DOI:
10.1039/C7SC05009G
(Edge Article)
Chem. Sci., 2018,
9, 3165-3176
Triplet state homoaromaticity: concept, computational validation and experimental relevance†
Received
22nd November 2017
, Accepted 16th February 2018
First published on 19th February 2018
Abstract
Cyclic conjugation that occurs through-space and leads to aromatic properties is called homoaromaticity. Here we formulate the homoaromaticity concept for the triplet excited state (T1) based on Baird's 4n rule and validate it through extensive quantum-chemical calculations on a range of different species (neutral, cationic and anionic). By comparison to well-known ground state homoaromatic molecules we reveal that five of the investigated compounds show strong T1 homoaromaticity, four show weak homoaromaticity and two are non-aromatic. Two of the compounds have previously been identified as excited state intermediates in photochemical reactions and our calculations indicate that they are also homoaromatic in the first singlet excited state. Homoaromaticity should therefore have broad implications in photochemistry. We further demonstrate this by computational design of a photomechanical “lever” that is powered by relief of homoantiaromatic destabilization in the first singlet excited state.
Introduction
Excited-state aromaticity is a concept describing the energetic stabilization of annulenes with 4n π electrons in the lowest ππ* electronically excited states of singlet (S1) and/or triplet (T1) multiplicity. Originally conceived on a theoretical basis by Baird in 1972![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 based on preliminary work by Dewar and Zimmerman,2 the concept has lately been applied experimentally to rationalize various excited state properties and reactivity.3 The findings on excited-state aromaticity are now conveniently summarized in Baird's rule: 4n π-electron annulenes are aromatic in their S1 and T1 states while (4n + 2)π-electron annulenes are antiaromatic. Thus, Baird's rule is the excited state counterpart of Hückel's rule for the electronic ground state (S0), yet, the electron counts for aromaticity and antiaromaticity in the T1 and S1 states are the exact opposite to those in the S0 state. Excited state aromaticity has been used to explain acid–base properties of polycyclic conjugated hydrocarbons,4 excitation energies of substituted fulvenes,5 and spectroscopic properties of expanded porphyrinoids.6–8 Recently, the concept has been applied to the development of new photochemical reactions such as the formation of benzofulvenes from enynes,9 and photohydrogenation and photo(hydro)silylation of small polycyclic aromatic hydrocarbons and graphene.10 Large conformational changes have been observed upon excitation of annulenes, leading to planarization for excited-state aromatic and puckering for antiaromatic molecules.7,11
1 based on preliminary work by Dewar and Zimmerman,2 the concept has lately been applied experimentally to rationalize various excited state properties and reactivity.3 The findings on excited-state aromaticity are now conveniently summarized in Baird's rule: 4n π-electron annulenes are aromatic in their S1 and T1 states while (4n + 2)π-electron annulenes are antiaromatic. Thus, Baird's rule is the excited state counterpart of Hückel's rule for the electronic ground state (S0), yet, the electron counts for aromaticity and antiaromaticity in the T1 and S1 states are the exact opposite to those in the S0 state. Excited state aromaticity has been used to explain acid–base properties of polycyclic conjugated hydrocarbons,4 excitation energies of substituted fulvenes,5 and spectroscopic properties of expanded porphyrinoids.6–8 Recently, the concept has been applied to the development of new photochemical reactions such as the formation of benzofulvenes from enynes,9 and photohydrogenation and photo(hydro)silylation of small polycyclic aromatic hydrocarbons and graphene.10 Large conformational changes have been observed upon excitation of annulenes, leading to planarization for excited-state aromatic and puckering for antiaromatic molecules.7,11
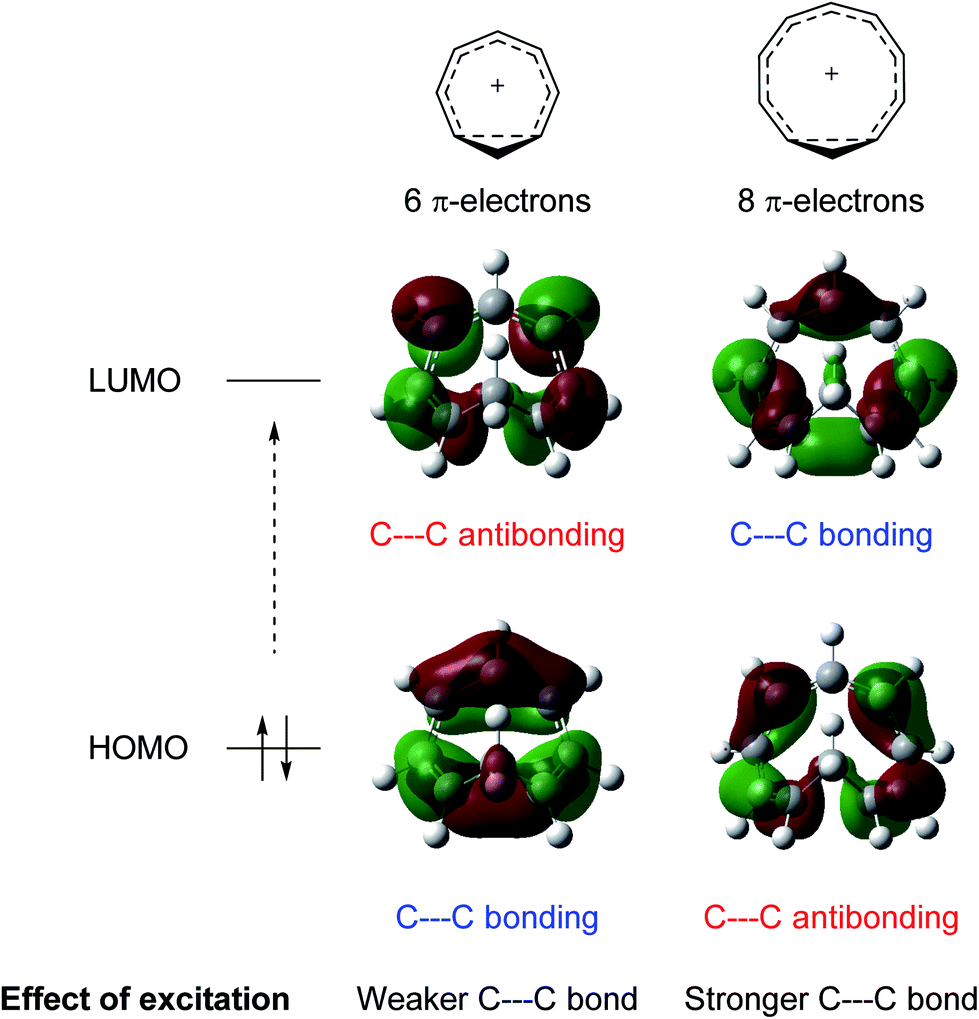
While excited-state aromaticity is a powerful concept for understanding properties and reactivity following excitation, its influence has almost exclusively been considered for conventional planar annulenes. One exception concerns Möbius aromaticity, where computations predicted that excited (4n)π-electron annulenes should be antiaromatic and (4n + 2)π-electron annulenes aromatic,12 and this was recently confirmed experimentally.8,13 In this study, we focus on homoaromaticity, i.e., aromaticity due to interaction of π orbitals over a formally saturated center,14 and show that this concept is applicable also in the excited triplet state. This can actually be anticipated from a simple analysis of the HOMO and LUMO orbitals of the aromatic homotropylium cation with six π-electrons and its larger analogue with eight π-electrons. Excitation should decrease the through-space conjugation of the six-electron system and enhance that of the eight-electron system, as electrons are excited from a bonding to an antibonding orbital for the former and from an antibonding to a bonding orbital for the latter (Fig. 1).
