
Hydrogenation of fourteen biomass-derived phenolics in water and in methanol: their distinct reaction behaviours†
Lijun
Zhang
a,
Guangzhi
Hu
 b,
Song
Hu
c,
Jun
Xiang
c,
Xun
Hu
*a,
Yi
Wang
*c and
Dongsheng
Geng
*d
b,
Song
Hu
c,
Jun
Xiang
c,
Xun
Hu
*a,
Yi
Wang
*c and
Dongsheng
Geng
*d
aSchool of Material Science and Engineering, University of Jinan, Jinan, 250022, P. R. China. E-mail: xun.hu@outlook.com; Fax: +86-531-89736201; Tel: +86-531-89736201
bKey Laboratory of Chemistry of Plant Resources in Arid Regions, State Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China
cSchool of Energy and Power Engineering, Huazhong University of Science and Technology, Wuhan, 430074, P. R. China. E-mail: alenwang@hust.edu.cn
dCenter for Green Innovation, Beijing Key Laboratory for Magneto-Photoelectrical Composite and Interface Science, School of Mathematics and Physics, University of Science and Technology Beijing, Beijing, 100083, P. R. China. E-mail: dgeng@ustb.edu.cn
First published on 23rd January 2018
Abstract
Phenolic compounds are an important component of pyrolysis oil (bio-oil). Understanding their reaction behaviours during hydrogenation helps to clarify the reaction network during hydrotreatment of bio-oil. This study investigated the hydrogenation of typical phenolic compounds in bio-oil in water, which is an important fraction of bio-oil, and in methanol, which is used to esterify bio-oil. The results indicated that hydrogenation of the phenolics is more efficient in water than in methanol. With water medium, both the unsaturated oxygen-containing functionalities and the benzene ring of the phenolics can be effectively hydrogenated. In comparison, with methanol medium, only the unsaturated oxygen-containing functionality can be hydrogenated, while the hydrogenation of the benzene ring is rather difficult. Stereoselectivity was also observed in the hydrogenation of phenolics with three or more hydroxyl groups/methoxyl groups. For phenolics with two hydroxyl groups/methoxyl groups, the hydrogenation produces both cis- and trans-configurations.
1. Introduction
Bio-oil, produced from pyrolysis of biomass, is a mixture of sugars, sugar derivatives and phenolic compounds.1,2 Bio-oil cannot be directly used as fuel for vehicles and it has to be upgraded via either acid-catalysis3–5 or/and hydrogenation to reduce its corrosiveness and high oxygen content.6–10 Phenolic compounds are a very important component of bio-oil. Understanding their reaction behaviors would contribute to the understanding of the reaction network during hydrotreatment of bio-oil.Hydrogenation of the phenolics or furans derived from bio-oil has been performed in many studies with a particular focus on the development of catalysts.11–18 Apart from catalysts, the reaction medium also plays important roles in hydrogenation reactions. In our previous study,19 we have observed the distinct effects of water and methanol media on the conversion of sugars such as levoglucosan and glucose in acid-catalysed reactions. In this study, we thus made an effort to focus particularly on the effect of water and methanol on the conversion of typical phenolics present in bio-oil in hydrogenation reactions. The reason for selecting water as the reaction medium is that the concentration of water in bio-oil can be up to 40 wt%.20 Furthermore, water under subcritical/critical conditions behaves like an acid catalyst,21,22 potentially affecting the hydrogenation of the phenolics. Methanol is often used to esterify bio-oil and is thus a major component of esterified bio-oil.23–25 Water and methanol may impact hydrogenation of the phenolics in bio-oil in various ways.
To gain insight into this, hydrogenation of bio-oil and 14 phenolic compounds with varied oxygen-containing functionalities was conducted over a Pd/C catalyst in water and in methanol medium, respectively. Our results demonstrated that hydrogenation of the phenolics in bio-oil was more efficient in water than in methanol. Further to this, stereoselectivity for the hydrogenation of phenolics with three oxygen-containing functionalities (–OCH3 or –OH but not –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) was observed.
O) was observed.
