
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New activation mechanism for half-sandwich organometallic anticancer complexes†
Samya
Banerjee‡
 a,
Joan J.
Soldevila-Barreda‡
a,
Juliusz A.
Wolny‡
b,
Christopher A.
Wootton
a,
Abraha
Habtemariam
a,
Isolda
Romero-Canelón
a,
Feng
Chen
a,
Guy J.
Clarkson
a,
Ivan
Prokes
a,
Lijiang
Song
a,
Peter B.
O'Connor
a,
Volker
Schünemann
b and
Peter J.
Sadler
*a
a,
Joan J.
Soldevila-Barreda‡
a,
Juliusz A.
Wolny‡
b,
Christopher A.
Wootton
a,
Abraha
Habtemariam
a,
Isolda
Romero-Canelón
a,
Feng
Chen
a,
Guy J.
Clarkson
a,
Ivan
Prokes
a,
Lijiang
Song
a,
Peter B.
O'Connor
a,
Volker
Schünemann
b and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
bDepartment of Physics, University of Kaiserslautern, Erwin-Schrödinger-Straße 46, 67663 Kaiserslautern, Germany
First published on 1st March 2018
Abstract
The Cpx C–H protons in certain organometallic RhIII half-sandwich anticancer complexes [(η5-Cpx)Rh(N,N′)Cl]+, where Cpx = Cp*, phenyl or biphenyl-Me4Cp, and N,N′ = bipyridine, dimethylbipyridine, or phenanthroline, can undergo rapid sequential deuteration of all 15 Cp* methyl protons in aqueous media at ambient temperature. DFT calculations suggest a mechanism involving abstraction of a Cp* proton by the Rh–hydroxido complex, followed by sequential H/D exchange, with the Cp* rings behaving like dynamic molecular ‘twisters’. The calculations reveal the crucial role of pπ orbitals of N,N′-chelated ligands in stabilizing deprotonated Cpx ligands, and also the accessibility of RhI–fulvene intermediates. They also provide insight into why biologically-inactive complexes such as [(Cp*)RhIII(en)Cl]+ and [(Cp*)IrIII(bpy)Cl]+ do not have activated Cp* rings. The thiol tripeptide glutathione (γ-L-Glu-L-Cys-Gly, GSH) and the activated dienophile N-methylmaleimide, (NMM) did not undergo addition reactions with the proposed RhI–fulvene, although they were able to control the extent of Cp* deuteration. We readily trapped and characterized RhI–fulvene intermediates by Diels–Alder [4+2] cyclo-addition reactions with the natural biological dienes isoprene and conjugated (9Z,11E)-linoleic acid in aqueous media, including cell culture medium, the first report of a Diels–Alder reaction of a metal-bound fulvene in aqueous solution. These findings will introduce new concepts into the design of organometallic Cp* anticancer complexes with novel mechanisms of action.
Introduction
Methylated cyclopentadienyl ligands, and especially pentamethyl-cyclopentadienyl (Cp*), are often used as η5–π-bonded ligands in transition metal complexes.1,2 Cp* and substituted Cp* (Cpx, e.g. tetramethyl(biphenyl)cyclopentadienyl, CpxPhPh) ligands form highly stable complexes with, for example, the low-spin d6 metal ions RhIII and IrIII.3–6 Such RhIII and IrIII complexes are of interest for their wide range of properties, including activity as catalysts and anticancer agents,3,4,7–12 and sometimes as both.13,14The biologically-active centre in half-sandwich [(η5-Cpx)Rh/Ir(N,N′/C,N)Cl] anticancer complexes containing N,N′- or C,N-chelated ligands is usually thought to be the metal ion itself.10–14 Initial displacement of chloride by water (aquation) can facilitate binding to target biomolecules, the possibility of direct reactions involving the Cpx ligand under biologically-relevant conditions has yet to be investigated.
In general C–H bonds in hydrocarbons are very stable and difficult to activate. They are only weakly acidic, and the available common bases are not strong enough.15,16 Such C–H bond activation can be promoted especially by the electronic effects of platinum group metals, and numerous examples have now been reported.17–21 However, reported Cpx activation reactions usually require harsh conditions.
In an early example, Kang and Maitlis deuterated the methyl groups in the dinuclear RhIII complex [(Cp*)Rh(μ2-OH)3Rh(Cp*)]Cl in D2O after addition of OD−, but the process was slow, requiring over 3 days at the elevated temperature of 343 K.22 At a similar time, Bercaw et al. used very strong bases in non-polar solvents to synthesize Me4CpCH2–bridged TiII complexes. These were sensitive to traces of water.23 Wei et al. have shown that deuteration of the Cp* in [(Cp*)RhD2(BPin)] (BPin = bis(pinacolato)diboron) occurs readily in MeOD-d4 and synthesized the deuterated compound [(D15Cp*)RhD2(BPin)], under reflux in D2O/OD− for 3 days.24 Ciancaleoni et al. showed that deuteration of [(Cp*)RhCl(PTA)2]+ (PTA = 7-phospha-1,3,5-triaza-adamantane) is assisted by the basic centers in the PTA ligand,25 and proposed a mechanism in which a Rh–OH species deprotonates the Cp* ring followed by formation of a RhI–fulvene species, but no experimental evidence for the fulvene complex was obtained.
Quintana et al.26 and Pitman et al.27 previously reported the direct involvement of Cp* ligands in hydride transfer reactions. In the former case, initial protonation of Cp* in the RhI catalyst [(Cp*)Rh(bpy)] can lead to formation of H2 in dry acetonitrile. In the latter case, [(Cp*H)Rh(bpy)Cl], generated by reduction of the RhIII complex [(Cp*)Rh(bpy)Cl]+ followed by acidification in ether, was isolated, and shown to reduce NAD+ to NADH. The Cp* methyl protons underwent exchange with deuterium. Peng et al. have recently characterized the RhI phenanthroline analogue containing protonated Cp*, [(Cp*H)Rh(phen)Cl].28 A recent report by Blakemore et al.29 introduced Cp*Rh complexes of substituted 2,2′-bipyridyl as effective hydrogen evolution catalysts involving the formation of unusual η4-pentamethylcyclopentadiene–RhI intermediates under protic conditions.
Our own investigations began with observations that in aqueous solutions at ambient temperature, methyl 1H NMR peaks of some [(Cp*)RhCl(N,N′)Cl]+ complexes which we had synthesized as potential anticancer agents and transfer hydrogenation catalysts, rapidly lost intensity, suggesting activation towards H/D exchange under mild conditions. We have investigated the conditions under which this occurs, including the influence of solvent, aquation, and pH, and monitored the reactions by both 1H NMR spectroscopy and high resolution FT ICR MS spectrometry. We have also investigated the influence of substituents on the Cp* ligand (Cp* versus CpxPh and CpxPhPh), influence of the N,N′-chelating ligand (aliphatic ethylenediamine (en) versus aromatic diamines bipyridyl (bpy), 4,4′-dimethyl-2,2′-bpyridine (mbpy), and phenanthroline (phen)), as well as the metal ion (RhIIIversus IrIII). The X-ray crystal structures of 8 new complexes are reported.