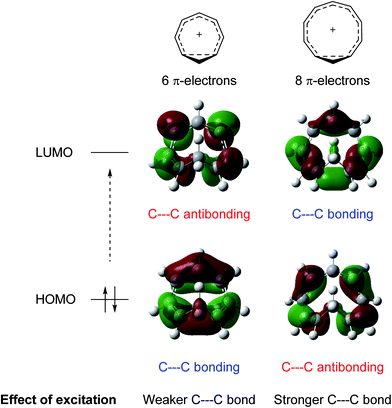 |
| | Fig. 1 Effect of excitation on cyclically homoconjugated systems with six and eight π-electrons. Molecular orbitals obtained at the B2PLYP/6-311+G(d,p) level (isosurface value = 0.02). | |
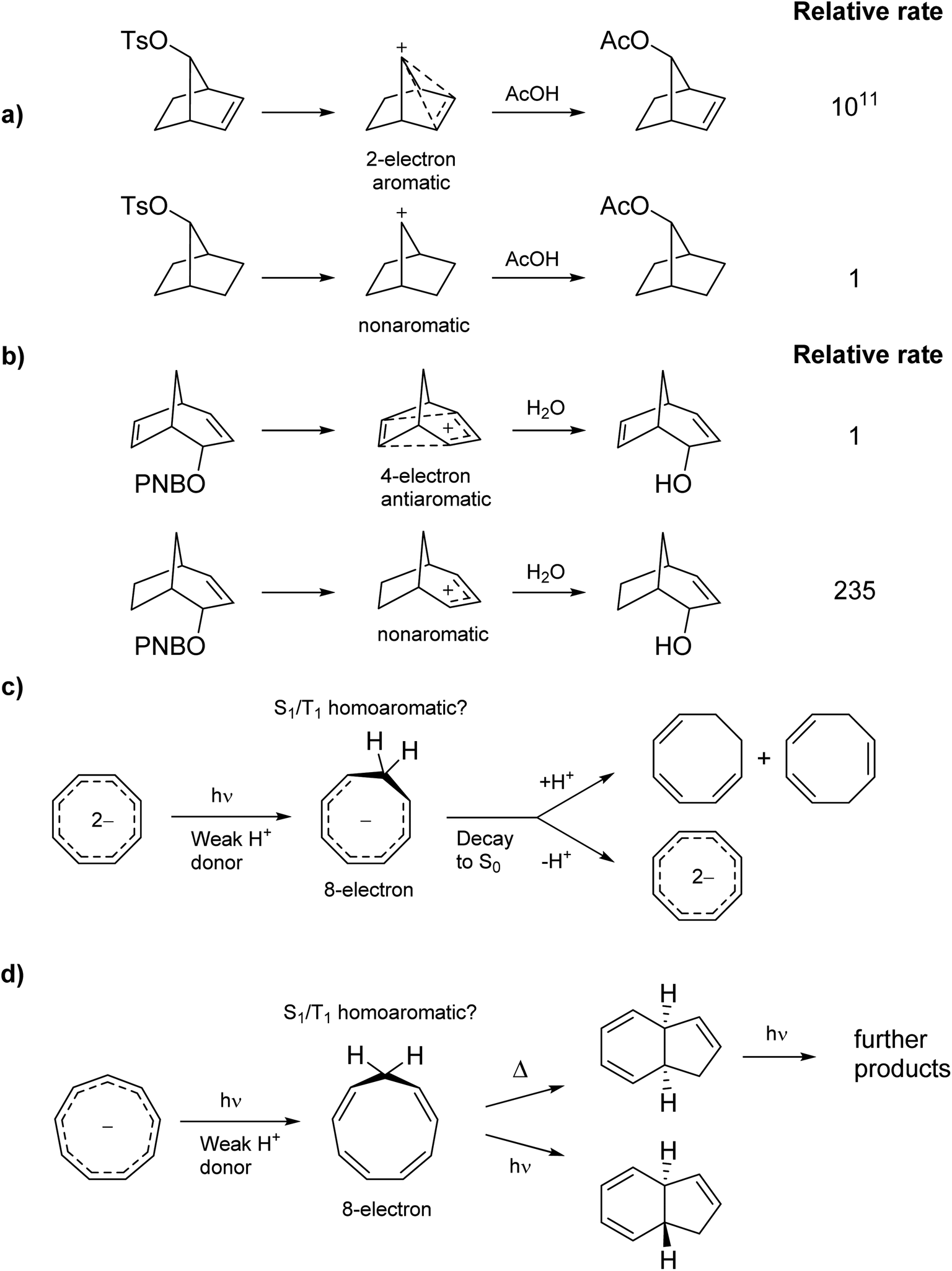
Homoaromaticity was coined by Winstein in 1959 to explain structures of non-classical carbocations.15 It refers to the appearance of aromatic properties through conjugation over saturated centers. One example is found in the acetolysis of the p-toluenesulfonate of 7-norborneol in which the cationic center of the intermediate conjugates through space with a double bond, creating a 2-electron aromatic cycle (Scheme 1a). The special stability of this cation is reflected in the rate enhancement of 1011 compared to the saturated analogue. Conversely, the special instability of homoantiaromatic 4π-electron cations was shown in the solvolysis of bicyclo[3.2.l]octa-2,6-dienyl p-nitrobenzoates where the saturated species reacted 235 times faster than the unsaturated (Scheme 1b).16,17 Perhaps the most well-known homoaromatic molecule is the homotropylium cation, which can be formed by protonation of cyclooctatetraene (COT) in strong acids such as H2SO4.18 Experimentally, homoaromaticity is also important for understanding the properties of substituted fullerenes19 and many inorganic molecules and clusters.20
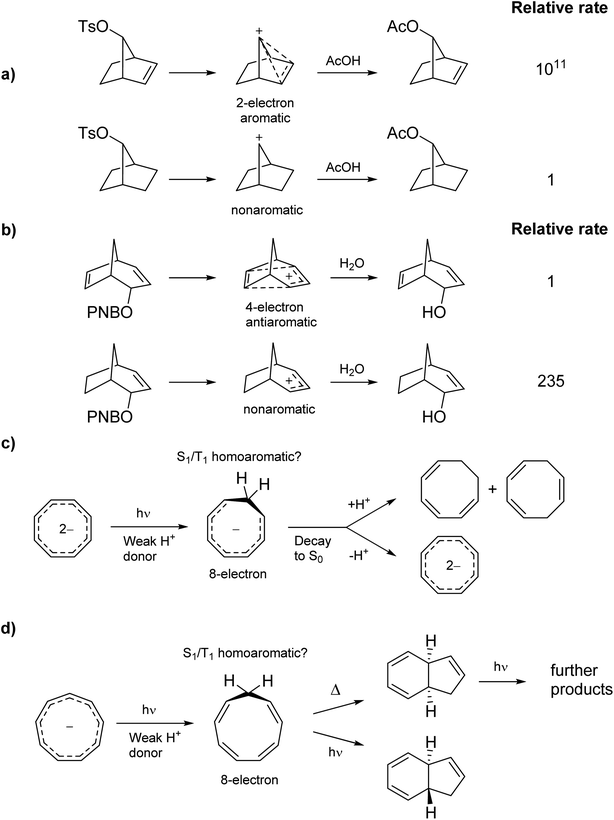 |
| | Scheme 1 Ground state (a) homoaromatic stabilization and (b) homoantiaromatic destabilization. Tentatively excited state homoaromatic intermediates (c and d). | |
While it is established that homo(anti)aromaticity can influence ground state reactions, its effect in the excited state is completely unexplored. However, we now postulate that many photochemical reactions can be influenced by homoaromaticity in S1/T1. For example, the photo-acidity of the cyclooctatetraene dianion is enhanced in the excited state, leading to the protonated cyclooctatrienyl anion as an excited state intermediate (Scheme 1c).21 Is this species excited-state homoaromatic? A similar reaction concerns the cyclononatetraenide anion which displays increased basicity in the excited state, being protonated to give cyclononatetraene as an intermediate to further products (Scheme 1d).22 In our earlier study of the norbornadiene–quadricyclane photo-switch system, we computationally found an excited-state minimum that we described as excited-state homoaromatic.23 These three examples indicate that excited state homoaromaticity may influence the outcome of photochemical reactions by stabilizing excited state intermediates. Now, through an extensive investigation of a number of a different species, possible in a facile manner only through quantum chemical computations, we find that homoaromaticity is indeed valid also for the excited state. A thorough understanding of excited state homoaromaticity could lead to the development of novel photochemical reactions, tuning of photophysical properties by appropriate choice of substrates and substituents, and design of new optically active molecular machinery.
Results and discussion
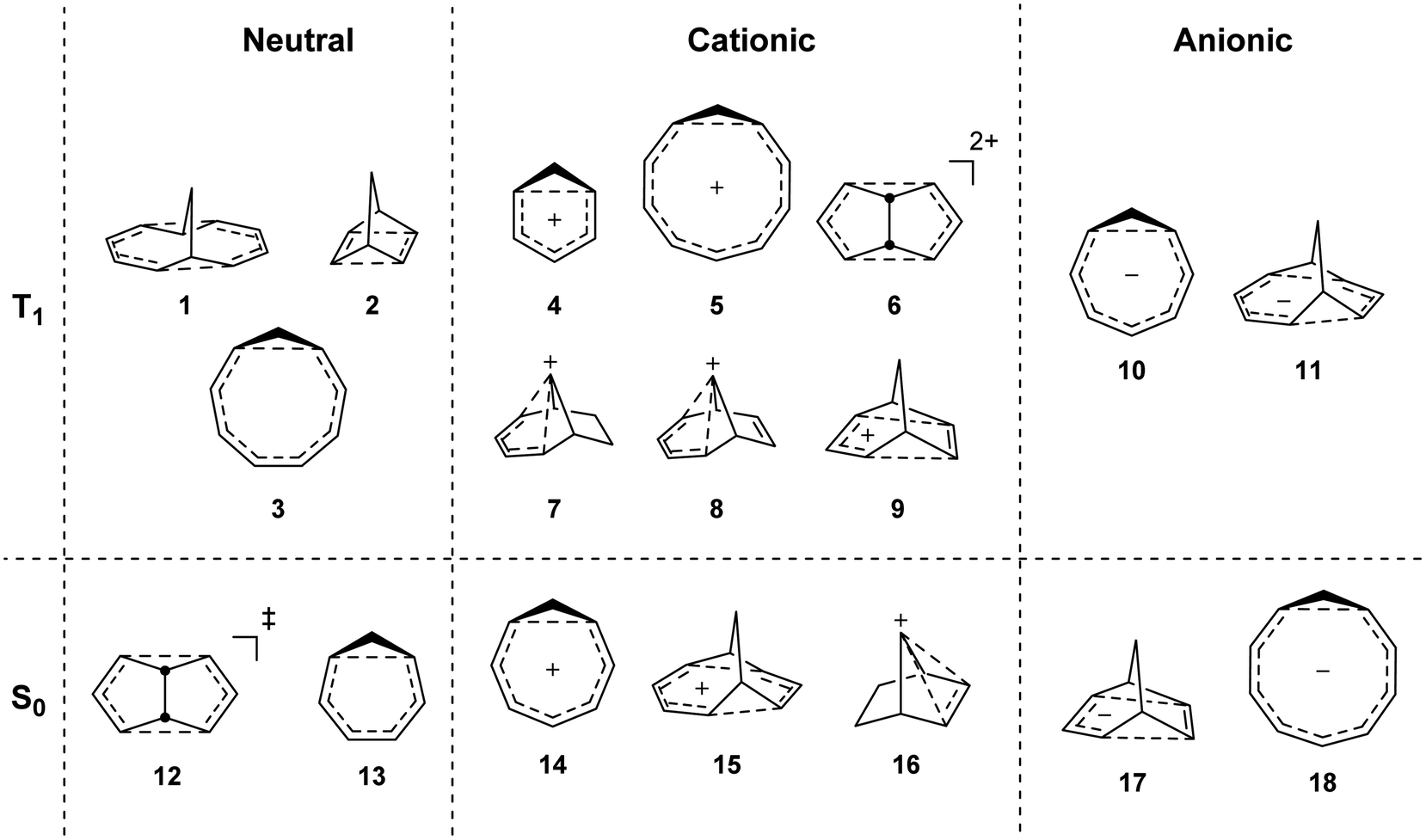
To investigate if homoaromaticity is influential in the T1 state, we computationally analyzed a series of neutral, cationic and anionic compounds (Fig. 2). We compared possible T1-homoaromatic structures (1–11) with compounds that are either experimentally established homoaromatics in S0 (12–16)14,24,25 or those whose homoaromaticity is so far only supported computationally (17–18).26,27 Although 12 is a transition state and not a stable compound, there have been extensive attempts to make derivatives in which this homoaromatic structure is a true energy minimum.28 While the T1-homoaromatic compounds all contain 4n π-electrons in the conjugated cycle, the S0 homoaromatic compounds contain (4n + 2)π-electrons.
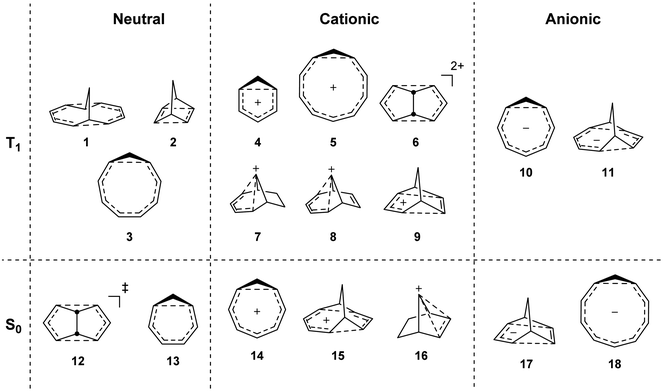 |
| | Fig. 2 Studied compounds in T1 (1–11) and in S0 (12–17). | |
Any rigorous assessment of homoaromaticity should include several different aspects.14 For the geometric aspect, we analyze the degree of the bond length alternation (BLA) and the distance of the though-space conjugative linkages (r(C⋯C)). For the electronic aspect, we look at the strength of the through-space and cyclic conjugation through the Wiberg bond indices29 and the multicenter indices,30 respectively. For the magnetic aspect, we use the anisotropy of the induced current density (ACID) plots31 and nucleus-independent shift (NICS)32 scans,33 and for the energetic aspect we look at aromatic stabilization energies using the ISE method.34 For the charged compounds we analyze charge delocalization and for the triplet state compounds also spin delocalization. We take care to compare the neutral, cationic and anionic compounds in S0 and T1 within each charge class as in the S0 state it is known that homoaromaticity is stronger in cationic than neutral or anionic compounds.14 Finally, we show how homoaromaticity could influence the photochemistry when extending the results to the S1 excited state.