2. Experimental
Hydrogenation of bio-oil and the phenolic compounds (1 or 2 g) in water or methanol (40 mL) with Pd/C (0.25 g) as the catalyst was conducted in an autoclave reactor. Before heating up, the reactor was pressurized with high-purity hydrogen (40 bar). When the reaction temperature reached 200 °C, more hydrogen was fed in to maintain a pressure of 70 bar, and the residence time at 200 °C was 100 min. The products were analyzed with a GCMS-QP2020 (Shimadzu) and a UV-fluorescence spectrometer (Shimadzu RF-6000). The elemental composition of the bio-oil and its composition are shown in Tables S1 and S2 in the ESI,† respectively. The configuration of the reactor for the production of the bio-oil is shown in Fig. S1.† Detailed experimental procedures are described in the ESI.†3. Results and discussion
3.1 Hydrogenation of bio-oil
Hydrogenation of bio-oil was investigated in methanol and in water medium, respectively. As shown in Fig. 1a, the distribution of the products in the hydrogenation of bio-oil with methanol as the reaction medium was similar to that in the original bio-oil. In comparison, the hydrogenation in water significantly affected the formation of the products. As only volatile compounds can be detected with a GC-MS, the products were further characterized with a UV-fluorescence spectrometer to measure the variation of the abundance of the aromatics in the products (Fig. 1b). It was confirmed that hydrogenation of the phenolics in bio-oil was more effective in water than in methanol, as some of the aromatic rings were saturated, reducing the amount of fluorescence.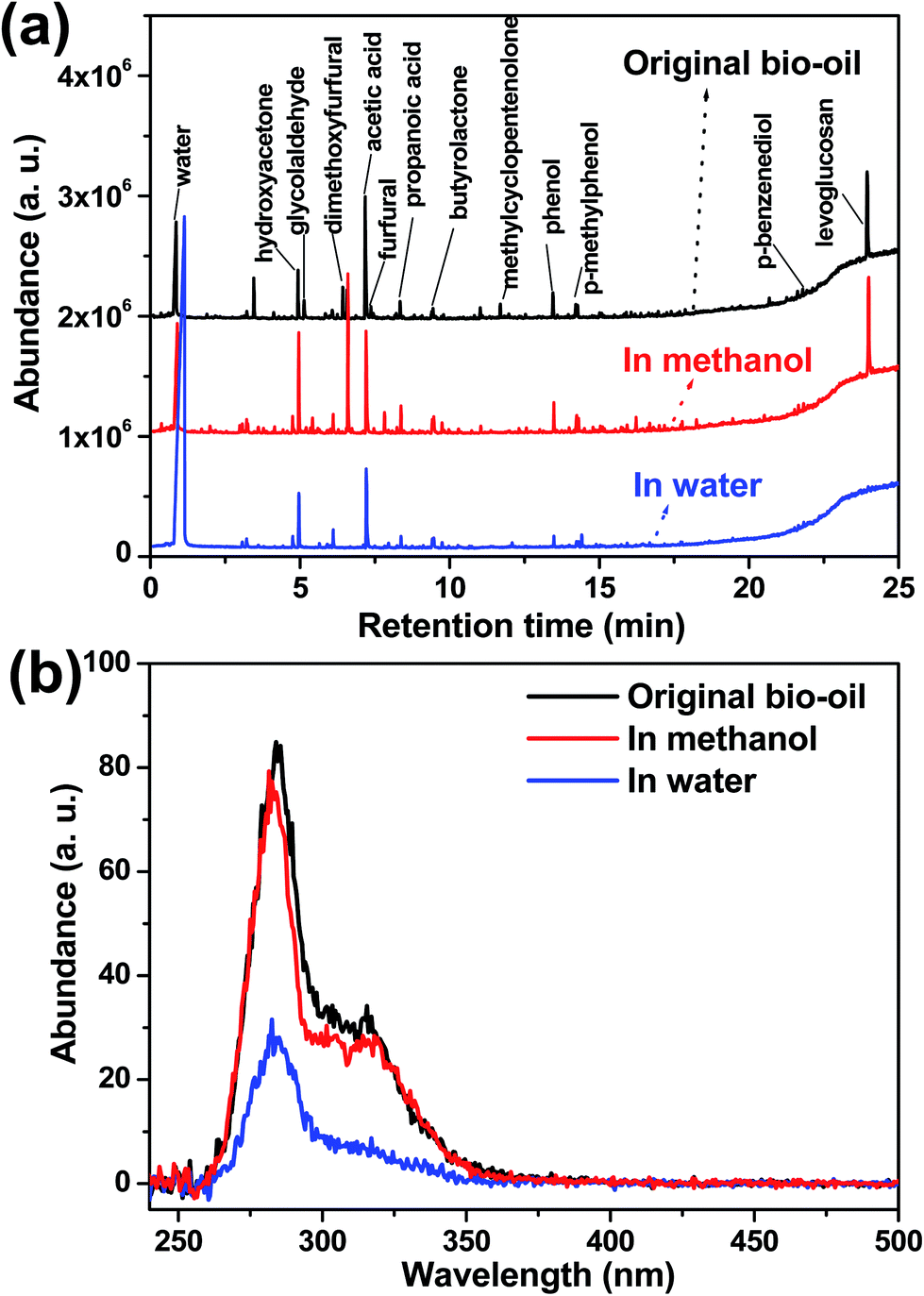 | ||
| Fig. 1 Hydrogenation of bio-oil in methanol and in water. (a) Distribution of the products from GC-MS characterization for the original bio-oil and that after hydrogenation in methanol and in water; T = 200 °C, t = 120 min, PH2 = 70 bar, Pd/C: 0.25 g, bio-oil: 2 g, and solvent: 40 mL. (b) Constant energy (−2800 cm−1) synchronous spectra of the original bio-oil and the ones after hydrogenation in methanol and in water. | ||
Phenolics in bio-oil have distinct oxygen-containing functionalities. It has not yet been made clear how methanol and water media affect the transformation of the oxygen-containing functionalities. Hydrogenation of the phenolics with distinct functionalities in water and in methanol was subsequently conducted.