We have used density functional theory (DFT) calculations to elucidate the mechanism of these Cpx C–H activation reactions. Importantly the DFT calculations led to the prediction of accessible RhI–fulvene states and so we investigated the trapping of such intermediates by addition reactions with thiols (glutathione), dienes (N-methyl maleimide, NMM) and dienophiles, Chart 1. The thiol tripeptide glutathione (γ-L-Glu-L-Cys-Gly) is present in cells at millimolar concentrations, and might be expected to undergo addition reactions with double bonds such as those in a fulvene, as might NMM which contains an activated double bond. Fulvene is known to undergo Diels–Alder reactions with both dienes and dienophiles.30 The exocyclic double bond of fulvene can also act as a dienophile in a Diels–Alder reaction.30,31 The presence of substituents on the exocyclic double bond shifts the Diels–Alder reaction to the endocyclic double bond.30,31 In attempts to trap adducts of the proposed fulvene intermediate as [4+2] cyclo-addition Diels–Alder adducts, we chose the natural biological conjugated dienes isoprene and (9Z,11E)-linoleic acid (Chart 1). Isoprene is the most abundant hydrocarbon in human breath, with an estimated human production of 17 mg per day.32,33 (9Z,11E)-Linoleic acid is a common dietary conjugated fatty acid, of particular interest for its potential role in the prevention and treatment of a variety of diseases,34 including roles in modulating the expression of oncogenes and cell cycle regulators.34,35
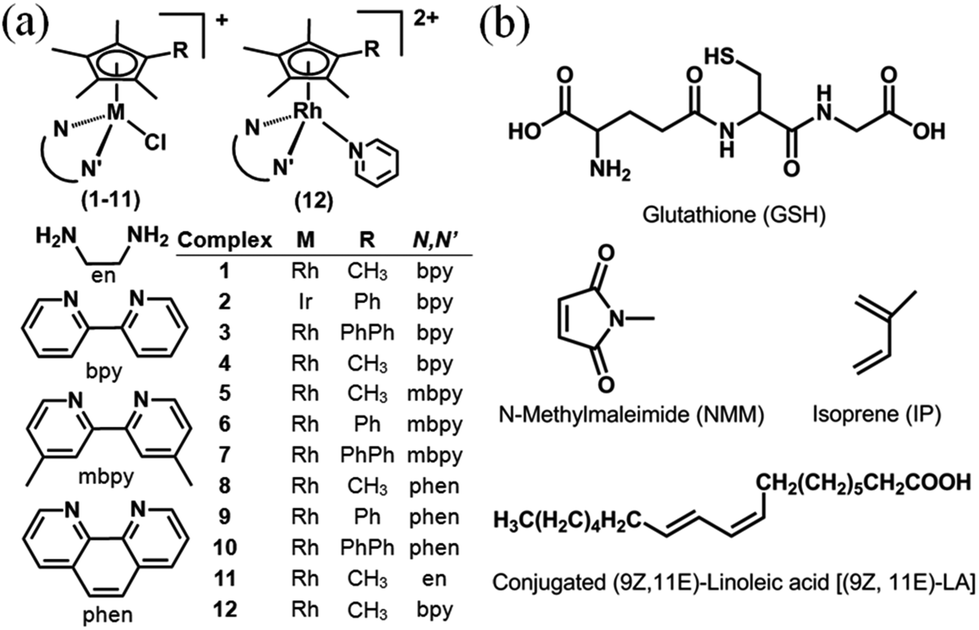 | ||
| Chart 1 (a) RhIII and IrIII complexes studied here as PF6− salts. (b) Structures of glutathione, dienes and dienophile used in attempts to trap fulvene intermediates. | ||
The surprising success of these RhI–fulvene trapping reactions under mild conditions of biological relevance is likely to open up new avenues of investigation into reactions of this class of half-sandwich organometallic complexes and influence their design as biologically-active agents. Our discovery of Diels–Alder reactions of a metal-bound fulvene under mild conditions in aqueous solutions, appears to be unprecedented.
Results and discussion
Synthesis and characterization of complexes
Organo–RhIII complexes 1 and 3–12 (Chart 1(a)) and IrIII complex 2 containing Cp*, CpxPh or CpxPhPh, together with an N,N′-chelated ligand (en, bpy, mbpy or phen), and a monodentate chloride ligand were synthesized by the reaction of the appropriate chloride-bridged dimer and chelating ligand, except complex 12 which was prepared by substitution of chloride in complex 1 by pyridine. They were fully characterized by 1H NMR spectroscopy, ESI-mass spectrometry, and elemental analysis (details in the ESI†). The X-ray crystal structures of complexes 1, 3, 4, 6, 7, 9, 10 and 12 were determined (Fig. S1, Tables S1 and S2†). All exhibit the typical ‘piano-stool’ geometry. Rh–Cl distances are in the range of 2.385–2.403 Å, with complexes containing extended Cpx rings having slightly longer bond lengths. The Rh–Cpx centroid distance of 1.778–1.795 Å shows little change with extension of the Cp* ring. Interestingly, complexes 6, 7 and 9 have one Rh–N distance slightly longer than the other, which might be due to the combined effects of crystal packing and ligand substituents. The bond lengths and angles for complexes 6 and 7 are similar to those reported for [(Cp*)Rh(phen)Cl]SO3CF3,36 and for complexes 1, 3 and 4 are similar to those reported for [(Cp*)Rh(bpy)Cl]SO3CF3.36 For the pyridine complex 12, the Rh–N(pyridine) bond (2.127(4) Å) is longer than other two Rh–N (bipyridine) bonds (ca. 2.105 Å).Hydrolysis
Aquation of chloride complexes 1 and 3–11 in 20% methanol-d4/80% D2O (1.4 mM, 310 K) was confirmed by comparing 1H NMR spectra before and after removal of the chloride ligand by reaction with AgNO3. Complexes 1, 3–11 show fast hydrolysis at 310 K, reaching equilibrium in <10 min. Complete conversion of chloride complex 11 to the aqua adduct was observed, whereas 1, and 3–10 reached equilibrium with 30–60% formation of the aqua species (Table S3†).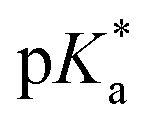 values of the aqua adducts of 1, 8, 11 were determined by 1H NMR. The
values of the aqua adducts of 1, 8, 11 were determined by 1H NMR. The 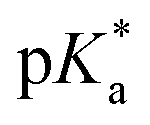 values for the RhIII complexes of 8.8–9.4 (Table S3†) are, ca. 1–2 pKa units higher than for the IrIII analogue 2.13 The pyridine complex 12 showed only ca. 10% hydrolysis even after 10 days under similar experimental conditions.
values for the RhIII complexes of 8.8–9.4 (Table S3†) are, ca. 1–2 pKa units higher than for the IrIII analogue 2.13 The pyridine complex 12 showed only ca. 10% hydrolysis even after 10 days under similar experimental conditions.
When rhodium complexes 1 and 3–10 were dissolved in 60% MeOD-d4/40% D2O and reacted with AgNO3 to generate the corresponding aqua adducts, the 1H NMR spectra after 24 h at 310 K showed the expected peaks for the chelated ligands (bipyridine, phenanthroline or 4,4′-dimethyl-2,2′-bipyridine), but resonances corresponding to the methyl protons in the Cp*, CpxPh and CpxPhPh ligands were not observed as the expected sharp singlets, but as broad signals, difficult to distinguish from the baseline (Fig. 1a, S2†). To identify the species formed, the FT-ICR MS spectrum of each product was recorded after 72 h (Fig. 1a). The MS peaks were assignable to the deuterated-Cpx forms of complexes 1 and 3–10. Hence, facile deuteration of the cyclopentadienyl ligands of these complexes occurs under very mild conditions. Notably however, we observed no deuteration of Ir–bpy complex 2, nor Rh–en complex 11 (Fig. S3†).
