
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization reactions†
Nastaran
Salehi Marzijarani
a,
Roozbeh
Yousefi
a,
Arvind
Jaganathan
b,
Kumar Dilip
Ashtekar
a,
James E.
Jackson
 *a and
Babak
Borhan
*a
*a and
Babak
Borhan
*a
aDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA. E-mail: babak@chemistry.msu.edu; jackson@chemistry.msu.edu
bDow AgroSciences LLC, 9330 Zionsville Road, Indianapolis, IN 46268, USA
First published on 2nd January 2018
Abstract
Though (DHQD)2PHAL-catalyzed chlorocyclizations of 1,1-disubstituted olefins show useful (and in some cases, reversible) asymmetric induction, stereochemically complete descriptions of these alkene additions have remained largely unknown. Herein, based on a combination of NMR, derivative, isotope labeling, and computational studies, we present detailed stereochemical analyses of chlorocyclizations of nucleophile-tethered 1,1-disubstituted styryl systems. The selectivities of the two asymmetric bond-forming processes, namely electrophilic chlorine attack and nucleophilic ring closure, are thus mapped out independently. Under the established optimal conditions, four related chlorocyclizations were subjected to this analysis. All showed a strong preference for Cl+ delivery from the same face of the alkene. However, depending on reaction conditions and substrate identity (carboxylic acid, amide or carbamate), the internal nucleophiles may close with a strong net preference for either syn or anti addition relative to the Cl atom. Studies of both uncatalyzed and (DHQD)2PHAL-catalyzed processes place new boundary conditions on the role of the catalyst in these reactions.
Introduction
With the advent of effective asymmetric catalytic methods, halocyclizations (and more broadly, electrophilic haloadditions to alkenes) have now emerged as useful tools for synthesis of chiral targets.1 Like the Nobel prize-winning asymmetric epoxidation and dihydroxylation reactions, halocyclization to alkenes had been known for many decades. But despite its obvious desirability and earlier valiant attempts, asymmetric control with significant enantiopreferences remained elusive. Reported in 2010,2 efforts from our group (and now many others) then discovered effective reagents, catalysts, and conditions to achieve chlorocyclizations with useful levels of stereocontrol, opening the floodgates of empirical exploration and synthetically valuable discovery, but leaving mechanistic understanding behind.Broadly, stereocontrol requires the reaction environment to be desymmetrized with a chiral catalyst that activates the reaction while guiding bond formation to prefer one face of the alkene. Many asymmetric halocyclizations of alkenes have now been described using a variety of chiral catalysts. Chiral phosphoric acid catalysts or related analogs were shown to catalyze bromo-amination and -etherification by Shi and coworkers;3 bromoetherification by Denmark and Burk;4 fluoro/bromo/iodo cyclization of amides by Toste and coworkers;5 bromo/iodo etherification by Hennecke and coworkers;1f,6 and iodolactonization by the Ishihara group.7 Fujioka and coworkers disclosed an asymmetric bromolactonization of olefins catalyzed by chiral benzene trimers.8 Yeung and coworkers showed that bromo-amination and etherification can be catalyzed by C2 symmetric seleno-THF analogs with high enantioselectivity,9 whereas asymmetric bromolactonization is achieved by chiral pyrrolidines possessing a thiocarbamate functionality.10 More recently the Johnston and Hansen groups reported asymmetric iodolactonizations mediated by chiral PBAM and squaramide based catalysts, respectively.11 An elegant report by Jacobsen and coworkers showed iodocyclizations mediated by urea/thiourea catalysts with high enantioinduction.12 New binaphthyl analogs reported by Martin and coworkers proved efficient as catalysts for asymmetric bromolactonization.13 Most directly relevant to the present report, several research groups including those of Sun, Yeung, Mukherjee, and Tang demonstrated highly enantioselective intramolecular halofunctionalization reactions catalyzed by monomeric cinchona alkaloid derivatives.2b,14 A number of excellent reviews can be consulted for a complete coverage of the literature in this area.1
Our own and others' studies have uncovered several stereoselective halofunctionalizations based on the organocatalyst (DHQD)2PHAL.2a,14b,15 This C2 symmetric cinchona alkaloid derivative efficiently catalyzes the reaction of substrates bearing hydrogen-bond donor groups such as unsaturated amides, carbamates, naphthols and carboxylic acids. In all these asymmetric halocyclization reactions, the olefin undergoes electrophilic attack by a halenium ion (X+) donor with intramolecular ring closure by a pendant nucleophile. Mechanistically, halofunctionalization of alkenes has been extensively studied since their discovery. The exclusive formation of anti-products from olefin halogenation led Kimball in 1937 to propose a stepwise mechanism, with symmetrically bridged haliranium ions as putative intermediates.16 However, the groups of Fahey, Sauers and others provided firm evidence for open β-halocarbenium ion intermediates in halofunctionalizations of unsymmetrical alkenes, especially those with aryl or other π-delocalizing substituents.17 Furthermore, seminal works by Fahey, Poutsma, Williams, and others concluded that neither the bridged nor the open cationic intermediates are completely compatible with the observed experimental outcomes.18 Specifically, reaction rates were accelerated by proximity of the intramolecular nucleophile to the alkene, pointing clearly to a concerted AdE3-type mechanism.17a,19 Our own recent work in this area has reaffirmed and further illustrated the critical role of the nucleophilic addition partner in activating olefins to abstract the electrophilic halenium ion from its donor reagent. Here, the intrinsic halenium ion affinity20 of the alkene pi system is boosted by contact with the nucleophile, favoring AdE3-type concerted halofunctionalization. This nucleophile assisted alkene activation (NAAA)21 pathway represents a less familiar but often dominant alternative to the usual textbook scheme of stepwise halofunctionalization wherein initial formation of a bridged halonium ion is followed by its anti opening with a nucleophile.
With asymmetric halocyclizations empirically established as useful synthesis tools, we turned to mechanistic studies of reactions developed in our labs over the past six years. Fig. 1 illustrates the four chlorocyclizations chosen for study.2a,15b,15d In the unlabeled systems as originally reported, the newly formed sp3 CH2Cl center lacked observable stereochemistry, leaving the face selectivity of chlorination unknown. To reveal the relative and absolute stereochemical outcomes of both the halenium ion attack and the nucleophilic closure in halocyclizations, we resorted to deuterium-labeling in the 1,1-disubstituted olefin substrates. In particular, we investigated the syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti selectivity of addition across the double bond, both for non-catalyzed reactions (to evaluate their intrinsic reactivity) and for their (DHQD)2PHAL-catalyzed analogues.
:anti selectivity of addition across the double bond, both for non-catalyzed reactions (to evaluate their intrinsic reactivity) and for their (DHQD)2PHAL-catalyzed analogues.
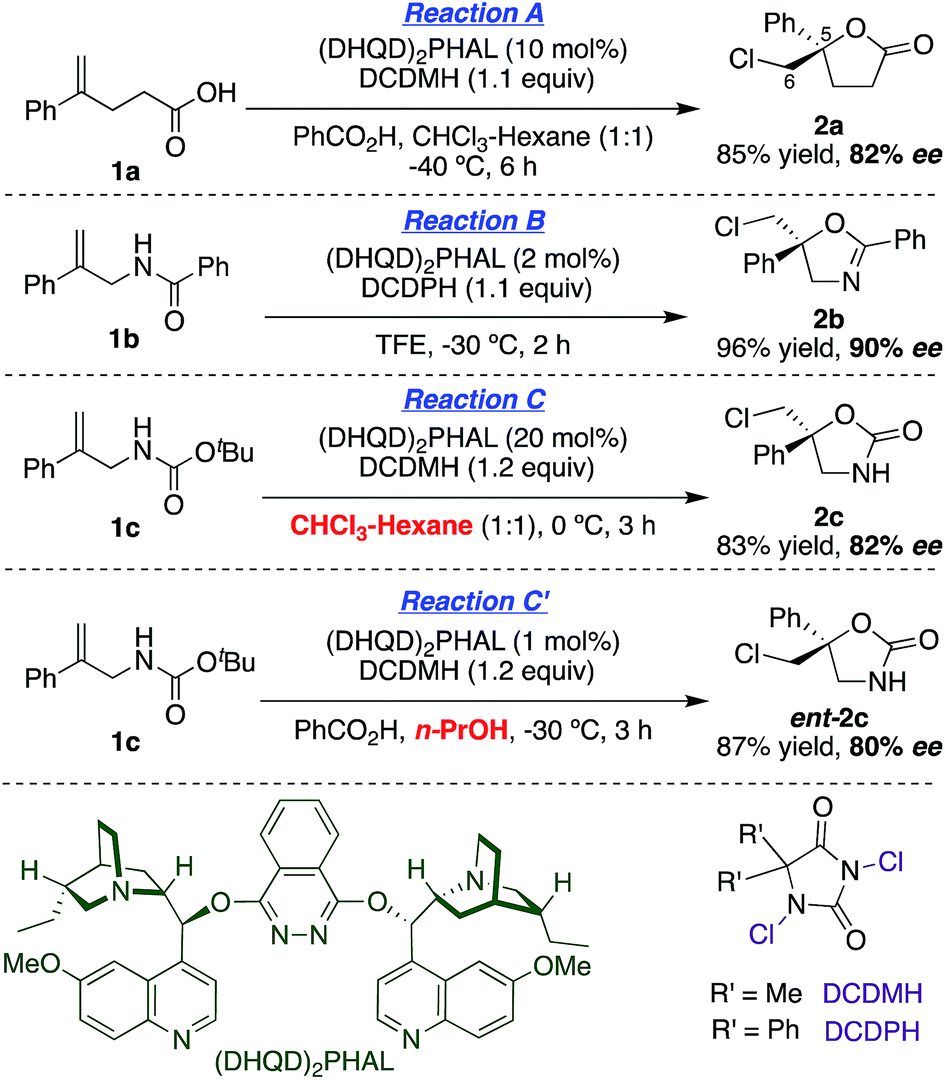 | ||
| Fig. 1 Asymmetric chlorocyclization of alkenoic acid 1a (Reaction A), unsaturated amide 1b (Reaction B), and carbamate 1c under two different conditions (Reactions C and C′). | ||
Results and discussion
This study sought to uncover and understand the stereochemical relationships between the chlorenium ion delivery and the nucleophilic ring closure events that form the adducts shown in Fig. 1. Some key similarities and differences among the reactions of carboxylic acid 1a, and those of amide 1b and carbamate 1c, should be noted at the outset. As earlier reported, (a) all these reactions employ (DHQD)2PHAL as the chiral catalyst and chlorinated hydantoins as the electrophilic chlorinating agents. (b) The configuration of the newly created stereogenic carbon (C5) in the product is nucleophile dependent. (c) In the case of the carbamate substrate 1c, the solvent can also modulate the C5 configuration.To understand these diverse behaviors, we asked the following questions: (a) is there a facial preference for electrophilic chlorine attack on the 1,1-disubstituted olefin? (b) If so, how strong is the preference, and how does it vary among substrates and conditions? (c) What patterns (if any) of stereochemical relationships are found between the chlorine atom and the nucleophile in the final adduct? Net syn or anti halocyclization would shed light on the nature of the reaction path. (d) Is the overall reaction concerted or stepwise; and if the latter, what sort of intermediate (e.g. bridged chloriranium ion, open benzylic carbocation, other?) might be formed? (e) How is the ultimate enantioselectivity set?
