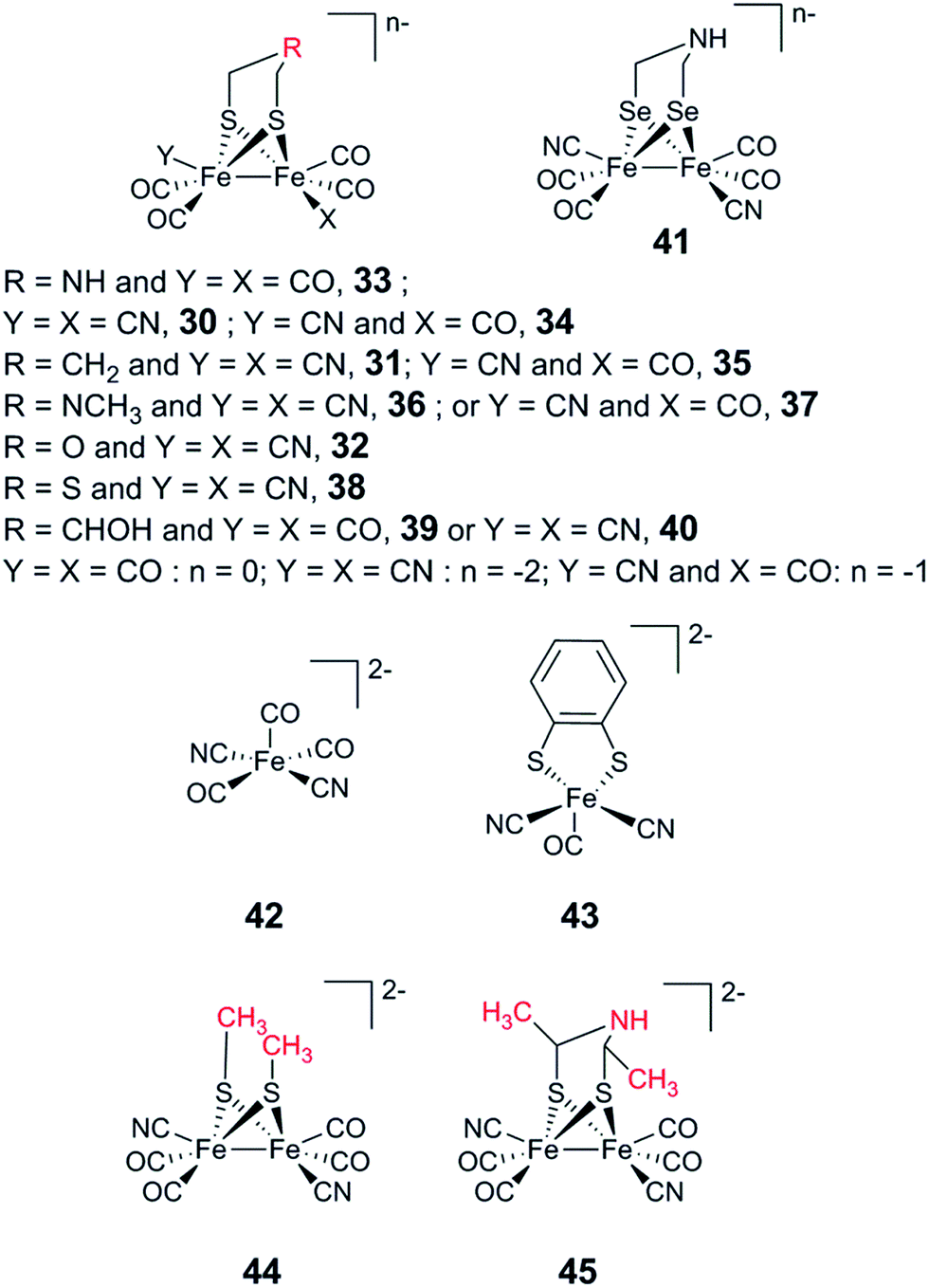 | ||
| Fig. 19 Overview of synthetic complexes screened for insertion into [FeFe] hydrogenases. | ||
In particular, 31-HydA attracted initial attention, as it appeared to generate a form of the H-cluster locked in a state highly similar to the Hox-state observed for the native enzyme.53,192 A combined spectroscopy and electrochemistry study of 31-HydA1 later verified that the [2Fe] subsite indeed was isolated in the [FeIFeII]-state, indicating a one-electron oxidation of the complex upon insertion into the protein.220 This modified H-cluster only appears to have one readily available redox transition, which involves the [4Fe–4S]H cluster rather than the [2Fe] subsite (Hox/H′red, see Fig. 3).62,220 This observation explains the low catalytic capacity of this semi-synthetic hydrogenase and is attributable to the lack of a protonatable bridgehead, which prevents stabilization of the reduced [2Fe] subsite by proton coupled electron transfer and instead favours reduction of the [4Fe–4S] cluster. Finally, it is noteworthy that 31-HydA1 appears to be completely unreactive towards CO binding.220
More recently, the 32-loaded form of the [FeFe] hydrogenase was explored in depth by Lubitz and co-workers.60 As isolated, the FTIR spectrum of 32-HydA1 displays strong similarities to the Htrans as well as the HHyd-state. As mentioned above (cf. Section 2.2); the latter state was recently reported and shown to accumulate under strongly reducing conditions.57,59 Both Htrans and HHyd have been described as consisting of a reduced [4Fe–4S]H cluster coupled to an oxidised [2Fe] subsite, i.e. [4Fe–4S]H+–[FeIIFeII]. However, while the Htrans-state is considered to be an inactive oxidised state, the HHyd-state is believed to represent an elusive terminal hydride-state in the catalytic cycle. In the native enzyme, the HHyd-state has been shown to accumulate in a mutant (Cys169Ser) with a disrupted H-bonding network. Arguably, replacing the amine bridgehead with an ether analogue can result in a similar situation. 57Fe labelling of the 32-cofactor, in combination with NRVS spectroscopy, allowed final confirmation of the presence of the hydride ligand, and DFT calculations were performed to verify that the electronic structure of the 32-HydA-hydride is virtually indistinguishable from that of the native HydA-hydride analogue previously observed by King and co-workers. Not only does the study of 32-HydA provide a direct observation of the terminal hydride state, but it also underscores the potential of the artificial maturation technique to provide mechanistic insight via the selective preparation of model systems for catalytically relevant states.
The most comprehensive screening study reported to date was by Siebel et al. in 2015, using the apo-form of CrHydA1.221 The enzyme was incubated with complexes 30–40 and 42–45 (Fig. 19), and their incorporation into the enzyme was determined by a combination of FTIR spectroscopy and Fe quantification. The study underscored the capacity of the active site pocket to harbour a wide range of [2Fe] subsite mimics, as the majority of the complexes assayed, including some of the mononuclear complexes, were successfully incorporated into the enzyme. However, the presence of two cyanide ligands and the avoidance of steric bulk appear to be crucial for the generation of a single well-defined product. Among the other trends observed, the study reinforced the notion that a secondary amine is strictly required in the bridgehead position for efficient catalysis. Still, many of the bridgehead-derivates did in fact show some minor residual enzymatic activity (e.g.31). Conversely, replacing one cyanide ligand with a CO ligand resulted in less well-defined semi-synthetic H-clusters, as determined by FTIR spectroscopy. However, this modification had relatively modest influence on the activity of the enzyme, and the mono-cyanide analogue [(μ-adt)Fe2(CO)5CN]− (34) (Fig. 19) retained almost 50% of the native activity. The latter complex also appears to generate catalytically competent semi-synthetic enzymes in vivo.212
The crystal structures of a series of semi-synthetic forms of HydA from C. pasterianum (X-CpI) were reported in 2016. Critically, this study showed that regardless of the nature of the bridgehead atom, the structural geometry of the complex remained unchanged, and all of the semi-synthetic hydrogenases studied generated structures similar to the Hox-state (Fig. 20).25 The striking structural similarities between the highly active 30-CpI and the modified cofactors underscore the strong influence of the active site on forming this specific geometry and reinforce the relevance of synthetically modified cofactors as spectroscopic models of the H-cluster. Moreover, these structural similarities appear to exclude the possibility to attribute the lack of activity of 31-, 32-, and 38-CpI to inhibition or steric hindrance and thus further highlight the importance of the bridgehead amine for activity. It is noteworthy that when CpI was assayed after loading with 31, no hydrogenase activity could be detected.25 It thus appears to be a variance in how well [FeFe] hydrogenases of different origin can accommodate and utilize modified cofactor mimics. Esselborn et al. raise the possibility that this difference in activity can be attributed to the extent of shielding of the active site from the surface of the protein.25 The deeply buried active site in CpI could be more sensitive to a disrupted proton transfer network as compared to the smaller CrHydA1 enzyme.