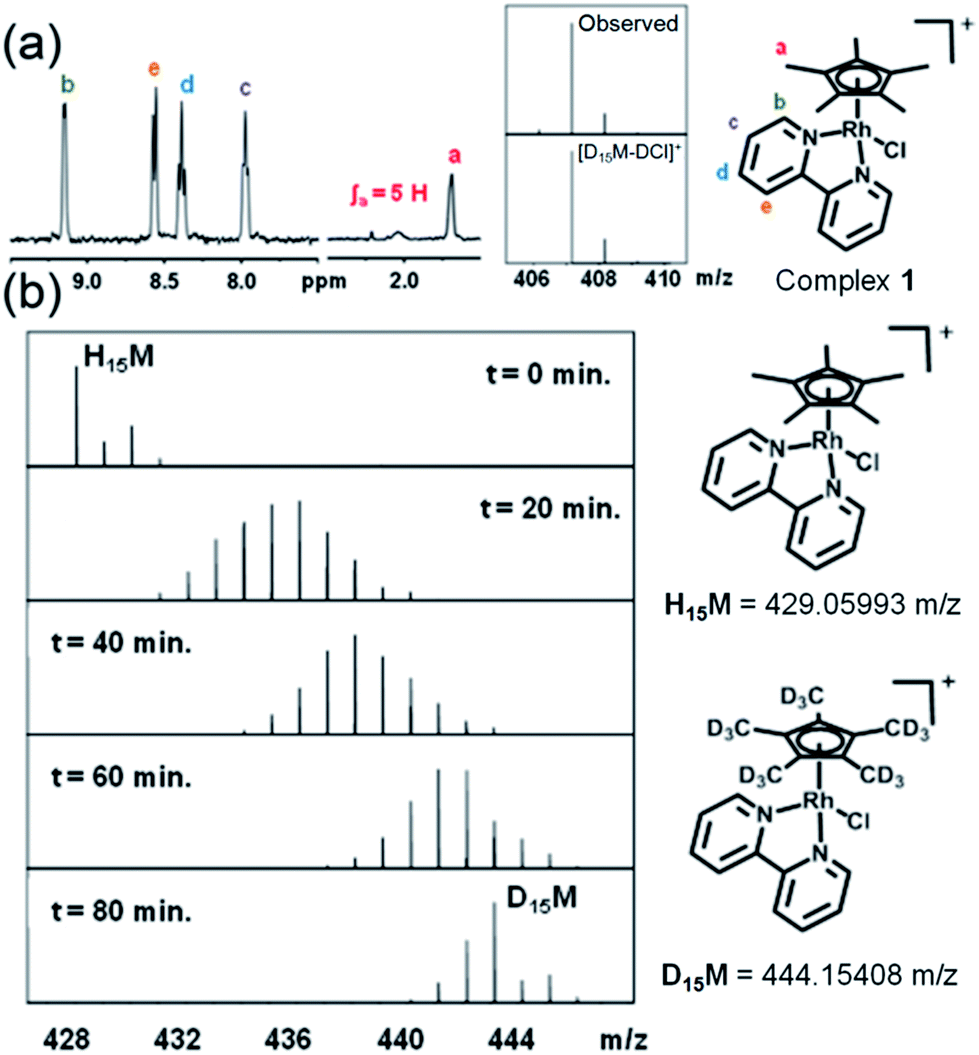 | ||
| Fig. 1 Sequential deuteration of complex 1 monitored by NMR and MS. (a) 1H NMR spectrum of 1 after reaction with AgNO3 in 60% MeOD-d4/40% D2O for 24 h, and FT-ICR mass spectrum after 72 h. (b) FT-ICR mass spectra of 1 treated with 0.95 mol equiv. AgNO3 and incubated at ambient temperature in 60% MeOD-d4/40% D2O. Isotopic pattern at t = 0 min corresponds to 1, and at t = 20, 40, 60 and 80 min to the sum of isotopic patterns for 1 at various stages of deuteration. | ||
Sequential deuteration
Experiments were then performed to investigate the conditions under which deuteration occurs, in particular the effect of time, pH, solvent, hydrolysis and use of AgI to remove the Cl− ligand. 1H NMR spectra of a 1.4 mM solution of complex 1 in 60% MeOD-d4/40% D2O treated with AgI were recorded at ambient temperature after 50 min and then every 410 s. A gradual decrease in intensity, with splitting and broadening of the Cpx methyl peaks was observed. FT-ICR MS spectra were recorded every 10 min for 80 min and then after 27 and 42 h. Gradual deuteration of the Cpx ring methyls was observed and it was evident that deuteration occurs via sequential replacement of methyl protons by deuterium from D1 to D15, complete deuteration (Fig. 1b).Deuteration of Cp* ring methyls of 1 occurred both in the presence and absence of AgI (Fig. S4†). In the absence of AgI, 30–50% hydrolysis was observed as a fast exchange on the NMR timescale. Addition of chloride suppressed both the hydrolysis and deuteration (Fig. S5†). Hence, we concluded that the formation of the Rh–aqua adduct is crucial for the deuteration.
The presence of methanol increased the rate of deuteration of 1 (Fig. S6†), increasing from 6% in D2O, to 20% in 40% D2O/60% MeOD-d4 to 66% in MeOD-d4 alone, after ca. 19 h at 293 K. Further evidence for involvement of the Rh–aqua complex in ring methyl deuteration was provided by [η5-(Cp*)Rh(bpy)(py)](PF6)2 (12). The X-ray crystal structure (Fig. S1, Table S2†) shows a strong Rh–N(pyridine) bond (2.127(4) Å), and even after 10 days at 310 K in 60% MeOD-d4/40% D2O, only 2 D had been incorporated into the Cp*, attributable to a small amount of aqua complex formed (Fig. S7†).
Since deprotonation of Rh–OH2 can provide a strategically-placed base for proton abstraction from Cp*, the effect of pH was investigated (Fig. S8†). Complex 1 in 60% MeOD-d4/40% D2O was treated with NaOD (in absence of AgI). Quantitative full deuteration was achieved at ambient temperature by the time the first 1H NMR spectrum was recorded (ca. 12 min). In contrast, when the sample of 1 was made acidic with DNO3, no deuteration was observed after 12 h. These results are compatible with Rh-OD acting as a base for proton abstraction and subsequent deuteration of the Cp*.
Back exchange
In order to study the reversibility of the deuterium/proton exchange, we prepared fully deuterated [(D15η5-Cp*)Rh(bpy)Cl]PF6 (1D15) and used 2H NMR to show that, as expected, back D/H exchange was slow (ca. 50%) after 3 days in 60% MeOH/40% H2O (Fig. S9†). Such facile deuterium labelling allows a wide range of reactions to be studied by 2H NMR, e.g. reaction with biologically-important molecules such as the tripeptide GSH or stability in complicated biological media such as cell culture medium, where 1H NMR peaks are heavily overlapped37 (exemplified in Fig. S9†).DFT calculations on H/D exchange
These were performed to gain insight into the mechanism of deuteration and the difference in behavior between the bpy and phen complexes, and inactive RhIIIen and IrIIIbpy complexes. Recently it was shown that the DFT modeling can be very helpful to elucidate the mechanism of hydrogen formation catalyzed by RhCp* (bipy) involving the Cp* protonation.38 We explored a mechanism for H/D exchange involving metal–ligand-assisted (M–OH) deprotonation of a Cp* ring methyl to generate a transition state with a bound dianionic ligand [Me4Cp![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2]2− which could then rapidly gain a deuteron from solvent to generate a –CH2D substituent. Rapid methyl and ring rotation can lead to the sequential deuteration of all 15 methyl protons. We modeled the hydroxido adducts using the CAM-B3LYP functional with a CEP-31G basis set. In the cases of both RhIII and IrIII bpyridine complexes [M(Cp*−)(bpy)OH]+, the optimized structures show a relatively close M–O(H)⋯HCH2 contact between the bound hydroxide and a Cp* ring methyl (Fig. 2a), being slightly shorter for Rh (2.165 Å) than Ir (2.227 Å), (Fig. 2b, Table S4†). In contrast, for the en–Rh complex (11), the hydroxide is oriented so as to optimize H-bonding with an en NH2 proton, Rh–O(H)⋯HNH(en) 2.053 Å, Fig. 2e.
CH2]2− which could then rapidly gain a deuteron from solvent to generate a –CH2D substituent. Rapid methyl and ring rotation can lead to the sequential deuteration of all 15 methyl protons. We modeled the hydroxido adducts using the CAM-B3LYP functional with a CEP-31G basis set. In the cases of both RhIII and IrIII bpyridine complexes [M(Cp*−)(bpy)OH]+, the optimized structures show a relatively close M–O(H)⋯HCH2 contact between the bound hydroxide and a Cp* ring methyl (Fig. 2a), being slightly shorter for Rh (2.165 Å) than Ir (2.227 Å), (Fig. 2b, Table S4†). In contrast, for the en–Rh complex (11), the hydroxide is oriented so as to optimize H-bonding with an en NH2 proton, Rh–O(H)⋯HNH(en) 2.053 Å, Fig. 2e.