As in our previous mechanistic investigations of Reaction A,22 the isotopic labeling enables full characterization of the addition stereochemistry in the chlorocyclization of unsaturated amides (Reaction B) and carbamates (Reactions C and C′, Fig. 1). The presence of the deuterium in 1a-D, 1b-D, and 1c-D leads to diastereomeric products that reveal not only the face selectivity of chlorenium ion attack on the olefin, but also the syn or anti relationship of addition between the delivered halogen and the captured nucleophile. The results of these studies show a diversity of relative and absolute stereochemical fates in both chlorenium ion delivery and ring closing processes. They also highlight the multiple ways that the catalyst can modulate reactivity.
Synthesis of labeled substrates
The synthesis of the E-deuterated substrates 1b-D and 1c-D was accomplished in four steps (Scheme 1). The deuterium was incorporated through palladium-catalyzed syn hydrophenylation of 4 with sodium tetraphenylborate in D2O/acetic acid to afford 5.23 Hydrolysis of imide 5 and subsequent derivatization of the deuterated alkenoic amine 6 with the appropriate acylating agent led to the formation of the desired substrates 1b-D and 1c-D with a high level of deuterium incorporation and good E/Z selectivity. For the final analysis of the chlorocyclization product ratios, the stereochemical impurity (∼94%) of the deuterated substrates 1b-D and 1c-D was taken into account by the mathematical treatment shown in the ESI.†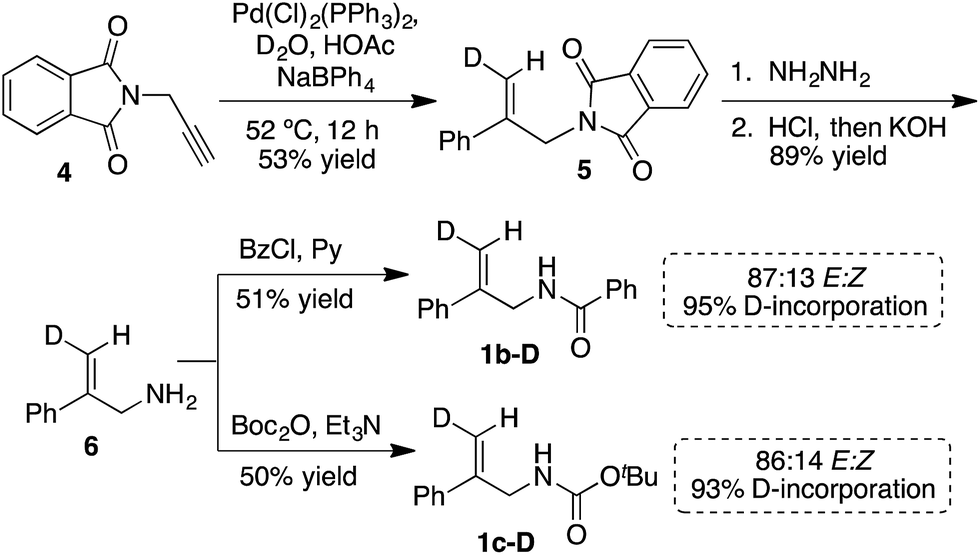 | ||
| Scheme 1 Synthesis of 1b-D and 1c-D. | ||
Absolute stereochemical determination
(DHQD)2PHAL catalyzed chlorocyclizations of 1b-D and 1c-D (vide infra) led to mixtures of diastereomeric products. The absolute configuration of the deuterated chlorocyclized products 2b-D and 2c-D at C6 was determined via transformation of the major product of both deuterated and non-deuterated substrates to an epoxide with known configuration (Scheme 2, top). Hydrolysis of oxazoline 5R-2b-D (absolute configuration at C5 was determined previously by X-ray crystallography)15d with HCl afforded the N-benzoyl β-amino alcohol 7-D (Scheme 2, top). The resulting halohydrin intermediate was treated with K2CO3 to afford the 1,1-disubstituted epoxide 3b-D under mild conditions. Non-deuterated epoxy amide 3b was synthesized similarly. ROESY and NOESY studies on the epoxy amide 3b established the relative stereochemistry of Ha (2.80 ppm, cis) and Hb (3.10 ppm, trans) with respect to the phenyl group. 1H NMR analysis of the epoxy amide 3b-D, obtained from the product of the chlorocyclization of 1b-Dvia Reaction B conditions, exhibits the peak at 3.10 ppm, establishing that the deuterium has a cis orientation with respect to the phenyl group. This leads to the assignment of R configuration for the carbon bearing the deuterium in epoxy amide 3b-D. Since the epoxy amide is formed through the SN2 closure of the corresponding chlorohydrin intermediate, the S configuration at C6 is assigned for amide 2b-D (major product of Reaction B, Scheme 2, top).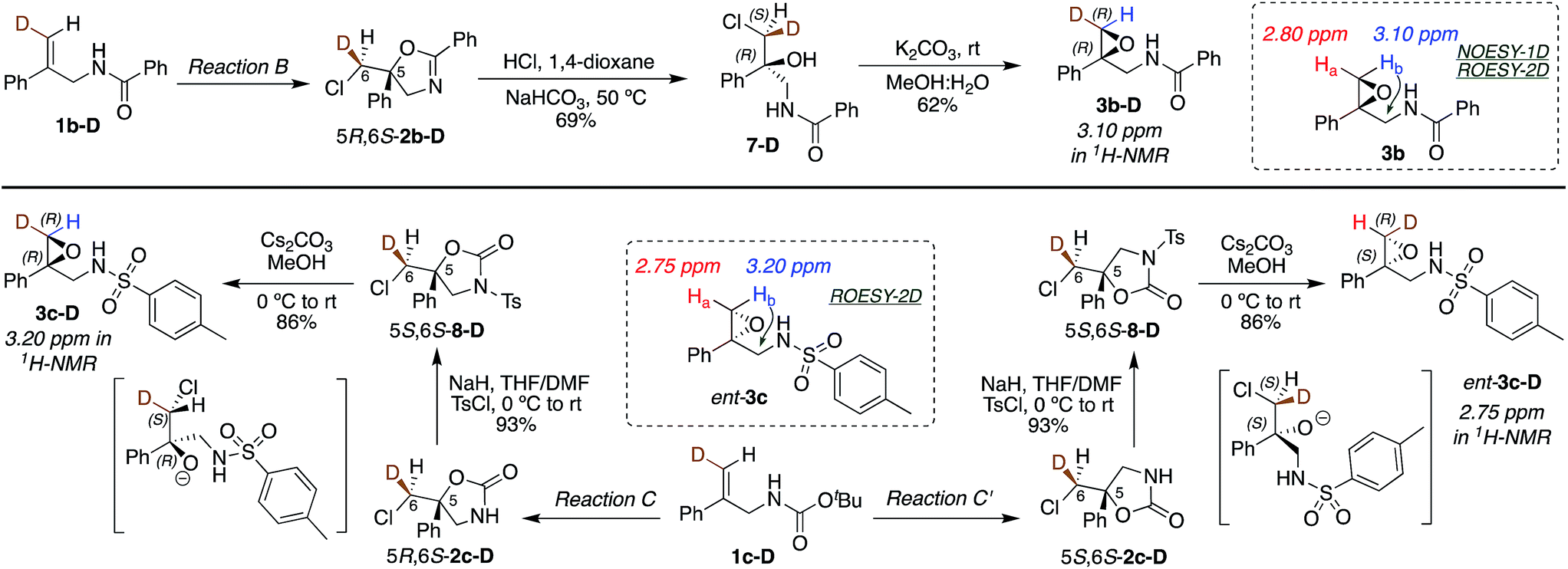 | ||
| Scheme 2 Absolute stereochemical assignment at the deuterated center (C6) for substrates 2b-D (top) and 2c-D and ent-2c-D (bottom). | ||
In an analogous study, the absolute configuration at C6 in products obtained from the chlorocyclization of the carbamate 1c-D under conditions denoted as Reaction C and C′ were determined (Scheme 2, bottom). Tosyl protection of oxazolidinone 5R-2c-D and 5S-2c-D (configuration of C5 was established previously via X-ray crystallography),15b followed by CsCO3 mediated ring opening of the resulting chlorohydrin intermediate, gave 1,1-disubstituted epoxy sulfonamide 3c-D and ent-3c-D. The non-deuterated epoxy amide ent-3c was synthesized from 5S-2c following the same reaction protocols. ROESY analysis of epoxy amide ent-3c indicated that Ha (2.75 ppm) is cis to the phenyl group, while Hb (3.20 ppm) is trans. 1H NMR data obtained for the epoxides derived from the major products of Reaction C and C′ chlorocyclization reveal that the absolute configuration on the labeled C6 center of the cyclized products 5R-2c-D and 5S-2c-D is S for both (Scheme 2, bottom).