Geometries
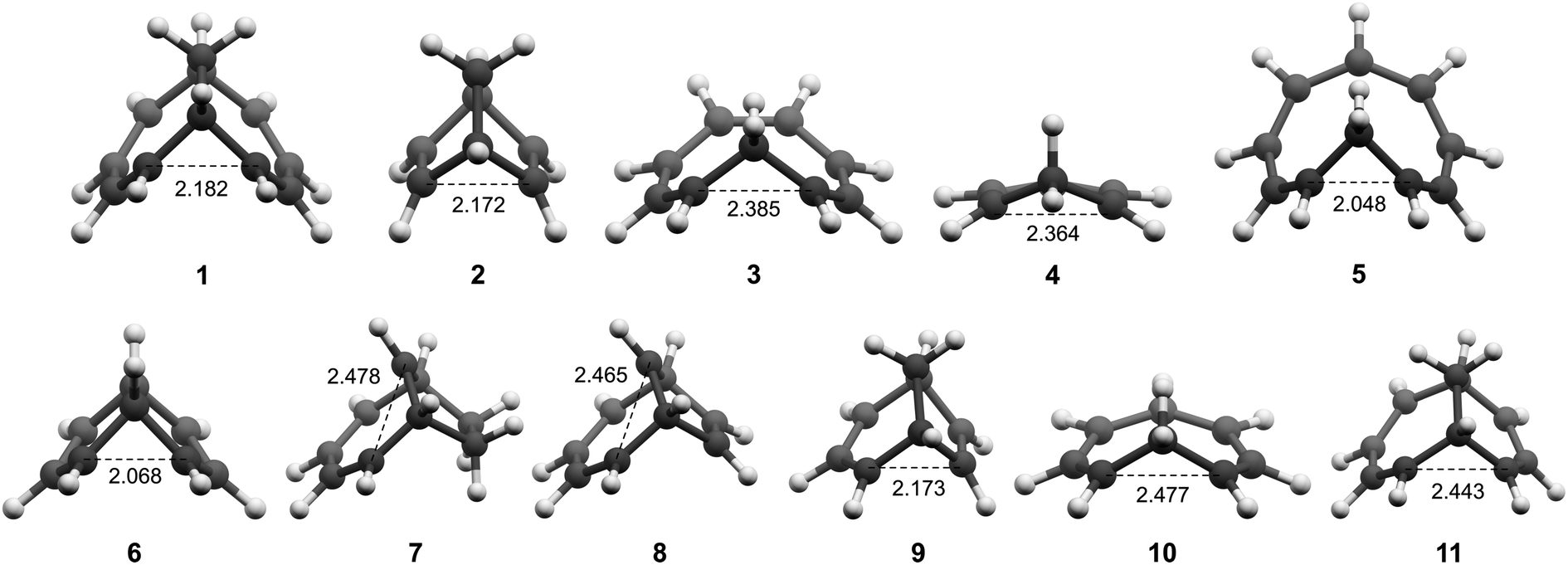
All molecules were optimized at the B2PLYP/6-311+G(d,p) level, and here we include only the homoaromatic structures for each molecule. For the bicyclic systems 1, 2, 7 and 8, we have found other conformers which are of similar or lower energy (see ESI,† Sections 5 and 12). For 5, we found two homoaromatic conformers. Here we discuss only the one which is lowest in energy. The optimized structures are non-planar with short r(C⋯C) in the range 2.048–2.478 Å (Fig. 3). Note that non-planarity does not preclude aromaticity as exemplified by 14, which is puckered and known to be strongly aromatic (see ESI,† Section 5). In particular, the optimized T1-structures of 1–11 show greater planarization and shorter r(C⋯C) in T1 than their optimized structures in S0.
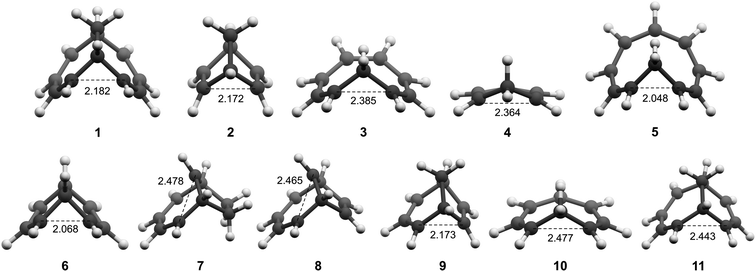 |
| | Fig. 3 Optimized structures of 1–11 in T1. | |
Bond length alternation
A low BLA is hallmark of both conventional aromaticity35 and homoaromaticity.14 The average and maximum BLAs (BLAavg and BLAmax, respectively) are given in Table 1 for the sp2-carbon framework of the monocyclic compounds. The BLAs for the T1 compounds are low and comparable in size to those in the S0 homoaromatic compounds (homoaromatics). Neutral 3 has a BLAavg of 0.045 Å and a BLAmax of 0.070 Å, which are even smaller than the 0.085 Å and 0.092 Å for 13 in S0. Among the charged T1 species, BLAavg and BLAmax are in the ranges 0.005–0.014 Å for cationic and 0.009–0.022 Å for anionic species, which is equal or lower to that of the established S0 homoaromatics (0.003–0.017 Å and 0.030–0.059 Å, respectively). Thus, the BLAs in the T1 monocyclic compounds are comparable or smaller than for the S0 homoaromatics, supporting their T1 homoaromaticity.
Table 1 Average and maximum bond length alternation (BLA) in Å for the monocyclic compounds. Results at the B2PLYP/6-311+G(d,p) level
| Compound |
Charge |
Electronic state |
BLAavg |
BLAmax |
|
3
|
0 |
T1 |
0.045 |
0.070 |
|
4
|
+1 |
T1 |
0.009 |
0.009 |
|
5
|
+1 |
T1 |
0.005 |
0.014 |
|
10
|
−1 |
T1 |
0.009 |
0.022 |
|
13
|
0 |
S0 |
0.085 |
0.092 |
|
14
|
+1 |
S0 |
0.003 |
0.017 |
|
18
|
−1 |
S0 |
0.030 |
0.059 |
C⋯C homoconjugative distances
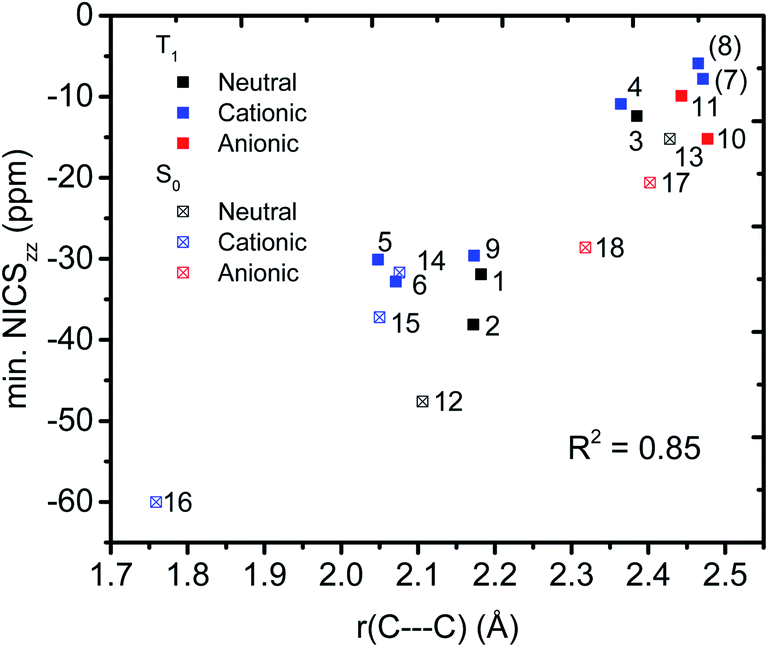
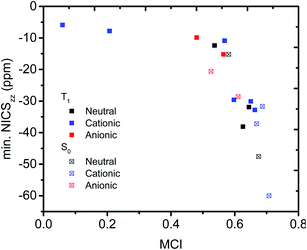
We then evaluated the r(C⋯C) distances as a measure of the strength of the homoconjugation, with shorter distances being expected for stronger homoconjugation. In Fig. 4 we plot r(C⋯C) against minimum NICSzz values along the NICS scan as a magnetic measure of aromaticity. A clear correlation is observed with both S0 and T1 and cationic compounds having shorter r(C⋯C) and more negative NICSzz than anionic or neutral compounds. Still, for all compounds r(C⋯C) is smaller than the sum of two carbon van der Waals radii (3.40 Å).36 The T1 compounds are fully comparable to their S0 analogues, with stronger conjugation and homoaromaticity expected for 1, 2, 5, 6 and 9 and weaker conjugation and homoaromaticity expected for 3, 4, 7, 8, 10 and 11. The exceptions are 7 and 8 that show much larger r(C⋯C) than their closest homoaromatic analogue in S0 (16).