3.2 Hydrogenation of vanillin
In methanol, hydrogenation of the carbonyl group of vanillin was easy (Fig. 2). The carbonyl group was mainly converted to a methyl group (compound 2, C2, the name of which is specified at the end of the ESI†) with the formation of compound 1 (C1) as the intermediate. The formation of C1 was confirmed by the formation of its ether, C8. Hydrogenation of the benzene ring in vanillin was difficult in methanol but not in water medium, as evidenced by the fluorescence spectra (Fig. S2†). | ||
| Fig. 2 Distribution of the products in the hydrogenation of vanillin in methanol and in water. The percentage mentioned below the compounds is their relative proportion in the products. Experimental conditions: T = 200 °C, t = 120 min, PH2 = 70 bar, Pd/C: 0.25 g, vanillin: 1 g, and solvent: 40 mL. The numbers in red colour are the yields of the product. | ||
During the hydrogenation of the benzene ring, C4 was the main intermediate. Hydrogenation of the carbon–carbon double bond (from C1 to C5 in water) proceeded in a stepwise manner. During this process, the carbon–carbon double bond together with the adjacent hydroxyl group underwent tautomerization to form ketones. In addition, the formation of stereoisomers confirmed the hydrogenation of the benzene ring in the phenolics in a stepwise manner, as evidenced by the formation of four stereoisomers of compound C5. The effect of vanillin loading on the hydrogenation reaction was also investigated and the results are presented in Fig. S3.† The varied vanillin loading did not impact much the distribution of the products. The difference in reactivity towards hydrogenation in water and in methanol was further confirmed through the hydrogenation of other phenolics.
3.3 Hydrogenation of guaiacol
Hydrogenation of guaiacol in methanol and in water generated a similar result to that of vanillin. Hydrogenation of the benzene ring of guaiacol was much easier in water than in methanol (Fig. 3 and 4). The hydroxyl group and the methoxy group are difficult to remove via the hydrogenation in methanol. C10 was the intermediate product for the formation of the fully hydrogenated product, C11, while C11 was further converted to the main product, C12, via etherification.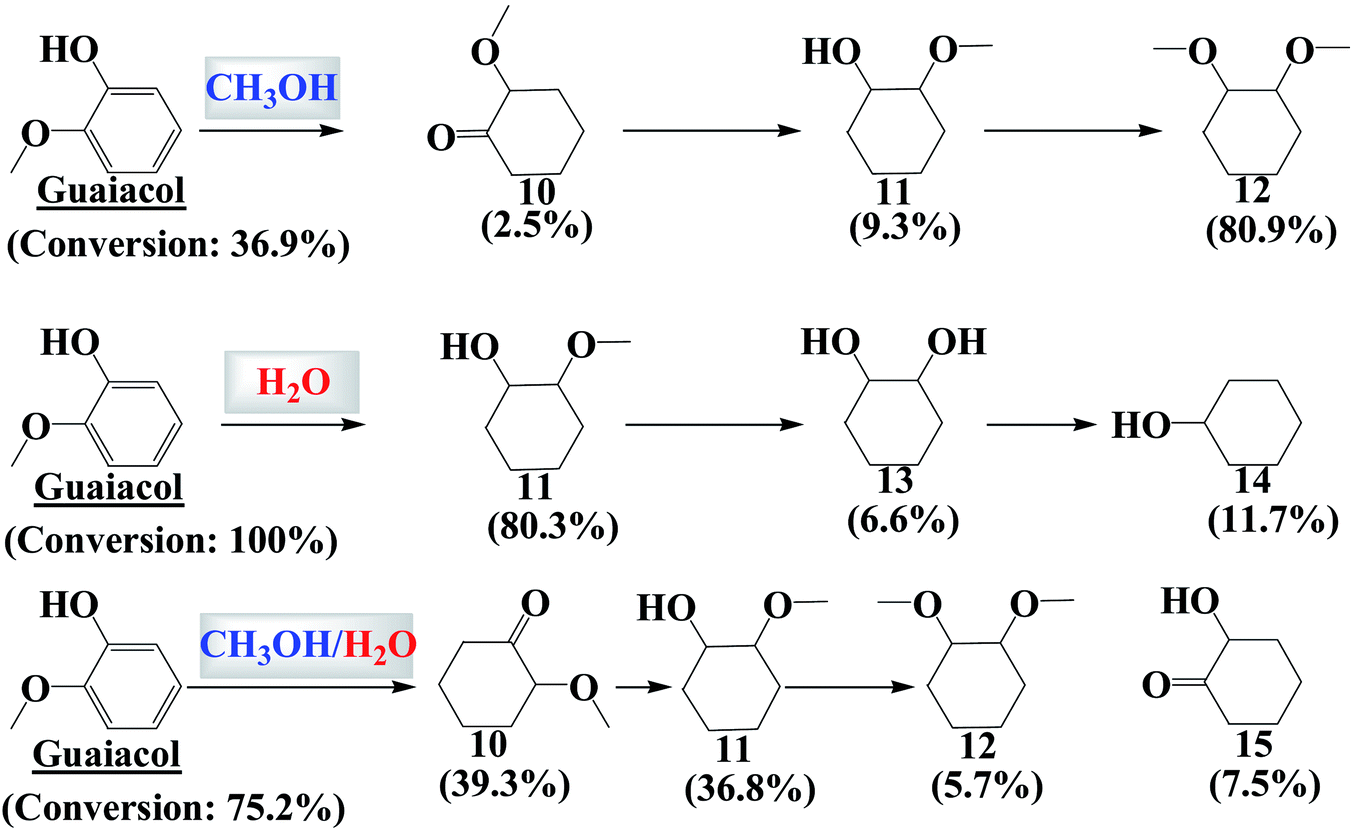 | ||
| Fig. 3 Distribution of the products in the hydrogenation of guaiacol in methanol and in water. The experimental conditions were the same as those in Fig. 2. | ||
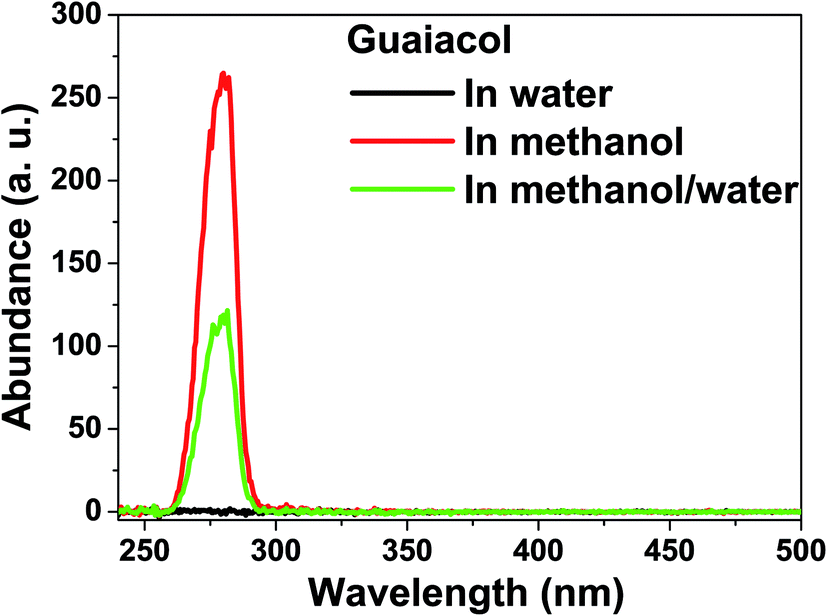 | ||
| Fig. 4 Constant energy (−2800 cm−1) synchronous spectra of the products after hydrogenation of guaiacol in water, in methanol and in methanol/water. | ||
Hydrogenation of guaiacol was further investigated in methanol/water (Vmethanol/Vwater = 1) to understand the reaction behaviors in the mixed reaction medium. As expected, hydrogenation of the benzene ring of guaiacol was more efficient in methanol/water than in methanol, while being less efficient than that in water. C10 was a major product produced from partial hydrogenation of the benzene ring and the tautomerization of the enol form of the intermediate. C10 was not detected in the products from the hydrogenation in water. It is probable that hydrogenation of the benzene ring in guaiacol was very quick in water and thus there was little chance for tautomerization to take place.