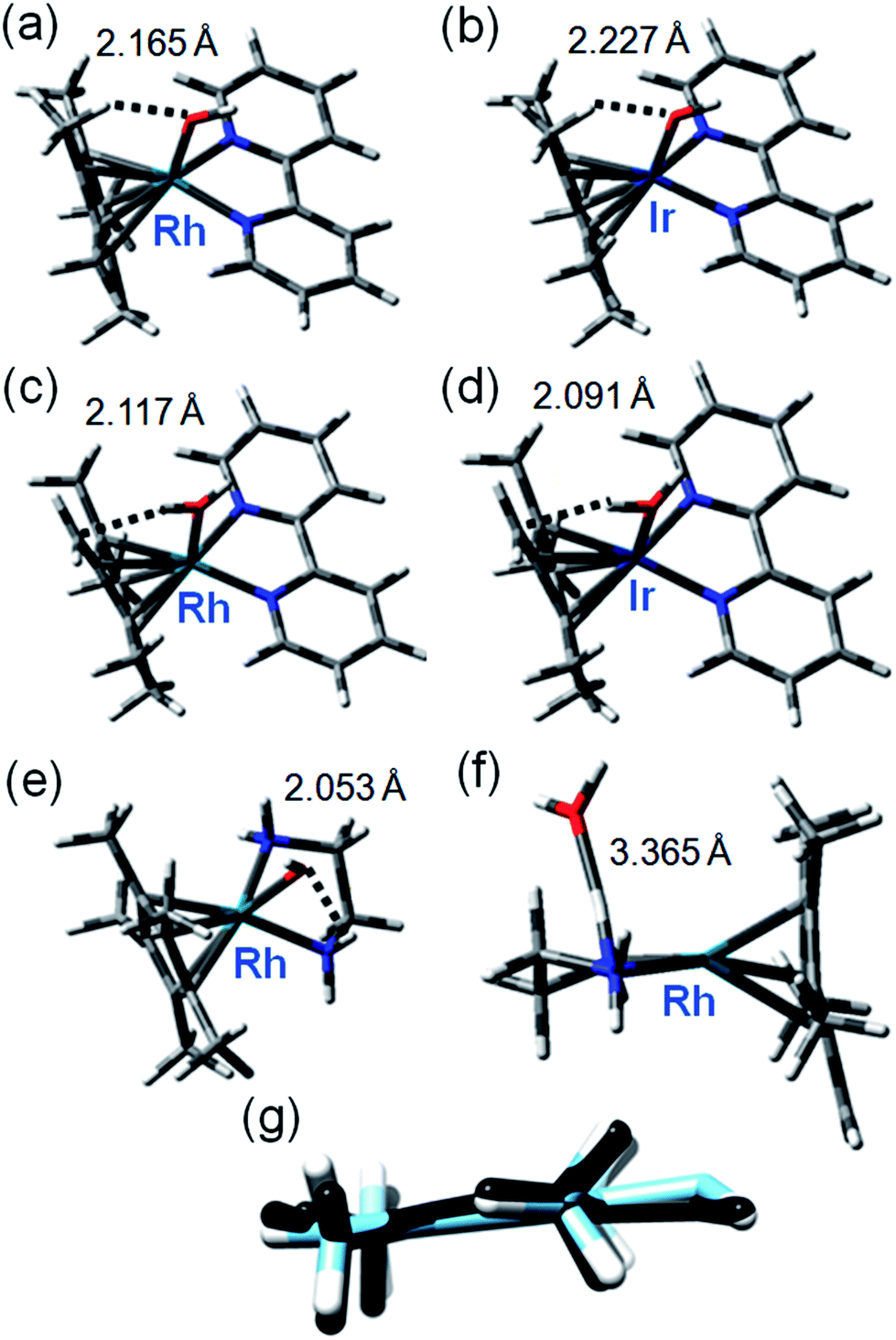 | ||
| Fig. 2 Key structures of hydroxo and aqua/fulvene complexes from DFT calculations. Optimized geometries with important weak interactions are shown. Calculated data for IrIII and RhIII systems are compared in Table S4,† and superposition of relevant OH and H2O complexes in Fig. S10.† (a) [Rh(Cp*)(bpy)OH]; (b) [Ir(Cp*)(bpy)OH]; (c) [Rh(Me4CpCH2)(bpy)OH2]; (d) [Ir(Me4CpCH2)(bpy)OH2]; (e) [Rh(Cp*)(en)OH]; (f) [Rh(Me4CpCH2)(en)OH2]; (g) superimposed structures of [Me4CpCH2]2− dianion in optimized structure of [Rh(Me4CpCH2)(bpy)OH2] (black) and optimized structure of free dianion (blue). | ||
Next we determined the energies of the structures after transfer of a proton from a Cp* methyl group to coordinated hydroxide, giving coordinated dianionic [Me4CpCH2]2− and H2O ligands, Fig. 2c and d. This state for Rh is only 35 kJ mol−1 higher in energy than the original R–OH/Cp*-state (Table S5, Fig. S10†, see the pdb files Rh_bipy_H2O.pdb and Rh_bipy_OH.pdb in ESI†). However, for the Ir complex it is 70 kJ mol−1 higher (Table S5†). The complementary results obtained with TPSSh functional and QZVP basis set were similar, revealing that the [Me4CpCH2]2−/H2O-coordinated chelates lay 28 kJ mol−1 for Rh and 62 kJ mol−1 for Ir above Cp*/M–OH. In the aqua adduct of the en Rh-complex, the H2O ligand is effectively outside the first coordination sphere (Rh–O 3.365 Å, Fig. 2f). In addition, the cyclopentadienyl-based dianion shifts “upwards”, with respect to the plane that approximately bisects that defined by the central ion and chelated nitrogens (Fig. 2f). The superimposed structures of both isomers of Rh and Ir bpy complexes, as well as the Rh en complex are shown in Fig. S11.† The energy barrier associated with the rotation of the cyclopentadienyl ring is low (calculated 6 kJ mol−1), which together with rapid methyl rotation, would allow sequential deprotonation and deuteration of each of the ring methyl protons.
Remarkably, the comparison of the geometries of (a) free deprotonated Cp* ligand (optimized with CAM-B3LYP/CEP-31G), (b) deprotonated Cp* in the optimized structures of the complex, and (c) that of the neutral fulvene Cp* derivative (optimized with CAM-B3LYP/CEP-31G) suggest that the latter two are close in energy to each other, while the former reveals an sp3 character of the CH2 carbon (Fig. S12 and S13†). This allows an alternative description of the complex containing deprotonated Cp* as [RhI/IrI(bpy)(H2O)(Me4CpCH2)]+. Thus, deprotonation of Cp* might introduce new pathways of activity including addition reactions of a bound fulvene, and redox reactions of rhodium.
Our calculations suggest that the π-acceptor strength of the N,N′-chelated ligand plays an important role in stabilizing the aqua/dianionic [Me4CpCH2]2− intermediate in which H/D exchange can readily occur. Within the series of chelates of varying π-character of the N,N′ ligands, the stability of the intermediates increases with increasing π-acceptor character of the ligand. When we increased the π-acceptor strength by adding strongly electron-withdrawing groups as in 1,2-diimino-4,5-dicyanobenzene, the energy difference decreased to only 13 kJ mol−1 above the OH adduct (Table S5).† Interestingly, the energy difference for each Ir system is systematically 35–40 kJ mol−1 higher in energy than that for the corresponding Rh complexes (Table S5†). Thus, DFT modeling implies that the stability of the dianion of the intermediate (or formation of RhI/IrI fulvene complex) is related to delocalization of the negative charge onto the N,N′-chelated ligand via the d-orbitals of the metal. This is facilitated by the π-acceptor character of the N,N′ ligand and is less effective for Ir than for Rh. Alternatively, the enhanced π-acceptor character of the N,N′-chelate stabilizes the lower RhI oxidation state.
Reactions with GSH
Since ene–thiol reactions are well known, and glutathione (GSH, γ-L-Glu-L-Cys-Gly) is prevalent in cells at millimolar concentrations, reactions of GSH were studied with 1 under conditions in which activation of Cp* ring methyls was expected. A solution of 1 and excess GSH at pH 7 gave rise only to MS peaks assignable to [(η5-Cp*)Rh(bpy)(SG)]. Interestingly, the presence of GSH restricted, but did not prevent, ring methyl deuteration (up to 7 days incorporated at pH 7 after 3 days at 310 K, Fig. S14†), DFT calculations on interactions with GSH and modelling (a) [(Cp*)Rh(bpy)(GS)]/H2O, (b) [(Cp*)Rh(bpy)(OH)]+/GSH, (c) [Rh(Me4CpCH2)(bpy)(OH2)]+/GSH, and (d) [Rh(Me4CpCH2)(bpy)]+/GSH/H2O, revealed [Rh(bpy)Cp*(SG)] as the most stable complex (Fig. S15†) formed by coordination of deprotonated GS− sulfur rather than an ene–thiol adduct. Interestingly, the adduct is further stabilized by specific interaction of a deprotonated GS carboxylate group with a Cp* methyl group and bpy 5-H and 6-H (2.50, 2.64 and 2.30 Å, respectively).