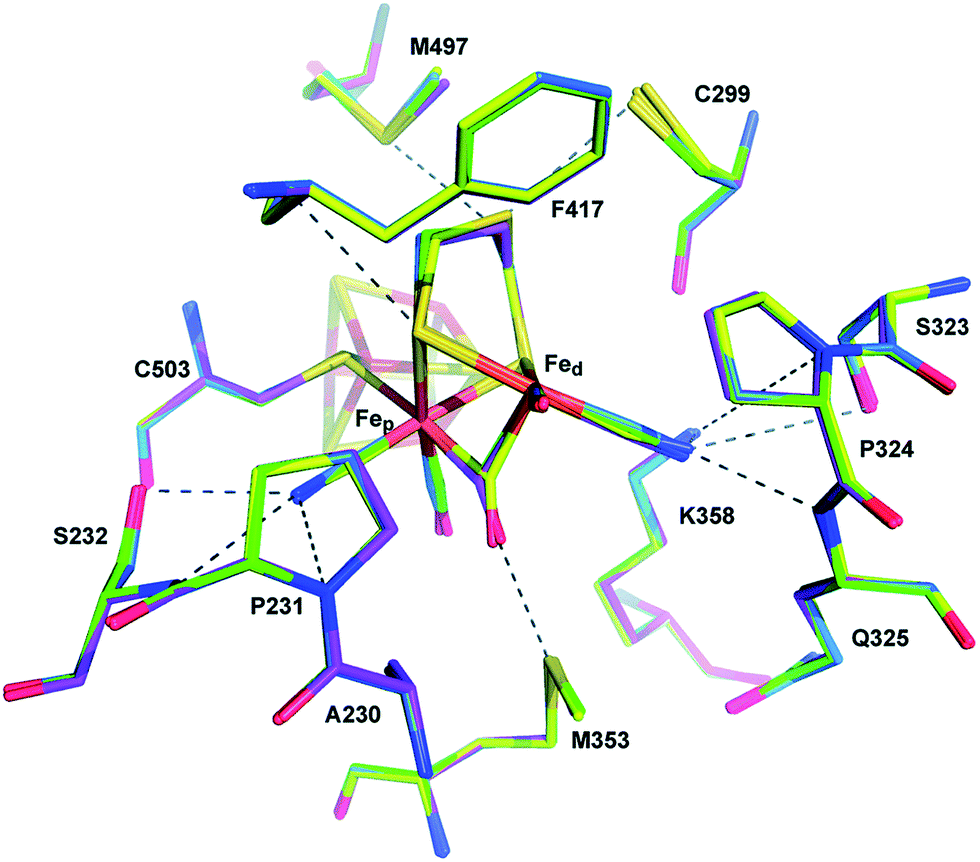 | ||
| Fig. 20 Comparison of 30-CpI (carbon atoms in marine) superposed on stick models of 31-CpI (magenta), 32-CpI (green) and 38-CpI (yellow). Dashed lines indicate potential proton transfer pathways or potential interactions of the [2Fe] subsite with the protein. Amino acid numbering as in the structure of native CpI. Adapted from Esselborn et al.25 with permission from the Royal Society of Chemistry. | ||
In parallel to modifications of the [2Fe] subsite, Happe and co-workers also explored the influence of chalcogen exchange of the [4Fe–4S]H-cluster.222 Chemical reconstitution was employed to generate the Se-derivate ([4Fe–4Se]H–[2Fe]) of the H-cluster in CrHydA1. Somewhat surprisingly, this Se analogue was shown to be practically indistinguishable from the native cofactor with regard to FTIR spectroscopic properties and enzymatic activities in standard chemical assays. In contrast, Se exchange of the adt-derived thiol ligands (Fig. 19, complex 41) was recently shown to result in increased electron density on the [2Fe] subsite.223 The initial characterisation suggests that this semi-synthetic hydrogenase displays increased bias towards H2 production as well as a decrease in O2 tolerance.
5.3 Insights into hydrogenase maturation gained from artificial maturation
In parallel to the direct application for the preparation of semi-synthetic hydrogenases, the artificial maturation technique has also started to provide valuable insights into the biosynthesis of the H-cluster.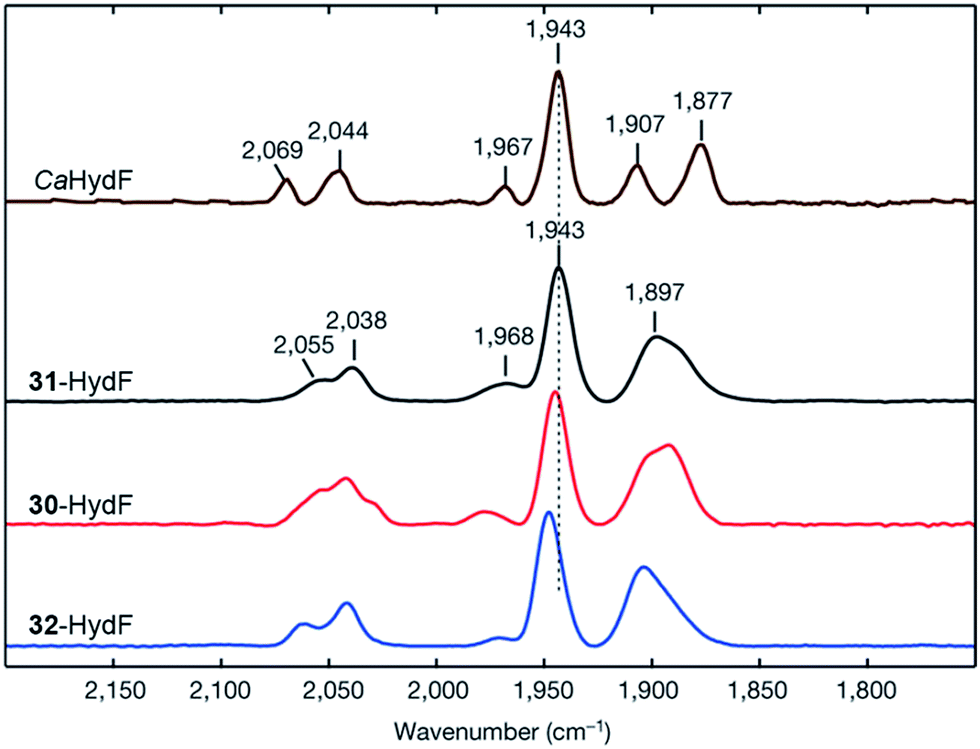 | ||
| Fig. 21 FTIR spectra with normalized intensities of selected forms of semi-synthetic HydF, loaded with synthetic co-factors 30–32 (30–32-HydF), compared to a form of HydF containing the native co-factor (CaHydF). The figure was adapted from Berggren et al., Nature, 2013.53 | ||
A recent X-ray crystallography study of HydF from Thermosipho melanesiensis containing its [4Fe–4S] cluster provides additional strong support for the involvement of this cluster in binding the synthetic pre-catalyst on HydF.233 The structure of holo-HydF containing also the pre-catalyst remains to be solved. However, the latter study revealed the presence of a labile glutamate ligand coordinating the [4Fe–4S] cluster, which arguably could become displaced upon binding the [2Fe] subsite precursor. Analogously to Thermotoga maritima HydF, wild-type T. melanesiensis HydF was capable of efficiently binding 30 and 31 and transferring these complexes to apo-HydA. Conversely, replacing the glutamate with potentially stronger ligands, i.e. histidine or cysteine, abolished the capacity of the protein to bind the synthetic cofactor.
