
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence modulation via photoinduced spin crossover switched energy transfer from fluorophores to FeII ions†
Jun-Li
Wang
a,
Qiang
Liu
a,
Yin-Shan
Meng
a,
Xin
Liu
 a,
Hui
Zheng
b,
Quan
Shi
b,
Chun-Ying
Duan
a and
Tao
Liu
*a
a,
Hui
Zheng
b,
Quan
Shi
b,
Chun-Ying
Duan
a and
Tao
Liu
*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Rd., 116024, Dalian, China. E-mail: liutao@dlut.edu.cn
bThermochemistry Laboratory, Liaoning Province Key Laboratory of Thermochemistry for Energy and Materials, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 5th February 2018
Abstract
Molecular materials possessing phototunable fluorescence properties have attracted great interest owing to their potential applications in optical switches and storage. However, most fluorescence modulation is realized through light-responsive structural isomerization in solution. It is a formidable challenge to achieve phototunable fluorescence emission with high fatigue resistance and a fast response rate in the solid state for the development of devices. Here, a mononuclear compound was constructed via the coordination of fluorophores with FeII ions, whose electronic configuration changed from low spin to high spin upon light irradiation. The photoinduced spin crossover of FeII ions was accompanied by a 20% increase in the fluorescence emission intensity. A temperature-dependent spectroscopic study together with time-dependent density functional theory calculations revealed that the effective spectral overlap between the emission of the fluorophores and the absorption band of the FeII ions differed between the low spin and high spin states. The photoinduced spin crossover switched the energy transfer from the fluorophore to the FeII ion, resulting in fluorescence modulation. The presented results provide a novel approach for developing optical memory and sensors via electron rearrangement of photoinduced spin crossover.
Introduction
The application of fluorescent materials for detection and tracer techniques is highly desirable because fluorescence signals provide superior contrast and sensitivity during remote detection at relatively high spatial and temporal resolution.1,2 The modulation of fluorescence signals via light is particularly attractive for noninvasive optical read-out technologies owing to the potential applications in optical storage, optical switches and sensors.3–7 Producing a coupling between a fluorophore unit and a light-responsive unit is a rational way to yield photoswitchable fluorescence emission. Previous strategies for achieving phototunable fluorescence emission for the development of new fluorescence switches mainly involved linking organic photochromic groups with fluorescent units via covalent bonds.7–12 Within the large family of photochromic groups, most are based on the photoisomerization of organic molecules, such as cis/trans or opened/closed ring isomerization.13–19 The fluorescence emission is modulated as a result of changes in the intramolecular energy transfer and/or electron transfer from the fluorescent donor to the photochromic acceptor during the process of a photochemical reaction under alternating ultraviolet and visible light irradiation.20 However, photoisomerization often results in the drastic movement of atoms, which are strongly restricted in the solid state due to tension and hindrance. Therefore, efficient photoisomerization reactions commonly occur in solution rather than the solid state. It is challenging to realize phototunable fluorescence in the solid state and thus develop applicable devices with high fatigue resistance and a fast response rate.When compared with the photoisomerization of organic molecules, the photoswitchable electron rearrangement of metal ions, such as spin crossover (SCO) and electron transfer, has been realized in the solid state with a fast response rate and good reversibility, accompanied by drastic changes in the electronic and crystal structure of the materials.21–23 Such drastic changes should sufficiently perturb the environment of the fluorophore and further allow a fluorescent response;24–34 thus providing a promising candidate for a light-responsive unit with which to construct photoswitchable fluorescent molecular devices. However, two challenges remain in achieving the goal in this system. One is how to preserve both the fluorescent function and the photoinduced electron rearrangement phenomenon when combining them in a single component because both are highly sensitive to minor external physical and internal chemical changes. The other is how to build an effective coupling pathway to realize the synergy between them, thereby obtaining photoswitchable fluorescence emission.