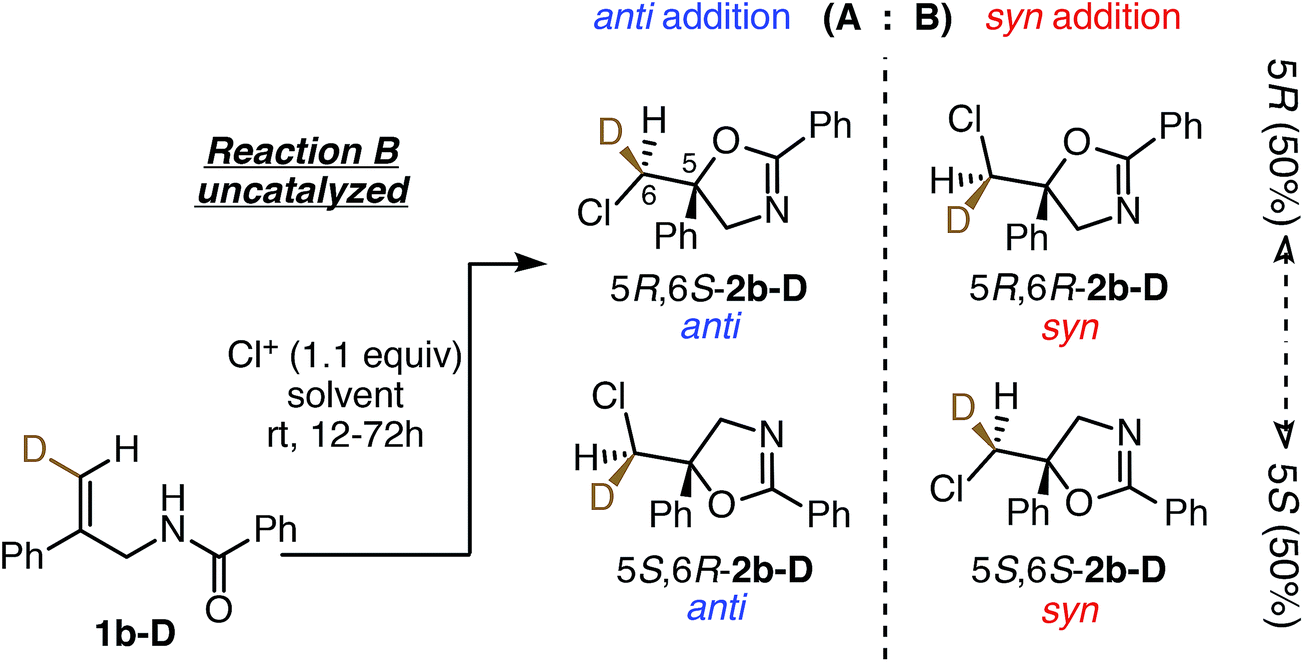
Stereochemical outcomes of uncatalyzed reactions
To understand the role of the catalyst in the enantiocontrolled reactions, the intrinsic diastereoselectivities of the uncatalyzed analogues of Reactions A–C and C′ were first investigated (Fig. 2).24 All favor anti addition; in Reaction A, chlorolactonization occurs with an anti:syn product ratio of 83:17.22 In Reaction B, amidoalkene 1b-D reacts with 1,3-dichloro-5,5-diphenylhydantoin (DCDPH) sluggishly in TFE at room temperature to afford an 85:15 mixture of the two diastereomers. Similarly, predominant anti addition is found for the uncatalyzed reaction of carbamate 1c-D with 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), both in CHCl3-hexanes and n-PrOH solvent systems; there, anti:syn ratios were 84:16 and 97:3, respectively. Formation of significant quantities of the syn isomer serves as evidence that these reactions do not simply proceed via one path through a stereochemically-defined intermediate (e.g. a cyclic chloriranium ion) able then to dictate stereospecific cyclization to the anti isomer; at least two pathways must contribute to yield the two distinct diastereomers. The substantial shift in anti:syn selectivity (84:16 to 97:3) between the solvent conditions corresponds to an increase of ∼1 kcal mol−1 in the relative barrier for syn vs. anti closure; evidently carbamate 1c-D interacts fairly strongly with its surroundings, as expected for such a polar substrate.
 | ||
| Fig. 2 Summary of intrinsic anti:syn product ratios for uncatalyzed Reactions A, B, C, and C′ (enantiomeric pairs are not shown for clarity). Reaction conditions mimic those shown in Fig. 1, except that the catalyst was omitted, and the reactions were run at room temperature. | ||
To probe the factors that determine anti:syn diastereoselectivity in the absence of catalyst, reaction parameters were systematically studied for chlorocyclization of the amidoalkene 1b-D. First, the effects of the chlorenium ion source were investigated (Table 1, entries 1–8); this factor significantly affects the stereochemical outcome; all yielded mainly anti product, but anti:syn ratios varied from 65:35 to 85:15. Though the most reactive chlorinating agent (TCCA) does show the lowest selectivity, no direct correlation is seen between chlorenium ion donor ability (assessed via HalA values)20 and anti:syn ratios. Thus it appears that the departing moiety of the chlorine donor reagent remains involved at the point of diastereoselection.25 What appears unlikely is simple Cl+ delivery followed by ring closure in an intermediate free of the leaving group from the chlorinating agent.
:syn ratios of 2b-D
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cl+ source | HalA or Solvent ε | Solvent | [Sub] M | [Cl+] M | dr (A:B) (anti:syn) |
|
a 58% conversion after 3 days.
b 82% conversion after 3 days.
c (1:1) ratio of the solvent; shown dielectric constant is the average of the ε of the two solvents.
|
||||||
| 1 | DiCh.T | 273.3 | TFE | 0.05 | 0.05 | 90:10 |
| 2 | NCSac | 265.0 | TFE | 0.05 | 0.05 | 86:14 |
| 3 | DCDPH | 270.1 | TFE | 0.05 | 0.05 | 85:15 |
| 4 | NCS | 290.1 | TFE | 0.05 | 0.05 | 80:20 |
| 5a | Ch.T | 268.2 | TFE | 0.05 | 0.05 | 80:20 |
| 6 | DCDMH | 275.7 | TFE | 0.05 | 0.05 | 79:21 |
| 7b | NCP | 286.7 | TFE | 0.05 | 0.05 | 78:22 |
| 8 | TCCA | 252.1 | TFE | 0.05 | 0.05 | 65:35 |
| 9 | DCDPH | 2.38 | Toluene | 0.05 | 0.05 | 98:2 |
| 10c | DCDPH | 3.35 | CHCl3:Hex |
0.05 | 0.05 | 97:3 |
| 11 | DCDPH | 8.93 | DCM | 0.05 | 0.05 | 97:3 |
| 12 | DCDPH | 4.81 | CHCl3 | 0.05 | 0.05 | 85:15 |
| 13 | DCDPH | 26.1 | TFE | 0.05 | 0.05 | 85:15 |
| 14 | DCDPH | 37.5 | CH3CN | 0.05 | 0.05 | 86:14 |
| 15 | DCDPH | — | TFE | 0.50 | 0.50 | 88:12 |
| 16 | DCDPH | — | TFE | 0.20 | 0.20 | 88:12 |
| 17 | DCDPH | — | TFE | 0.10 | 0.10 | 88:12 |
| (13) | DCDPH | — | TFE | 0.05 | 0.05 | 85:15 |
| 18 | DCDPH | — | TFE | 0.01 | 0.01 | 69:31 |
| 19 | DCDPH | — | TFE | 0.005 | 0.005 | 62:38 |
| 20 | DCDPH | — | TFE | 0.0025 | 0.0025 | 56:44 |
| 21 | DCDPH | — | TFE | 0.10 | 0.11 | 84:16 |
| 22 | DCDPH | — | TFE | 0.05 | 0.11 | 82:18 |
| 23 | DCDPH | — | TFE | 0.01 | 0.11 | 66:34 |
| 24 | DCDPH | — | TFE | 0.005 | 0.11 | 61:39 |
| 25 | DCDPH | — | TFE | 0.01 | 0.055 | 62:38 |
| 26 | DCDPH | — | TFE | 0.01 | 0.022 | 61:39 |
| 27 | DCDPH | — | TFE | 0.01 | 0.011 | 69:31 |
The effect of the reaction solvent on anti:syn ratios was next studied (Table 1, entries 9–14). The dr values showed significant solvent dependence; broadly, reaction in solvents of low polarity such as PhCH3 and DCM gave high anti:syn ratios (98:2 and 97:3) whereas relatively polar, hydrogen bonding solvents eroded the anti selectivities. Likewise, running the reaction in a 1:1 mixture of CHCl3-hexanes gave a much higher 97:3 dr than in CHCl3 alone (86:14). These results suggest contributions from mechanisms that are differentially affected by solvent polarity and hydrogen bonding ability. For instance, nonpolar solvents would favor substrate self-association via hydrogen bonds between the amide moieties,26 enabling a structurally defined, concerted path to form anti products from reagent + substrate dimers (see Fig. 4a). Polar, hydrogen bonding solvents would disrupt such associations, promoting syn product formation via the more statistically probable 1:1 reaction complex (Fig. 4b). We note here that CHCl3 should disrupt substrate aggregation more than CH2Cl2; CHCl3 has long been recognized as the stronger hydrogen bond donor solvent, despite its lower polarity as measured via dielectric constants.27 Notably, no intermolecular adducts were seen even in neat solvents capable of serving as nucleophiles (CH3CN and TFE), or when the amide substrate itself was present in concentrations as high as 0.5 M. Thus, any electrophilic intermediates, if formed, must have lifetimes shorter than trapping times in these neat nucleophilic solvents. And even the most aggressive chlorenium ion donors, expected to require little or no nucleophilic assistance, did not trigger intermolecular product formation.