Water is more acidic than methanol, which possibly accounts for the high activity for the hydrogenation reactions. To confirm this hypothesis, hydrogenation of guaiacol in the presence of Amberlyst 70 (A70), a strong solid acid catalyst, was performed in methanol, as shown in Fig. S4 in the ESI.† The conversion of guaiacol increased but that increase did not match the conversion in water. The different acidities of water and methanol were thus not the main reason for the distinct impact of water and methanol on the hydrogenation of guaiacol and vanillin. In addition, the added acid catalyst (A70 catalyst) mainly impacted the acid-catalyzed reactions such as the etherification of compound C16 to C17 (Fig. S4†). The above results clearly indicated that the hydrogenation of the benzene ring with the oxygen-containing functionalities was difficult in methanol. But what would happen if there were no oxygen-containing functionalities attached on the benzene ring of the compound?
3.4 Hydrogenation of allylbenzene and eugenol
Hydrogenation of allylbenzene in methanol completely saturated the propenyl functionality via hydrogenation, as shown in Fig. 5. Unexpectedly, the benzene ring can also be hydrogenated to a significant extent. The propenyl group in allylbenzene is an electron donating group, which can increase the electron density in the benzene ring. The higher electron density enhances the interaction of the electron with the d-orbital of Pd metal, which favors hydrogenation of the benzene ring. Compared with allylbenzene molecules, eugenol molecules have an additional hydroxyl group and a methoxyl group. In methanol, hydrogenation of the propenyl group in eugenol was easy, but not for the benzene ring in eugenol (Fig. 6). The hydroxyl group and methoxyl group are electron-withdrawing functionalities, which lowers the electron density of the benzene ring, which probably makes subsequent hydrogenation of the benzene ring more difficult. | ||
| Fig. 5 Distribution of the products in the hydrogenation of allylbenzene and eugenol. The experimental conditions were the same as those in Fig. 2. The numbers in red colour are the yields of the product. | ||
 | ||
| Fig. 6 Constant energy (−2800 cm−1) synchronous spectra of the products after hydrogenation of eugenol in water and methanol. | ||
In water, however, the benzene ring of eugenol could be effectively hydrogenated. The structural difference of water and methanol must be responsible. Water and methanol can form hydrogen bonds with the phenolic compounds. π–n electron conjugation between π-bonds in the benzene ring and the isolated electron pair (n-electrons) in the oxygen atom in water might exist. The conjugation effect may increase the electron density of the benzene ring and favor the hydrogenation reaction. Methanol could have the same effect, but methanol molecules are much bigger than water molecules in terms of size. The resulting steric hindrance makes the conjugation less effective; this, however, needs further verification.