Photoinduced SCO of FeII ions is a typical example of photoswitchable electron rearrangement,35–41 which could serve as a light-responsive unit for coupling with an organic fluorophore unit via a coordination bond linkage. To retain fluorescence and SCO behavior, the fluorescent ligand should exhibit sufficiently strong fluorescence emission and overcome the quenching effect when coordinated with FeII ions. Meanwhile, the FeII ions must be located within a suitable ligand field to support the spin transition triggered by light. Moreover, to achieve synergy, the emission or excitation band of the ligand should have an effective spectral overlap with the absorption band of the FeII ions at a given spin state. Considering the above, we designed a fluorophore ligand (pyrene-1-yl)-N-(3,5-di(pyridin-2-yl)-4H-1,2,4-triazol-4-yl)methanimine (L). This ligand shows strong fluorescence emission at 516 nm, thus overlapping with the typical absorption region of FeII complexes.38 Therefore, the fluorescence emission is expected to be quenched or increased depending on whether the resonant energy transfer from the fluorescent donor to the FeII acceptor occurs in the photoinduced spin transition process (Scheme 1). According to this strategy, we successfully obtained a mononuclear complex [Fe(L)2(NCS)2] (1), which exhibited significant enhancement in both fluorescence emission intensity and magnetization upon light irradiation at 671 nm. These findings provide direct evidence for photoswitchable fluorescence emission via photoinduced electron rearrangement in a fluorescence-coupled SCO complex.
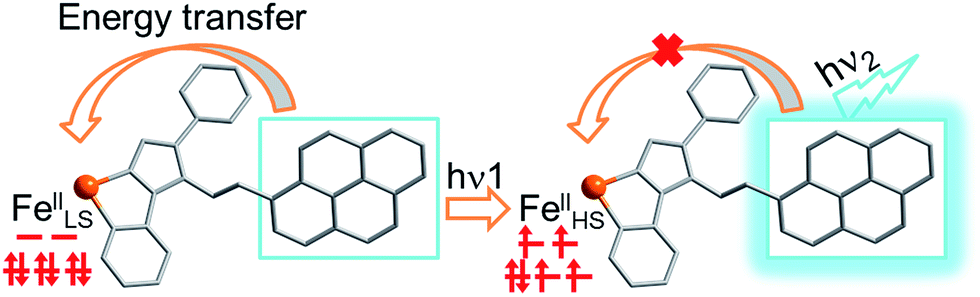 | ||
| Scheme 1 Modulation of fluorescence emission via energy transfer with a photoinduced spin transition from FeLSII to FeHSII. | ||
Results and discussion
Green crystals of 1 were obtained by the diffusion of diethyl ether into a DMF solution of 1. Complex 1 was crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The iron(II) ion adopted an N6 octahedral coordination configuration, surrounded by two nitrogen atoms from two trans-isothiocyanate anions and four nitrogen atoms from two bidentate ligands (Fig. 1). The solvent-free complex 1 showed high thermal stability up to 200 °C (Fig. S1†). At 277 K, the Fe–Npyridyl bond length (2.197(2) Å) was slightly longer than the Fe–Ntriazole (2.124(2) Å) and Fe–Nisothiocyanate (2.077(3) Å) bond lengths, which were close to that of FeHSII–N (HS = high spin).42 The bond lengths were in the range of 2.014(2)–1.932(2) Å at 90 K (Table S3†), which is in agreement with related FeLSII (LS = low spin) complexes containing bidentate abpt ligands.43,44 These temperature-dependent structural variations suggest the occurrence of thermal-induced SCO. The spin transition from HS to LS was also verified by the change in the distortion of the N6 octahedron. The Σ parameter, defined as the total deviation of the 12 cis N–Fe–N angles from 90°, decreased from 72° to 51° when samples were cooled from 277 to 90 K. When compared with the HS state structure, the LS state structure showed a smaller deformation. This phenomenon is consistent with that previously observed in other SCO complexes.45–47
. The iron(II) ion adopted an N6 octahedral coordination configuration, surrounded by two nitrogen atoms from two trans-isothiocyanate anions and four nitrogen atoms from two bidentate ligands (Fig. 1). The solvent-free complex 1 showed high thermal stability up to 200 °C (Fig. S1†). At 277 K, the Fe–Npyridyl bond length (2.197(2) Å) was slightly longer than the Fe–Ntriazole (2.124(2) Å) and Fe–Nisothiocyanate (2.077(3) Å) bond lengths, which were close to that of FeHSII–N (HS = high spin).42 The bond lengths were in the range of 2.014(2)–1.932(2) Å at 90 K (Table S3†), which is in agreement with related FeLSII (LS = low spin) complexes containing bidentate abpt ligands.43,44 These temperature-dependent structural variations suggest the occurrence of thermal-induced SCO. The spin transition from HS to LS was also verified by the change in the distortion of the N6 octahedron. The Σ parameter, defined as the total deviation of the 12 cis N–Fe–N angles from 90°, decreased from 72° to 51° when samples were cooled from 277 to 90 K. When compared with the HS state structure, the LS state structure showed a smaller deformation. This phenomenon is consistent with that previously observed in other SCO complexes.45–47