Further mechanistic clues emerged from studies of the effects of reactant concentration on diastereoselectivity. Decreasing the concentration of both the substrate and DCDPH in TFE led to an increase in syn product formation (Table 1, entries 15–20). This is graphically illustrated in Fig. 3, where a steep dropoff in anti:syn ratio occurs at ∼0.05 M. Further explorations varying the individual component concentrations showed little change with varying [DCDPH] but significant falloff in anti selectivity as [1b-D] was lowered. These findings support the above suggestion that syn and anti products may arise via mechanisms of different reactant molecularities. More succinctly stated, at high concentrations, an anti-forming transition state could be composed of two (or more) molecules of olefin substrate and one molecule of the DCDPH reactant (Fig. 4a). An obvious mode of interaction involving two substrate molecules would be amide dimerization via NH–O hydrogen bonding, altering conformational preferences and enhancing both the effective amide group size and the nucleophilicity of the carbonyl oxygen. Such a complex would be expected to prefer anti addition as depicted in Fig. 4a. Here, the non-reacting amide serves as both the hydrogen bond relay and activator between the DCDPH and the N–H site of the reacting amide, enabling chlorenium ion transfer, ring closure, and proton transfer to take place concertedly with minimal charge separation.
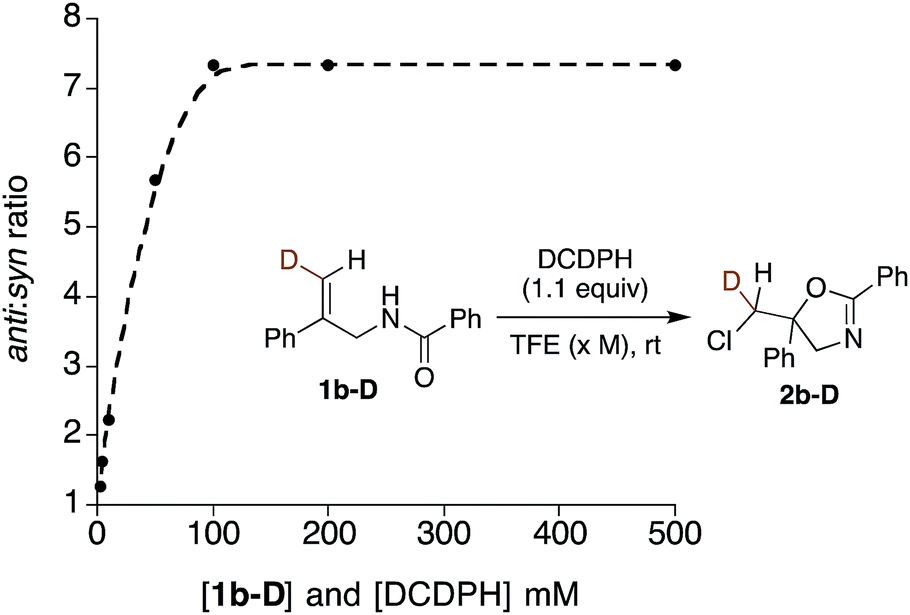 | ||
| Fig. 3 The plot of anti:syn ratios vs. concentration of DCDPH reagent and 1b-D substrate reaction mixtures. | ||
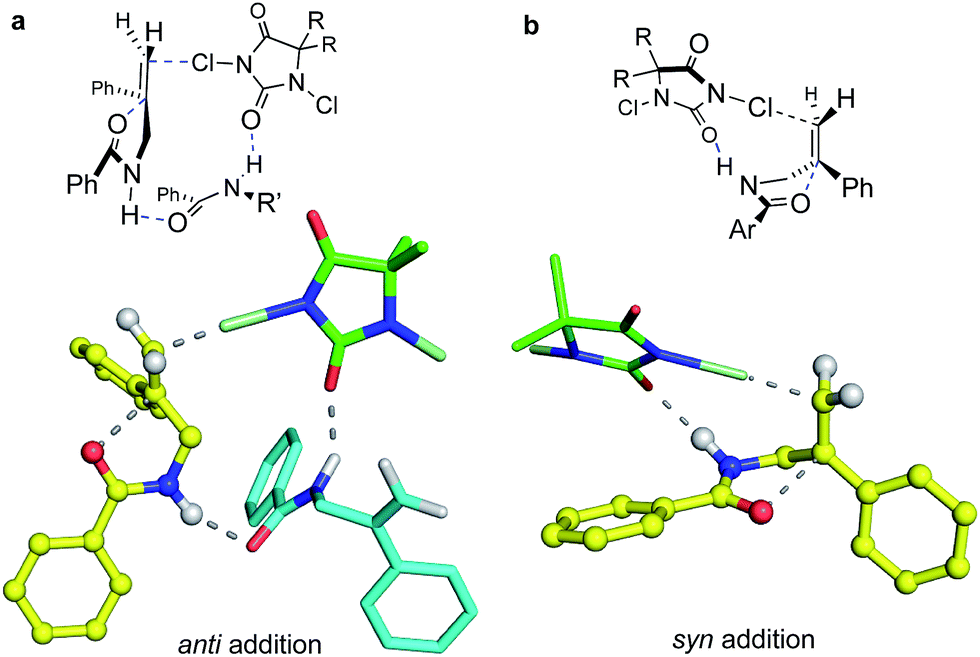 | ||
| Fig. 4 DFT-computed models for anti (a) and syn (b) cyclo addition of 1b. For computational efficiency, the dimethyl hydantoin was modeled in place of the diphenyl reagent used in the experiments. The higher propensity for anti addition at higher concentrations is consistent with the ca. 15 kcal mol−1 lower activation free energy calculated for the termolecular addition TS (structure a) in which a second amide substrate molecule bridges from the chlorine-donating hydantoin to the N–H site of the cyclizing amide moiety. At lower substrate concentration, the more strained bimolecular path (structure b) leading to syn addition is favored. | ||
In the 1:1 DCDPH:substrate complex that would be entropically favored at lower concentrations, the DCDPH may function as both the base and the chlorenium ion source (Fig. 4b), delivering the halogen from the same face as the nucleophile. The amide NH pyramidalization, and the twisting of the alkene seen in the calculated syn transition structure suggests that this species may suffer from substantial strain, consistent with its higher calculated barrier to reaction. On the other hand, the geometries of the hydrogen bonding interactions in the anti TS also appear non-ideal. Nonetheless, these modeled reaction paths are qualitatively consistent with the drop in anti:syn selectivity observed when the concentration of substrate in TFE was decreased from 0.10 M to 0.005 M with [DCDPH] held constant at 0.11 M (Table 1, entries 21–24). Conversely, when the concentration of substrate was kept constant at 0.01 M in TFE and concentration of DCDPH was lowered from 0.11 M (10.0 equiv.) to 0.011 M (1.1 equiv.), no significant changes were observed in diastereoselectivity (Table 1, entries 25–28). These findings point to a scenario with more than one substrate molecule but only one chlorinating agent in the transition state for anti cyclization (Fig. 4a).
Quantum chemical modeling of the above bi- and tri-molecular complexes at the T1//EDF2/6-31G* level, with TFE “solvation” simulated with the SMD solvent model,28 did indeed find a lower energy path for the termolecular than for the bimolecular process, as seen in Fig. 4; the energetics of these species are summarized in the ESI.† The free energy barriers from separated starting materials via these respective TS structures place the termolecular complex ca. 15 kcal mol−1 lower in energy than the bimolecular case. Solvation in TFE and in chloroform, as simulated at the SMD/EDF2/6-31G* level, modulates both paths but does little to change their relative energies. Aggregation via hydrogen bonded interactions, as seen in the termolecular complex, might seem unexpected in a hydroxylic medium such as TFE; however, this solvent is well known to promote polypeptide folding.29 On the other hand, entropy would strongly disfavor this higher molecularity structure at the dilute concentrations where syn products emerge.
Lastly, we investigated the effect of altering the electronic properties of the amide aroyl group on the diastereoselectivity of the chloro amidoalkene cyclization (Fig. 5). The largest change is seen with the 3,5-dinitro substituted benzamide 11b-D, which yields a near 1:1 anti:syn ratio of products 14b-D. Here, we speculate that reduced carbonyl group nucleophilicity in this substrate weakens dimer formation via hydrogen bonding, enhancing the contribution from the syn-favoring 1:1 reaction. On the other hand, the increased N–H acidity could favor interaction with the DCDMH, activating less selective chlorenium ion delivery reaction.
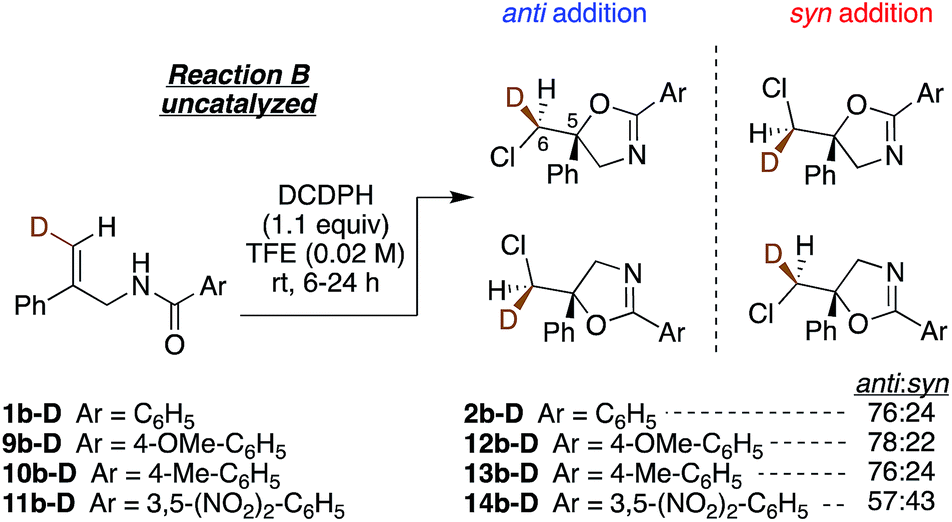 | ||
| Fig. 5
Anti:syn product ratios of electronically perturbed aryl amides. | ||
Diastereoselectivity in catalytic asymmetric reactions
Having probed the intrinsic diastereoselectivity of uncatalyzed Reaction B, we turned to the stereochemical analysis of the (DHQD)2PHAL catalyzed asymmetric reactions. As reported earlier, chlorolactonization of alkenoic acid 1a-D (Fig. 1, Reaction A) effects syn-selective carboxylate/chlorenium ion addition with an overall 88:12 syn:anti preference (Fig. 6, top).22,30 In Reaction B, the (DHQD)2PHAL-catalyzed chlorocyclization of deuterated amide 1b-D yields four stereoisomers under the previously reported catalytic asymmetric conditions (Fig. 6, bottom).15d The facial selectivity of chlorination, as determined via1H NMR analysis of the HPLC purified diastereomers (see ESI† for full details), reveals that the anti product is the major component of the mixture (94%). Thus, (DHQD)2PHAL controls the facial selectivity of chlorenium ion attack, forming the major epimer with a 99:1 preference for the 6S configuration, but only moderate 6S selectivity in forming the minor (5S) product (73:27). The nucleophilic closure occurs with high selectivity (93:7 ratio) favoring the R configuration at C5. As such, the two bond-forming events appear to be independently controlled by the catalyst. Noteworthy is the fact that in Reactions A and B (chlorolactonization and chloroamidocyclization, respectively), the chlorenium ion delivery occurs to the same face of the olefin, but ring closure occurs on opposite faces of the cyclizing carbon (note that differing substituent priorities designate both products 5R, while differing C6 configurations in 1a-Dvs.1b-D and 1c-D lead to opposite C6 configurations in the respective products 2 for the same facial preference of Cl attack). The overall process, therefore, is syn for chlorolactonization (Reaction A) and anti for chloroamidocyclization (Reaction B).