In addition, both methanol and water can be strongly adsorbed on activated carbon, via the formation of hydrogen bonds with the carboxylic group and the hydroxyl groups on activated carbon.26,27 This will inevitably affect the adsorption/activation of the phenolics and hydrogen. However, Lee et al. reported that methanol is less competitive than water for the adsorption of H2 on Pd/SiO2.28 Nevertheless, hydrogenation of the phenolics was more effective in water than in methanol, and hence competitive adsorption was not the main reason for the distinct reactivity of the phenolics in methanol and in water.
The adsorption of water on the surface of the activated carbon was stronger, due to its high capacity to form hydrogen bonds with functionalities of activated carbon.29 This would make the surface of the catalyst more hydrophilic, facilitating adsorption of the polar phenolics, which might aid the hydrogenation. In order to prove this, a Pd/SiO2 catalyst was prepared via an impregnation method and the temperature-programmed reduction profiles are shown in Fig. S5.† The results for the hydrogenation of vanillin are shown in Table S3.† The results were similar to those over the Pd/C catalyst. The hydrogenation in methanol could not effectively hydrogenate the benzene ring while in water it could. The result obtained herein further confirmed that the carbon support did not play an essential role in the hydrogenation of the benzene ring in the phenolics while the solvent (water or methanol) did. In addition, hydrogenation of the benzene ring possibly needs the co-operation of several metal sites. The higher affinity of the phenolics for the catalyst in water medium would facilitate the hydrogenation of the benzene ring.
3.5 Hydrogenation of p-cresol, o-cresol, m-cresol and their derivatives
Hydrogenation of typical phenolics such as p-cresol was also preformed in methanol and in water medium, as shown in Fig. S6.† In methanol, the conversions of p-cresol and m-cresol were much higher than that of o-cresol. The fully hydrogenated product, methyl cyclohexanol, was the initial main product, but it was soon converted to the corresponding ethers in methanol and further to methyl cyclohexane. Although the conversion of o-cresol was lower, the relative proportion of 2-methyl-cyclohexanone and its acetal was much higher (18.7%) than that in p-cresol (2.3%) and m-cresol (3.7%). 2-Methyl-cyclohexanone was produced via the partial hydrogenation of the benzene ring and the subsequent tautomerisation. The adjacent methyl group negatively affected the hydrogenation of 2-methyl-cyclohexanone in two ways. Firstly, in the enol form of 2-methyl-cyclohexanone, the electrons in the π bonds could delocalize to the methyl group due to hyperconjugation, which made it more stable. Secondly, the methyl group also created some steric hindrance for the hydrogenation of the ketones.In water, steric hindrance was not an issue. Even phenolics such as 3,4,5-trimethylphenol with four substituted functionalities could be effectively hydrogenated (Fig. S3†). Other bigger phenolics such as 4-ethyl guaiacol, 2-methoxyl-4-propyl phenol, and 4-sec-butylphenol could also be fully converted via hydrogenation. However, stereoselectivity was encountered in the hydrogenation of other phenolics.
3.6 Hydrogenation of 3,4,5-trimethoxybenzaldehyde, syringaldehyde and other phenolics
During the hydrogenation of vanillin, guaiacol or other substituted phenols, isomers were always formed (Fig. S7 and S8†). The molecules could access the metal sites via different orientations and the hydrogenation could proceed via multiple adsorption–desorption steps, resulting in the formation of products with different steric configurations. However, during the hydrogenation of “heavier” phenolics such as 3,4,5-trimethoxybenzaldehyde and syringaldehyde, the product distribution did not follow this rule (Fig. S9 and S10†).Similar to the case of vanillin, the carbonyl group in 3,4,5-trimethoxybenzaldehyde could be completely hydrogenated. Nevertheless, hydrogenation of the benzene ring was difficult in methanol (Fig. 7). However, one remarkable difference was that the hydrogenation of C63 produced C64 with only a cis-configuration. No other isomers of C64 were detected although subsequent products such as C65 and C66 still had isomers. The hydrogenation of syringaldehyde, 1,2,4-trimethoxybenzene and 3,4,5-trimethoxy toluene in either water or in methanol initially produced the fully hydrogenated product with only a cis-configuration. The common feature of these “heavier” phenolics is that they all have three oxygen-containing functionalities in the form of either –OH or –OCH3, which is the intrinsic reason for the stereoselectivity. The adsorption–hydrogenation–desorption of the reactants was probably finalized in one step, leaving no chance for isomer formation.