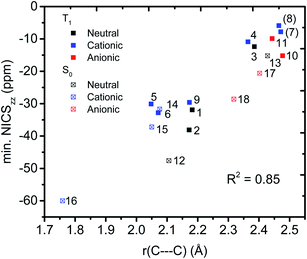 |
| | Fig. 4 Correlation between r(C⋯C) and minimum NICSzz values along the NICS scan. | |
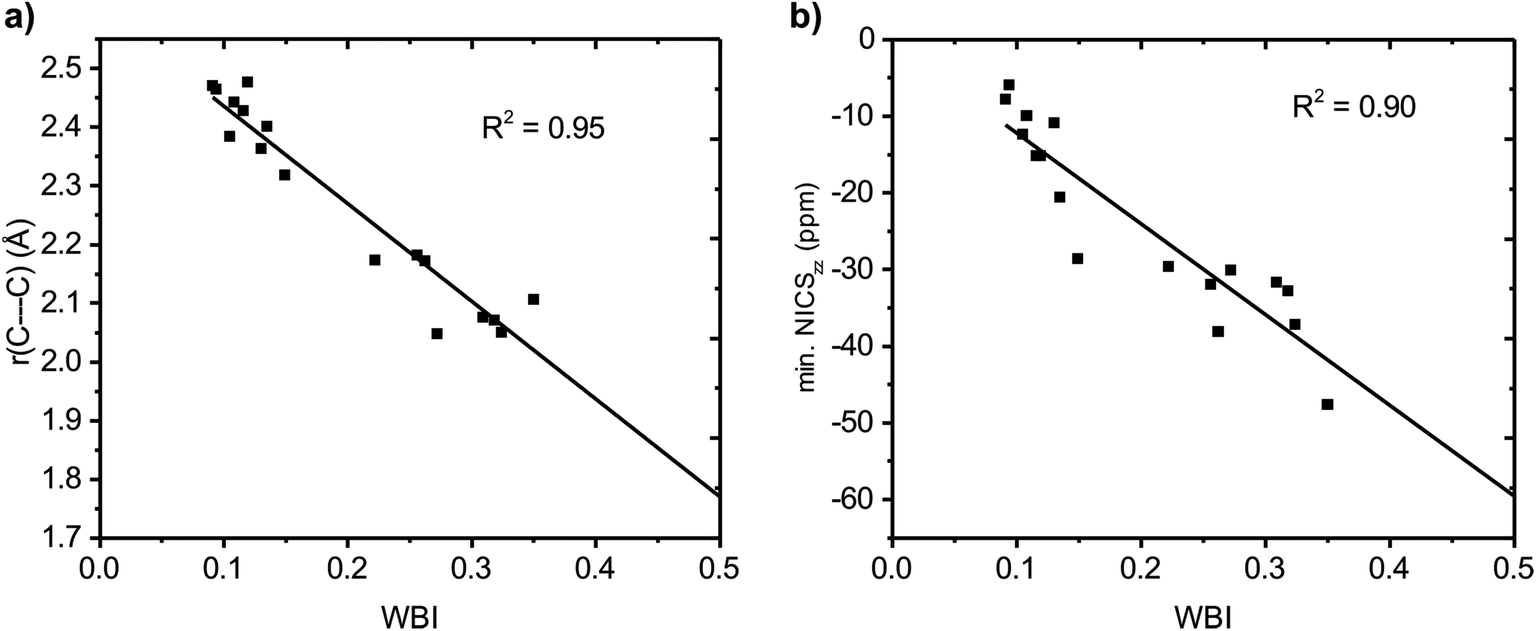
Wiberg bond indices
We further quantified the strength of the through-space conjugation by the Wiberg bond indices (WBI).29 The WBI values (Table 2) correlate well with r(C⋯C) for all compounds (1–18), showing that smaller r(C⋯C) are indeed associated with increased through-space conjugation (Fig. 5a).
Table 2 C⋯C distances, Wiberg bond indices, MCI, standard deviation of NPA charges and spin densities for C atoms (with attached hydrogens) in the homoaromatic circuit, ring current directions according to the ACID plots and minimum NICS values
|
|
State |
Charge |
r(C⋯C)a (Å) |
WBIa |
MCIa |
σ
Q
|
σ
SD
|
Ring currentb |
Min. NICSb |
|
At the B2PLYP/6-311+G(d,p) level.
At the B3LYP/6-311+G(d,p)//B2PLYP/6-311+G(d,p) level.
|
|
1
|
T1 |
0 |
2.182 |
0.256 |
0.645 |
— |
0.115 |
Diatropic |
−31.9 |
|
2
|
T1 |
0 |
2.172 |
0.262 |
0.626 |
— |
0.000 |
Diatropic |
−38.1 |
|
3
|
T1 |
0 |
2.385 |
0.105 |
0.537 |
— |
0.363 |
Diatropic |
−12.4 |
|
4
|
T1 |
+1 |
2.364 |
0.130 |
0.568 |
0.116 |
0.248 |
Not clear |
−10.9 |
|
5
|
T1 |
+1 |
2.048 |
0.272 |
0.650 |
0.084 |
0.140 |
Diatropic |
−30.1 |
|
6
|
T1 |
+1 |
2.071 |
0.318 |
0.663 |
0.044 |
0.013 |
Diatropic |
−32.8 |
|
7
|
T1 |
+1 |
2.471/2.478 |
0.091 |
0.207 |
0.138 |
0.373 |
Not clear |
−7.8 |
|
8
|
T1 |
+1 |
2.465 |
0.094 |
0.060 |
0.141 |
0.384 |
Not clear |
−5.9 |
|
9
|
T1 |
+1 |
2.173 |
0.222 |
0.597 |
0.083 |
0.222 |
Diatropic |
−29.6/−28.6 |
|
10
|
T1 |
−1 |
2.477 |
0.119 |
0.565 |
0.090 |
0.264 |
Diatropic |
−15.2 |
|
11
|
T1 |
−1 |
2.443 |
0.108 |
0.481 |
0.095 |
0.326 |
Not clear |
−9.9 |
|
12
|
S0 |
0 |
2.106 |
0.350 |
0.675 |
— |
— |
Diatropic |
−47.6 |
|
13
|
S0 |
0 |
2.428 |
0.116 |
0.581 |
— |
— |
Diatropic |
−15.2 |
|
14
|
S0 |
+1 |
2.076 |
0.309 |
0.687 |
0.074 |
— |
Diatropic |
−31.7 |
|
15
|
S0 |
+1 |
2.050 |
0.324 |
0.669 |
0.099 |
— |
Diatropic |
−37.2 |
|
16
|
S0 |
+1 |
1.759 |
0.518 |
0.708 |
0.015 |
— |
Diatropic |
−60.0 |
|
17
|
S0 |
−1 |
2.402 |
0.135 |
0.525 |
0.135 |
— |
Not clear |
−20.6 |
|
18
|
S0 |
−1 |
2.318 |
0.149 |
0.611 |
0.062 |
— |
Diatropic |
−28.6 |
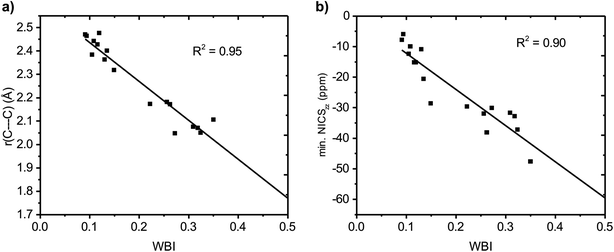 |
| | Fig. 5 Correlation between Wiberg bond index (WBI) and (a) C⋯C distance and (b) minimum NICSzz value along the NICS scan. | |
The positively charged S0 state homoaromatics have WBI values in the range 0.309–0.518, indicating very strong homoconjugation, while the anionic compounds have significantly lower values of 0.135–0.149. For comparison, π-WBIs of fully aromatic bonds lie in the range 0.220–0.428 (S0 values; see Table S1, ESI†). For neutral 13, a rather small value of 0.116 is obtained, indicating only weak homoconjugation. Among the positively charged T1 homoaromatics, 5, 6 and 9 show strong homoconjugation on par with 14 in S0. Cation 4 has moderate conjugation, while that in 7 and 8 is weak. The anionic 10 and 11 show moderate homoconjugation only slightly lower than that of 17 and 18 in S0.
Multicenter indices
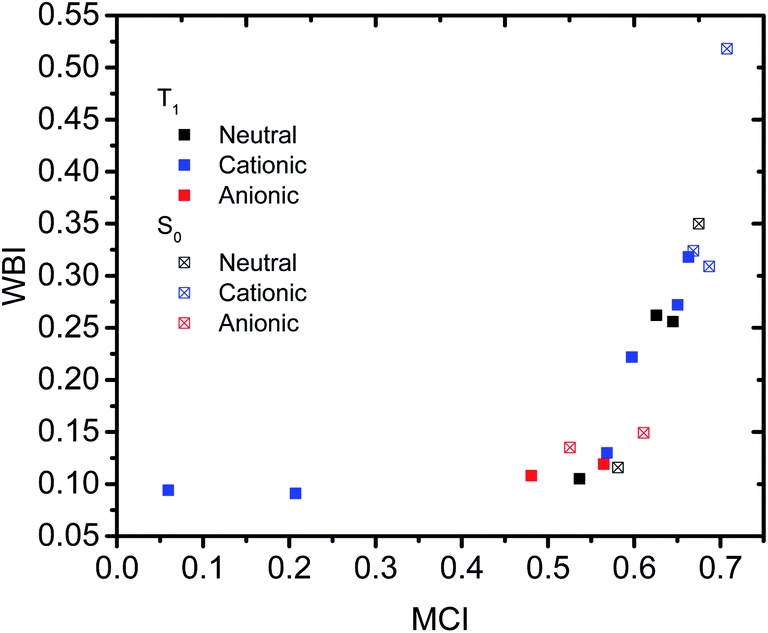
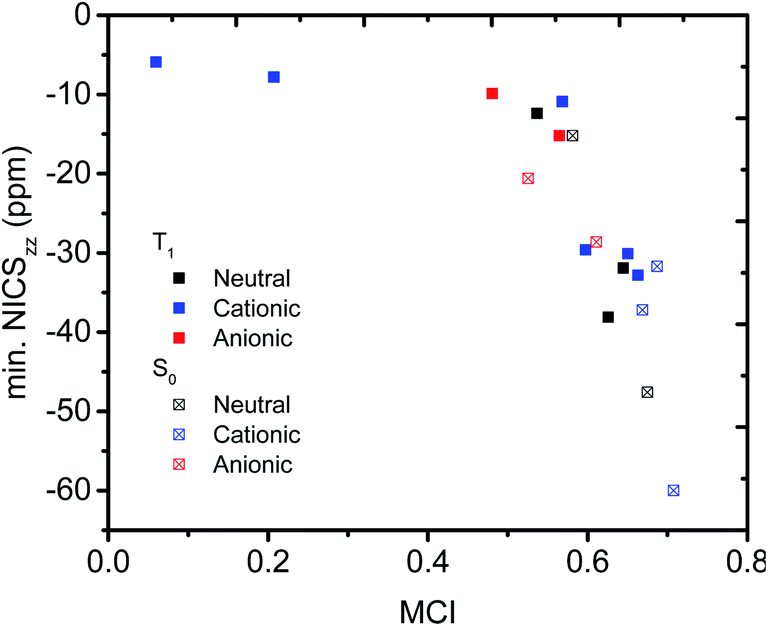
Having established that a short r(C⋯C) is indeed related to stronger through-space conjugation, as evidenced by the WBI(C⋯C), we went on and calculated the multicenter indices (MCI)30 to verify the existence of larger cyclic conjugation. The MCI quantifies the extent of delocalized cyclic bonding and has been used previously to assess homoaromatic species in the S0 state.37 To compare the homoaromaticity of rings of different size, we employed the normalization procedure by Mandado et al.38 Henceforth, MCI therefore refers to the normalized MCI index. For full MCI values, see Table S2, ESI.† The MCI values correlate with the WBI(C⋯C) ones in a non-linear fashion with no clear difference between S0 and T1 compounds (Fig. 6). However, according to MCI the aromatic character of 7 (0.207) and 8 (0.060) is much lower than the rest of studied compounds (range of 0.481–0.708) and they are best considered non-aromatic. The correlation of MCI with NICS is similarly non-linear (Fig. 7). In summary, the MCI calculations support varying extent of homoaromatic character of all compounds except 7 and 8 and allow a quantitative ordering that will be discussed further in the Conclusions section.