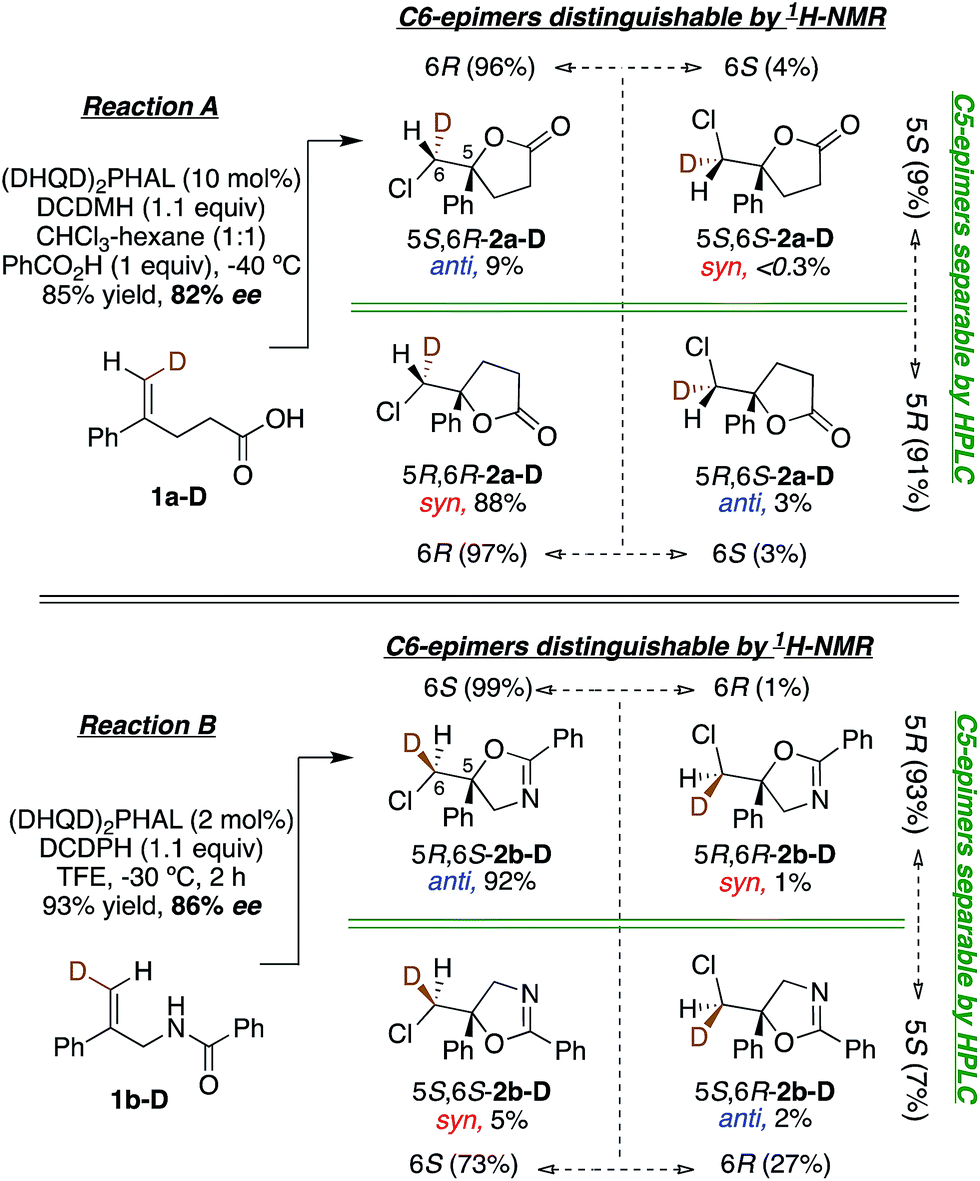 | ||
| Fig. 6 (DHQD)2PHAL catalyzed chlorocyclization of 1-D leads to four isomeric products. Ratios of each isomer are quantified by 1H NMR and HPLC analysis. Reaction A (top, previously reported) shows syn selectivity in net addition of the chlorenium cation and carboxylate anion to olefin 1a-D.22 Noting that deuterium differs in configuration between 1a-D and 1b-D, and in CIP group priorities about C5 in the product, Reaction B, (bottom) shows predominant anti addition with C6-pro-S/C5-pro-R selectivity in this catalyst templated addition of the chlorine electrophile and the amide nucleophile across the olefin. | ||
In Reactions C and C′, carbamate 1c shows a switch in the enantiopreference of (DHQD)2PHAL-catalyzed chlorocyclization depending on reaction conditions – primarily the reaction solvent.15b The overall stereochemistry of both these asymmetric alkene additions is now revealed via deuterated probe 1c-D. Chlorocyclization of 1c-D catalyzed with (DHQD)2PHAL in 1:1 CHCl3:hexane (Reaction C, Fig. 7, top) yields the anti product 5R,6S-2c-D as the major isomer (85%); this behavior is thus stereochemically the same as for amide 1b-D. The chlorenium ion delivery to yield products with 6S configuration occurs with high selectivity (93:7) for the major (5R) diastereomers. As in the above cases, although the 6S selectivity is still predominant for the minor diastereomer, it occurs with reduced discrimination (61:39). Thus, the overall face selectivity for chlorination on the pro-S face of the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHD site (i.e. 6S:6R) is 91:9. This C6 stereoselectivity incidentally has the same numerical value as the enantioselectivity of the reaction (91:9 5R:5S).
CHD site (i.e. 6S:6R) is 91:9. This C6 stereoselectivity incidentally has the same numerical value as the enantioselectivity of the reaction (91:9 5R:5S).
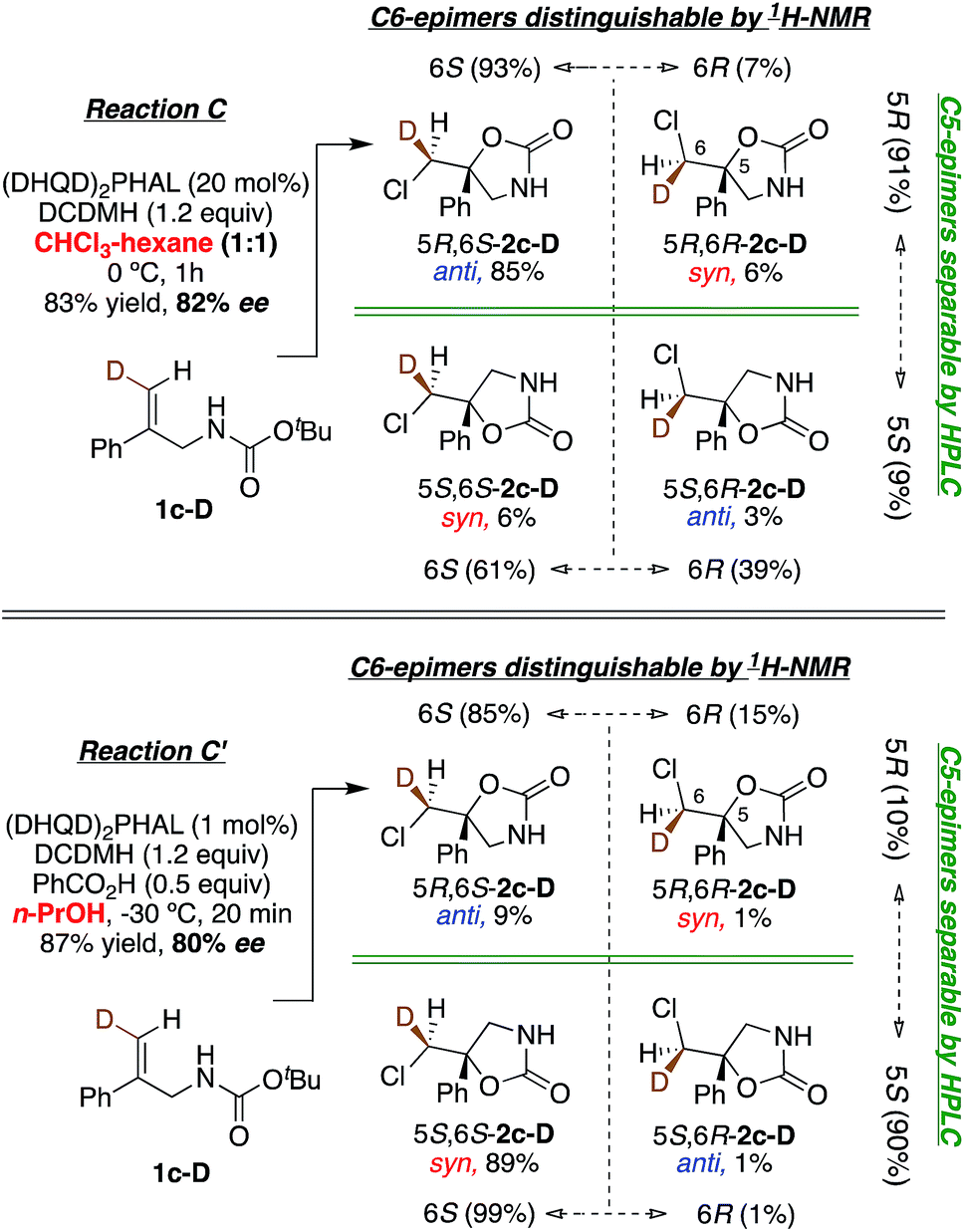 | ||
| Fig. 7 Chlorocyclization of carbamate 1c-D yields the anti product as the major isomer in 1:1 chloroform:hexanes (Reaction C, top), and the syn product as the major isomer in n-PrOH (Reaction C′, bottom). Both reactions are catalyzed by (DHQD)2PHAL. | ||
When applied to Reaction C′ (Fig. 7, bottom), the above analysis reveals a contrast relative to both the non-catalyzed reaction in n-PrOH and the catalyzed Reaction C in CHCl3-hexanes where anti additions predominate (97:3 and 88:12, respectively). Instead, the (DHQD)2PHAL catalyzed reaction in n-PrOH effects syn addition (net anti:syn = 10:90). However, as in all the other reactions discussed so far, chlorenium ion delivery occurs to the same face, forming the 6S epimers (with this substrate) in high (99:1) and good (85:15) enantioselectivity for the major and minor diastereomers, respectively. The net selectivity for chlorination of the C6 pro-S face of 1c-D is 98:2 whereas the C5 face selectivity is somewhat lower, at 90:10 pro-S.
