
Reaction-based ratiometric fluorescent probe for selective recognition of sulfide anions with a large Stokes shift through switching on ESIPT†
Parthasarathi
Karmakar
a,
Srimanta
Manna
a,
Syed Samim
Ali
a,
Uday Narayan
Guria
a,
Ripon
Sarkar
b,
Pallab
Datta
b,
Debasish
Mandal
c and
Ajit Kumar
Mahapatra
 *a
*a
aDepartment of Chemistry, Indian Institute of Engineering Science and Technology, Shibpur, Howrah-711103, West Bengal, India. E-mail: akmahapatra@chem.iiests.ac.in; Fax: +913326684564
bCentre for Healthcare Science and Technology, Indian Institute of Engineering Science and Technology, Shibpur, India
cIndian Institute of Toxicology Research, Lucknow, India
First published on 10th November 2017
Abstract
An ESIPT-based, highly selective, ratiometric fluorescent probe BNPT has been synthesized and characterized, which shows turn-on fluorescence response in the presence of S2−, attributed to the removal of a protective 2,4-dinitrobenzene (DNB) moiety, and the resulting fluorescent product has been used for the selective detection of Zn2+. UV-vis and fluorescence spectroscopy analysis, and DFT and TDDFT calculations were carried out to understand the sensing mechanism. The probe BNPT displays a rapid response time and good sensitivity. The probe can detect quantitatively in a concentration range of 0–6.0 μM. The detection limit is 31.3 nM. The sensing ability of BNPT has been successfully applied in real water samples. Applicability as an in-field test kit has been demonstrated by sensing with a BNPT-coated TLC plate. The probe BNPT has been successfully used for visualization of intracellular environment in living cells. The uniqueness lies in the fact that starting with the probe BNPT, sensing can be achieved by two different mechanisms – first, by selective de-protection of the probe BNPT and second, by reacting with the Zn2+ ensemble with S2−.
Introduction
Developing fluorescent chemosensors for chemical species that can be used in detecting biologically and environmentally important anions and cations has received tremendous attention due to their crucial roles in a variety of biological and chemical processes.1 In particular, Zn2+ detection has aroused immense interest, because it plays critical roles in the fundamental physiological processes of organisms. In contrast, sulfide (S2−), as a toxic, hazardous, and traditional pollutant, is widely spread in the environment.2 It is produced naturally and as a result of human activity. Natural sources include non-specific and anaerobic bacteria reduction of sulfates and sulfur-containing amino acids in meat proteins. S2− is found naturally in volcanic eruptions, crude petroleum and hot springs. It is released from stagnant or polluted waters and manure or coal pits. S2− may be produced by a variety of commercial methods as a byproduct in industrial processes, for example, conversion into sulfur and sulfuric acid, dyes and cosmetic manufacturing, production of an agricultural disinfectant, paper and wood pulp, etc.3 Therefore, it is possible to bio-concentrate and bio-magnify in the food-chain. So continuous and high concentration exposure of S2− would lead to various physiological and biochemical problems such as irritation in mucous membranes, unconsciousness, and respiratory paralysis.4 Once a sulfide anion is protonated, it becomes even more toxic. Therefore, the detection of S2− has become very important from an industrial, environmental and biological point of view through newer and quicker analytical methodologies.5 To date, a variety of detection strategies have been developed for S2−, such as spectroscopy,6 electrochemical methods,7 titration,8 ion chromatography9 and chemiluminescence methods.10 Among the techniques employed, S2− sensing by fluorescence spectrometry has received much attention due to its easy operability and high sensitivity.11 On the other hand, Zn2+ is one of the most abundant transition-metal ions present in living cells, owing to its rich coordination chemistry and it plays a variety of fundamental roles in the physiological processes in organisms ranging from bacteria to mammals.12 Pools of labile Zn2+ are found in the cells of mammalian organs, including the brain,13 pancreas and prostate.14 In contrast, overloading of Zn2+ in the neuronal cytoplasm and the metabolic process are associated with some serious diseases, such as Wilson's Menke's disease and various infectious diseases.15 Zn2+ is also associated with various anions, especially with sulfides and phosphates, by showing strong affinity.16 Recently, metal ion complexes have been used as receptors for sulfides and phosphates, and this emerged as one of the most successful strategies, since it provides specific metal ion–anion interactions.17 Among the metal complex-based receptors, the ones that exhibit fluorescence change in the presence of Zn2+ have attracted considerable attention because of the strong affinity of Zn2+ toward sulfides and phosphates.18 Thus, considerable attention has been devoted to the development