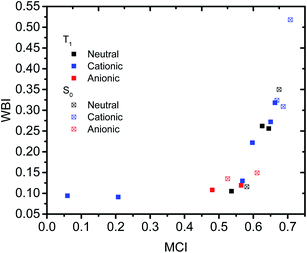 |
| | Fig. 6 Relationship between MCI and WBI(C⋯C). | |
 |
| | Fig. 7 Relationship between MCI and minimum NICSzz value along the NICS scan. | |
Charge and spin distribution
Delocalization of charge is often employed to assess the homoaromaticity of charged species in the S0 ground state.14 The prime example is the homotropylium cation (14), in which charge delocalization is evidenced by the small variation in 13C NMR shifts.39 To assess this property we calculated atomic charges using the natural population analysis (NPA) scheme40 and computed the standard deviation of the charge (σQ) for the unsaturated carbon atoms in the homoaromatic circuit. The results in Table 2 show that 4–6 and 9–11 in their T1 states have similar charge delocalization as the S0 state homoaromatics. Compounds 7 and 8 show high charge localization and should thus be only weakly homoaromatic or non-aromatic. The charge localization in 17 (σQ = 0.135) is also quite high. For the cationic species in the T1 state there is a clear correlation between σQvs. WBI(C⋯C) (Fig. S2a, ESI,†R2 = 0.93) and minimum NICSzz values (Fig. S2c, ESI,†R2 = 0.88), but if one considers all positively and negatively charged compounds in both the S0 and T1 states, the correlations become worse. The correlation between MCI and σQ is non-linear (Fig. S2b, ESI†).
For the T1 state we can also quantify the delocalization of excess spin, and we expect that more homoaromatic molecules show larger spin delocalization with smaller standard deviations (σSD). Indeed, σSD (Table 2) is correlated to both WBI(C⋯C) (Fig. S3a, ESI,†R2 = 0.88) and the minimum NICSzz values (Fig. S3b, ESI,†R2 = 0.86), showing that the delocalization of spin in the T1 state is strongly associated with the extent of homoconjugation and homoaromaticity. Correlation between MCI and σSD is again non-linear (Fig. S3b, ESI†). Figures with the spin densities are given in the ESI,† Section 10.
ACID plots
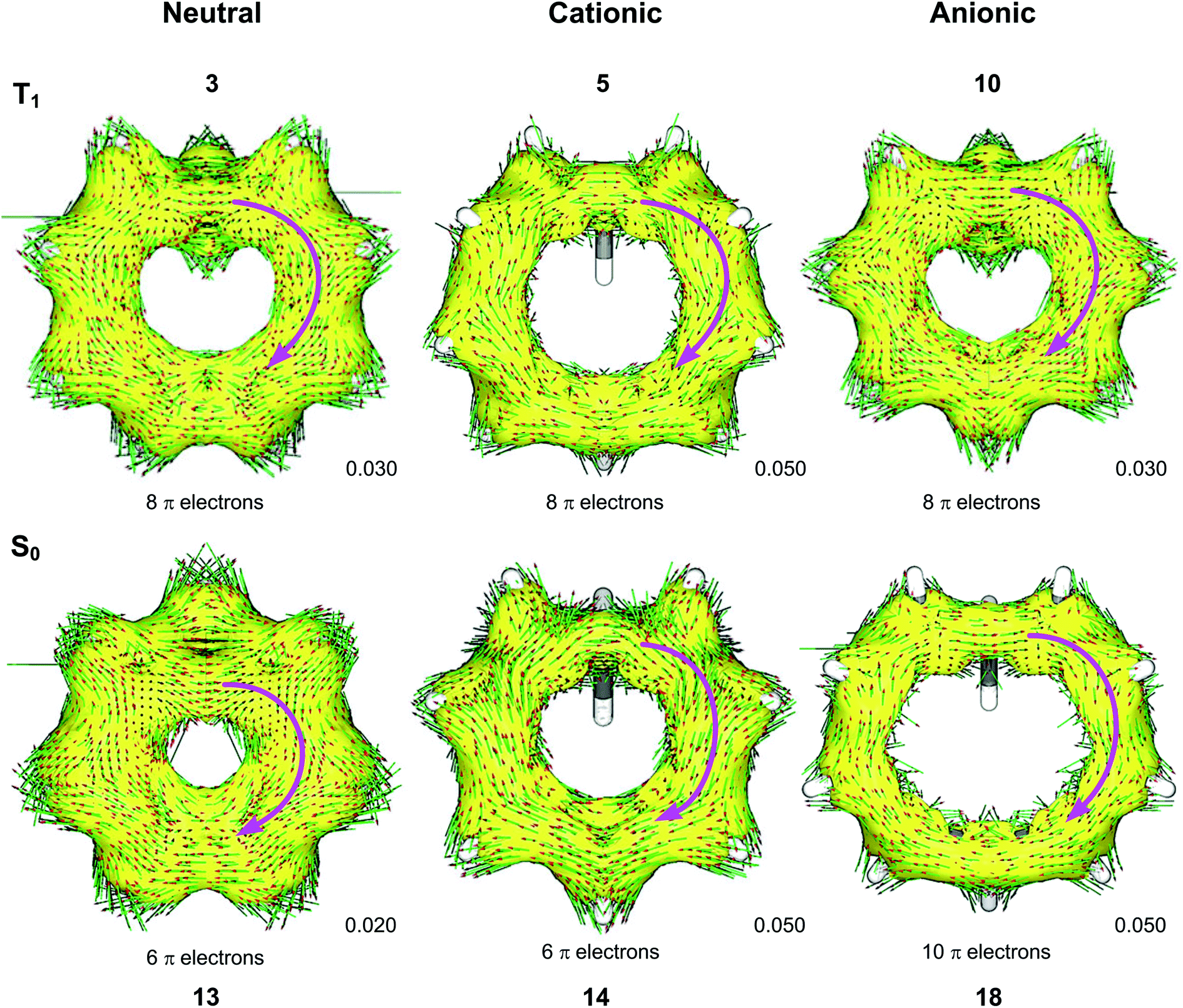
We used the ACID method to visualize the ring current associated with homoaromaticity. Ring currents have previously been analyzed by Sundholm41 and Schleyer25 for homoaromatic compounds in the S0 ground state. In our plots, diatropic currents run clockwise and indicate aromaticity, while paratropic currents run counter-clockwise and indicate antiaromaticity. In Fig. 8, the ACID plots for a selection of compounds in T1 and S0 are shown. ACID plots for all compounds are given in Section 9 of the ESI,† and the direction of the ring current (diatropic or paratropic) is given in Table 2. The ACID plots in Fig. 8 identify that 3, 5 and 10 are T1 homoaromatic, while 13, 14 and 18 are S0 homoaromatic. Considering the strength of the ring currents (through the length of the arrows in the ACID plots), 14 appears to be most aromatic, while 3 and 13 have lower aromaticity. Notably, 5 displays a strong ring current and is clearly aromatic. All T1 compounds except 4, 7, 8 and 11 have visible ring currents in the ACID plots, supporting their T1 homoaromaticity.
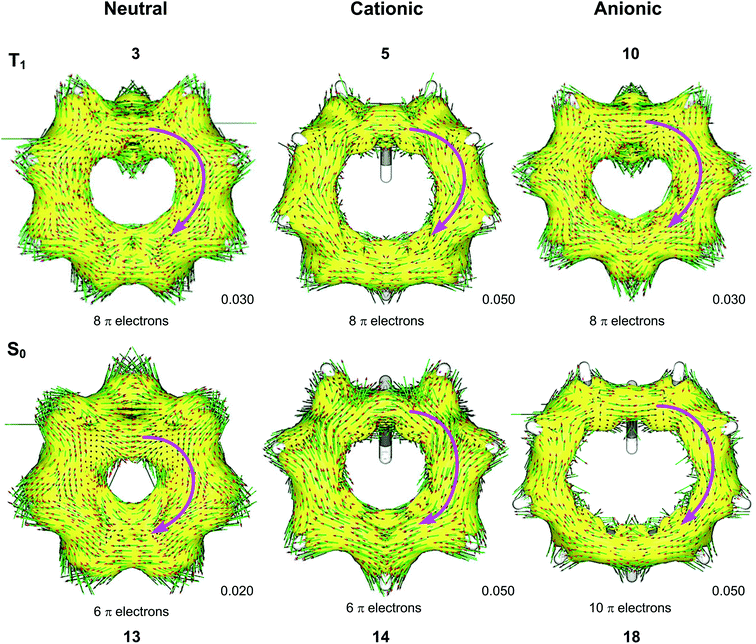 |
| | Fig. 8 ACID plots with B3LYP/6-311+G(d,p) for selected compounds in T1 and S0. Clockwise arrows indicate a diatropic ring current indicative of aromaticity. ACID isosurface values are indicated in the figure. | |
NICS scans
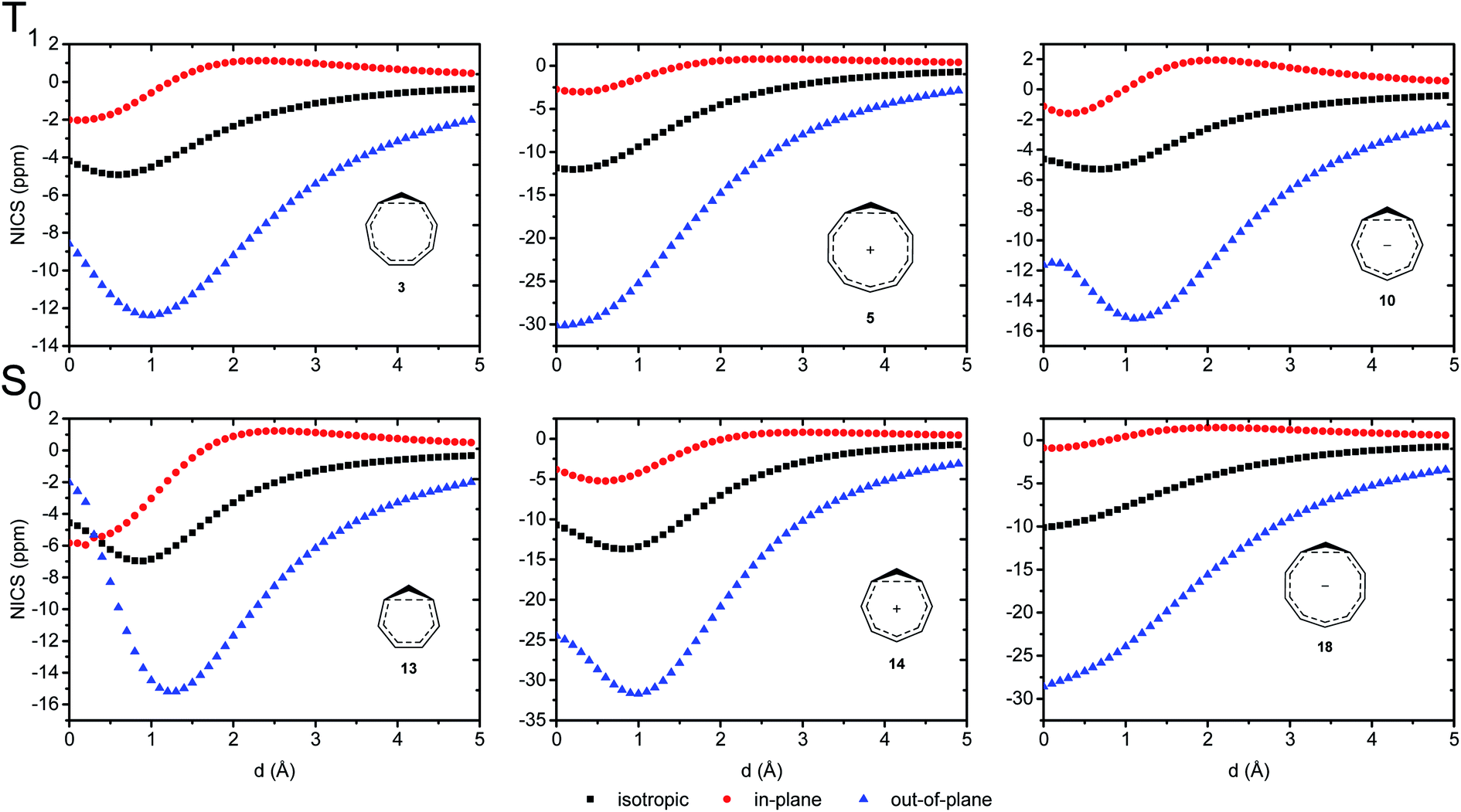
We further probed the magnetic properties with the NICS scan method.33 Aromatic compounds are characterized by a relatively deep minimum in the out-of-plane component of the magnetic shielding tensor, while antiaromatic compounds have a maximum at 0 Å that goes to zero with increasing distance. Non-aromatic compounds have a shallow minimum. Although NICS has shown limitations for non-planar and polycyclic compounds,42 a previous study on homoaromatic compounds found a very good correlation between NICS and multicenter indices (R2 = 0.96).37 We also complement the NICS scans with ring current density plots (using ACID), as recommended in the recent literature.43 The NICS scans for a selection of compounds are given in Fig. 9, while NICS scans for all compounds are given in the ESI† and the minimum NICSzz values along the scan are given in Table 2. As the direction of the NICS scan is not unambiguous for non-planar homoaromatic compounds, the placement of the bq probe atoms (along the z-axis of the coordinate system) are given graphically in Section 6 of the ESI† and also as separate coordinate files. As shown for the Bind aromaticity index, the direction of the z-axis in magnetic calculations is important.44 We have therefore also calculated three-dimensional iso-chemical shielding surfaces (ICSSs)45 for a selection of compounds (Section 15 of the ESI†), and the results show that small deviations in scan direction or origin do not affect the interpretations. From Fig. 9 we see that 3 has a minimum of nearly the same depth as 13 (−12.4 vs. −15.2 ppm), 5 has a minimum nearly as deep as 14 (−30.1 vs. −31.7 ppm) and 10 has a shallower minimum than 18 (−15.2 vs. −28.6 ppm). Overall, the NICS scans support strong homoaromaticity of 1, 2, 5, 6 and 9 in the T1 state as well as 12 and 14–18 in the S0 state. Compounds 3, 4, 10, 11 and 13 have weaker aromaticity and 7 and 8 are best characterized as non-aromatic. As noted above, the minimum NICS values along the scan are well-correlated to the WBI(C⋯C) and MCI values, indicating that it is indeed the increased through-space conjugation and aromaticity that leads to the lower NICS values (Fig. 5b and 7).