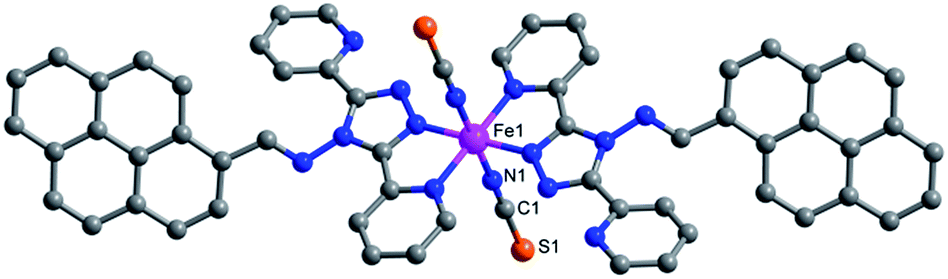 | ||
| Fig. 1 The crystal structure of complex 1. The pink, gray, blue and orange spheres represent FeII, C, N and S atoms, respectively. Hydrogen atoms are omitted for clarity. | ||
Adjacent molecules were connected via π⋯π stacking interactions involving pyridine rings to form a one-dimensional (1D) supramolecular chain along the crystallographic b axis. At 277 K, the π⋯π interactions are characterized by a centroid-to-centroid distance of 3.91 Å. Such one-dimensional chains are associated through four face-to-face π⋯π interactions between pyrene rings generating a 3D supramolecular network. The centroid-to-centroid distances are 3.66 Å, 3.86 Å and 3.85 Å (Fig. S2†).
To confirm the photo-responsive properties of the SCO unit, the magnetic susceptibilities of 1 were measured before and after irradiation. The temperature-dependent magnetic measurement revealed that complex 1 underwent complete SCO with a transition temperature (T1/2) of 267 K (Fig. S4†). As the temperature increased from 2 K after irradiation at 671 nm, the χT value increased from 0.05 cm3 mol−1 K to a maximum value of 0.68 cm3 mol−1 K at 12 K due to the light-induced excited spin state trapping (LIESST) effect, and this corresponds to the trapping of approximately 21.3% of the metastable FeHSII ions (Fig. S5†). Moreover, the irradiation-time dependence of the infrared (IR) spectra further verifies the trapping of the photogenerated metastable FeHSII ions by irradiation at 20 K. The νNCS− stretching bands (2080 cm−1) in the HS state began to appear after irradiation at 671 nm for 10 min and the intensity of the HS mode increased with increasing irradiation time (Fig. S6†). This result can be attributed to the photoinduced spin transition from the LS state to the metastable HS state. The photogenerated magnetization relaxed to the initial value upon heating to 60 K. Relaxation of the photoinduced metastable state was monitored at 10 K to probe the stability of the photoinduced phase, which can be determined based on the time decay of the magnetization after irradiation (Fig. S7†). The photoinduced phase showed considerable stability with a relaxation time of 3331.75 s at 10 K, which was sufficiently long to monitor the phototunable fluorescence emission at 10 K.
To verify the fluorescent properties, the fluorescence emission spectra of 1 and the ligand were obtained. At room temperature, ligand L showed an emission band at 516 nm under excitation at 380 nm (Fig. S8†). At the same excitation wavelength, the fluorescence emission spectrum of 1 displayed a maximum emission at 513 nm at 300 K, indicating that the fluorescence property of the ligand was well retained when coordinated with FeHSII ions. The fluorescence emission of 1 before and after irradiation was recorded to further examine the photoswitchable fluorescence emission behavior at 10 K. Interestingly, a significant increase in fluorescence emission intensity was observed after 2 h of irradiation. The increased degree (ΔR) before and after irradiation is defined as ΔR = (R1 − R0)/R0, where R0 and R1 are the emission intensities before and after 2 h of irradiation at 498 nm, respectively. The ΔR value of complex 1 reached up to 19.8% at 10 K, which is in agreement with the increased degree of magnetic susceptibility before and after irradiation. Thus, the above results demonstrate that fluorescence emission intensity can be controlled by photoinduced SCO from FeLSII to FeHSII ions. The fluorescence emission intensity of the photogenerated metastable state decayed rapidly upon heating (Fig. 2), which is consistent with the temperature-dependent magnetization behavior of the photogenerated metastable state. When the temperature was increased to 40 K, the emission spectrum almost overlapped with that obtained before irradiation at 10 K, demonstrating that the emission intensity of 1 could be increased by light irradiation and decreased by thermal treatment. This increase is possibly related to a photoinduced spin transition from the LS state to the HS state of FeII ions, whereas the decrease was caused by thermal relaxation of the photogenerated metastable HS state.