of fluorescent probes for highly selective recognition of Zn2+. ESIPT-based fluorophores such as benzimidazole,19 benzoxazole,20 benzothiazole21 and thiazole22 have been explored for the detection of Zn2+.Although a great number of selective fluorescent probes for S2− have been developed,23 fluorescent sensing of S2− in water solution still remains a challenging task because of the strong hydration nature of anions, which weakens the interaction of probes with the target anions.24 A number of sensing mechanisms have been employed to overcome this issue for S2− detection, such as metal displacement (ensemble),25 Michael addition reaction,26 cleavage reactions of 2,4-dinitrobenzenesulfonyl (DNBS) with S2−,27 nucleophilic substitution reactions,28 and disulfide exchange reactions.29 These studies greatly advanced the research on the fluorescent detection of S2−; however, many of these probes suffer from relatively low sensitivity due to the inefficiency of the quenching group or inadequate selectivity due to the cross reactivity of the reactive unit utilized. A ratiometric method measuring the ratio of fluorescence intensities at two wavelengths provides an alternative approach, which can overcome the drawbacks of intensity-based measurements by built-in correction of two emission bands and seems to be more favorable for sensing target ions in comparison with fluorescence intensity-based probes. Therefore, there still has been a large demand to develop a new fluorescent probe that features high reactivity and specificity, excellent sensitivity and fast response toward S2−.
In this paper, we identify the 2,4-dinitrobenzene (DNB) moiety as a novel reactive group and reaction switch specific for S2−. Integration of this unique moiety into the phenylthiazole and benzothiazole fluorophores afforded a ratiometric fluorescence “off–on” probe 2-[3-benzothiazol-2-yl-4-(2,4-dinitrophenoxy)phenyl]-4-methylthiazole-5-carboxylic ethyl ester, BNPT, for highly specific and sensitive detection of sulfide. In BNPT, the DNB group is especially preferable as an efficient reactive site for S2− and the benzothiazole moiety used as recognition unit for signal transducer through excited state intramolecular proton transfer (ESIPT) resulted in a ratiometric fluorescent probe. We envisioned that the probe itself gave almost no fluorescence emission in the absence of S2− due to the dual-quenching effect of DNB via the intramolecular charge transfer (ICT) and the thiazole can quench the fluorescence via a photoinduced electron transfer (PET) effect. We predicted that fluorescence will be heavily quenched through the combined usage of PET–ICT dual-quenching effects.
Upon reaction with S2−, the fluorophore BNPT-OH was released and showed significant fluorescence enhancement attributing to the ESIPT process (Scheme 1).
 | ||
| Scheme 1 Structure and sensing mechanism of BNPT against S2−. | ||
Results and discussion
The precursor hydroxyphenylbenzothiazole derivative 5 has been synthesized in four steps on the basis of our previously reported literature method30 starting from p-hydroxybenzonitrile. All the precursor and probe molecules were fully characterized by 1H NMR, 13C NMR, and ESI-MS spectroscopy. The S2−-promoted aromatic nucleophilic substitution reaction cleaves the specific O–C(DNB) bond of probe BNPT, which liberates precursor 5 and an intramolecular proton transfer gives ratiometric emission from the excited state.The reactivity of probe BNPT toward different anions and their preferential selectivity toward S2− over the other ions have been studied by fluorescence titrations. We found that the addition of trace amounts of S2− causes the absorption and fluorescence signal to change rapidly, which is very important for real-time detection. To find the suitability of the solvent system of BNPT to sense S2− in environmental and biological samples, we first investigated the emission properties of probe BNPT in pure CH3CN and pure aqueous solution; disappointingly, in pure aqueous solutions, the probe showed no response to S2−. We speculate that this phenomenon was probably due to the hydrogen bond between S2− and water in an aqueous solution that weakened the nucleophilicity of S2− to probes and then decreased the reaction speed. Finally, the fluorescence signaling behavior of the probe BNPT was investigated in an optimized aqueous acetonitrile (CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) solution at room temperature.
:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) solution at room temperature.
Spectral response and detection of S2− in solutions
We examined the absorption spectra of probe BNPT in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) at room temperature. The absorption spectra of free probe BNPT displayed a maximum absorbance at 312 nm. Upon the addition of S2− (Na2S has been used as a source of S2− in all the experiments), the color of the solution turned from colorless to slight greenish yellow, which was clearly recognizable by the naked eye.