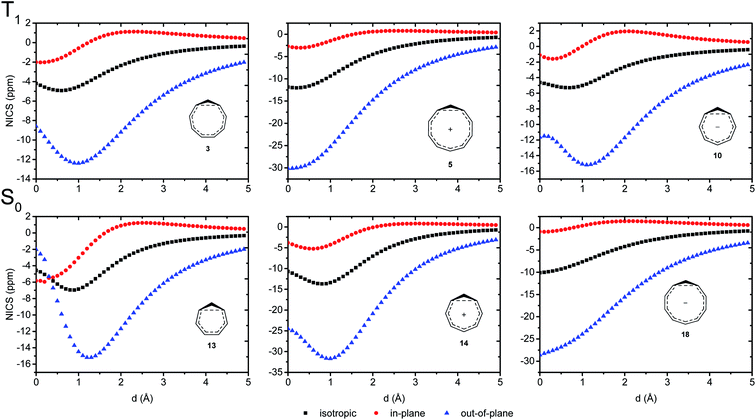 |
| | Fig. 9 NICS scans of selected compounds in the T1 and S0 states. Deep minima in the out-of-plane component of the NICS indicate aromatic character. | |
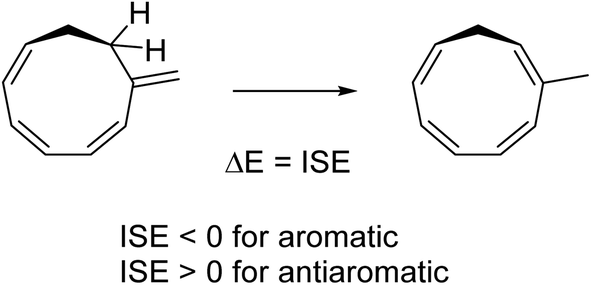
ISE values
To assess the energetic stabilization due to homoaromaticity we used the isomerization stabilization energy (ISE) method.34 The ISE method computes the aromatic stabilization energy by comparing a methylated aromatic compound to its non-aromatic exocyclic methylene isomer (Scheme 2). Typical homoaromatic stabilization energies in the ground state are small and in the range 2–10 kcal mol−1 and therefore easily offset by destabilizing strain.14b Therefore, we focus on the difference ΔISE = ISE(T1) − ISE(S0) for which strain effects should largely cancel between the two electronic states. We expect positive ΔISE values for S0-homoaromatic species (S0-homoaromatic → T1-nonaromatic) and negative ΔISE values for T1-homoaromatic species (S0-nonaromatic → T1-homoaromatic).
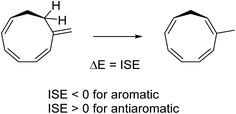 |
| | Scheme 2 Example reaction for determining the ISE values. | |
Indeed, the S0-homoaromatic compounds 13–18 display positive ΔISE values as expected (Table 3). Compounds 1–11 show the expected negative values except for 4 and 7. The homoaromaticity of 7 is questionable and weak at best according to the geometric, electronic and magnetic criteria (vide supra). It is therefore not surprising that it is not aromatic according to the energetic criterion. For 4 the unexpected positive value is due to hyperconjugative aromaticity which stabilizes the S0 state (the electrons of the C–H bonds of the saturated center take part in the conjugation, giving a 6π system).46 Interestingly, 4 could be seen as an aromatic chameleon47 which is 6 π-electron hyperconjugatively aromatic in the S0 state and 4 π-electron homoaromatic in the T1 state. In summary, we conclude that the ISE method supports the homoaromatic character of 1–3, 5, 6 and 8–11 while 7 is considered non-aromatic and the situation is not clear for 4.
Table 3 ISE values in kcal mol−1 for the S0 and T1 states at the B2PLYP/6-311+G(d,p) levela
| Compound |
ISE(S0) |
ISE(T1) |
ΔISEa |
|
Negative values indicate homoaromatic stabilization in T1 while positive values indicate loss of homoaromatic stabilization in S0. Values close to zero are characteristic of non-aromaticity.
12 was not included as it is a TS structure that cannot be treated with the method.
|
|
1
|
−1.3 |
−6.8 |
−5.5 |
|
2
|
+6.6 |
+3.4 |
−3.2 |
|
3
|
−0.2 |
−4.0 |
−3.8 |
|
4
|
−11.6 |
−5.0 |
+6.6 |
|
5
|
−3.0 |
−6.5 |
−3.5 |
|
6
|
−0.7 |
−6.5 |
−5.8 |
|
7
|
−10.0 |
−7.7 |
+2.3 |
|
8
|
−0.4 |
−4.0 |
−3.6 |
|
9
|
+2.9 |
−12.7 |
−15.5 |
|
10
|
+4.1 |
−4.3 |
−8.4 |
|
11
|
+2.2 |
−3.2 |
−5.4 |
|
12
|
n/ab |
n/ab |
n/ab |
|
13
|
−4.0 |
−3.2 |
+0.8 |
|
14
|
−15.0 |
−1.6 |
+13.3 |
|
15
|
−13.8 |
−1.7 |
+12.1 |
|
16
|
−21.6 |
−5.1 |
+16.5 |
|
17
|
−2.1 |
+2.2 |
+4.3 |
|
18
|
−5.9 |
+4.3 |
+10.3 |
Extension to the S1 state
After having thoroughly assessed homoaromaticity in T1, we extend the concept also to the S1 state. We turn to 3 and 10 that were identified as possible excited state intermediates (Scheme 1c and d). Although we have shown that they are homoaromatic in the T1 state, it is likely that the photoreactions described occur in the S1 state. Therefore, we optimized 3 and 10 in the S1 state using CASSCF/6-31G(d). For 10 we could also employ TD-B3LYP/6-31+G(d,p), while this was not possible for 3 due to the doubly excited character of the S1 state. The optimized structure of 3 in S1 is similar to that in T1 but with a shorter r(C⋯C) of 2.119 Å in S1 with CASSCF compared to 2.385 Å in T1 with B2PLYP.
The C2v-symmetric optimized structure of 10 in S1 with r(C⋯C) = 2.379 Å is not stable with CASSCF and distorts to a more charge-localized C1-symmetric structure with r(C⋯C) = 2.360 Å. In T1 the structure is C2v-symmetric with r(C⋯C) = 2.477 Å according to B2PLYP. In contrast to CASSCF, TD-B3LYP favors the C2v-symmetric geometry with a r(C⋯C) = 2.495 Å which is more like the T1 value (2.477 Å). We conclude that 3 and 10 have significant homoconjugation in S1 based on their geometries.