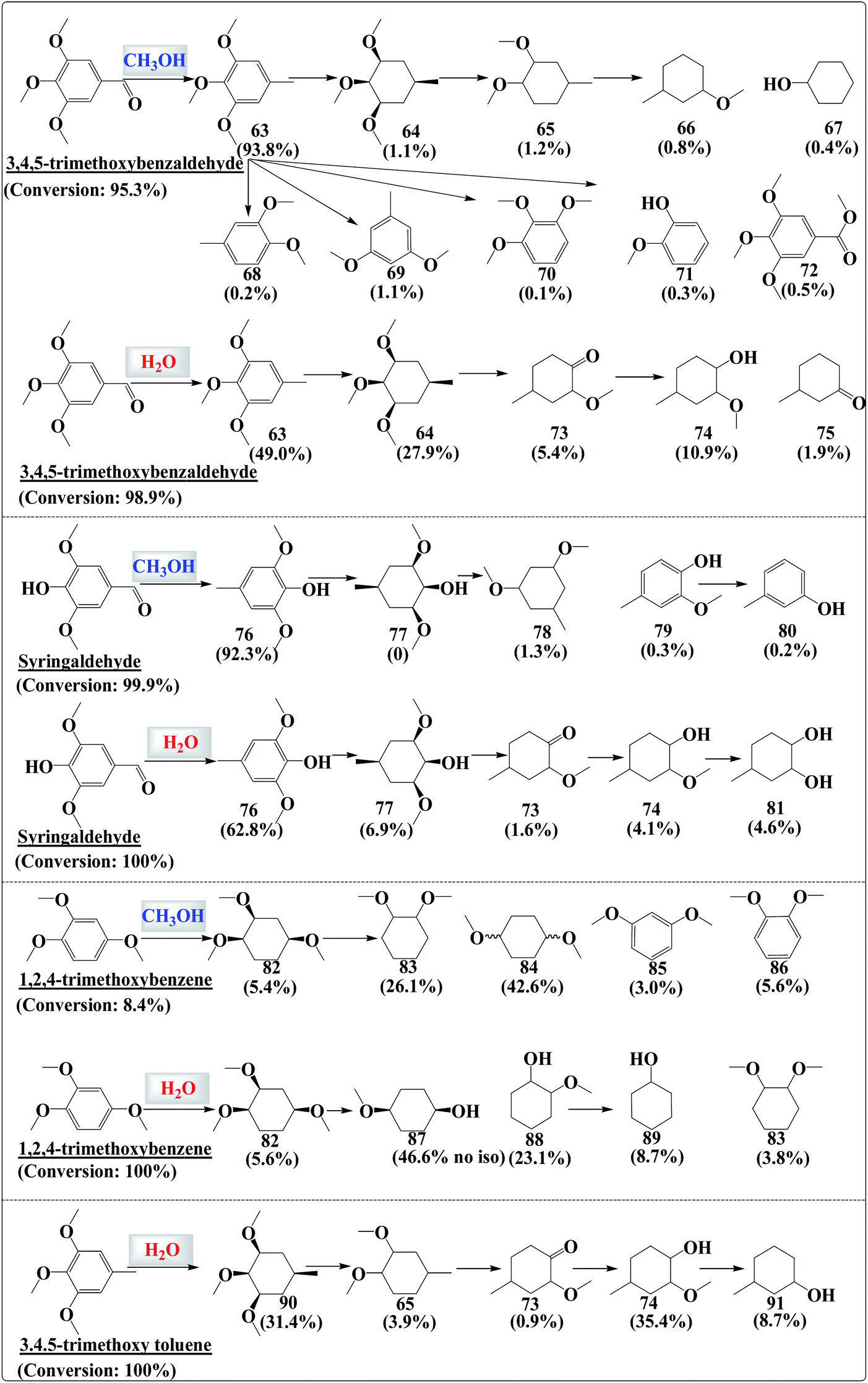 | ||
| Fig. 7 Distribution of the products in the hydrogenation of the phenolics in methanol and in water. The experimental conditions were the same as those in Fig. 2. | ||
If the molecules contained one or two oxygen-containing functionalities, isomers would be formed even if the molecules had three to four substituted functionalities (such as 4-ethyl guaiacol and 3,4,5-trimethylphenol). It needs to be noted here that some compounds such as vanillin also contain three oxygen-containing functionalities (hydroxyl group, methoxy group, and carbonyl group). Nevertheless, the carbonyl group in vanillin could be readily hydrogenated and eliminated either in methanol or in water medium. The resulting product, 2-methoxy-4-methyl-phenol, still has two oxygen-containing groups and its hydrogenation produced isomers (Fig. 2).
Activated carbon is the support for the Pd/C catalyst, and has many surface functionalities such as hydroxyl groups and carboxyl groups.30 The oxygen-containing functionalities of the phenolics can form hydrogen bonds with the hydroxyl groups and/or carboxyl groups on the activated carbon surface, as shown in Fig. 8. If the phenolics contain two oxygen-containing functionalities, and they form two hydrogen bonds on the catalyst surface (anchor sites), the benzene ring could still rotate backwards and forwards. This means that the compound could access the metal sites from two directions (Fig. 8). However, if three oxygen-containing functionalities could bind to the surface of the activated carbon via hydrogen bonds, the force of the hydrogen bonds would be tripled. More importantly, the molecule would not be able to rotate easily. The benzene ring could then access the active sites from only one orientation, producing the product with only one cis-configuration. Thus, stereoselectivity is an important factor to be considered when manufacturing fine chemicals via hydrogenation of phenolic compounds with multiple hydroxyl groups/methoxy groups.