Reaction with N-methylmaleimide
Attempts were made to trap a RhI–bound fulvene intermediate via [4+2] cyclo-addition between the dienophile, N-methylmaleimide, and complex 1. Even after 5 days at 310 K no such adduct was observed by 1H NMR or MS, presumably due to steric crowding by the Cp* ring methyls hindering approach of the dienophile. However, N-methylmaleimide clearly interacted with complex 1, [(η5-Cp*)Rh(bpy)Cl]PF6, since with 2 mol equiv. of N-methylmaleimide present in 60% MeOD-d4/40% D2O, complete deuteration was not observed even after 7 days, rather MS peaks for all D1 to D15 sequential steps (Fig. 3a) were present. With 4 mol equiv. of N-methylmaleimide, only D1–D9 species were observed after 7 days (Fig. 3b). A DFT study suggested that N-methylmaleimide interacts strongly with the coordinated water in the fulvene intermediate through a short maleimide CO⋯H–OH bond of 1.68 Å rather than attacking the fulvene double bonds (Fig. S16†). The difference in energy of the complex–maleimide pair and the sum of the energies of the isolated complex and maleimide is 50 kJ mol−1. Thus the fulvene is too crowded to allow a strong interaction with the dienophile, but the strong interaction of N-methylmaleimide with coordinated H2O may result in lower acidity, thus explaining the lack of deuteration.
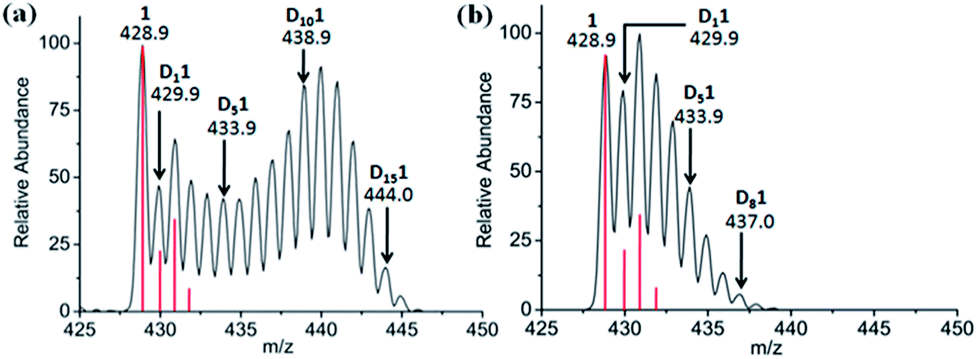 | ||
| Fig. 3 Effect of N-methylmaleimide on deuteration of [(η5-Cp*)Rh(bpy)Cl]+, 1, in 60% MeOD-d4/40% D2O after 7 days at 310 K monitored by MS. (a) 2 mol equiv. and (b) 4 mol equiv. N-methylmaleimide. The red lines show the isotopic distribution for the non-deuterated complex. | ||
Trapping the RhI–fulvene intermediates
[4+2] Diels–Alder reactions with the exocyclic double bond of fulvene as dienophile have been reported in the literature.30,31 The conjugated dienes, isoprene and (9Z,11E)-linoleic acid, were used in attempts to trap the possible fulvene intermediate as [4+2] cyclo-addition Diels–Alder adducts. These are both biologically-important, natural dienes. Isoprene is the most abundant hydrocarbon in human breath, with an estimated human production of 17 mg per day.32,33 (9Z,11E)-Linoleic acid is a common dietary conjugated fatty acid of much interest because of its potential health effects.34,35First, the reaction between complex 1 in 60% MeOD-d4/40% D2O at 310 K and isoprene was studied, since isoprene was expected to offer minimal steric hindrance for the reaction with the proposed exocyclicfulvene CCH2. The ESI-MS spectra clearly indicated formation of a [4+2] cyclo-addition adduct, with the peaks at m/z = 497.1225 assignable to the RhI complex (13+H)+ (Fig. 4). HR-MS data confirmed the formulations C25H30ClN2Rh, Table S6.† Therefore, the data provide strong evidence for the formation of a fulvene intermediate during Cp*–CH3 activation. As expected, adduct formation hinders deuteration. ESI-MS data suggested that the RhI–Cl bond in the fulvene adduct is weak, giving rise to the peak at 461.1459 assignable to [13-Cl]+. This is consistent with the report of a very long RhI–Cl bond (ca. 2.55 Å) in a RhI–(Cp*H) complex.27 DFT calculations indicated lengthening of the Rh–Cl bond by 0.2 Å in the RhI system compared to RhIII, making a square-planar RhI (4d8) adduct accessible (Fig. S17†).
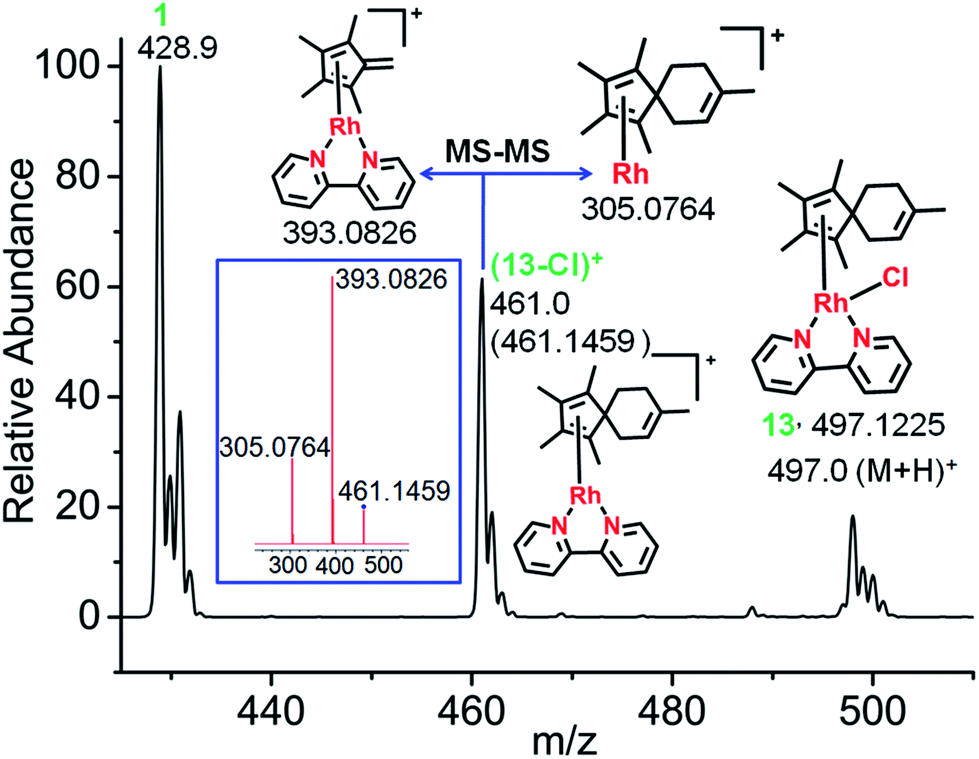 | ||
| Fig. 4 Characterization of the Diels–Alder adduct of complex 1 with isoprene (complex 13) by ESI-MS and MS-MS. The mass spectrum shows the formation of the fulvene-isoprene [4+2] cyclo-addition adduct with coordination of η4-1,2,3,4,8-pentamethylspiro[4,5]deca-1,3,7-triene to RhI, and its fragmentation by MS-MS (blue box). | ||
Adduct formation was also evident from LC-MS data (Fig. S18†), showing a reverse-phase HPLC peak at 25.6 min with an m/z of 461.16 assignable to [13-Cl]+, as was also observed by ESI-MS.
The formation such a [4+2] cyclo-addition adduct was further confirmed by 1H NMR studies. Time-dependent 600 MHz 1H NMR spectra of the reaction mixture of complex 1 and isoprene (10 mol equiv.) in 60% MeOD-d4/40% D2O showed a decrease in the intensity of the isoprene peaks with time, Fig. S19,† accompanied by an increase in the intensity of peaks arising from the formation of the adduct (complex 13). The new peaks due to formation of the [4+2] cyclo-addition adduct are assigned in Fig. 5. The assignments to a Diels–Alder cyclo-addition adduct were made on the basis of 1H COSY and NOESY data (Fig. S20 and S21†).