Given the various syn:anti addition ratios seen as a function of starting material and reaction conditions, it might be suggested that cis–trans isomerization in the starting olefin could explain the observed stereo-randomized products (see Scheme 3 for this hypothetical pathway). This scenario was ruled out (a) implicitly, by the large C6 stereoselectivities seen in some cases (e.g. 99:1 in Reaction B), and (b) explicitly, by verification of the stereochemical integrity of labeled substrates 1b-D and 1c-D recovered during the course of Reactions B, C, and C′. Reactions quenched at various extents of conversion yield recovered alkenes with E/Z isomeric ratios identical within measurement uncertainties to the starting ratios for all three reactions (see Table S3†). Besides showing that isomerized reactant is not the source of the disfavored addition products, these results agree with earlier findings that chlorenium ion transfer is not reversible.22,31
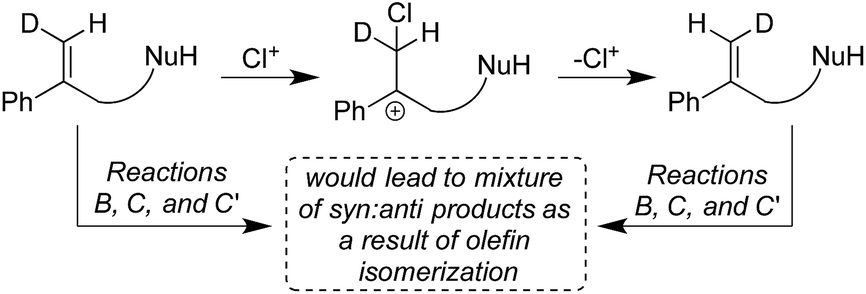 | ||
| Scheme 3 Possibility of the formation of the isomerized starting material, ruled out by recovery studies, see Table S3.† | ||
A summary of the stereochemical outcomes of the various asymmetric chlorocyclization reactions disclosed here is presented in Fig. 8. Most striking is the uniform and selective delivery of chlorine to the same face of C6 in all the alkene substrates. On the other hand, the net syn addition of halogen and nucleophile across the olefin that we had first observed in the initial chlorolactonization chemistry is by no means general. Despite the use of the same organocatalyst and similar hydantoin chlorine sources, different cyclization stereopreferences dominate with different substrates and reaction conditions. Related work from other laboratories5d,7,8,10–14,15a,32 seems likely to exhibit a similar diversity of syn and anti addition across 1,1-disubstituted alkenes.
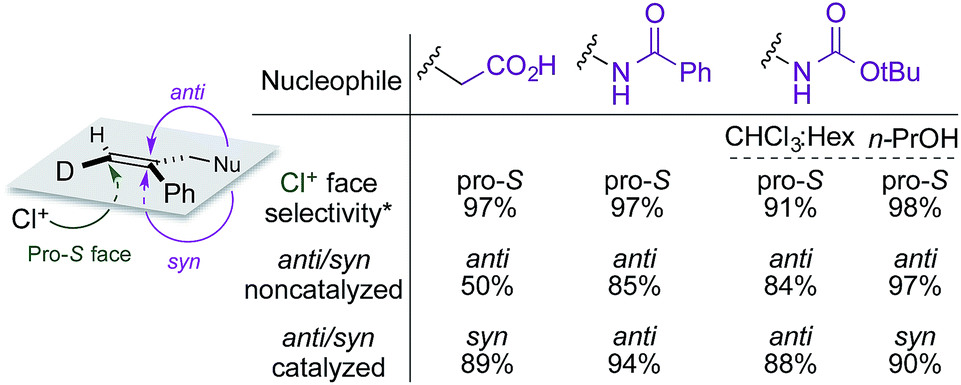 | ||
| Fig. 8 Summary of stereochemical outcomes for Reactions A, B, C, and C′. *Configuration designators are listed as shown in the image at left. | ||
Though a comprehensive quantum chemical simulation of interacting substrates, organocatalyst, reagents and solvents is beyond the scope of this report, limited simulation studies of the complexation and reaction processes of carbamate substrate 1c were pursued using the Spartan'16 software.33 As noted earlier, the ring closure stereochemistry of 1c switches from syn to anti when the medium is changed from n-PrOH to CHCl3/hexane. Based on preliminary NMR studies that suggest a 2:1 binding of substrates in the (DHQD)2PHAL organocatalyst, broad conformational searches on such 2:1 complexes were performed with the MMFF94 (ref. 34) and Sybyl35 force fields. These simulations typically explored 1–2000 conformations and were repeated multiple times from various arbitrary starting geometries. They consistently turned up low energy structures that exposed the pro-R alkene face, the experimentally preferred site of chlorine attack. Conformations in the lowest 10 kcal mol−1 range were then re-optimized using the PM6 semiempirical molecular orbital model.36 Again, the lowest energy conformations found were orientations with the pro-R face of the alkene exposed. A second series of conformational searches of similar breadth was generated by placing the PM6 calculated transition structures (TSs) for anti and syn chlorocyclizations into one (DHQD)2PHAL pocket, together with a partner non-reacting substrate complexed to the other face. In these cases, the lengths of the partial (reacting) bonds (N–Cl, Cl–C, and C–O), were held constant at the gas-phase TS values, but all other degrees of freedom were allowed to vary in the conformational search. Again, the lowest energy set of resulting conformations was reoptimized (still constrained) using PM6, after which full TS optimizations were completed. The resulting syn and anti TSs (each with a single imaginary vibrational frequency corresponding to the alkene addition trajectory) were reevaluated via single-point B3LYP-D3/6-31G* energy calculations,28a,37 with solvation corrections for chloroform and n-PrOH solvents (dielectric constants of 4.8 and 20.3) computed via the C-PCM method.38
The final complexes identified via the above procedures are displayed in Fig. 9. Given the relatively low level of theoretical models used to develop them, these structures must be understood mainly as proposed guides for visualization. Nonetheless, we were encouraged by the predominance of low energy catalyst–substrate complex conformations (Fig. 9a) that orient the alkene to expose the face that is actually chlorinated. Likewise, the close energies calculated for the anti and syn transition state structures correspond well with the fact that the difference between CHCl3 and n-PrOH media can switch the anti/syn addition selectivities of Reactions C/C′. Notably, the relaxed precursor complex (Fig. 9b) positions the carbonyl oxygen close to the alkene plane, suggesting that its rotations to approach to either side might face similar barriers. Whether closing in anti or syn modes (Fig. 9c and d, respectively), the extended carbamate backbone lying in the catalyst groove must fold up to a TS conformation that brings the carbonyl into contact with the alkene, activating the chlorine transfer and closing the ring. Both TS structures have thus lost factors that stabilized the bound GS, specifically van der Waals interactions between substrate t-butyl groups and the phthalazine ring, and among substrate phenyl groups and the quinoline side “walls” of the catalyst. The amide N–H site, however, remains associated with catalyst nitrogen atoms by hydrogen bonding, which also activates the nucleophilicity of the carbonyl oxygen as it closes to form the oxazolinone ring.