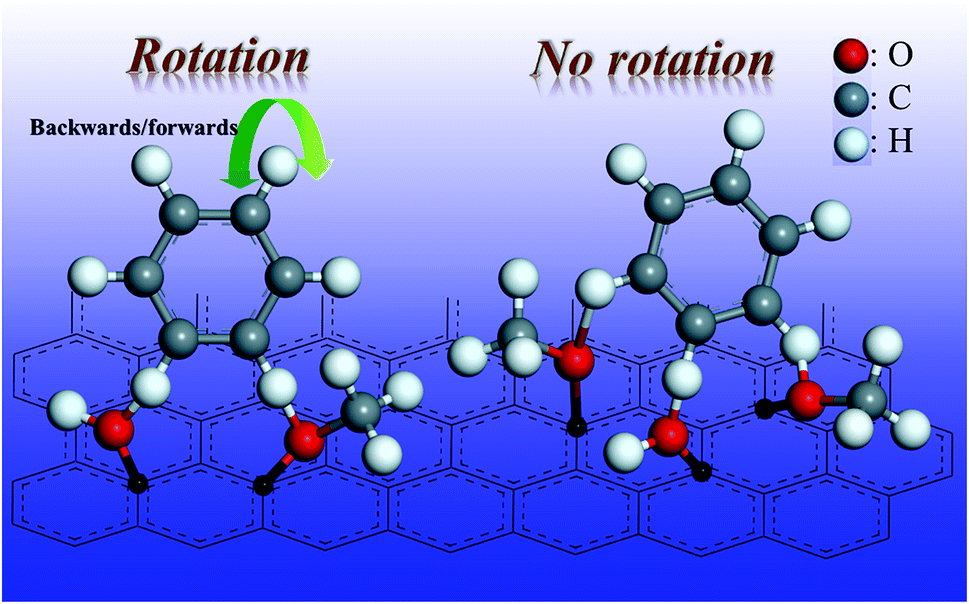 | ||
| Fig. 8 Adsorption and rotation of the phenolics on the surface of the Pd/C catalyst. | ||
4. Conclusions
In summary, the hydrogenation of phenolics in bio-oil was more efficient in water than in methanol medium, since in methanol hydrogenation of the benzene ring of the phenolics was relatively difficult. The hydrogenation of phenolics with three or more oxygen-containing functionalities (i.e. –OCH3 or –OH, but not –CO) resulted in the formation of a product with only a cis-configuration. If the phenolics contain two oxygen-containing functionalities, then the hydrogenation product would have both cis- and trans-configurations. The distinct reactivity of the phenolics towards hydrogenation in water and in methanol and stereoselectivity have to be considered during upgrading of the phenolics in bio-oil to value-added chemicals and biofuels.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Strategic International Scientific and Technological Innovation Cooperation Special Funds of the National Key R&D Program of China (No. 2016YFE0204000), the Program for Taishan Scholars of Shandong Province Government, the Recruitment Program of Global Young Experts (Thousand Youth Talents Plan) and the Natural Science Fund of Shandong Province (ZR2017BB002).References
- D. Mohan, C. U. Pittman Jr and P. H. Steele, Pyrolysis of wood/biomass for bio-oil: a critical review, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- D. Carpenter, T. L. Westover, S. Czernik and W. Jablonski, Biomass feedstocks for renewable fuel production: a review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors, Green Chem., 2014, 16, 384–406 RSC.
- J. C. Manayil, C. V. M. Inocencio, A. F. Lee and K. Wilson, Mesoporous sulfonic acid silicas for pyrolysis bio-oil upgrading via acetic acid esterification, Green Chem., 2016, 18, 1387–1394 RSC.
- M. Zhou, P. Liu, K. Wang, J. Xu and J. Jiang, Catalytic hydrogenation and one step hydrogenation-esterification to remove acetic acid for bio-oil upgrading: model reaction study, Catal. Sci. Technol., 2016, 6, 7783–7792 CAS.
- J. A. Melero, J. Iglesias and A. Garcia, Biomass as renewable feedstock in standard refinery units. Feasibility, opportunities and challenges, Energy Environ. Sci., 2012, 5, 7393–7420 CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, A review and perspective of recent bio-oil hydrotreating research, Green Chem., 2014, 16, 491–515 RSC.
- M. Patel and A. Kumar, Production of renewable diesel through the hydroprocessing of lignocellulosic biomass-derived bio-oil: a review, Renewable Sustainable Energy Rev., 2016, 58, 1293–1307 CrossRef CAS.
- J. Horáček and D. Kubička, Bio-oil hydrotreating over conventional CoMo & NiMo catalysts: the role of reaction conditions and additives, Fuel, 2017, 15, 49–57 CrossRef.