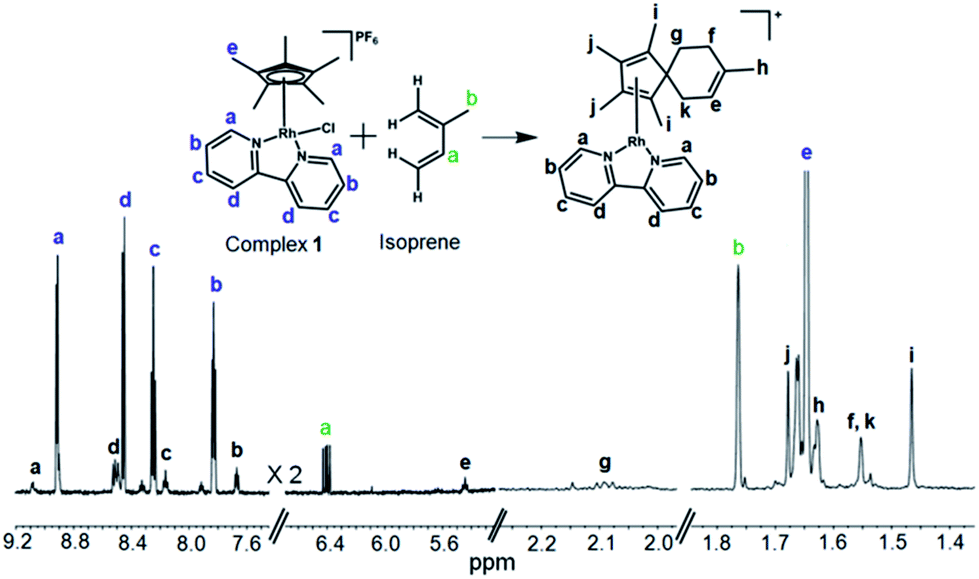 | ||
| Fig. 5 600 MHz 1H NMR characterization of the isoprene adduct complex 13 in 60% MeOD-d4/40% D2O after 3 days at 310 K. δH 9.09 (d, 2H, J = 6.0 Hz), 8.51 (d, 2H, J = 6.1 Hz), 8.17 (t, 2H, J = 8.0 Hz), 7.67 (dd, 2H, J1 = 8.1 Hz, J2 = 6.2 Hz), 5.44 (t, 1H, J = 8.6 Hz), 2.10 (t, 2H, J = 7.0 Hz), 1.68 (s, 6H), 1.63 (s, 3H), 1.55 (s, 4H), 1.47 (s, 6H). The peaks form excess isoprene (10 mol equiv.) from 4.9–5.2 ppm are shown in Fig. S19.†. 2D COSY NMR data (Fig. S20†) indicated the coupling of peak “f” (1.55 ppm) with peak “g” (2.10 ppm), and peak “k” (1.55 ppm) with “e” (5.43 ppm). The 2D NOESY spectrum (Fig. S21†) indicated cross-correlation of peak “g” (2.10 ppm) with peaks “h” (1.47 ppm) and “k” (1.55 ppm) suggesting that these protons are closer than ca. 5 Å (for a model see Fig. S21†). | ||
The MS and NMR data provide the first experimental evidence for the formation of a fulvene intermediate trapped during the course of Cp*–CH3 activation under such mild conditions in aqueous media, and this appears to be the first report of a Diels–Alder reaction of a metal-bound fulvene in aqueous solution. There appears to be only one previous report on the promotion of a Diels–Alder reaction through enhancement of diene reactivity through metal coordination.39 Interestingly, we detected the same Diels–Alder adduct by HRMS when complex 1 was incubated with isoprene (10 mol equiv.) in RPMI-1640 cell culture medium supplemented with 10% fetal calf serum, a typical medium used for culture of cancer cells (Fig. S22†).
The reaction of [(Cp*)Rh(bpy)Cl]+ with isoprene in water was modeled using DFT calculations (Fig. 6). The results indicate the relative stability of the initial RhIII complex with the lowest energy corresponding to [(Cp*)Rh(bpy)OH]+. The formation of the Diels–Alder adduct from the fulvene species [(Me4CpCH2)RhI(bpy)OH2]+ is energetically very favourable (106 kJ mol−1), while the corresponding end product [(adduct)RhI(bpy)]+ is presumably driven by entropic effects, with a calculated energy difference between it and the [(adduct)RhIII(bpy)](OH2) of only 3 kJ mol−1. The change of the oxidation state is suggested by the calculated Rh–N bond lengths of 2.141 and 2.192 Å for the species with coordinated water, and 2.106 and 2.102 Å for the final product.
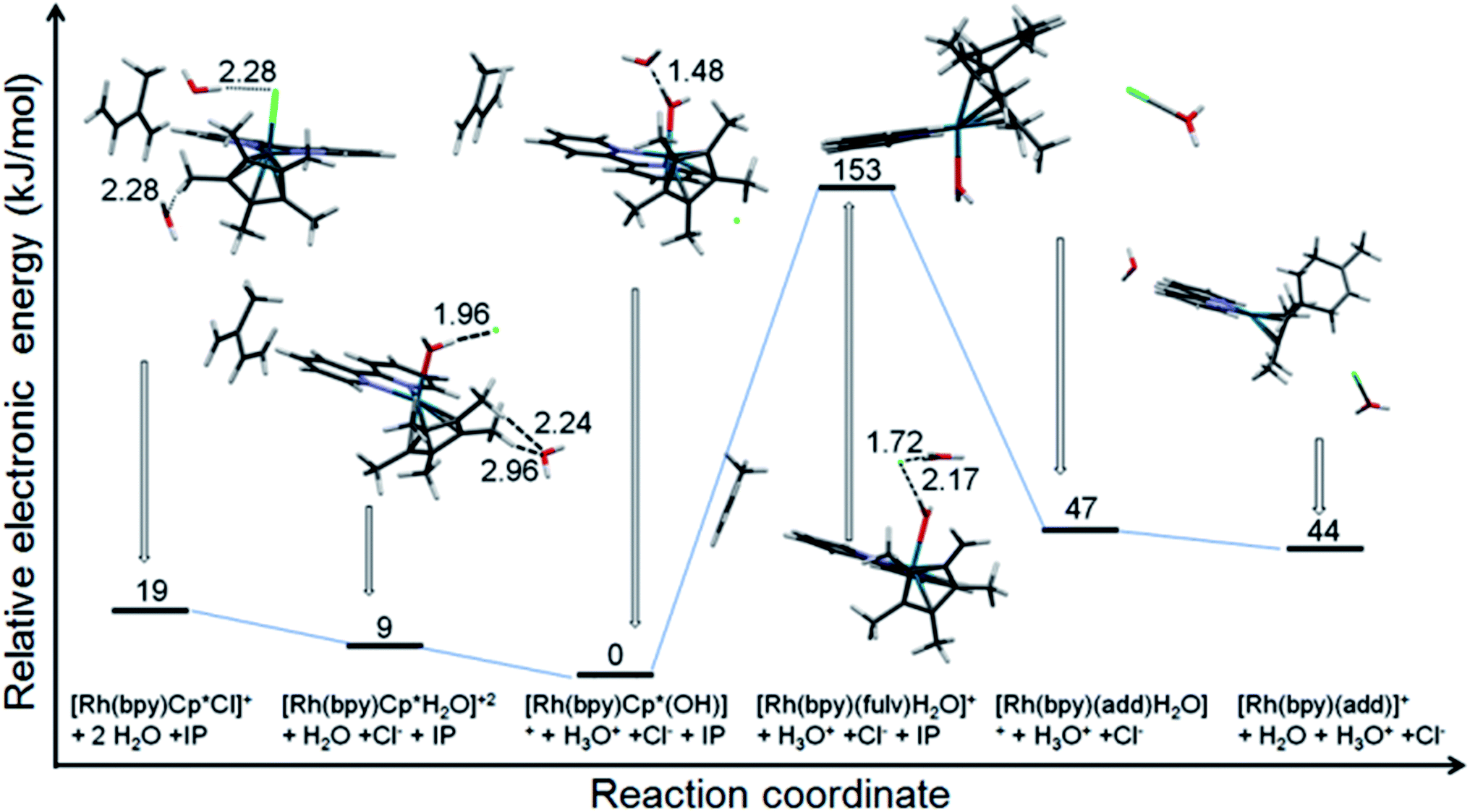 | ||
| Fig. 6 Electronic energy profile for the reaction of [(Cp*)Rh(bpy)Cl]+ with isoprene in water at neutral pH (calculated using CAM-B3LYP/CEP-31g, solvent = water, IEFPCM model). The pdb files (pdb files bp1-6.pdb) of the optimised structures of all six species are supplied as ESI.† | ||
Next, the reactions of complex 1 with the conjugated fatty acid, (9Z,11E)-linoleic acid, under similar experimental conditions in water, and in RPMI-1640 cell culture medium supplemented with 10% of fetal calf serum, were studied. The HRMS and MS-MS data again clearly indicated the formation of the fulvene-diene cyclo-addition adduct (Fig. 7 and S23†), with peaks at m/z = 673.3235 and 674.3298 assignable to the RhI complexes [14-Cl]+ and [D114-Cl]+, respectively. HR-MS data confirmed the formulations as C38H54N2O2Rh and C38H53DN2O2Rh, Table S7.† Interestingly, adduct formation is reversible, as shown in Fig. S24.† Dilution of the reaction mixture leads to the regeneration of the parent RhIII complex suggesting possible use as a drug delivery vehicle.