5.4 Artificial maturation of [Fe] hydrogenases
The concept of artificial enzyme activation has also been applied to two other gas-processing enzymes featuring complex inorganic cofactors, namely [Fe] hydrogenases and nitrogenases.236,237 In contrast to [FeFe] hydrogenases, the mono-iron FeGP cofactor found in [Fe] hydrogenases can be extracted from the enzyme and re-introduced when the apo-form of the enzyme is mixed with an ultrafiltrate from the denatured active enzyme.238 The successful artificial activation of a Methanocaldococcus jannaschii [Fe] hydrogenase with synthetic analogues of the native FeGP cofactor was reported by Hu and co-workers in 2015 (Fig. 22).237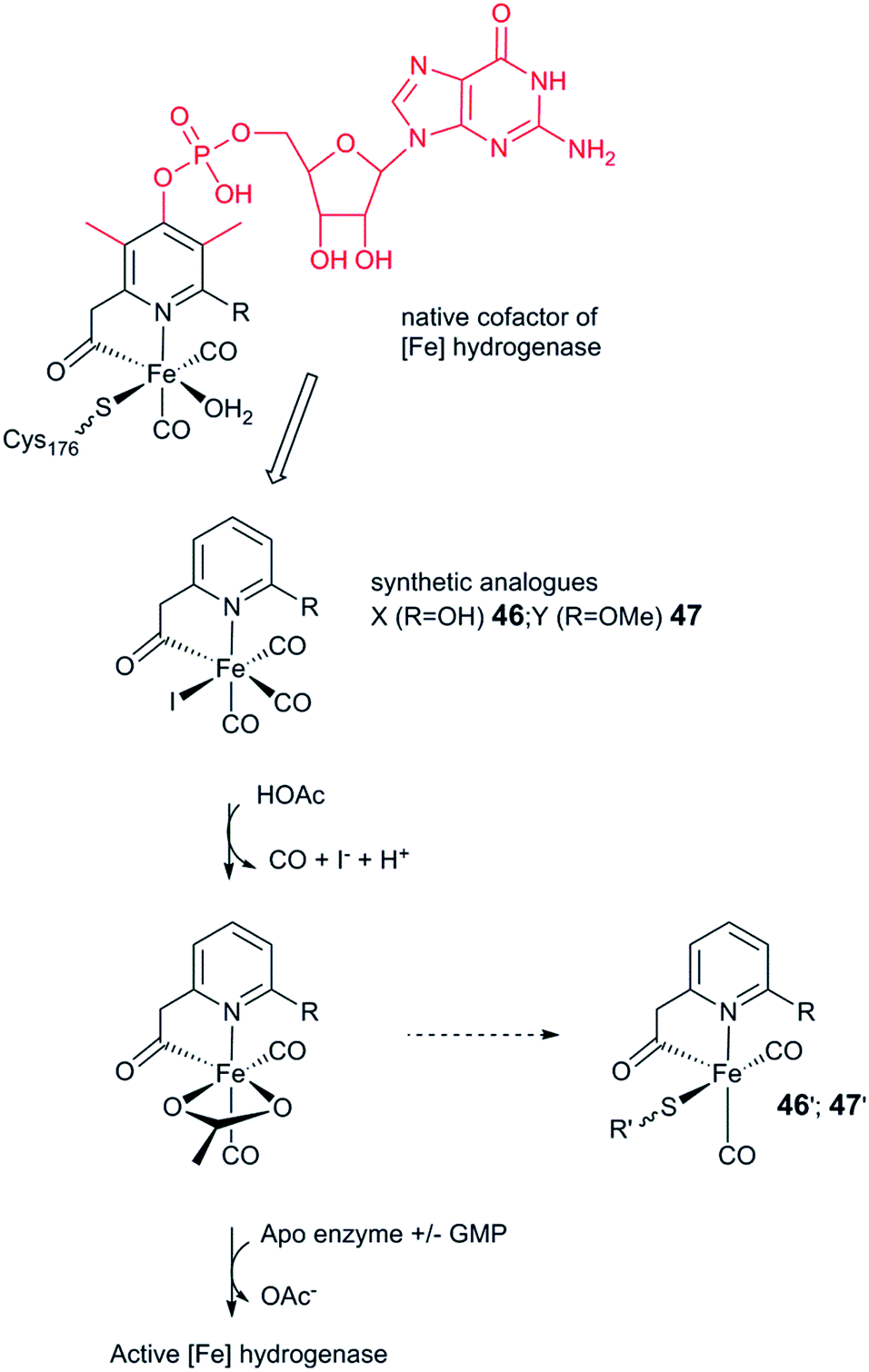 | ||
| Fig. 22 Complexes 46 and 47 are inserted into the apo-[Fe] hydrogenase following the removal of two monodentate ligands (CO and I−). Substituents on the pyridine ring lacking in 46 and 47 as compared to the native FeGP cofactor indicated in red. | ||
The synthetic complexes employed (46 and 47) represent truncated versions of the native FeGP cofactor, lacking the two methyl substituents as well as the GMP group on the pyridine ligand. Still, the primary ligand sphere of the Fe centre is highly similar to that of the enzymatic cofactor (Fig. 22). When the acetate ligand is replaced with a thiol ligand, mimicking the coordination environment of the cofactor (46′ and 47′), both complexes are incapable of H2 activation in solution.239,240 Conversely, introduction of complex 46 into the apo-enzyme resulted in a semi-synthetic hydrogenase with 1% activity as compared to the native enzyme, further underscoring the importance of a protein environment for the activity of hydrogenase model complexes. The successful incorporation of 46 into the active site of the enzyme was further supported by FTIR spectroscopy, as the 46–[Fe] hydrogenase displayed two dominant bands at ∼2020 and 1945 cm−1, similar to samples reconstituted with native FeGP (2011 and 1944 cm−1). In the former case, additional minor peaks were also clearly discernible, indicative of non-specific binding. The reconstitution of the apo-enzyme using 46 in the presence of GMP resulted in a decrease in the number of minor peaks and a small shift of the dominant bands to ∼2016 and 1944 cm−1; moreover, the specific activity increased to ∼2%. The introduction of the methylated cofactor analogue, 47, into the enzyme resulted in FTIR spectra similar to those observed for 46–[Fe] hydrogenase, but no enzymatic activity was observed.
This highlights the importance of the 2-hydroxy group for the activity of the enzyme, in agreement with prior theoretical work.69,70 Moreover, the improved incorporation of 46 in the presence of GMP suggests that the synthesis of closer structural FeGP analogues is the key challenge to address artificial maturation of [Fe] hydrogenases.
5.5 Summary and outlook
The concept of “artificial enzyme maturation” is derived from the idea of artificial enzymes developed in the 1970s and made possible by our understanding of the biological maturation processes. Despite its relatively recent inception, the artificial maturation method has proven to be an extremely powerful tool for studying [FeFe] hydrogenases and has accelerated research in this field.The decoupling of the preparation of active hydrogenases from the biological maturation system, both in vitro and in vivo, has facilitated the study of these enzymes in a variety of ways. The technique greatly simplifies heterologous expression enabling the large scale preparation of active enzymes as well as the exploration of different subclasses of [FeFe] hydrogenases, two critical aspects for the implementation of these enzymes in (bio-)technological applications. In combination with the possibility to selectively label the [2Fe] subsite, it can certainly also be expected to continue to improve our understanding of these enzymes. The introduction of modified, or non-native, [2Fe] cofactors has primarily proven to be a powerful method for unravelling mechanistic details so far. However, this field is still in its infancy. Looking forward, it provides synthetic chemists with the possibility of designing more robust enzymes as well as a tool for exploring the interplay between synthetic [2Fe] cofactors and the protein cavity.