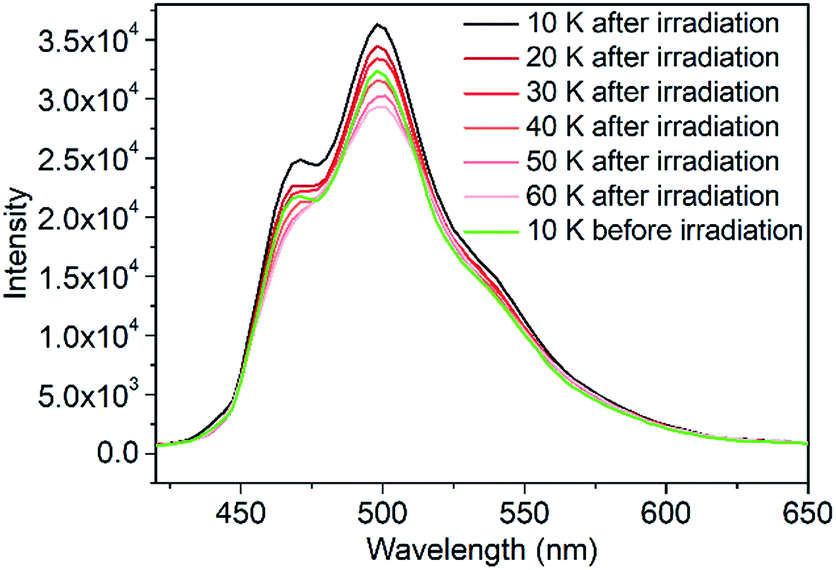 | ||
| Fig. 2 Thermal variations in the fluorescence emission of 1 after irradiation in the heating mode. | ||
The correlation between the fluorescence and the spin state of the FeII ions was studied to verify the photoinduced SCO tuning of the fluorescence emission intensity. As mentioned earlier, complex 1 was in the HS state at room temperature and the LS state at 90 K. With a temperature decrease from 300 to 200 K, the χT value decreased abruptly from 2.78 to 0.20 cm3 mol−1 K, gradually reaching a minimum value of 0.08 cm3 mol−1 K at 100 K, which is indicative of complete SCO with a transition temperature (T1/2) of 267 K (Fig. S4†). The first-order phase transition was further confirmed by heat capacity measurements over the SCO range with T1/2 = 265 K and this was in agreement with the transition temperature obtained from the magnetic data for 1 (Fig. S9†). The entropy and enthalpy changes associated with the spin transition were ΔS = 43.97 (±0.73) J K−1 mol−1 and ΔH = 11.34 (±0.16) kJ mol−1, respectively, indicating that the thermal-induced SCO is an entropy-driven process. In addition, the temperature-dependent Raman spectra of 1 revealed that a significant increase in the stretching intensity at 2120 cm−1 (LS) and a clear decrease at 2075 cm−1 (HS) were caused by a further reduction in temperature. The band (2075 cm−1) of the HS state disappeared at 100 K providing further evidence of complete spin transition behavior (Fig. S10†). Similar to Raman spectra, temperature-dependent IR spectra are also very useful to clarify the thermal-induced SCO process. Both the decrease of the HS mode at 2076 cm−1 and the increase of the LS mode at 2121 cm−1 in the intensity of νNCS− stretching bands were caused by decreasing temperature (Fig. S11†), which further confirms the occurrence of thermal-induced SCO behavior.
Temperature-dependent fluorescence emission spectra were also obtained to further elucidate the correlation between fluorescence and SCO from 300 to 10 K. Upon cooling, the emission intensity of the ligand gradually increased as a result of the suppression of thermal fluorescence quenching (Fig. S8†). However, when compared with the free ligand, complex 1 exhibited significantly different temperature-dependent fluorescence emission behavior (Fig. 3a). The emission intensity of 1 initially decreased with decreasing temperature, then reached the lowest value at 250 K. Upon further cooling, the emission intensity started to gradually increase and finally reached a maximum at 10 K (Fig. 3b). In addition, the maximum emission exhibited a marked hypsochromic shift of 15 nm as 1 was cooled from 300 to 100 K. A comparison of the magnetic and fluorescent properties shows that abnormal regions only occurred in the abrupt spin transition process from FeHSII to FeLSII ions. Moreover, the emission intensity of the HS state was stronger than that of the LS state in the narrow spin transition temperature range, demonstrating that fluorescence is indeed affected by the spin state of FeII ions more than the thermal treatment in the spin transition process. The results not only provide direct evidence for the synergy between fluorescence and thermal-induced SCO, but also confirm the earlier conclusion that the photoinduced fluorescence increase in emission intensity was controlled by photoinduced SCO from FeLSII to FeHSII ions.