So are they aromatic? Unfortunately, ACID plots are not available for the S1 state. However, qualitative NICSiso scans in S1 show the characteristic aromatic minima, indicating that they are indeed aromatic (Fig. S9 and S10, ESI†).48 We therefore tentatively conclude that 3 and 10 are both T1- and S1-homoaromatic, although we defer an extended investigation into S1 homoaromaticity to a later study.
Application to design of photomechanical materials
Compound 16 is especially interesting as it can function as a photomechanical lever. In S0 it prefers the conformation with the bridging carbon coordinated to the double bond due to homoaromatic stabilization. From the results above, we expect that this cycle with 2π electrons will become homoantiaromatic in the S1 and T1 states and that the bridging carbon will swing away from the ethylene segment. Indeed, optimization in T1 leads to a minimum with a considerably larger distance between the bridge and the bridging carbon (Fig. 10a). A minimum energy path calculation in S1 with CASSCF/6-31G(d) leads to a similar structure which is now a S0/S1 conical intersection (CI) leading back to the S0 minimum (Fig. 10b). The C–C–C angle is opened from 72° in S0 to 101° at the T1 minimum and S0/S1 CI. At the same time, r(C⋯C) increases from 1.759 Å in S0 to 2.299 Å in T1 and 2.316 Å at the CI. The S0, S1 and T1 states are almost degenerate at the CI as judged by MS-CASPT2 calculations (Fig. S6, ESI†).
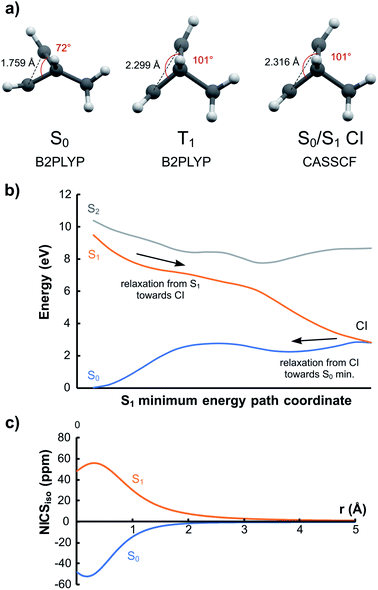 |
| | Fig. 10 (a) Optimized geometries for 16 for S0, T1 and S0/S1 conical intersection. (b) CASSCF energies on minimum energy path in S1 following vertical excitation. (c) NICSiso scans for S0 and S1 at the S0 geometry. | |
The S1/T1 relaxation represents a substantial geometrical change that could be utilized as a photochemical “lever”. Exchanging the hydrogen substituent at the bridging carbon for larger groups would lead to an even higher torque. To confirm that homoantiaromaticity is indeed responsible for this conformational change, we calculated the NICS scan for the vertically excited S1 state. It shows a clear antiaromatic profile that is a mirror image of the aromatic scan in S0 (Fig. 10c). The T1 NICS scans at the vertically excited and relaxed T1 geometries shows that also the T1 state is antiaromatic but that the antiaromaticity is alleviated by de-coordination of the bridging carbon (Fig. S7 and S8, ESI†). Compound 16 could thus be a starting point for design of new photomechanical materials based on the concept of excited state homoantiaromaticity.
Synopsis and outlook
The analysis of the different aromaticity criteria for 1–11 is summarized in Table 4, together with an overall assessment of the extent of T1 homoaromaticity. We find that 1, 2, 5, 6 and 9 are strongly T1 homoaromatic, while 3, 4, 10 and 11 are weakly T1 homoaromatic. Compounds 7 and 8 are non-aromatic. It is thus established that homoaromaticity is important in the T1 excited state, and our calculations also indicated that this extends to the S1 state. Further investigations in this direction are currently ongoing in our laboratory. Electronic excitation emerges as a second way, besides a Möbius orbital topology,49 for 4nπ systems to become homoaromatic. Based on the selected reactions from the literature with probable excited state homoaromatic intermediates (3 and 10), we expect that there are more photochemical mechanisms that can be interpreted using homoaromaticity. Furthermore, the concept should aid in the discovery and development of new photochemical reactions, as well as tailoring of photophysical properties. One example that we highlighted are the predicted photomechanical properties of 16 that are due to the homoantiaromatic destabilization in the excited state of the preferred ground state conformation. Thus, excited state homo(anti)aromaticity could be a new tool for design of novel optically active molecular machines.
Table 4 Summary of aromatic criteria and overall assessment of T1 state homoaromaticity of 1–11. Full circle means that criterion is fulfilled, empty circle that it is not, dash that it could not be evaluated. Ranking of homoaromatic character is giving according to the MCI
| # |
BLA |
r(C⋯C) |
WBI(C⋯C) |
σ
Q
|
ACID |
NICS |
ΔISE |
MCI |
T1 homoaromaticity |
|
1
|
— |
● |
● |
● |
● |
● |
● |
3 |
Strong |
|
2
|
— |
● |
● |
● |
● |
● |
● |
4 |
Strong |
|
3
|
● |
● |
○ |
● |
● |
● |
● |
8 |
Weak |
|
4
|
● |
● |
● |
● |
○ |
● |
— |
6 |
Weak |
|
5
|
● |
● |
● |
● |
● |
● |
● |
2 |
Strong |
|
6
|
— |
● |
● |
● |
● |
● |
● |
1 |
Strong |
|
7
|
— |
○ |
○ |
○ |
○ |
○ |
○ |
10 |
Nonaromatic |
|
8
|
— |
○ |
○ |
○ |
○ |
○ |
● |
11 |
Nonaromatic |
|
9
|
— |
● |
● |
● |
● |
● |
● |
5 |
Strong |
|
10
|
● |
● |
● |
● |
● |
● |
● |
7 |
Weak |
|
11
|
— |
● |
● |
● |
○ |
● |
● |
9 |
Weak |
Computational methods
All calculations in the S0 and T1 states were performed using Gaussian 09 Revision D.01 and E.0150 with the B2PLYP51 and B3LYP52 functionals and the 6-311+G(d,p) basis set.53 Restricted calculations were used for singlet states and unrestricted for triplet states. All compounds were optimized with B2PLYP/6-311+G(d,p) as it performed well against CCSD(T),54 BD(T)55 and CASPT256 results for a selection of compounds (see Section 11 of the ESI† for an extended discussion). Analysis of the CCSD T1 diagnostics and CASSCF configurational weights show that the triplet states are single-configurational and the use of DFT should therefore be appropriate (see ESI,† Section 13). Spin contamination of the T1 states is small at 2.01–2.05 except for two cases with 2.09 and 2.11. B2PLYP is insensitive to spin contamination of this magnitude.57 We did not include dispersion corrections beyond what is given by the MP2 component of B2PLYP, and calculations on a selection of the compounds show a very small effect on the obtained geometries (see Section 14 of the ESI†). Stationary points were confirmed by frequency calculations with no negative frequencies except for the TS structure of 12 with one negative frequency. The wavefunctions were checked for instabilities using the “stable” keyword in Gaussian 09. All NICS scans in T1 were generated with the Aroma β.4 package,58 using the GIAO method59 at the B3LYP/6-311+G(d,p) level as these open-shell magnetic calculations are not possible at the B2PLYP level. The placement of the ghost atoms is given in figures and included in the atomic coordinates in the ESI.† ACID calculations were performed using the AICD 2.0.0 program at the B3LYP/6-311+G(d,p) level using the CSGT method.60 We have chosen this level of theory to be consistent with the previous literature,25 and because ACID cannot be calculated at the B2PLYP level. Note that different ACID isosurface values were used to clearly identify the direction of the ring currents. ISE values were calculated at the B2PLYP/6-311+G(d,p) level. No anti–syn diene mismatch corrections were made. Atomic charges and spin densities were calculated with the natural population analysis scheme40 using NBO 6.0.61 Bond length alternation (BLA) was calculated according to the formula:
CASSCF optimization in the S1 states were performed with an active space of all π orbitals and electrons, a two-state (for 3 and 10) or three-state (for 16) average procedure and the 6-31G(d) basis set62 using a development version of Molcas 8.1.63 The smaller 6-31G(d) basis set was used to allow numerical frequency calculations in the S1 state. TD-DFT optimizations were done at the B3LYP/6-31+G(d,p) level with Gaussian 16 Rev. A.03.64 In both cases minima were confirmed by frequency calculations. NICS scans in S1 were calculated with Dalton 2016.0.48 WBI and multicenter indices were computed according to the equations reported previously30,65,66 and applied for the study of ground state homoaromaticity,37 including all permutations over the atomic labels as in several cases the sequential ordering was not the largest contributor to the MCI value. Bader's atoms in molecules approach,67 known as quantum chemical topology was used to distinguish the atoms in the molecule and to yield atomic overlap matrices using AIMAll.68 Orbitals from the DFT Kohn–Sham scheme were used in the same way as Hartree–Fock orbitals as experience has shown that this reveals the relevant chemistry properly.69 Normalization of the resulting indices was done as proposed by Mandado et al.38 for both spin contributions separately after which both were summed to yield a normalized total MCI. σ and π separation was done for planar conventional aromatics by limiting the sums over all orbitals to those relevant irreducible representations per contribution.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Prof. R. Herges is kindly acknowledged for providing the AICD 2.0.0 program. H. O. and K. J. acknowledge the Swedish Research Council for financial support (grant number 2015-04538). B. O. J. is grateful to the Wenner-Gren Foundations for a postdoctoral fellowship. P. B. acknowledges continuous support from the Fund for Scientific Research in Flanders (FWO-Vlaanderen). K. J. acknowledges a travel grant from the Royal Swedish Academy of Sciences. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and HPC2N.
Notes and references
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
-
(a) M. J. S. Dewar, Tetrahedron, 1966, 22(suppl. 8), 75–92 CrossRef;
(b) H. E. Zimmerman, J. Am. Chem. Soc., 1966, 88, 1564–1565 CrossRef CAS.
-
(a) H. Ottosson, Nat. Chem., 2012, 4, 969–971 CrossRef CAS PubMed;
(b) M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed;
(c) R. Papadakis and H. Ottosson, Chem. Soc. Rev., 2015, 44, 6472–6493 RSC.
-
(a) P. Wan and E. Krogh, J. Chem. Soc., Chem. Commun., 1985, 1207–1208 RSC;
(b) P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef CAS.
-
(a) H. Ottosson, K. Kilså, K. Chajara, M. Piqueras, R. Crespo, H. Kato and D. Muthas, Chem.–Eur. J., 2007, 13, 6998–7005 CrossRef CAS PubMed;
(b) M. Rosenberg, H. Ottosson and K. Kilså, Phys. Chem. Chem. Phys., 2011, 13, 12912–12919 RSC.