6. Concluding remarks
Tremendous progress has been made in our understanding of hydrogenases and our capacity to mimic their reactivity with synthetic systems over the last few years. In particular, work on [NiFe] hydrogenases has revealed that evolution has developed a number of solutions to influence O2 tolerance and catalytic bias of these enzymes. Our capacity to introduce these properties via enzyme design is still limited. Nevertheless, our attempts to improve on Nature's solution to these issues are providing critical information for the implementation of these enzymes in (bio-)technological applications and guiding us in the preparation of synthetic catalysts.The development of artificial hydrogenases has proven that encapsulation of catalysts in a variety of different scaffolds results in systems with remarkable catalytic activities. Moreover, as shown by the work on Dubois catalysts in particular, we can greatly enhance the reactivity of synthetic systems by modulating the immediate vicinity of the catalytic site. The impact of the catalyst environment, and by extension the potential of artificial hydrogenases, is underscored by the success of the artificial maturation technique. Indeed, the introduction of [2Fe] subsite mimic 30 into the active site of an [FeFe] hydrogenase transforms this inactive and fragile complex into an outstanding catalyst. In the context of catalyst design it is noteworthy that we now have the possibility to study [2Fe] subsite models ranging from isolated complexes in organic solvents all the way to their reactivity inside the enzyme under in vivo conditions.
Undoubtedly, the combined efforts of molecular biologists, biophysicists and chemists will continue to improve both our understanding of these different systems and their applicability in an industrial context. Considerable challenges remain, but nevertheless there are strong reasons to believe that we will be able to harness this knowledge in the development of a hydrogen economy over the next few decades.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research performed in the authors' laboratories is supported by grants from the Swedish Research Council, VR (G. B. contract no. 621-2014-5670), the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning, Formas (G. B. contract no. 213-2014-880), Stiftelsen Olle Engkvist Byggmästare (G. B.), the Wenner-Gren Foundations (C. E. and G. B.) and the ERC (G. B. contract no. 714102).Notes and references
- G. Lauermann, P. Häussinger, R. Lohmüller and A. M. Watson, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. 1–15, DOI:10.1002/14356007.a13_297.pub3.
- J. M. Ogden, Annu. Rev. Energy, 1999, 24, 227–279 CrossRef.
- S. P. S. Badwal, S. Giddey and C. Munnings, Wiley Interdiscip. Rev.: Energy Environ., 2013, 2, 473–487 CrossRef CAS.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10017 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Nature, 2009, 460, 814–822 CrossRef CAS PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew. Chem., Int. Ed. Engl., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- C. Baffert, K. Sybirna, P. Ezanno, T. Lautier, V. Hajj, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Anal. Chem., 2012, 84, 7999–8005 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- F. A. Armstrong, R. M. Evans, S. V. Hexter, B. J. Murphy, M. M. Roessler and P. Wulff, Acc. Chem. Res., 2016, 49, 884–892 CrossRef CAS PubMed.
- C. Wombwell, C. A. Caputo and E. Reisner, Acc. Chem. Res., 2015, 48, 2858–2865 CrossRef CAS PubMed.
- N. Khanna and P. Lindblad, Int. J. Mol. Sci., 2015, 16, 10537 CrossRef CAS PubMed.
- M. Ghirardi, Photosynth. Res., 2015, 125, 383–393 CrossRef CAS PubMed.
- V. Artero, G. Berggren, M. Atta, G. Caserta, S. Roy, L. Pecqueur and M. Fontecave, Acc. Chem. Res., 2015, 48, 2380–2387 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- S. Kaur-Ghumaan and M. Stein, Dalton Trans., 2014, 43, 9392–9405 RSC.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS PubMed.
- J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, Joule, 2017, 1, 61–76 CrossRef.
- M. Stephenson and L. H. Stickland, Biochem. J., 1931, 25, 205–214 CrossRef CAS PubMed.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, ISME J., 2015, 10, 761–777 CrossRef PubMed.
- D. W. Mulder, E. S. Boyd, R. Sarma, R. K. Lange, J. A. Endrizzi, J. B. Broderick and J. W. Peters, Nature, 2010, 465, 248–251 CrossRef CAS PubMed.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- H. Ogata, P. Kellers and W. Lubitz, J. Mol. Biol., 2010, 402, 428–444 CrossRef CAS PubMed.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS PubMed.
- M. Teixeira, G. Fauque, I. Moura, P. A. Lespinat, Y. Berlier, B. Prickril, H. D. Peck, A. V. Xavier, J. Le Gall and J. J. G. Moura, Eur. J. Biochem., 1987, 167, 47–58 CrossRef CAS PubMed.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS PubMed.
- P. A. Ash, R. Hidalgo and K. A. Vincent, ACS Catal., 2017, 7, 2471–2485 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414–4435 CrossRef CAS PubMed.
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571–574 CrossRef PubMed.
- A. Abou Hamdan, B. Burlat, O. Gutiérrez-Sanz, P.-P. Liebgott, C. Baffert, A. L. De Lacey, M. Rousset, B. Guigliarelli, C. Léger and S. Dementin, Nat. Chem. Biol., 2012, 9, 15 CrossRef PubMed.
- S. J. George, S. Kurkin, R. N. F. Thorneley and S. P. J. Albracht, Biochemistry, 2004, 43, 6808–6819 CrossRef CAS PubMed.
- C. Fan, M. Teixeira, J. Moura, I. Moura, H. Huynh Boi, J. Le Gall, H. D. Peck and B. M. Hoffman, J. Am. Chem. Soc., 1991, 113, 20–24 CrossRef CAS.
- J. P. Whitehead, R. J. Gurbiel, C. Bagyinka, B. M. Hoffman and M. J. Maroney, J. Am. Chem. Soc., 1993, 115, 5629–5635 CrossRef CAS.
- J. W. van der Zwaan, S. P. J. Albracht, R. D. Fontijn and E. C. Slater, FEBS Lett., 1985, 179, 271–277 CrossRef CAS PubMed.
- S. Foerster, M. Stein, M. Brecht, H. Ogata, Y. Higuchi and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 83–93 CrossRef CAS PubMed.
- B. J. Murphy, R. Hidalgo, M. M. Roessler, R. M. Evans, P. A. Ash, W. K. Myers, K. A. Vincent and F. A. Armstrong, J. Am. Chem. Soc., 2015, 137, 8484–8489 CrossRef CAS PubMed.
- R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Angew. Chem., Int. Ed. Engl., 2015, 54, 7110–7113 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- Y. Nicolet, A. L. de Lacey, X. Vernede, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- M. Calusinska, T. Happe, B. Joris and A. Wilmotte, Microbiology, 2010, 156, 1575–1588 CrossRef CAS PubMed.
- G. J. Schut and M. W. W. Adams, J. Bacteriol., 2009, 191, 4451–4457 CrossRef CAS PubMed.
- M. F. J. M. Verhagen, T. O'Rourke and M. W. W. Adams, Biochim. Biophys. Acta, Bioenerg., 1999, 1412, 212–229 CrossRef CAS.
- B. L. Greene, G. J. Schut, M. W. W. Adams and R. B. Dyer, ACS Catal., 2017, 7, 2145–2150 CrossRef CAS.
- J. A. Birrell, K. Wrede, K. Pawlak, P. Rodriguez-Maciá, O. Rüdiger, E. J. Reijerse and W. Lubitz, Isr. J. Chem., 2016, 56, 852–863 CrossRef CAS.
- A. Silakov, E. J. Reijerse, S. P. J. Albracht, E. C. Hatchikian and W. Lubitz, J. Am. Chem. Soc., 2007, 129, 11447–11458 CrossRef CAS PubMed.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, J. Biol. Inorg Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- M. Winkler, J. Esselborn and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 974–985 CrossRef CAS PubMed.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- S. Morra, S. Maurelli, M. Chiesa, D. W. Mulder, M. W. Ratzloff, E. Giamello, P. W. King, G. Gilardi and F. Valetti, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 98–106 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- S. Mebs, M. Senger, J. Duan, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, S. T. Stripp and M. Haumann, J. Am. Chem. Soc., 2017, 139, 12157–12160 CrossRef CAS PubMed.
- R. K. Thauer, A. R. Klein and G. C. Hartmann, Chem. Rev., 1996, 96, 3031–3042 CrossRef CAS PubMed.