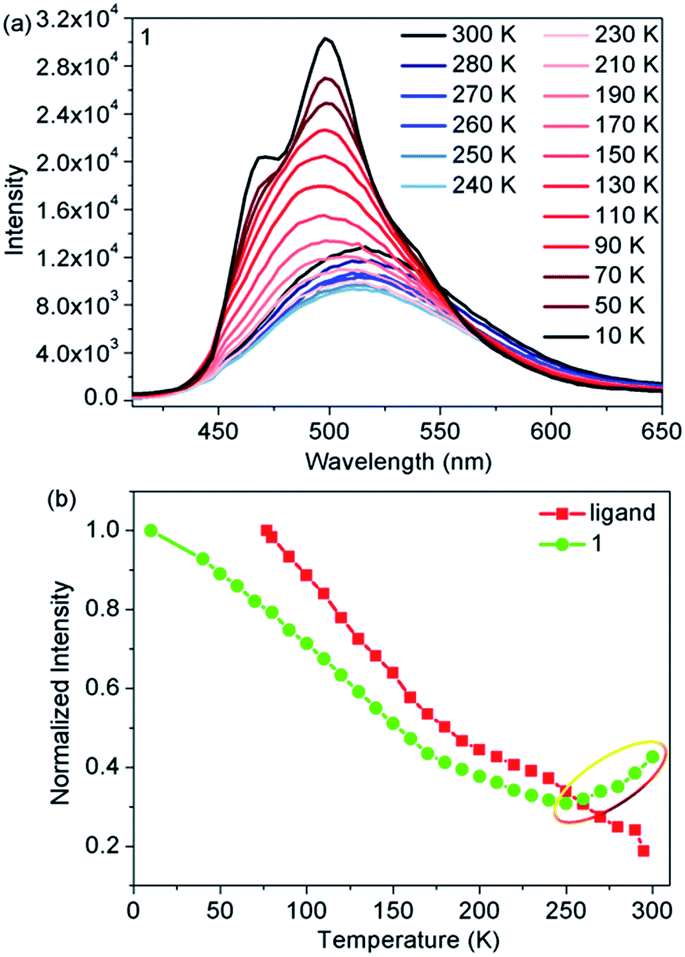 | ||
| Fig. 3 (a) Temperature-dependent fluorescence emission spectra for complex 1; (b) normalized maximum fluorescence emission intensity as a function of temperature for the ligand (λem = 516 nm) and 1 (λem = 513 nm at 300–170 K and λem = 498 nm at 160–10 K). | ||
To better understand the modulation mechanism, the variable UV-vis absorption spectra were recorded and they showed that the absorption intensity gradually increased upon cooling (Fig. 4). A new absorption band started to appear at around 700 nm at 260 K and the intensity increased with a lowering of the temperature, which was ascribed to the 1A1 → 1T1 transitions of the FeLSII ion.38 Furthermore, the time-dependent density functional theory (TD-DFT) calculated UV-vis absorption spectra of complex 1 in its HS and LS states were also obtained to help determine the possible electronic transition processes. The structures of the ground states and the low-lying excited states of the HS and LS complexes were fully optimized and verified using frequency calculations. The optimized LS and HS ground state structures of 1 compared well with their experimental counterparts. A calculated absorption peak at 388 nm for the HS state can be assigned to the intrapyrene π → π* transition and this is in agreement with the experimentally observed peaks at around 410 nm (Fig. S13†). Another calculated absorption peak at 476 nm was derived from the d(Fe) + π(SCN) → π*(pyrene) transitions and is ascribed to a metal-to-ligand charge transition (MLCT) and ligand-to-ligand charge transition (LLCT). As for the LS state, the calculated peaks centred around 392 and 509 nm. The former mainly arose from the π → π* excitation within the pyrene groups with a small MLCT contribution, and the latter mainly corresponded to d(Fe) + π(SCN) → π*(adpt) transitions (Fig. S15†). For the calculated MLCT bands, the charge acceptor is the pyrene group in the HS state, whereas it is mainly the adpt group in the LS state (Fig. 5). This discrepancy likely caused the bathochromic shift (33 nm) of the MLSLCT absorption band in the LS state. Compared with that in the HS state, the fluorescence emission band of the LS state shows opposing hypsochromic shifts (15 nm) (Fig. S16†). These phenomena together lead to an increase in the spectral overlap between MLSLCT absorption and fluorescence emission in the LS state, indicating that the energy level of the MLSLCT excitation closely matches that of the excited state related to the fluorescent ligand. Therefore, the excited state energy of the ligand may be transferred to the FeLSII ions via the resonant energy transfer process, which further leads to quenching of the fluorescence during spin transition from the HS state to the LS state.