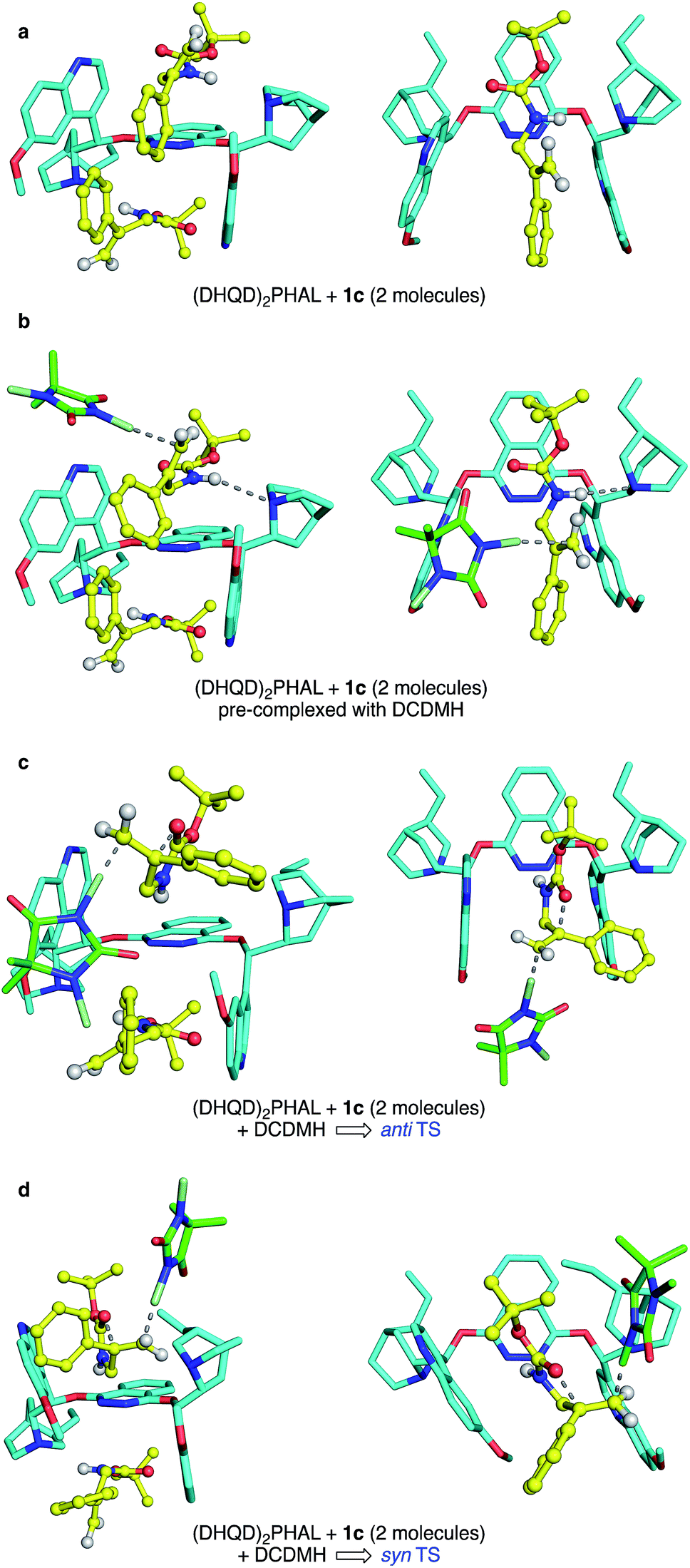 | ||
| Fig. 9 Side (left) and top (right) view of (DHQD)2PHAL complexed with 1c and DCDMH. Hydrogen atoms are not shown in all structures except for the substrate amide and vinylidene protons. In the top view, the second bound substrate is omitted for sake of clarity. (a) (DHQD)2PHAL bound with 1c. (b) Ground state, pre-complexed interaction of DCDMH with 1c, bound to catalyst, showing the preferred olefin face selectivity as a result of the orientation of the substrate. (c) The anti TS, illustrating a concerted path to product formation. (d) The syn TS, also in a concerted path toward product. | ||
Energetically, the TS structures for anti and syn (Fig. 9c and d) are calculated to be almost 40 kcal mol−1 higher than the relaxed (DHQD)2PHAL-carbamate-DCDMH complex (Fig. 9b) at the B3LYP-D3/6-31G*//PM6 level. These high DFT-based barriers change surprisingly little upon extension of the calculations to include CPCM simulated n-PrOH and CHCl3 solvation environments. Interestingly, the pure PM6//PM6 barriers fall in the much more reasonable 20 kcal mol−1 range (see SI for summary) with a small (4.1 kcal mol−1) free energy preference for syn. We suggest that the more intimately bound complexes in Fig. 9a and b are over-stabilized by the well-known overestimation of van der Waals interactions in B3LYP and related DFT methods, augmented by the basis set superposition errors of the relatively small 6-31G* basis set. Unfortunately, the solvation calculations, which include no geometry relaxation or explicit solvent interactions, offer little insight into the solvent-switched selectivities of Reactions C/C′.
The work presented above highlights one of the fundamental challenges in developing highly enantioselective halocyclizations and more generally, additions across alkenes. Here, exceptional face-selectivity in the alkene chlorination is no guarantee of a strong enantiopreference for the newly created C5 sp3 stereocenter; syn and anti addition paths are in close competition. Conversely, a finding of poor enantioselectivity at C5 does not imply poor face-selectivity in the halogen-alkene bond formation.
With the present array of data, our working mechanistic interpretation is that reaction occurs via intramolecular versions of the AdE3 olefin addition.39 Here, preorganization of the catalyst and substrate expose the preferred alkene face to the incoming chlorenium ion donor, while still allowing the nucleophilic moiety the conformational flexibility to fold in to contact either face of the alkene. The calculations suggest that the chlorinating agent may form a weak preassociation with the complexed alkene, but the actual bond-forming addition of the halogen to the alkene requires the nucleophile to fold to a conformation that contacts and activates the pi bond for the concerted addition. We recently explored such concerted paths in the substrate framework of Reaction A, where nucleophile-assisted alkene activation (NAAA) promoted non-catalyzed chlorolactonization of 1,1-disubstituted alkenoic acids. That both syn and anti paths can selectively occur among reactions A–C rules out bridged chloronium ions as stereocontrolling intermediates, as expected for these conjugated, 1,1-disubstituted alkenes.
What remains difficult to explain is the observed variability in the ring closure stereochemistry for the four highlighted reactions. As noted above, the complexed alkenes in our calculated structures (see Fig. 9b) place the carbonyl oxygen nearly in the plane of the alkene, establishing no obvious preference for which alkene face would be more easily accessed. Yet amide and carbamate reacting in a non-polar solvent system (Reactions B and C′) proceed with anti ring closure in contrast to the syn mode seen for carboxylic acids and for carbamates reacting in polar protic solvents. We propose that the amides and carbamates 1b and 1c (both more nucleophilic and, with only two freely rotating bonds, less conformationally flexible than 1a) most readily fold “inward” to achieve the pro-anti conformation. In contrast, carboxylic acid 1a is less conformationally restricted and potentially capable of hydrogen bond formation, which enables a preference for it to fold outward and activate the alkene from the same face approached by the chlorine-donating hydantoin. Likewise, folding of the polar carbamate moiety of 1c out into the solvent could be supported by the more polar n-PrOH medium, favoring approach from the same face as the chlorenium ion delivery. The carbamate in n-PrOH (Reaction C′) thus behaves much like the carboxylic acid 1a, yielding mainly syn addition product.
Conclusions
For a family of chlorocyclizations, both uncatalyzed and mediated by (DHQD)2PHAL, the relative and absolute stereochemical outcomes have been fully analyzed. In four distinct (DHQD)2PHAL—catalyzed processes—chlorocyclizations of carboxylic acid 1a, of amide 1b, and of carbamate 1c2a,15b,15d under two sets of conditions—the chlorine attacks the same face of the olefin. This high facial selectivity for chlorenium ion delivery presumably reflects catalyst-mediated pre-organization of the styrene substrate, directing chlorine donor access to only one alkene face. Cyclization by nucleophilic bond closure can show high syn or anti selectivity depending on the nature of the nucleophile and the medium. Thus, the net stereoselectivities at the two new stereocenters appear to be related only in the sense that one of these concerted paths is strongly preferred over others in the optimized reactions. The resulting structural insights place boundary conditions on any mechanistic hypothesis proposed to further refine and generalize this synthetically versatile class of transformations. Detailed kinetic analyses and simulation efforts are ongoing to probe molecularity, catalyst-substrate-reagent binding, preferred conformations in different settings, and reaction rate effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the NIH (GM110525) and the NSF (CHE-1362812) for funding. The authors thank Dr Daniel Holmes (NMR), and Professor Daniel Jones (mass spec).Notes and references
- (a) A. Castellanos and S. P. Fletcher, Chem.–Eur. J., 2011, 17, 5766 CrossRef CAS PubMed; (b) G. F. Chen and S. M. Ma, Angew. Chem., Int. Ed., 2010, 49, 8306 CrossRef CAS PubMed; (c) Y. A. Cheng, W. Z. Yu and Y. Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333 RSC; (d) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed; (e) U. Hennecke, Chem.–Asian J., 2012, 7, 456 CrossRef CAS PubMed; (f) U. Hennecke and M. Wilking, Synlett, 2014, 25, 1633 CrossRef; (g) C. K. Tan and Y. Y. Yeung, Chem. Commun., 2013, 49, 7985 RSC; (h) C. K. Tan, L. Zhou and Y.-Y. Yeung, Synlett, 2011, 2011, 1335 CrossRef; (i) S. Q. Zheng, C. M. Schienebeck, W. Zhang, H. Y. Wang and W. P. Tang, Asian J. Org. Chem., 2014, 3, 366 CrossRef CAS; (j) J. R. Wolstenhulme and V. Gouverneur, Acc. Chem. Res., 2014, 47, 3560 CrossRef CAS PubMed; (k) S. R. Chemler and M. T. Bovino, ACS Catal., 2013, 3, 1076 CrossRef CAS PubMed; (l) J. Chen and L. Zhou, Synthesis, 2014, 46, 586 CrossRef; (m) U. Hennecke, Angew. Chem., Int. Ed. Engl., 2012, 51, 4532 CrossRef CAS PubMed; (n) K. Murai and H. Fujioka, Heterocycles, 2013, 87, 763 CrossRef CAS; (o) A. Rouf and S. C. Taneja, Chirality, 2014, 26, 63 CrossRef CAS PubMed; (p) S. A. Snyder, D. S. Treitler and A. P. Brucks, Aldrichimica Acta, 2011, 44, 27 CAS; (q) C. K. Tan, W. Z. Yu and Y. Y. Yeung, Chirality, 2014, 26, 328 CrossRef CAS PubMed.
- (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed; (b) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed.
- (a) D. Huang, X. Liu, L. Li, Y. Cai, W. Liu and Y. Shi, J. Am. Chem. Soc., 2013, 135, 8101 CrossRef CAS PubMed; (b) D. Huang, H. Wang, F. Xue, H. Guan, L. Li, X. Peng and Y. Shi, Org. Lett., 2011, 13, 6350 CrossRef CAS PubMed.
- S. E. Denmark and M. T. Burk, Chirality, 2014, 26, 344 CrossRef CAS PubMed.
- (a) K. Hiramatsu, T. Honjo, V. Rauniyar and F. D. Toste, ACS Catal., 2015, 6, 151 CrossRef PubMed; (b) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed; (c) H. P. Shunatona, N. Fruh, Y. M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed. Engl., 2013, 52, 7724 CrossRef CAS PubMed; (d) Y. M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928 CrossRef CAS PubMed.
- C. H. Müller, C. Rösner and U. Hennecke, Chem.–Asian J., 2014, 9, 2162 CrossRef PubMed.
- H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6974 CrossRef CAS PubMed.