- C. Zirngibl, R. Hedderich and R. K. Thauer, FEBS Lett., 1990, 261, 112–116 CrossRef CAS.
- S. Shima and R. K. Thauer, Chem. Rec., 2007, 7, 37–46 CrossRef CAS PubMed.
- S. Shima, E. J. Lyon, M. Sordel-Klippert, M. Kauß, J. Kahnt, R. K. Thauer, K. Steinbach, X. Xie, L. Verdier and C. Griesinger, Angew. Chem., Int. Ed. Engl., 2004, 43, 2547–2551 CrossRef CAS PubMed.
- T. Hiromoto, K. Ataka, O. Pilak, S. Vogt, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer, S. Shima and U. Ermler, FEBS Lett., 2009, 583, 585–590 CrossRef CAS PubMed.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 10901–10908 CrossRef CAS PubMed.
- A. R. Finkelmann, H. M. Senn and M. Reiher, Chem. Sci., 2014, 5, 4474–4482 RSC.
- T. Hiromoto, E. Warkentin, J. Moll, U. Ermler and S. Shima, Angew. Chem., Int. Ed. Engl., 2009, 48, 6457–6460 CrossRef CAS PubMed.
- A. Abou Hamdan, S. Dementin, P.-P. Liebgott, O. Gutierrez-Sanz, P. Richaud, A. L. De Lacey, M. Rousset, P. Bertrand, L. Cournac and C. Léger, J. Am. Chem. Soc., 2012, 134, 8368–8371 CrossRef CAS PubMed.
- A. S. Ouattara, B. K. C. Patel, J.-L. Cayol, N. Cuzin, A. S. Traore and J.-L. Garcia, Int. J. Syst. Evol. Microbiol., 1999, 49, 639–643 CAS.
- P. Bertrand, F. Dole, M. Asso and B. Guigliarelli, J. Biol. Inorg Chem., 2000, 5, 682–691 CrossRef CAS PubMed.
- D. W. Bak and S. J. Elliott, Curr. Opin. Chem. Biol., 2014, 19, 50–58 CrossRef CAS PubMed.
- J. A. Birrell, C. Laurich, E. J. Reijerse, H. Ogata and W. Lubitz, Biochemistry, 2016, 55, 4344–4355 CrossRef CAS PubMed.
- J. M. Moulis, V. Davasse, M.-P. Golinelli, J. Meyer and I. Quinkal, J. Biol. Inorg Chem., 1996, 1, 2–14 CrossRef CAS.
- K. Sybirna, P. Ezanno, C. Baffert, C. Léger and H. Bottin, Int. J. Hydrogen Energy, 2013, 38, 2998–3002 CrossRef CAS.
- T. Maeda, V. Sanchez-Torres and T. K. Wood, Appl. Microbiol. Biotechnol., 2008, 79, 77–86 CrossRef CAS PubMed.
- S. Dementin, V. Belle, P. Bertrand, B. Guigliarelli, G. Adryanczyk-Perrier, A. L. De Lacey, V. M. Fernandez, M. Rousset and C. Léger, J. Am. Chem. Soc., 2006, 128, 5209–5218 CrossRef CAS PubMed.
- A. Petrenko and M. Stein, J. Phys. Chem. B, 2015, 119, 13870–13882 CrossRef CAS PubMed.
- P. Raleiras, P. Kellers, P. Lindblad, S. Styring and A. Magnuson, J. Biol. Chem., 2013, 288, 18345–18352 CrossRef CAS PubMed.
- A. Petrenko and M. Stein, Int. J. Mol. Sci., 2017, 18(1), 100 CrossRef PubMed.
- M. Rousset, Y. Montet, B. Guigliarelli, N. Forget, M. Asso, P. Bertrand, J. C. Fontecilla-Camps and E. C. Hatchikian, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11625–11630 CrossRef CAS.
- I. T. Yonemoto, C. W. Matteri, T. A. Nguyen, H. O. Smith and P. D. Weyman, J. Biol. Eng., 2013, 7, 17 CrossRef CAS PubMed.
- I. T. Yonemoto, B. R. Clarkson, H. O. Smith and P. D. Weyman, BMC Biochem., 2014, 15, 10 CrossRef PubMed.
- P. Raleiras, N. Khanna, H. Miranda, L. S. Mészáros, H. Krassen, F. Ho, N. Battchikova, E.-M. Aro, A. Magnuson, P. Lindblad and S. Styring, Energy Environ. Sci., 2016, 9, 581–594 CAS.
- L. A. Flanagan, J. J. Wright, M. M. Roessler, J. W. Moir and A. Parkin, Chem. Commun., 2016, 52, 9133–9136 RSC.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS PubMed.
- E. C. Hatchikian, N. Forget, V. M. Fernandez, R. Williams and R. Cammack, Eur. J. Biochem., 1992, 209, 357–365 CrossRef CAS PubMed.
- J. Cohen, K. Kim, P. King, M. Seibert and K. Schulten, Structure, 2005, 13, 1321–1329 CrossRef CAS PubMed.
- A. S. Bingham, P. R. Smith and J. R. Swartz, Int. J. Hydrogen Energy, 2012, 37, 2965–2976 CrossRef CAS.
- V. H. Teixeira, A. M. Baptista and C. M. Soares, Biophys. J., 2006, 91, 2035–2045 CrossRef CAS PubMed.
- Y. Montet, P. Amara, A. Volbeda, X. Vernede, E. C. Hatchikian, M. J. Field, M. Frey and J. C. Fontecilla-Camps, Nat. Struct. Mol. Biol., 1997, 4, 523–526 CAS.
- P.-P. Liebgott, F. Leroux, B. Burlat, S. Dementin, C. Baffert, T. Lautier, V. Fourmond, P. Ceccaldi, C. Cavazza, I. Meynial-Salles, P. Soucaille, J. C. Fontecilla-Camps, B. Guigliarelli, P. Bertrand, M. Rousset and C. Léger, Nat. Chem. Biol., 2010, 6, 63–70 CrossRef CAS PubMed.
- T. Buhrke, O. Lenz, N. Krauss and B. Friedrich, J. Biol. Chem., 2005, 280, 23791–23796 CrossRef CAS PubMed.
- O. Duché, S. Elsen, L. Cournac and A. Colbeau, FEBS J., 2005, 272, 3899–3908 CrossRef PubMed.
- M. Ludwig, J. A. Cracknell, K. A. Vincent, F. A. Armstrong and O. Lenz, J. Biol. Chem., 2009, 284, 465–477 CrossRef CAS PubMed.
- S. Dementin, F. Leroux, L. Cournac, A. L. d. Lacey, A. Volbeda, C. Léger, B. Burlat, N. Martinez, S. Champ, L. Martin, O. Sanganas, M. Haumann, V. M. Fernández, B. Guigliarelli, J. C. Fontecilla-Camps and M. Rousset, J. Am. Chem. Soc., 2009, 131, 10156–10164 CrossRef CAS PubMed.
- P.-P. Liebgott, A. L. de Lacey, B. Burlat, L. Cournac, P. Richaud, M. Brugna, V. M. Fernandez, B. Guigliarelli, M. Rousset, C. Léger and S. Dementin, J. Am. Chem. Soc., 2011, 133, 986–997 CrossRef CAS PubMed.
- A. Volbeda and J. C. Fontecilla-Camps, Coord. Chem. Rev., 2005, 249, 1609–1619 CrossRef CAS.
- Y. Shomura, K. S. Yoon, H. Nishihara and Y. Higuchi, Nature, 2011, 479, 253–256 CrossRef CAS PubMed.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. T. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, C. Darnault, J.-M. Mouesca, A. Parkin, M. M. Roessler, F. A. Armstrong and J. C. Fontecilla-Camps, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS PubMed.
- P. Wulff, C. C. Day, F. Sargent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6606–6611 CrossRef CAS PubMed.
- M. J. Lukey, M. M. Roessler, A. Parkin, R. M. Evans, R. A. Davies, O. Lenz, B. Friedrich, F. Sargent and F. A. Armstrong, J. Am. Chem. Soc., 2011, 133, 16881–16892 CrossRef CAS PubMed.