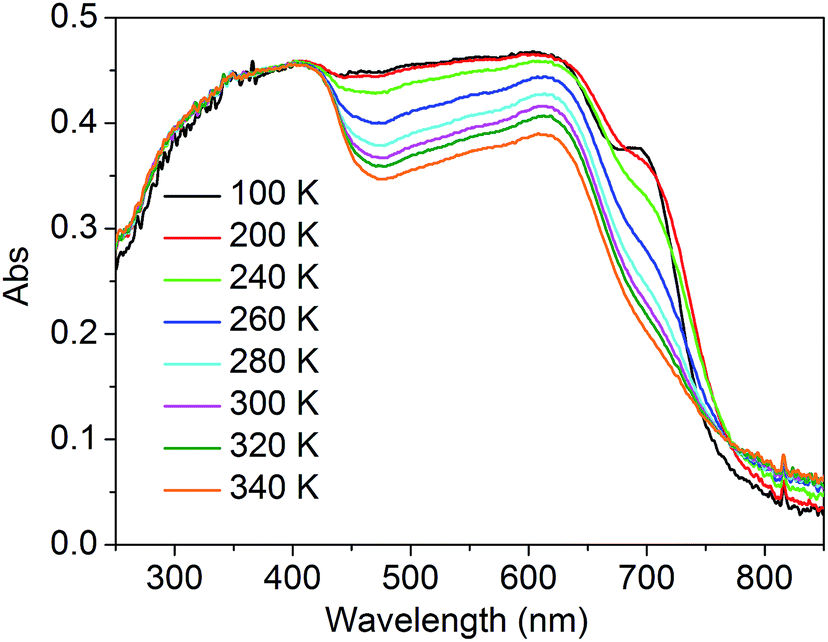 | ||
| Fig. 4 Temperature-dependent absorption spectra of 1. | ||
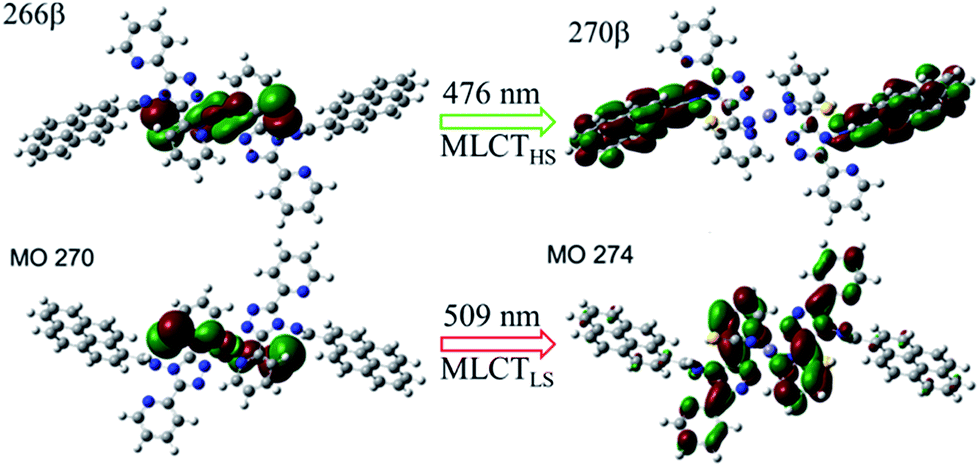 | ||
| Fig. 5 Characteristic orbitals related to the MLCT transitions of 1 for the HS and LS states. | ||
In contrast to the LS complex, the HS complex exhibits a small and insignificant spectral overlap between fluorescence emission and MHSLCT absorption. This result further suggests that the energy level of the MHSLCT is slightly higher than the excited energy level of the ligand. Therefore, the excited energy of the ligand cannot be effectively transferred to the FeHSII center and the fluorescence is only slightly decreased in the HS state. After light irradiation, complex 1 underwent an incomplete light-induced SCO from the LS state to the metastable HS state. The energy transfer process is partly deactivated due to a decrease of the LS species, causing a slight increase in the emission intensity after irradiation. Therefore, fluorescence modulation via the LIESST effect is related to the energy transfer between the excited state of the ligand and the energy level of MLSLCT excitation.