At the same time, a concomitant increase in a new absorption band at 426 nm was observed with two isosbestic points at 275 nm and 347 nm (Fig. 1). The isosbestic points observed at 275 nm and 347 nm were indicative of the transition between the reactant and the product through the formation of aryl-σ-complex species.
 | ||
| Fig. 1 (a) UV-Vis spectral change of BNPT (1 × 10−5 M) in the presence of increasing amounts of S2− (1 × 10−4 M) in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) at room temperature. Inset: Photograph of visible color under ambient light in the absence and presence of S2−. (b) The linear relationship between the absorbance ratio (I426/I312) and the concentration of S2−. | ||
Due to the specific O–C (DNB) cleavage, a distinct 114 nm red shift in absorbance was observed. Since the phenolate group is a much stronger electron donor than the ether group in BNPT, the ICT efficiency should be significantly enhanced by the interaction of probe BNPT with S2− and thus shifting the absorption to a longer wavelength.
Subsequently, the fluorescence spectra of probe BNPT in the absence and presence of S2− were recorded (Fig. 2). The probe BNPT exhibits very weak emission at ∼426 nm when excited at 347 nm in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4). This negligible emission of BNPT has been explained in terms of photoinduced electrontransfer (PET) as well as prohibitions of the ESIPT process produced by the 2,4-dinitrobenzene moiety. Besides these two effects, the electron-withdrawing nature of the nitro group may also be contributing toward the non-fluorescent nature of BNPT.
 | ||
| Fig. 2 (a) Fluorescence emission spectra (λex = 347 nm) of BNPT (1 × 10−5 M) in the presence of increasing amounts of S2− (1 × 10−4 M) in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) at room temperature. Inset: Enlarged view of the isobestic point (b) concentration-dependent fluorescence intensity changes of BNPT (1 × 10−5 M) in the presence of increasing amounts of S2− (1 × 10−4 M). Inset: Visual fluorescence color of BNPT before and after the addition of 5 equiv. of S2−. | ||
However, upon the progressive additions of S2− (0–5 equiv.) to the solution of BNPT, distinct enhancement of the fluorescent intensity at 503 nm was observed and a simultaneous decrease in emission at 426 nm, which indicated that the chemical reaction between S2− and the DNB group could be attributed to the regeneration of a hydroxyl group by effective cleavage of the 2,4-dinitrobenzene moiety through the ArSN reaction and restoration of the ESIPT process.
The observation is in good agreement with the aforementioned design concept. Accordingly, the emission changes also resulted in obvious fluorescence color changes from colorless to green as well as a well-defined isoemissive point at 437 nm. The emission intensity ratio, I503/I426, increases with the increase in the S2− concentrations, which allows the detection of S2− ratiometrically. It is noteworthy that the difference in the two emission wavelengths is very large (emission shift = 77 nm), which not only contributes to the accurate measurement of the intensities of the two emission peaks, but also results in a huge ratiometric value (I503/I426 = 450 fold). Under the same conditions, the ratio changes of the two fluorescence peaks (I503/I426) produced a good linear relationship with the concentration of S2− between 0 and 0.6 μM, and the detection limit for S2− was determined as 31.3 nM based on S/N = 2, which was sufficiently low for the detection of S2− in environmental samples (Fig. S13, ESI†).
Based on the spectral changes, we propose that a cleavage of ether group by S2− with BNPT enables it to turn-on fluorescence. Furthermore, the ratiometric emission by BNPT with S2− could be explained as a consequence of ICT and ESIPT. The ratiometric emissions in the case of BNPT may be attributed to the photo tautomer of excited normal isomer (N* emission) and tautomer (T* emission) forms, which result from an ESIPT (N* ↔ T*) process (Scheme 3).
Molecules having both acidic and basic moieties in close proximity often undergo ESIPT in the excited state. In the present case, transfer of a phenolic-OH proton to the benzothiazole nitrogen through keto–enol tautomerization leads to significant differences in structure and electronic configuration from its corresponding normal species.31 If the phenolic-OH of the probe is protected by a suitable group, the ESIPT process is inhibited resulting in fluorescence switch off. The probe BNPT shows only local fluorescence emissions at 426 nm. The emission of BNPT at lower wavelength (426 nm) is assigned to the enol form, i.e., normal isomer (N* emission), while the keto form emits (T* emission) at the higher wavelength (503 nm). Thus, the ESIPT process is much more favorable in the probe BNPT after the S2− reaction and it is highly dependent on the chemical structure.