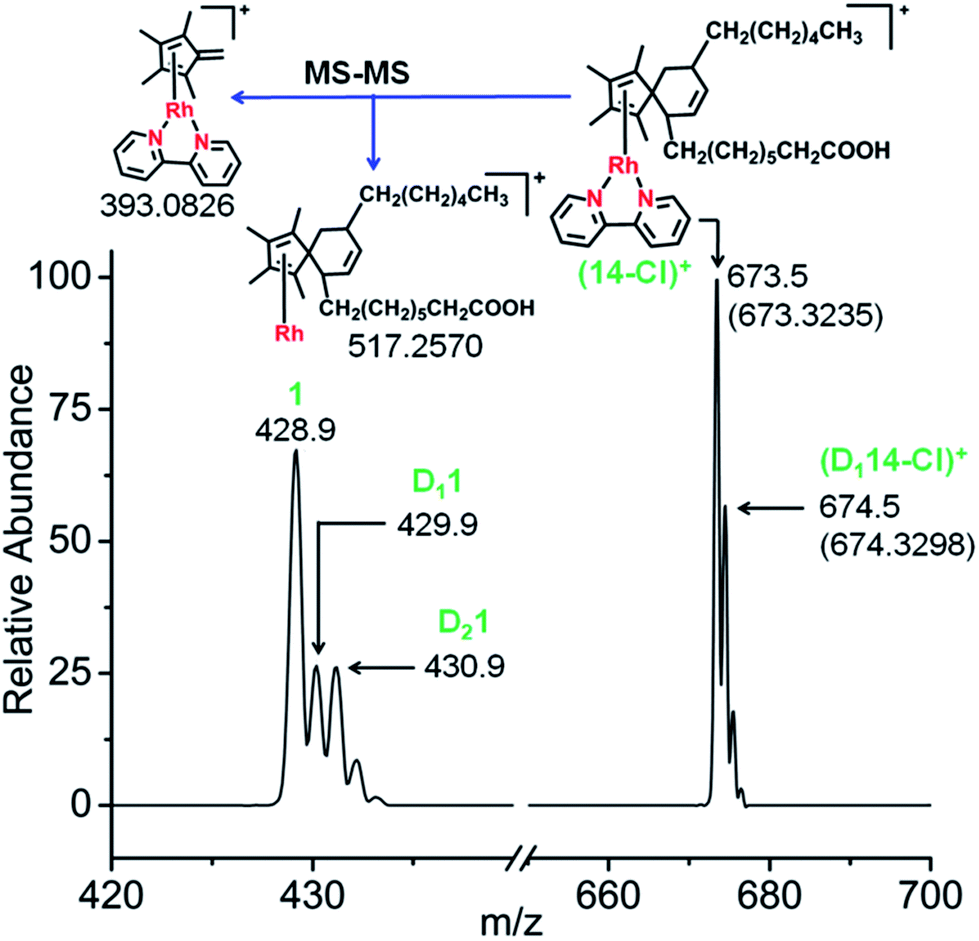 | ||
| Fig. 7 Mass spectra of complex 1 after reaction with conjugated fatty acid, (9Z,11E)-linoleic acid showing the formation of the fulvene-linoleic acid [4+2] cyclo-addition adduct, complex 14. | ||
We investigated the formation of [4+2] cyclo-addition adducts between the biphenyl-substituted Cp* complex 10, an active anticancer complex, with isoprene in 60% MeOD-d4/40% D2O at 310 K. This gave rise to a product complex 15, as evidenced by ESI-MS, HRMS and LC-MS data (Fig. S25 and S26†), which suggested that a similar cyclo-addition adduct was formed. The peaks at m/z = 623.1928, 624.1980 and 625.2030 are assignable to the RhI complexes [15-Cl]+, [D115-Cl]+ and [D215-Cl]+ respectively (Fig. S25†). HR-MS data confirmed the formulations C38H36N2Rh, C38H35DN2Rh and C38H34D2N2Rh. The presence of isomers in the product was evident from LC-MS data (Fig. S26†), and reflect the non-equivalence of methyl deprotonation sites on the bound CpxPhPh. Two reverse-phase HPLC peaks were detected at 32.3 min with an m/z of 623.49 assignable to [15-Cl]+ and 35.2 min with an m/z 623.48. These data are in good agreement with the observed ESI-MS results.
They, therefore, establish the formation of RhI–fulvene intermediates during Cp*–CH3 activation in reactions with conjugated dienes which are abundant in the body. These observations are of particular interest for the future design of organometallic half-sandwich anticancer complexes.13 For example, it might be possible to use such conjugates to deliver and release complexes in specific cellular organelles.
Conclusions
It is difficult to activate C–H bonds of hydrocarbons, especially CH3 groups, in water under mild conditions, but we show that this is facile for certain RhIII cyclopentadienyl complexes. DFT calculations revealed the role of strategically-placed Rh-OD groups in abstracting protons from coordinated Cpx rings aided by the presence of chelated π-acceptor diamine ligands such as bpy or phen (Fig. 8). These ligands help to stabilize the deprotonated Cpx–H dianion. The Cpx ring behaves like a ‘molecular twister’ with low barriers to rotation about the Cpx–Rh and ring-CH3 axes, allowing rapid sequential deuteration. The readily prepared deuterated products are useful for 2H NMR studies in situations where overlap in 1H NMR spectra hinders studies, for example in cell culture media (Fig. S9†).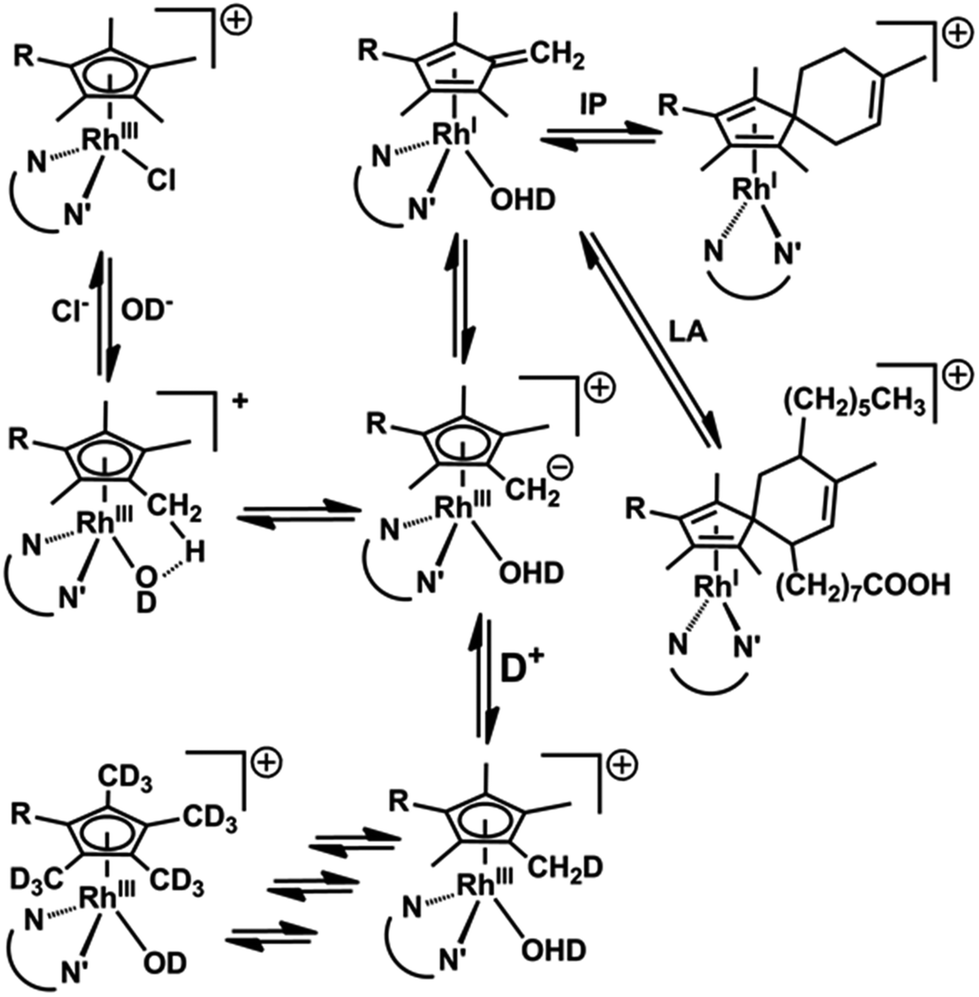 | ||
| Fig. 8 Reaction pathways for Cpx methyl deuteration and RhI fulvene-diene adduct formation. | ||
The DFT calculations predicted the accessibility of RhI–fulvene intermediates formed via 2-electron transfer from the dianionic Cpx2− ligand. We were not able to generate addition products between the thiol of glutathione and the exocyclic CCH2, or with the dienophile N-methylmaleimide, although they can control the extent of ring deuteration (Fig. 3 and S14†). However, the trapping of the RhI–fulvene intermediates was surprisingly facile in Diels–Alder [4+2]-cycloaddition reactions with natural conjugated dienes. This appears to be the first report of metal-assisted in situ formation of a dieneophile, and trapping of that dieneophile by a Diels–Alder cyclo-addition reaction of a metal-bound fulvene in aqueous solution.