Conclusions
In summary, a photoinduced FeII SCO unit has been coupled with a fluorescent ligand via coordination bonds, and the fluorescence emission can be modulated by their synergy. Theoretical and experimental analyses showed that the modulation mechanism arises mainly from the energy transfer from the excited state of the fluorescent ligand to the MLCT energy level of FeLSII ions. These findings demonstrate that photoinduced FeII SCO can serve as a light-responsive unit for coupling with a fluorophore, providing a potential approach for producing switched devices with photoinduced fluorescence.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was partly supported by the NSFC (Grants 21421005, 21322103, 91422302, 21771029, 21373036 and 21573034), the Open Fund of the National Laboratory of Molecular Science (20140116), the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) under Grant No. U1501501, and the Chinese Scholarship Council (201706060254). The supercomputer time was supported by the National Supercomputing Center in Guangzhou, China and the High Performance Computing Center at Dalian University of Technology.Notes and references
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- S. V. Eliseeva and J.-C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- K. M. Chan, D. K. Kölmel, S. Wang and E. T. Kool, Angew. Chem., Int. Ed., 2017, 56, 6497–6501 CrossRef CAS PubMed.
- J. Su, T. Fukaminato, J.-P. Placial, T. Onodera, R. Suzuki, H. Oikawa, A. Brosseau, F. Brisset, R. Pansu, K. Nakatani and R. Métivier, Angew. Chem., Int. Ed., 2016, 55, 3662–3666 CrossRef CAS PubMed.
- S.-J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS PubMed.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- Y. Hashimoto, T. Nakashima, D. Shimizu and T. Kawai, Chem. Commun., 2016, 52, 5171–5174 RSC.
- Q. Zhang, H. Sun, X. Wang, X. Hao and S. An, ACS Appl. Mater. Interfaces, 2015, 7, 25289–25297 CAS.
- I. L. Medintz, S. A. Trammell, H. Mattoussi and J. M. Mauro, J. Am. Chem. Soc., 2004, 126, 30–31 CrossRef CAS PubMed.
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672–687 RSC.
- S. Ishida, T. Fukaminato, D. Kitagawa, S. Kobatake, S. Kim, T. Ogata and S. Kurihara, Chem. Commun., 2017, 53, 8268–8271 RSC.
- K. Jeong, S. Park, Y.-D. Lee, C.-K. Lim, J. Kim, B. H. Chung, I. C. Kwon, C. R. Park and S. Kim, Adv. Mater., 2013, 25, 5574–5580 CrossRef CAS PubMed.
- J. W. Chung, S.-J. Yoon, S.-J. Lim, B.-K. An and S. Y. Park, Angew. Chem., Int. Ed., 2009, 48, 7030–7034 CrossRef CAS PubMed.
- K. A. Green, M. P. Cifuentes, T. C. Corkery, M. Samoc and M. G. Humphrey, Angew. Chem., Int. Ed., 2009, 48, 7867–7870 CrossRef CAS PubMed.
- S.-J. Lim, B.-K. An, S. D. Jung, M.-A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS PubMed.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS PubMed.
- S. Castellanos, A. Goulet-Hanssens, F. Zhao, A. Dikhtiarenko, A. Pustovarenko, S. Hecht, J. Gascon, F. Kapteijn and D. Bléger, Chem.–Eur. J., 2016, 22, 746–752 CrossRef CAS PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem.–Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- A. Bousseksou, G. Molnár, J. A. Real and K. Tanaka, Coord. Chem. Rev., 2007, 251, 1822–1833 CrossRef CAS.
- S. Ohkoshi, S. Takano, K. Imoto, M. Yoshikiyo, A. Namai and H. Tokoro, Nat. Photonics, 2013, 8, 65–71 CrossRef.
- D.-P. Dong, T. Liu, S. Kanegawa, S. Kang, O. Sato, C. He and C.-Y. Duan, Angew. Chem., Int. Ed., 2012, 51, 5119–5123 CrossRef CAS PubMed.
- M. Engeser, L. Fabbrizzi, M. Licchelli and D. Sacchi, Chem. Commun., 1999, 1191–1192 RSC.
- M. Masaki, I. Hikaru and T. Hiroyuki, Chem. Lett., 2008, 37, 374–375 CrossRef.