-
(a) H. Ottosson and K. E. Borbas, Nat. Chem., 2015, 7, 373–375 CrossRef CAS PubMed;
(b) Y. M. Sung, M.-C. Yoon, J. M. Lim, H. Rath, K. Naoda, A. Osuka and D. Kim, Nat. Chem., 2015, 7, 418–422 CrossRef CAS PubMed;
(c) Y. M. Sung, J. Oh, W. Kim, H. Mori, A. Osuka and D. Kim, J. Am. Chem. Soc., 2015, 137, 11856–11859 CrossRef CAS PubMed;
(d) Y. M. Sung, J. Oh, W.-Y. Cha, W. Kim, J. M. Lim, M.-C. Yoon and D. Kim, Chem. Rev., 2017, 117, 2257–2312 CrossRef CAS PubMed.
- Y. M. Sung, J. Oh, K. Naoda, T. Lee, W. Kim, M. Lim, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2016, 55, 11930–11934 CrossRef CAS PubMed.
- M. Izawa, T. Kim, S. Ishida, T. Tanaka, T. Mori, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 3982–3986 CrossRef CAS PubMed.
- R. K. Mohamed, S. Mondal, K. Jorner, T. Faria Delgado, V. V. Lobodin, H. Ottosson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 15441–15450 CrossRef CAS PubMed.
- R. Papadakis, H. Li, J. Bergman, A. Lundstedt, K. Jorner, R. Ayub, S. Haldar, B. O. Jahn, A. Denisova, B. Zietz, R. Lindh, B. Sanyal, H. Grennberg, K. Leifer and H. Ottosson, Nat. Commun., 2016, 7, 12962 CrossRef CAS PubMed.
-
(a) D. Shukla and P. Wan, J. Am. Chem. Soc., 1993, 115, 2990–2991 CrossRef CAS;
(b) M. Ueda, K. Jorner, Y. M. Sung, T. Mori, Q. Xiao, D. Kim, H. Ottosson, T. Aida and Y. Itoh, Nat. Commun., 2017, 8, 346 CrossRef PubMed;
(c) M. Hada, S. Saito, S. Tanaka, R. Sato, M. Yoshimura, K. Mouri, K. Matsuo, S. Yamaguchi, M. Hara, Y. Hayashi, F. Röhricht, R. Herges, Y. Shigeta, K. Onda and R. J. D. Miller, J. Am. Chem. Soc., 2017, 139, 15792–15800 CrossRef CAS PubMed;
(d) J. Oh, Y. M. Sung, H. Mori, S. Park, K. Jorner, H. Ottosson, M. Lim, A. Osuka and D. Kim, Chem, 2017, 3, 870–880 CrossRef CAS.
-
(a) J. Aihara, Bull. Chem. Soc. Jpn., 1978, 51, 1788 CrossRef CAS;
(b) C. J. Kastrup, S. P. Oldfield and H. S. Rzepa, Chem. Commun., 2002, 642–643 RSC.
-
(a) J. Oh, Y. M. Sung, W. Kim, S. Mori, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2016, 55, 6487–6491 CrossRef CAS PubMed;
(b) Y. Hong, J. Oh, Y. M. Sung, Y. Tanaka, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2017, 56, 2932–2936 CrossRef CAS PubMed.
-
(a) R. V. Williams, Chem. Rev., 2001, 101, 1185–1204 CrossRef CAS PubMed;
(b)
D. Cremer, R. F. Childs and E. Kraka, in Patai's Chemistry of Functional Groups, John Wiley & Sons, 2009 Search PubMed;
(c)
R. F. Childs, D. Cremer and G. Elia, in Patai's Chemistry of Functional Groups, John Wiley & Sons, 2009 Search PubMed.
- S. Winstein, J. Am. Chem. Soc., 1959, 81, 6524–6525 CrossRef CAS.
- A. F. Diaz, M. Sakai and S. Winstein, J. Am. Chem. Soc., 1970, 92, 7477–7480 CrossRef CAS.
-
(a) H. Jiao and P. v. R. Schleyer, AIP Conf. Proc., 1995, 330, 107–128 CrossRef CAS;
(b) J.-H. Shin, Bull. Korean Chem. Soc., 1982, 3, 66–70 CAS;
(c) H. Volz and J.-H. Shin, J. Org. Chem., 2006, 71, 2220–2226 CrossRef CAS PubMed.
- J. L. von Rosenberg, J. E. Mahler and R. Pettit, J. Am. Chem. Soc., 1962, 84, 2842–2843 CrossRef CAS.
- M. Garcia-Borràs, S. Osuna, J. M. Luis, M. Swart and M. Solà, Chem. Soc. Rev., 2014, 43, 5089–5105 RSC.
- Q. Zhang, S. Yue, X. Lu, Z. Chen, R. Huang, L. Zheng and P. v. R. Schleyer, J. Am. Chem. Soc., 2009, 131, 9789–9799 CrossRef CAS PubMed.
- J. I. Brauman, J. Schwartz and E. E. van Tamelen, J. Am. Chem. Soc., 1968, 90, 5328–5329 CrossRef CAS.
- J. Schwartz, J. Chem. Soc. D, 1969, 833–834 RSC.
- K. Jorner, A. Dreos, R. Emanuelsson, O. El Bakouri, I. Fernández Galván, K. Börjesson, F. Feixas, R. Lindh, B. Zietz, K. Moth-Poulsen and H. Ottosson, J. Mater. Chem. A, 2017, 5, 12369–12378 CAS.
-
(a) M. Roberts, H. Hamberger and S. Winstein, J. Am. Chem. Soc., 1970, 92, 6346–6348 CrossRef CAS;
(b) G. Schröder, U. Prange, N. S. Bowman and J. F. M. Oth, Tetrahedron Lett., 1970, 11, 3251–3254 CrossRef.
-
(a) Z. F. Chen, H. J. Jiao, J. I. Wu, R. Herges, S. B. Zhang and P. v. R. Schleyer, J. Phys. Chem. A, 2008, 112, 10586–10594 CrossRef CAS PubMed;
(b) R. V. Williams, W. D. Edwards, P. Zhang, D. J. Berg and R. H. Mitchell, J. Am. Chem. Soc., 2012, 134, 16742–16752 CrossRef CAS PubMed.
- J. M. Brown, Aust. J. Chem., 2014, 67, 1296–1300 CAS.
- J. Jusélius, M. Patzschke and D. Sundholm, J. Mol. Struct.: THEOCHEM, 2003, 633, 123–136 CrossRef.
- P. R. Griffiths, D. E. Pivonka and R. V. Williams, Chem.–Eur. J., 2011, 17, 9193–9199 CrossRef CAS PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS.
-
(a) R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS;
(b) D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
-
(a) A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed;
(b) J. O. C. Jiménez-Halla, E. Matito, J. Robles and M. Solà, J. Organomet. Chem., 2006, 691, 4359–4366 CrossRef.
-
(a) P. v. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4, 2873–2876 CrossRef CAS PubMed;
(b) J. Zhu, K. An and P. v. R. Schleyer, Org. Lett., 2013, 15, 2442–2445 CrossRef CAS PubMed.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. Ponec, P. Bultinck and A. G. Saliner, J. Phys. Chem. A, 2005, 109, 6606–6609 CrossRef CAS PubMed.
- M. Mandado, A. M. Graña and I. Pérez-Juste, J. Chem. Phys., 2008, 129, 164114 CrossRef PubMed.
- L. A. Paquette, M. J. Broadhurst, P. Warner, G. A. Olah and G. Liang, J. Am. Chem. Soc., 1973, 95, 3386–3387 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- H. Fliegl, D. Sundholm, S. Taubert, J. Jusélius and W. Klopper, J. Phys. Chem. A, 2009, 113, 8668–8676 CrossRef CAS PubMed.
-
(a) S. Fias, S. Van Damme and P. Bultinck, J. Comput. Chem., 2008, 29, 358–366 CrossRef CAS PubMed;
(b) S. Fias, S. Van Damme and P. Bultinck, J. Comput. Chem., 2010, 31, 2286–2293 CAS.
-
(a) S. Van Damme, G. Acke, R. W. A. Havenith and P. Bultinck, Phys. Chem. Chem. Phys., 2016, 18, 11746–11755 RSC;
(b) G. Acke, S. Van Damme, R. W. A. Havenith and P. Bultinck, J. Comput. Chem., 2018, 39, 511–519 CrossRef CAS PubMed.
-
(a) G. Merino, T. Heine and G. Seifert, Chem.–Eur. J., 2004, 10, 4367–4371 CrossRef CAS PubMed;
(b) R. Islas, T. Heine and G. Merino, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS PubMed.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
-
(a) S. Sieber, P. v. R. Schleyer and J. Gauss, J. Am. Chem. Soc., 1993, 115, 6987–6988 CrossRef CAS;
(b) D. A. Lawlor, D. E. Bean, P. W. Fowler, J. R. Keeffe, J. S. Kudavalli, R. A. More O'Ferrall and S. N. Rao, J. Am. Chem. Soc., 2011, 133, 19729–19742 CrossRef CAS PubMed.
- H. Möllerstedt, M. C. Piqueras, R. Crespo and H. Ottosson, J. Am. Chem. Soc., 2004, 126, 13938–13939 CrossRef PubMed.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CrossRef CAS PubMed.
- C. S. M. Allan and H. S. Rzepa, J. Chem. Theory Comput., 2008, 4, 1841–1848 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01 and E.01, Gaussian, Inc., Wallingford CT, 2009. For full reference, see ESI.†.
- S. Grimme, J. Chem. Phys., 2006, 124, 34108 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- N. C. Handy, J. A. Pople, M. Head-Gordon, K. Raghavachari and G. W. Trucks, Chem. Phys. Lett., 1989, 164, 185–192 CrossRef CAS.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- A. S. Menon and L. Radom, J. Phys. Chem. A, 2008, 112, 13225–13230 CrossRef CAS PubMed.
-
A. Rahalkar and A. Stanger, Aroma, http://schulich.technion.ac.il/Amnon_Stanger.htm Search PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- T. A. Keith and R. F. W. Bader, Chem. Phys. Lett., 1993, 210, 223–231 CrossRef CAS.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin-Madison, 2013 Search PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fernández Galván, N. Ferre, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. A. Malmqvist, T. Muller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Marti, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016. For full reference, see ESI.†.
- P. Bultinck, D. L. Cooper and R. Ponec, J. Phys. Chem. A, 2010, 114, 8754–8763 CrossRef CAS PubMed.
- R. F. W. Bader and M. E. Stephens, J. Am. Chem. Soc., 1975, 97, 7391–7399 CrossRef CAS.
-
(a)
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed;
(b) R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
-
T. A. Keith, AIMAll, Version 17.01.25, TK Gristmill Software, Overland Park KS, USA, 2017, http://aim.tkgristmill.com Search PubMed.
- J. Poater, M. Duran, M. Solà and B. Silvi, Chem. Rev., 2005, 105, 3911–3947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05009g |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Burkhard O.
Jahn
a,
Burkhard O.
Jahn