Adducts with isoprene, an abundant diene in human breath, and with the common dietary diene conjugated (9Z,11E)-linoleic acid formed readily in aqueous solution. They might easily form in cells, and influence the redox state of cells and biological activity of this class of organo–rhodium anticancer complexes. The Diels–Alder adducts formed readily in biological media, e.g. cell culture medium (Fig. S22 and S23†) and since they are reversible (Fig. S24†), open up the possibility of designing novel drug targeting and delivery systems. Interestingly, the most active anticancer complexes in the limited set we have studied here, are those that have activated Cpx rings (Table S3†). The possible involvement of such Diels–Alder adducts in the biological activity of this class of complexes will be studied in future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC (grant no. 247450), EPSRC (grant no. EP/F034210/1), Warwick Institute of Advanced Studies, Alexander von Humboldt scholarship for J. J. S. B., The Royal Society and SERB (Newton International Fellowship for S. B.), China Scholarship Council (studentship for F. C.) and Bruker UK Ltd (Warwick Collaborative Studentship for C. A. W.). V. S. and J. A. W. acknowledge support by the research initiative NANOKAT and the German Federal Ministry of Education and Research (BMBF), as well as Allianz für Hochleistungsrechnen Rheinland-Pfalz (AHRP) for providing CPU-time within the project TUK-SPINPLUSVIB. We also thank Dr David Fox (University of Warwick) and members of COST Action CM1105 for stimulating discussions.Notes and references
- J. M. O'Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- D. L. Huang, D. J. Vinyard, J. D. Blakemore, S. M. Hashmi and R. H. Crabtree, Organometallics, 2017, 36, 199–206 CrossRef CAS.
- A. J. Bloomfield, A. J. Matula, B. Q. Mercado, V. S. Batista and R. H. Crabtree, Inorg. Chem., 2016, 55, 8121–8129 CrossRef CAS PubMed.
- R. Pettinari, F. Marchetti, C. Pettinari, F. Condello, A. Petrini, R. Scopelliti, T. Riedel and P. J. Dyson, Dalton Trans., 2015, 44, 20523–20531 RSC.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC.
- N. Schröder, J. Wencel-Delord and F. Glorius, J. Am. Chem. Soc., 2012, 134, 8298–8301 CrossRef PubMed.
- C. Li and J. Xiao, J. Am. Chem. Soc., 2008, 130, 13208–13209 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- Z. Almodares, S. J. Lucas, B. D. Crossley, A. M. Basri, C. M. Pask, A. J. Hebden, R. M. Phillips and P. C. McGowan, Inorg. Chem., 2014, 53, 727–736 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, A. Habtemariam, I. Romero-Canelón and P. J. Sadler, J. Inorg. Biochem., 2015, 153, 322–333 CrossRef CAS PubMed.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., 2014, 126, 4022–4027 CrossRef.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed.
- J. E. McMurry, Organic Chemistry, Brooks-Cole, Pacific Grove, CA, 5th edn, 2000 Search PubMed.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed.
- S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538–12542 CrossRef CAS PubMed.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS PubMed.
- S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544–6556 RSC.
- D. C. McAteer, E. Javed, L. Huo and S. Huo, Org. Lett., 2017, 19, 1606–1609 CrossRef CAS PubMed.
- J. W. Kang and P. M. Maitlis, J. Organomet. Chem., 1971, 30, 127–133 CrossRef CAS.
- J. E. Bercaw, R. H. Marvich, L. G. Bell and H. H. Brintzinger, J. Am. Chem. Soc., 1972, 94, 1219–1238 CrossRef CAS.
- C. S. Wei, C. A. Jiménez-Hoyos, M. F. Videa, J. F. Hartwig and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 3078–3091 CrossRef CAS PubMed.
- G. Ciancaleoni, S. Bolano, J. Bravo, M. Peruzzini, L. Gonsalvi and A. Macchioni, Dalton Trans., 2010, 39, 3366–3368 RSC.
- L. M. A. Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard III, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6409–6414 CrossRef PubMed.
- C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Chem. Commun., 2016, 52, 9105–9108 RSC.
- Y. Peng, M. V. Ramos-Garcés, D. Lionetti and J. D. Blakemore, Inorg. Chem., 2017, 56, 10824–10831 CrossRef CAS PubMed.
- W. C. Henke, D. Lionetti, W. N. G. Moore, J. A. Hopkins, V. W. Day and J. D. Blakemore, ChemSusChem, 2017, 10, 4589–4598 CrossRef CAS PubMed.
- P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930–3989 CrossRef CAS PubMed.
- A. Z. Bimanand, Y. N. Gupta, M. J. Doa, T. A. Eaton, K. N. Houk and F. R. Fronczek, J. Org. Chem., 1983, 48, 403–405 CrossRef CAS.
- D. Gelmont, R. A. Stein and J. F. Mead, Biochem. Biophys. Res. Commun., 1981, 99, 1456–1460 CrossRef CAS PubMed.
- J. King, H. Koc, K. Unterkofler, P. Mochalski, A. Kupferthaler, G. Teschl, S. Teschl, H. Hinterhuber and A. Amann, J. Theor. Biol., 2010, 267, 626–637 CrossRef CAS PubMed.
- G. Fuke and J. L. Nornberg, Crit. Rev. Food Sci. Nutr., 2017, 57, 1–7 CrossRef CAS PubMed.
- A. A. Hennessy, P. R. Ross, G. F. Fitzgerald and C. Stanton, Lipids, 2016, 51, 377–397 CrossRef CAS PubMed.
- M. A. Scharwitz, I. Ott, Y. Geldmacher, R. Gust and W. S. Sheldrick, J. Organomet. Chem., 2008, 693, 2299–2309 CrossRef CAS.
- J. L. Wagstaff, R. J. Masterton, J. F. Povey, C. M. Smales and M. J. Howard, PLoS One, 2013, 8, e77195 CAS.
- S. I. Johnson, H. B. Gray, J. D. Blakemore and W. A. Goddard III, Inorg. Chem., 2017, 56, 11375–11386 CrossRef CAS PubMed.
- W. Liu, F. You, C. J. Mocella and W. D. Harman, J. Am. Chem. Soc., 2006, 128, 1426–1427 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1420046–1420052 and 1569283. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05058e |
| ‡ Equal contributions, joint first authors. |
| This journal is © The Royal Society of Chemistry 2018 |