- G.-F. Huang, X.-B. Wu, L.-P. Bai, M.-N. Long and Q.-X. Chen, Int. J. Hydrogen Energy, 2014, 39, 18604–18611 CrossRef CAS.
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. J. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg Chem., 2005, 10, 239–249 CrossRef CAS PubMed.
- T. Goris, A. F. Wait, M. Saggu, J. Fritsch, N. Heidary, M. Stein, I. Zebger, F. Lendzian, F. A. Armstrong, B. Friedrich and O. Lenz, Nat. Chem. Biol., 2011, 7, 310–318 CrossRef CAS PubMed.
- M. C. Marques, C. Tapia, O. Gutierrez-Sanz, A. R. Ramos, K. L. Keller, J. D. Wall, A. L. De Lacey, P. M. Matias and I. A. C. Pereira, Nat. Chem. Biol., 2017, 13, 544–550 CrossRef CAS PubMed.
- B. Ginovska-Pangovska, M.-H. Ho, J. C. Linehan, Y. Cheng, M. Dupuis, S. Raugei and W. J. Shaw, Biochim. Biophys. Acta, 2014, 1837, 131–138 CrossRef CAS PubMed.
- A. J. Cornish, K. Gärtner, H. Yang, J. W. Peters and E. L. Hegg, J. Biol. Chem., 2011, 286, 38341–38347 CrossRef CAS PubMed.
- G. Hong, A. J. Cornish, E. L. Hegg and R. Pachter, Biochim. Biophys. Acta, 2011, 1807, 510–517 CrossRef CAS PubMed.
- S. Morra, A. Giraudo, G. Di Nardo, P. W. King, G. Gilardi and F. Valetti, PLoS One, 2012, 7, e48400 CAS.
- A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, J. Am. Chem. Soc., 2008, 130, 4533–4540 CrossRef CAS PubMed.
- V. H. Teixeira, C. M. Soares and A. M. Baptista, Proteins: Struct., Funct., Bioinf., 2008, 70, 1010–1022 CrossRef CAS PubMed.
- I. F. Galván, A. Volbeda, J. C. Fontecilla-Camps and M. J. Field, Proteins: Struct., Funct., Bioinf., 2008, 73, 195–203 CrossRef PubMed.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306–307 CrossRef CAS.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- Y.-W. Lin, Coord. Chem. Rev., 2017, 336, 1–27 CrossRef CAS.
- T. K. Hyster and T. R. Ward, Angew. Chem., Int. Ed. Engl., 2016, 55, 7344–7357 CrossRef CAS PubMed.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- C. Marchi-Delapierre, L. Rondot, C. Cavazza and S. Ménage, Isr. J. Chem., 2015, 55, 61–75 CrossRef CAS.
- I. Drienovská and G. Roelfes, Isr. J. Chem., 2015, 55, 21–31 CrossRef.
- P. Saint-Martin, P. A. Lespinat, G. Fauque, Y. Berlier, J. LeGall, I. Moura, M. Teixeira, A. V. Xavier and J. J. G. Moura, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9378–9380 CrossRef CAS.
- J. W. Slater and H. S. Shafaat, J. Phys. Chem. Lett., 2015, 6, 3731–3736 CrossRef CAS PubMed.
- D. J. Sommer, M. D. Vaughn and G. Ghirlanda, Chem. Commun., 2014, 50, 15852–15855 RSC.
- D. J. Sommer, M. D. Vaughn, B. C. Clark, J. Tomlin, A. Roy and G. Ghirlanda, Biochim. Biophys. Acta, 2016, 1857, 598–603 CrossRef CAS PubMed.
- B. Kandemir, S. Chakraborty, Y. Guo and K. L. Bren, Inorg. Chem., 2016, 55, 467–477 CrossRef CAS PubMed.
- M. Bacchi, G. Berggren, J. Niklas, E. Veinberg, M. W. Mara, M. L. Shelby, O. G. Poluektov, L. X. Chen, D. M. Tiede, C. Cavazza, M. J. Field, M. Fontecave and V. Artero, Inorg. Chem., 2014, 53, 8071–8082 CrossRef CAS PubMed.
- K. A. Vincent, G. J. Tilley, N. C. Quammie, I. Streeter, B. K. Burgess, M. R. Cheesman and F. A. Armstrong, Chem. Commun., 2003, 20, 2590–2591 RSC.
- S. Roy, M. Bacchi, G. Berggren and V. Artero, ChemSusChem, 2015, 8, 3632–3638 CrossRef CAS PubMed.
- L. I. Simándi, Z. Szeverényi and É. Budó-Záhonyi, Inorg. Nucl. Chem. Lett., 1975, 11, 773–777 CrossRef.
- M. Bacchi, E. Veinberg, M. J. Field, J. Niklas, T. Matsui, D. M. Tiede, O. G. Poluektov, M. Ikeda-Saito, M. Fontecave and V. Artero, ChemPlusChem, 2016, 81, 1083–1089 CrossRef CAS.
- E. A. Brucker, J. S. Olson, G. N. Phillips, Y. Dou and M. Ikeda-Saito, J. Biol. Chem., 1996, 271, 25419–25422 CrossRef CAS PubMed.
- C. M. Bianchetti, G. C. Blouin, E. Bitto, J. S. Olson and G. N. Phillips, Proteins: Struct., Funct., Bioinf., 2010, 78, 917–931 CrossRef CAS PubMed.
- S. R. Soltau, J. Niklas, P. D. Dahlberg, O. G. Poluektov, D. M. Tiede, K. L. Mulfort and L. M. Utschig, Chem. Commun., 2015, 51, 10628–10631 RSC.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. G. Poluektov, K. L. Mulfort and L. M. Utschig, Chem. Sci., 2016, 7, 7068–7078 RSC.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- S. R. Soltau, J. Niklas, P. D. Dahlberg, K. L. Mulfort, O. G. Poluektov and L. M. Utschig, ACS Energy Lett., 2017, 2, 230–237 CrossRef CAS.
- Y. Sano, A. Onoda and T. Hayashi, Chem. Commun., 2011, 47, 8229–8231 RSC.
- A. Onoda, Y. Kihara, K. Fukumoto, Y. Sano and T. Hayashi, ACS Catal., 2014, 4, 2645–2648 CrossRef CAS.
- A. K. Jones, B. R. Lichtenstein, A. Dutta, G. Gordon and P. L. Dutton, J. Am. Chem. Soc., 2007, 129, 14844–14845 CrossRef CAS PubMed.
- U.-P. Apfel, M. Rudolph, C. Apfel, C. Robl, D. Langenegger, D. Hoyer, B. Jaun, M.-O. Ebert, T. Alpermann, D. Seebach and W. Weigand, Dalton Trans., 2010, 39, 3065–3071 RSC.
- Y. Sano, A. Onoda and T. Hayashi, J. Inorg. Biochem., 2012, 108, 159–162 CrossRef CAS PubMed.
- S. Roy, S. Shinde, G. A. Hamilton, H. E. Hartnett and A. K. Jones, Eur. J. Inorg. Chem., 2011, 2011, 1050–1055 CrossRef.
- A. Roy, C. Madden and G. Ghirlanda, Chem. Commun., 2012, 48, 9816–9818 RSC.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- S. Roy, T.-A. D. Nguyen, L. Gan and A. K. Jones, Dalton Trans., 2015, 44, 14865–14876 RSC.
- A. Dutta, G. A. Hamilton, H. E. Hartnett and A. K. Jones, Inorg. Chem., 2012, 51, 9580–9588 CrossRef CAS PubMed.
- A. Jain, S. Lense, J. C. Linehan, S. Raugei, H. Cho, D. L. DuBois and W. J. Shaw, Inorg. Chem., 2011, 50, 4073–4085 CrossRef CAS PubMed.
- M. L. Reback, B. Ginovska-Pangovska, M.-H. Ho, A. Jain and T. C. Squier, Chem.–Eur. J., 2013, 19, 1928–1941 CrossRef CAS PubMed.