- (a) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174 CrossRef CAS PubMed; (b) K. Murai, A. Nakamura, T. Matsushita, M. Shimura and H. Fujioka, Chem.–Eur. J., 2012, 18, 8448 CrossRef CAS PubMed.
- (a) F. Chen, C. K. Tan and Y.-Y. Yeung, J. Am. Chem. Soc., 2013, 135, 1232 CrossRef CAS PubMed; (b) Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627 CrossRef CAS PubMed.
- X. Jiang, C. K. Tan, L. Zhou and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2012, 51, 7771 CrossRef CAS PubMed.
- (a) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068 CrossRef CAS PubMed; (b) J. E. Tungen, J. M. J. Nolsoe and T. V. Hansen, Org. Lett., 2012, 14, 5884 CrossRef CAS PubMed.
- G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332 CrossRef CAS PubMed.
- D. H. Paull, C. Fang, J. R. Donald, A. D. Pansick and S. F. Martin, J. Am. Chem. Soc., 2012, 134, 11128 CrossRef CAS PubMed.
- (a) C. B. Tripathi and S. Mukherjee, Angew. Chem., Int. Ed., 2013, 52, 8450 CrossRef CAS PubMed; (b) W. Zhang, N. Liu, C. M. Schienebeck, X. Zhou, I. I. Izhar, I. A. Guzei and W. P. Tang, Chem. Sci., 2013, 4, 2652 RSC.
- (a) A. Armstrong, D. C. Braddock, A. X. Jones and S. Clark, Tetrahedron Lett., 2013, 54, 7004 CrossRef CAS; (b) A. Garzan, A. Jaganathan, N. Salehi Marzijarani, R. Yousefi, D. C. Whitehead, J. E. Jackson and B. Borhan, Chem.–Eur. J., 2013, 19, 9015 CrossRef CAS PubMed; (c) K. Ikeuchi, S. Ido, S. Yoshimura, T. Asakawa, M. Inai, Y. Hamashima and T. Kan, Org. Lett., 2012, 14, 6016 CrossRef CAS PubMed; (d) A. Jaganathan, A. Garzan, D. C. Whitehead, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2011, 50, 2593 CrossRef CAS PubMed; (e) L. J. Li, C. X. Su, X. Q. Liu, H. Tian and Y. A. Shi, Org. Lett., 2014, 16, 3728 CrossRef CAS PubMed; (f) M. Wilking, C. Muck-Lichtenfeld, C. G. Daniliuc and U. Hennecke, J. Am. Chem. Soc., 2013, 135, 8133 CrossRef CAS PubMed; (g) Q. Yin, S. G. Wang, X. W. Liang, D. W. Gao, J. Zheng and S. L. You, Chem. Sci., 2015, 6, 4179 RSC; (h) Q. Yin and S. L. You, Org. Lett., 2013, 15, 4266 CrossRef CAS PubMed; (i) Q. Yin and S. L. You, Org. Lett., 2014, 16, 2426 CrossRef CAS PubMed; (j) A. Jaganathan, R. J. Staples and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14806 CrossRef CAS PubMed; (k) B. Soltanzadeh, A. Jaganathan, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2015, 54, 9517 CrossRef CAS PubMed; (l) A. Jaganathan and B. Borhan, Org. Lett., 2014, 16, 3616 CrossRef CAS PubMed; (m) R. Yousefi, D. C. Whitehead, J. M. Mueller, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 608 CrossRef CAS PubMed; (n) O. Lozano, G. Blessley, T. Martinez del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS PubMed; (o) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134 CrossRef CAS PubMed.
- I. Roberts and G. E. Kimball, J. Am. Chem. Soc., 1937, 59, 947 CrossRef CAS.
- (a) R. C. Fahey, in Topics in Stereochemistry, ed. E. L. Eliel and N. L. Allinger, John Wiley & Sons, Inc., New York, NY, 1968, vol. 3, p. 237 Search PubMed; (b) R. C. Fahey and C. McPherso, J. Am. Chem. Soc., 1969, 91, 3865 CrossRef CAS; (c) R. C. Fahey and M. W. Monahan, J. Am. Chem. Soc., 1970, 92, 2816 CrossRef CAS; (d) H. Haubenstock and R. R. Sauers, Tetrahedron, 2004, 60, 1191 CrossRef CAS; (e) H. Haubenstock and R. R. Sauers, Tetrahedron, 2005, 61, 8358 CrossRef CAS.
- (a) R. C. Fahey, J. Am. Chem. Soc., 1966, 88, 4681 CrossRef CAS; (b) R. C. Fahey and C. Schubert, J. Am. Chem. Soc., 1965, 87, 5172 CrossRef CAS; (c) M. L. Poutsma, J. Am. Chem. Soc., 1965, 87, 4285 CrossRef CAS; (d) B. B. Snider and M. I. Johnston, Tetrahedron Lett., 1985, 26, 5497 CrossRef CAS; (e) D. L. H. Williams, E. Bienvenue-Goetz and J. E. Dubois, J. Chem. Soc. B, 1969, 517 RSC.
- Due to the intramolecularity of the process, the typical AdE3 mechanism, with three participating particles in the transition state, is here formally AdE2 (bimolecular), with two components in the transition state. However, common parlance would still refer to it as AdE3, just as closure of a β-haloalkoxide to epoxide + halide would commonly be described as an SN2 process.
- K. D. Ashtekar, N. S. Marzijarani, A. Jaganathan, D. Holmes, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2014, 136, 13355 CrossRef CAS PubMed.
- K. D. Ashtekar, M. Vetticatt, R. Yousefi, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2016, 138, 8114 CrossRef CAS PubMed.
- R. Yousefi, K. D. Ashtekar, D. C. Whitehead, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14524 CrossRef CAS PubMed.
- H. Zeng and R. Hua, J. Org. Chem., 2008, 73, 558 CrossRef CAS PubMed.
- The uncatalyzed reactions were carried in a similar fashion as those detailed in Fig. 2 and 3 except at RT and in the absence of (DHQD)2PHAL. Although the products were obtained as a racemic mixture, the syn:anti ratios show the intrinsic selectivity for each reaction.
- To explain the observed anti:syn ratios, we envision a concerted AdE3-like mechanism (formally AdE2).19 This nucleophile-activated alkene addition (NAAA) scheme21 involves concerted bond formation to the alkene by both nucleophile oxygen and chlorine, a mechanism by which the structure of the chlorine donor could modulate selectivity. Presumably, different substrate conformers or aggregates (vide infra) in solution would have variable reactivity with the different chlorinating agents, yielding different proportions of anti vs. syn products.
- H. L. Schenck and K. W. Hui, J. Chem. Educ., 2011, 88, 1158 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, J. Chem. Soc., Perkin Trans. 2, 1979, 337 RSC.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS; (b) C. Y. Lin, M. W. George and P. M. W. Gill, Aust. J. Chem., 2004, 57, 365 CrossRef CAS; (c) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CAS; (d) W. S. Ohlinger, P. E. Klunzinger, B. J. Deppmeier and W. J. Hehre, J. Phys. Chem. A, 2009, 113, 2165 CrossRef CAS PubMed.
- (a) J. F. Povey, C. M. Smales, S. J. Hassard and M. J. Howard, J. Struct. Biol., 2007, 157, 329 CrossRef CAS PubMed; (b) K. Shiraki, K. Nishikawa and Y. Goto, J. Mol. Biol., 1995, 245, 180 CrossRef CAS PubMed.
- A referee has suggested that KIEs due to substrate alkene deuteration might perturb the labelled experiments enough that they are not representative of the parent reactions. The values of asymmetric induction at C5 now show little if any difference between perprotio and monodeuterated cases for Reactions A, C and C′, as can be seen by comparing data in Fig. 1vs. those in Fig. 6 and 7.
- S. E. Denmark, M. T. Burk and A. J. Hoover, J. Am. Chem. Soc., 2010, 132, 1232 CrossRef CAS PubMed.
- (a) C. S. Brindle, C. S. Yeung and E. N. Jacobsen, Chem. Sci., 2013, 4, 2100 RSC; (b) K. Murai, T. Matsushita, A. Nakamura, N. Hyogo, J. Nakajima and H. Fujioka, Org. Lett., 2013, 15, 2526 CrossRef CAS PubMed; (c) D. W. Tay, G. Y. C. Leung and Y. Y. Yeung, Angew. Chem., Int. Ed., 2014, 53, 5161 CAS; (d) Y. Zhao, X. J. Jiang and Y. Y. Yeung, Angew. Chem., Int. Ed., 2013, 52, 8597 CrossRef CAS PubMed; (e) L. Zhou, J. Chen, C. K. Tan and Y. Y. Yeung, J. Am. Chem. Soc., 2011, 133, 9164 CrossRef CAS PubMed; (f) L. Zhou, C. K. Tan, X. J. Jiang, F. Chen and Y. Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474 CrossRef CAS PubMed.
- Spartan'16, Wavefunction, Inc., Irvine, CA Search PubMed.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 730 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 720 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (c) P. c. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- An alternative interpretation is that reaction follows a stepwise mechanism via an intermediate (presumably the benzylic carbocation), where catalyst in independent events rigorously controls the facial selectivity of chlorenium ion delivery to the olefin, and then, less precisely, the enantiodetermining cyclization step. The catalyst would then need to be able to consistently guide attacks from the same face of the olefin in every case, but then specifically favor either syn or anti relative closure trajectories of the nucleophile, depending on conditions. As noted in the text, a key strike against this scenario is that no intermolecular cation trapping products (alkenes, ethers or esters) are seen in the product mixtures, even in cases such as Reaction A, where an equivalent of benzoic acid is available, or Reaction C′, where the solvent is the alcohol n-PrOH. In addition, to the extent that chlorenium ion transfer might be reversible, the absence of isomerized alkenes is further evidence against a cation formed with any significant lifetime. Another concern is the notion of concerted syn addition across an alkene, but actually such events are familiar as in the Diels-Alder and related but more polar cycloadditions. Likewise, pyrolytic carboxylic acid loss from esters is a well-known concerted syn elimination, the microscopic reverse of syn addition.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04430e |
| This journal is © The Royal Society of Chemistry 2018 |