- L. Salmon, G. Molnar, D. Zitouni, C. Quintero, C. Bergaud, J.-C. Micheau and A. Bousseksou, J. Mater. Chem., 2010, 20, 5499–5503 RSC.
- S. Titos-Padilla, J. M. Herrera, X.-W. Chen, J. J. Delgado and E. Colacio, Angew. Chem., Int. Ed., 2011, 50, 3290–3293 CrossRef CAS PubMed.
- C. M. Quintero, I. y. A. Gural’skiy, L. Salmon, G. Molnar, C. Bergaud and A. Bousseksou, J. Mater. Chem., 2012, 22, 3745–3751 RSC.
- C.-F. Wang, R.-F. Li, X.-Y. Chen, R.-J. Wei, L.-S. Zheng and J. Tao, Angew. Chem., Int. Ed., 2015, 54, 1574–1577 CrossRef CAS PubMed.
- C.-F. Wang, M.-J. Sun, Q.-J. Guo, Z.-X. Cao, L.-S. Zheng and J. Tao, Chem. Commun., 2016, 52, 14322–14325 RSC.
- Y. Garcia, F. Robert, A. D. Naik, G. Zhou, B. Tinant, K. Robeyns, S. Michotte and L. Piraux, J. Am. Chem. Soc., 2011, 133, 15850–15853 CrossRef CAS PubMed.
- M. Estrader, J. Salinas Uber, L. A. Barrios, J. Garcia, P. Lloyd-Williams, O. Roubeau, S. J. Teat and G. Aromi, Angew. Chem., Int. Ed., 2017, 56, 15622–15627 CrossRef CAS PubMed.
- Y. Jiao, J. Zhu, Y. Guo, W. He and Z. Guo, J. Mater. Chem. C, 2017, 5, 5214–5222 RSC.
- B. Schäfer, T. Bauer, I. Faus, J. A. Wolny, F. Dahms, O. Fuhr, S. Lebedkin, H. C. Wille, K. Schlage, K. Chevalier, F. Rupp, R. Diller, V. Schunemann, M. M. Kappes and M. Ruben, Dalton Trans., 2017, 46, 2289–2302 RSC.
- I.-R. Jeon, C. Mathoniere, R. Clerac, M. Rouzieres, O. Jeannin, E. Trzop, E. Collet and M. Fourmigue, Chem. Commun., 2017, 53, 10283–10286 RSC.
- C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2013, 135, 19083–19086 CrossRef PubMed.
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Angew. Chem., Int. Ed., 2015, 54, 823–827 CrossRef CAS PubMed.
- S. Ohkoshi, K. Imoto, Y. Tsunobuchi, S. Takano and H. Tokoro, Nat. Chem., 2011, 3, 564–569 CrossRef CAS PubMed.
- J.-F. Létard, J. Mater. Chem., 2006, 16, 2550–2559 RSC.
- S. Decurtins, P. Gütlich, C. P. Köhler and H. Spiering, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef CAS.
- A. Hauser, Chem. Phys. Lett., 1986, 124, 543–548 CrossRef CAS.
- L. J. Kershaw Cook, H. J. Shepherd, T. P. Comyn, C. Baldé, O. Cespedes, G. Chastanet and M. A. Halcrow, Chem.–Eur. J., 2015, 21, 4805–4816 CrossRef CAS PubMed.
- C.-F. Sheu, S.-M. Chen, S.-C. Wang, G.-H. Lee, Y.-H. Liu and Y. Wang, Chem. Commun., 2009, 7512–7514 RSC.
- C.-H. Shih, C.-F. Sheu, K. Kato, K. Sugimoto, J. Kim, Y. Wang and M. Takata, Dalton Trans., 2010, 39, 9794–9800 RSC.
- R. W. Hogue, H. L. C. Feltham, R. G. Miller and S. Brooker, Inorg. Chem., 2016, 55, 4152–4165 CrossRef CAS PubMed.
- J.-L. Wang, Q. Liu, Y.-S. Meng, H. Zheng, H.-L. Zhu, Q. Shi and T. Liu, Inorg. Chem., 2017, 56, 10674–10680 CrossRef CAS PubMed.
- W. Phonsri, P. Harding, L. Liu, S. G. Telfer, K. S. Murray, B. Moubaraki, T. M. Ross, G. N. L. Jameson and D. J. Harding, Chem. Sci., 2017, 8, 3949–3959 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and physical measurement details. Crystal data in CIF format and additional figures (Fig. S1–S14). CCDC 1447594 and 1447595. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05221a |
| This journal is © The Royal Society of Chemistry 2018 |