- L. J. Snowden-Swan, K. A. Spies, G. J. Lee and Y. Zhu, Life cycle greenhouse gas emissions analysis of catalysts for hydrotreating of fast pyrolysis bio-oil, Biomass Bioenergy, 2016, 86, 136–145 CrossRef CAS.
- M. R. Rover, P. H. Hall, P. A. Johnston, R. G. Smith and R. C. Brown, Stabilization of bio-oils using low temperature, low pressure hydrogenation, Fuel, 2015, 153, 224–230 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Highly selective catalytic conversion of phenolic bio-oil to alkanes, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- S. Fehn, M. Zaheer, C. E. Denner, M. Friedrich and R. Kempe, Robustly supported rhodium nanoclusters: synthesis and application in selective hydrogenation of lignin derived phenolic compounds, New J. Chem., 2016, 40, 9252–9256 RSC.
- S. Shylesh, A. A. Gokhale, K. Sun, A. M. Grippo, D. Jadhav, A. Yeh, C. R. Ho and A. T. Bell, Integrated catalytic sequences for catalytic upgrading of bio-derived carboxylic acids to fuels, lubricants and chemical feedstocks, Sustainable Energy Fuels, 2017, 1, 1805–1809 CAS.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Solvent free depolymerization of Kraft lignin to alkyl-phenolics using supported NiMo and CoMo catalysts, Green Chem., 2015, 17, 4921–4930 RSC.
- N. Siddiqui, A. S. Roy, R. Goyal, R. Khatun, C. Pendem, A. N. Chokkapu, A. Bordoloi and R. Bal, Hydrogenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran over nickel supported tungsten oxide nanostructured catalyst, Sustainable Energy Fuels, 2018, 2, 191–198 CAS.
- S. De, B. Saha and R. Luque, Hydrodeoxygenation processes: advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- M. S. Talmadge, R. M. Baldwin, M. J. Biddy, R. L. McCormick, G. T. Beckham, G. A. Ferguson, S. Czernik, K. A. Magrini-Bair, T. D. Foust, P. D. Metelski, C. Hetrick and M. R. Nimlos, A perspective on oxygenated species in the refinery integration of pyrolysis oil, Green Chem., 2014, 16, 407–453 RSC.
- H. Wang, J. Male and Y. Wang, Recent advances in hydrotreating of pyrolysis bio-oil and its oxygen-containing model compounds, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- X. Hu and C.-Z. Li, Levulinic esters from the acid-catalysed reactions of sugars and alcohols as part of a bio-refinery, Green Chem., 2011, 13, 1676–1679 RSC.
- S. Ren, X. P. Ye and A. P Borole, Separation of chemical groups from bio-oil water-extract via sequential organic solvent extraction, J. Anal. Appl. Pyrolysis, 2017, 123, 30–39 CrossRef CAS.
- A. Kruse and N. Dahmen, Water–a magic solvent for biomass conversion, J. Supercrit. Fluids, 2015, 96, 36–45 CrossRef CAS.
- D. A. Cantero, Á. S. Tapia, M. D. Bermejo and M. J. Cocero, Pressure and temperature effect on cellulose hydrolysis in pressurized water, Chem. Eng. J., 2015, 276, 145–154 CrossRef CAS.
- X. Hu, R. Gunawan, D. Mourant, M. D. M. Hasan, L. Wu, Y. Song, C. Lievens and C.-Z. Li, Upgrading of bio-oil via acid-catalyzed reactions in alcohols—a mini review, Fuel Process. Technol., 2017, 155, 2–19 CrossRef CAS.
- M. Milina, S. Mitchell and J. Pérez-Ramírez, Prospectives for bio-oil upgrading via esterification over zeolite catalysts, Catal. Today, 2014, 235, 176–183 CrossRef CAS.
- T. Sundqvist, A. Oasmaa and A. Koskinen, Upgrading fast pyrolysis bio-oil quality by esterification and azeotropic water removal, Energy Fuels, 2015, 29, 2527–2534 CrossRef CAS.
- Y. I. Tarasevich, S. V. Bondarenko and A. I. Zhukova, Gas-chromatographic studies of the interaction between water and methanol molecules and the surface of carbon materials, Adsorption, 2005, 11, 385–391 CrossRef CAS.
- R. Tsunoda, Adsorption of water vapor on active carbon preadsorbed with methanol and benzene, J. Colloid Interface Sci., 1997, 188, 224–228 CrossRef CAS.
- D. W. Lee, C. Y. Yu and K. H. Lee, Competitive adsorption-driven separation of water/methanol mixtures using hydrogen as a third competitor, J. Colloid Interface Sci., 2009, 340, 62–66 CrossRef CAS PubMed.
- A. V. Shevade, S. Jiang and K. E. Gubbins, Molecular simulation study of water–methanol mixtures in activated carbon pores, J. Chem. Phys., 2000, 113, 6933 CrossRef CAS.
- P. J. M. Carrott and M. M. L. R. Carrott, Lignin–from natural adsorbent to activated carbon: a review, Bioresour. Technol., 2007, 98, 2301–2312 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00006a |
| This journal is © The Royal Society of Chemistry 2018 |