- M. L. Reback, B. Ginovska-Pangovska, M.-H. Ho, A. Jain, T. C. Squier, S. Raugei, J. A. S. Roberts and W. J. Shaw, Chem.–Eur. J., 2013, 19, 1928–1941 CrossRef CAS PubMed.
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed.
- A. Dutta, J. A. S. Roberts and W. J. Shaw, Angew. Chem., Int. Ed. Engl., 2014, 53, 6487–6491 CrossRef CAS PubMed.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS PubMed.
- A. Dutta, B. Ginovska, S. Raugei, J. A. S. Roberts and W. J. Shaw, Dalton Trans., 2016, 45, 9786–9793 RSC.
- N. Priyadarshani, A. Dutta, B. Ginovska, G. W. Buchko, M. O'Hagan, S. Raugei and W. J. Shaw, ACS Catal., 2016, 6, 6037–6049 CrossRef CAS.
- M. L. Reback, G. W. Buchko, B. L. Kier, B. Ginovska-Pangovska, Y. Xiong, S. Lense, J. Hou, J. A. S. Roberts, C. M. Sorensen, S. Raugei, T. C. Squier and W. J. Shaw, Chem.–Eur. J., 2014, 20, 1510–1514 CrossRef CAS PubMed.
- M. L. Reback, B. Ginovska, G. W. Buchko, A. Dutta, N. Priyadarshani, B. L. Kier, M. L. Helm, S. Raugei and W. J. Shaw, J. Coord. Chem., 2016, 69, 1730–1747 CrossRef CAS.
- B. Kandemir, L. Kubie, Y. Guo, B. Sheldon and K. L. Bren, Inorg. Chem., 2016, 55, 1355–1357 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef CAS PubMed.
- C. Travaglini-Allocatelli, S. Gianni, V. K. Dubey, A. Borgia, A. Di Matteo, D. Bonivento, F. Cutruzzolà, K. L. Bren and M. Brunori, J. Biol. Chem., 2005, 280, 25729–25734 CrossRef CAS PubMed.
- P. W. J. M. Frederix, R. Kania, J. A. Wright, D. A. Lamprou, R. V. Ulijn, C. J. Pickett and N. T. Hunt, Dalton Trans., 2012, 41, 13112–13119 RSC.
- H.-Y. Wang, W.-G. Wang, G. Si, F. Wang, C.-H. Tung and L.-Z. Wu, Langmuir, 2010, 26, 9766–9771 CrossRef CAS PubMed.
- F. Wang, W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung and L.-Z. Wu, ACS Catal., 2012, 2, 407–416 CrossRef CAS.
- F. Quentel, G. Passard and F. Gloaguen, Chem.–Eur. J., 2012, 18, 13473–13479 CrossRef CAS PubMed.
- X. Ru, X. Zeng, Z. Li, D. J. Evans, C. Zhan, Y. Tang, L. Wang and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2410–2417 CrossRef CAS.
- C. Zhan, X. Wang, Z. Wei, D. J. Evans, X. Ru, X. Zeng and X. Liu, Dalton Trans., 2010, 39, 11255–11262 RSC.
- S. Ibrahim, P. M. Woi, Y. Alias and C. J. Pickett, Chem. Commun., 2010, 46, 8189–8191 RSC.
- J. A. Wright, L. Webster, A. Jablonskyte, P. M. Woi, S. K. Ibrahim and C. J. Pickett, Faraday Discuss., 2011, 148, 359–371 RSC.
- K. N. Green, J. L. Hess, C. M. Thomas and M. Y. Darensbourg, Dalton Trans., 2009, 4344–4350 RSC.
- F. Wang, W.-J. Liang, J.-X. Jian, C.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed. Engl., 2013, 52, 8134–8138 CrossRef CAS PubMed.
- W.-J. Liang, F. Wang, M. Wen, J.-X. Jian, X.-Z. Wang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2015, 21, 3187–3192 CrossRef CAS PubMed.
- T. Yu, Y. Zeng, J. Chen, Y.-Y. Li and G. Yang, Angew. Chem., Int. Ed. Engl., 2013, 52, 5631–5635 CrossRef CAS PubMed.
- M. L. Singleton, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2010, 132, 8870–8871 CrossRef CAS PubMed.
- M. L. Singleton, D. J. Crouthers, R. P. Duttweiler, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2011, 50, 5015–5026 CrossRef CAS PubMed.
- X. Li, M. Wang, D. Zheng, K. Han, J. Dong and L. Sun, Energy Environ. Sci., 2012, 5, 8220–8224 CAS.
- J.-X. Jian, Q. Liu, Z.-J. Li, F. Wang, X.-B. Li, C.-B. Li, B. Liu, Q.-Y. Meng, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Nat. Commun., 2013, 4, 2695 Search PubMed.
- F. Wang, M. Wen, K. Feng, W.-J. Liang, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2016, 52, 457–460 RSC.
- F. Quentel, G. Passard and F. Gloaguen, Energy Environ. Sci., 2012, 5, 7757–7761 CAS.
- C. Orain, F. Quentel and F. Gloaguen, ChemSusChem, 2014, 7, 638–643 CrossRef CAS PubMed.
- R. Fritzsch, O. Brady, E. Adair, J. A. Wright, C. J. Pickett and N. T. Hunt, J. Phys. Chem. Lett., 2016, 7, 2838–2843 CrossRef CAS PubMed.
- S. Pullen and S. Ott, Top. Catal., 2016, 59, 1712–1721 CrossRef CAS.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- S. Pullen, S. Roy and S. Ott, Chem. Commun., 2017, 53, 5227–5230 RSC.
- S. Roy, V. Pascanu, S. Pullen, G. Gonzalez Miera, B. Martin-Matute and S. Ott, Chem. Commun., 2017, 53, 3257–3260 RSC.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Chem. Commun., 2014, 50, 10390–10393 RSC.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- Ö. F. Erdem, L. Schwartz, M. Stein, A. Silakov, S. Kaur-Ghumaan, P. Huang, S. Ott, E. J. Reijerse and W. Lubitz, Angew. Chem., Int. Ed. Engl., 2011, 50, 1439–1443 CrossRef PubMed.
- S. Ott, M. Kritikos, B. Åkermark, L. Sun and R. Lomoth, Angew. Chem., Int. Ed. Engl., 2004, 43, 1006–1009 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- D. W. Mulder, D. O. Ortillo, D. J. Gardenghi, A. V. Naumov, S. S. Ruebush, R. K. Szilagyi, B. Huynh, J. B. Broderick and J. W. Peters, Biochemistry, 2009, 48, 6240–6248 CrossRef CAS PubMed.
- J. K. Rubach, X. Brazzolotto, J. Gaillard and M. Fontecave, FEBS Lett., 2005, 579, 5055–5060 CrossRef CAS PubMed.
- X. Brazzolotto, J. K. Rubach, J. Gaillard, S. Gambarelli, M. Atta and M. Fontecave, J. Biol. Chem., 2006, 281, 769–774 CrossRef CAS PubMed.
- P. W. King, M. C. Posewitz, M. L. Ghirardi and M. Seibert, J. Bacteriol., 2006, 188, 2163–2172 CrossRef CAS PubMed.
- M. C. Posewitz, P. W. King, S. L. Smolinski, L. Zhang, M. Seibert and M. L. Ghirardi, J. Biol. Chem., 2004, 279, 25711–25720 CrossRef CAS PubMed.
- S. E. McGlynn, S. S. Ruebush, A. Naumov, L. E. Nagy, A. Dubini, P. W. King, J. B. Broderick, M. C. Posewitz and J. W. Peters, JBIC, J. Biol. Inorg. Chem., 2007, 12, 443–447 CrossRef CAS PubMed.
- E. M. Shepard, S. E. McGlynn, A. L. Bueling, C. S. Grady-Smith, S. J. George, M. A. Winslow, S. P. Cramer, J. W. Peters and J. B. Broderick, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10448–10453 CrossRef CAS PubMed.
- S. E. McGlynn, E. M. Shepard, M. A. Winslow, A. V. Naumov, K. S. Duschene, M. C. Posewitz, W. E. Broderick, J. B. Broderick and J. W. Peters, FEBS Lett., 2008, 582, 2183–2187 CrossRef CAS PubMed.
- I. Czech, S. Stripp, O. Sanganas, N. Leidel, T. Happe and M. Haumann, FEBS Lett., 2011, 585, 225–230 CrossRef CAS PubMed.
- I. Czech, A. Silakov, W. Lubitz and T. Happe, FEBS Lett., 2010, 584, 638–642 CrossRef CAS PubMed.
- D. L. M. Suess, J. M. Kuchenreuther, L. De La Paz, J. R. Swartz and R. D. Britt, Inorg. Chem., 2016, 55, 478–487 CrossRef CAS PubMed.
- E. M. Shepard, F. Mus, J. N. Betz, A. S. Byer, B. R. Duffus, J. W. Peters and J. B. Broderick, Biochemistry, 2014, 53, 4090–4104 CrossRef CAS PubMed.
- C. Esmieu and G. Berggren, Dalton Trans., 2016, 45, 19242–19248 RSC.
- A. Adamska-Venkatesh, S. Roy, J. F. Siebel, T. R. Simmons, M. Fontecave, V. Artero, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2015, 137, 12744–12747 CrossRef CAS PubMed.
- R. Gilbert-Wilson, J. F. Siebel, A. Adamska-Venkatesh, C. C. Pham, E. Reijerse, H. Wang, S. P. Cramer, W. Lubitz and T. B. Rauchfuss, J. Am. Chem. Soc., 2015, 137, 8998–9005 CrossRef CAS PubMed.
- J. M. Kuchenreuther, Y. Guo, H. Wang, W. K. Myers, S. J. George, C. A. Boyke, Y. Yoda, E. E. Alp, J. Zhao, R. D. Britt, J. R. Swartz and S. P. Cramer, Biochemistry, 2013, 52, 818–826 CrossRef CAS PubMed.
- A. S. Pereira, P. Tavares, I. Moura, J. J. G. Moura and B. H. Huynh, J. Am. Chem. Soc., 2001, 123, 2771–2782 CrossRef CAS PubMed.
- J. Noth, R. Kositzki, K. Klein, M. Winkler, M. Haumann and T. Happe, Sci. Rep., 2015, 5, 13978 CrossRef CAS PubMed.
- G. Caserta, A. Adamska-Venkatesh, L. Pecqueur, M. Atta, V. Artero, S. Roy, E. Reijerse, W. Lubitz and M. Fontecave, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1734–1740 CrossRef CAS PubMed.
- N. Khanna, C. Esmieu, L. S. Meszaros, P. Lindblad and G. Berggren, Energy Environ. Sci., 2017, 10, 1563–1567 CAS.
- L. S. Meszaros, B. Nemeth, C. Esmieu, P. Ceccaldi and G. Berggren, Angew. Chem., Int. Ed. Engl. DOI:10.1002/anie.201710740.
- Y. Qiao, H. Molina, A. Pandey, J. Zhang and P. A. Cole, Science, 2006, 311, 1293–1297 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- J. A. Zorn and J. A. Wells, Nat. Chem. Biol., 2010, 6, 179–188 CrossRef CAS PubMed.
- J. J. Woodward, N. I. Martin and M. A. Marletta, Nat. Methods, 2007, 4, 43–45 CrossRef CAS PubMed.
- T. Mathes, C. Vogl, J. Stolz and P. Hegemann, J. Mol. Biol., 2009, 385, 1511–1518 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- J. Noth, J. Esselborn, J. Güldenhaupt, A. Brünje, A. Sawyer, U.-P. Apfel, K. Gerwert, E. Hofmann, M. Winkler and T. Happe, Angew. Chem., Int. Ed. Engl., 2016, 55, 8396–8400 CrossRef CAS PubMed.
- L. Kertess, F. Wittkamp, C. Sommer, J. Esselborn, O. Rudiger, E. J. Reijerse, E. Hofmann, W. Lubitz, M. Winkler, T. Happe and U. P. Apfel, Dalton Trans., 2017, 46, 16947–16958 RSC.
- G. Berggren, R. Garcia-Serres, X. Brazzolotto, M. Clemancey, S. Gambarelli, M. Atta, J.-M. Latour, H. L. Hernandez, S. Subramanian, M. K. Johnson and M. Fontecave, JBIC, J. Biol. Inorg. Chem., 2014, 19, 75–84 CrossRef CAS PubMed.
- M. Albertini, P. Berto, F. Vallese, M. Di Valentin, P. Costantini and D. Carbonera, J. Phys. Chem. B, 2015, 119, 13680–13689 CrossRef CAS PubMed.
- M. Albertini, L. Galazzo, L. Maso, F. Vallese, P. Berto, E. De Rosa, M. Di Valentin, P. Costantini and D. Carbonera, Top. Catal., 2015, 58, 708–718 CrossRef CAS.
- A. M. Lunsford, C. C. Beto, S. Ding, O. F. Erdem, N. Wang, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2016, 7, 3710–3719 RSC.
- B. S. Lim and R. H. Holm, Inorg. Chem., 1998, 37, 4898–4908 CrossRef CAS PubMed.
- M. J. Scott and R. H. Holm, J. Am. Chem. Soc., 1994, 116, 11357–11367 CrossRef CAS.
- M. Shatruk, A. Dragulescu-Andrasi, K. E. Chambers, S. A. Stoian, E. L. Bominaar, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2007, 129, 6104–6116 CrossRef CAS PubMed.
- E. Coronado, M. C. Giménez-López, G. Levchenko, F. M. Romero, V. García-Baonza, A. Milner and M. Paz-Pasternak, J. Am. Chem. Soc., 2005, 127, 4580–4581 CrossRef CAS PubMed.
- A. Geiss and H. Vahrenkamp, Inorg. Chem., 2000, 39, 4029–4036 CrossRef CAS PubMed.
- G. Caserta, L. Pecqueur, A. Adamska-Venkatesh, C. Papini, S. Roy, V. Artero, M. Atta, E. Reijerse, W. Lubitz and M. Fontecave, Nat. Chem. Biol., 2017, 13, 779–784 CrossRef CAS PubMed.
- C. F. Megarity, J. Esselborn, S. V. Hexter, F. Wittkamp, U.-P. Apfel, T. Happe and F. A. Armstrong, J. Am. Chem. Soc., 2016, 138, 15227–15233 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, E. Reijerse, W. Lubitz, J. A. Birrell and O. Rüdiger, J. Phys. Chem. Lett., 2017, 8(16), 3834–3839 CrossRef PubMed.
- K. Tanifuji, C. C. Lee, Y. Ohki, K. Tatsumi, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed. Engl., 2015, 54, 14022–14025 CrossRef CAS PubMed.
- S. Shima, D. Chen, T. Xu, M. D. Wodrich, T. Fujishiro, K. M. Schultz, J. Kahnt, K. Ataka and X. Hu, Nat. Chem., 2015, 7, 995–1002 CrossRef CAS PubMed.
- G. Buurman, S. Shima and R. K. Thauer, FEBS Lett., 2000, 485, 200–204 CrossRef CAS PubMed.
- B. Hu, D. Chen and X. Hu, Chem.–Eur. J., 2014, 20, 1677–1682 CrossRef CAS PubMed.
- D. Chen, R. Scopelliti and X. Hu, Angew. Chem., Int. Ed. Engl., 2011, 50, 5671–5673 CrossRef CAS PubMed.
Footnotes |
| † The terms “O2 tolerance” and “O2 resistance” are used interchangeably in the current literature. |
| ‡ The correct specific epithet for this bacterium is fructosivorans.73 (A. S. Ouattara, B. K. C. Patel, J.-L. Cayol, N. Cuzin, A. S. Traore and J.-L. Garcia, Int. J. Syst. Evol. Microbiol., 1999, 49, 639–643). The epithet fructosovorans is nevertheless more common in the literature, and we use it here for the sake of clarity. |
| § Now renamed to Cupriavidus necator; the name Ralstonia eutropha is nevertheless still common in the literature, and we use it here for the sake of clarity. |
| This journal is © The Royal Society of Chemistry 2018 |