Sensing of S2− in the presence of other anions
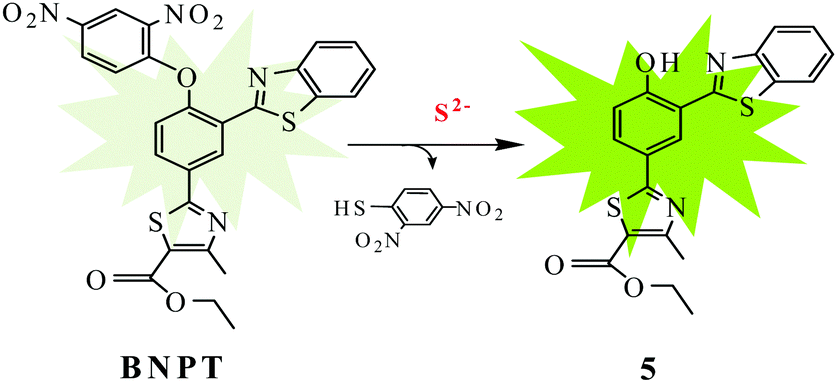
In order to check whether the probe BNPT is sensitive to only S2− or even to the other competitive anions, similar fluorescence titrations were carried out in the same medium with 16 other different anions, viz. N3−, AcO−, SCN−, PO43−, H2PO4−, HCO3−, CO32−, F−, Cl−, Br−, I−, NO3−, SO42−, HSO4−, SO32−, and S2O32− as well as a thiol species (cysteine, homocysteine, and glutathione), and no significant fluorescence enhancement was found in the presence of any of these ions (Fig. 3). Visual fluorescent color change experiments have been carried out to look at the behavior of probe BNPT in the presence of various anions. Under UV light, the solution of BNPT is weakly fluorescent, whereas in the presence of S2−, it shows an intense green fluorescent color that is otherwise not present in the case of the other ions studied (Fig. S9, ESI†). Therefore, S2− can easily be differentiated by visual color change among the other anions.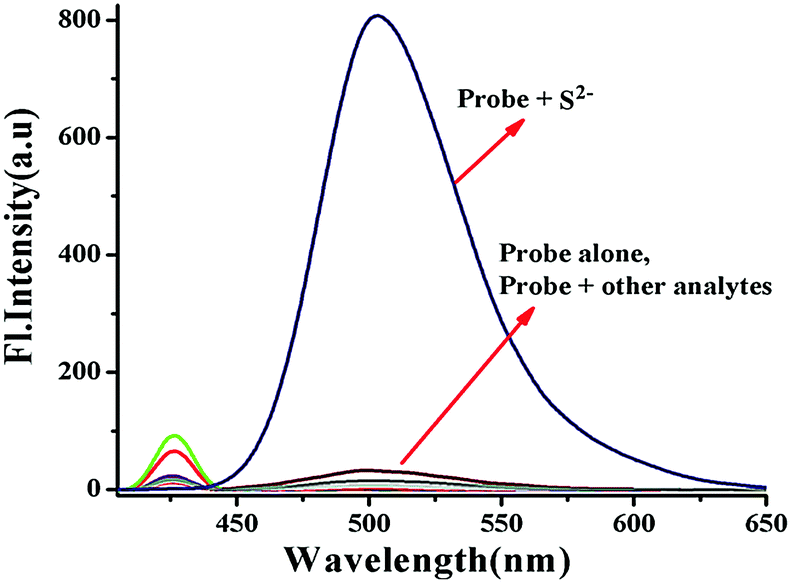 | ||
| Fig. 3 Fluorescence spectra of probe BNPT (10 μM) in the presence of various anions (50 μM) in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4). | ||
In order to show the practical utility of the probe BNPT to detect S2− selectively even in the presence of other anions, competitive allied anion titrations were carried out. As can be seen, these competitive species, including N3−, AcO−, SCN−, PO43−, H2PO4−, HCO3−, CO32−, NO3−, SO42−, F−, Cl−, Br−, I−, HSO4−, SO32−, S2O32−, Cys, Hcy, and GSH, which often show strong interference to S2− detection, did not lead to any significant ratiometric fluorescence changes at all, and the fluorescence emission spectrum of BNPT remained almost undisturbed (Fig. S8, ESI†). Therefore, it can be concluded that probe BNPT selectively reacts with S2− even in the presence of other analytes.
Time courses of the BNPT and S2− system
We further moved forward to study the reaction kinetics of the probe BNPT in the presence of S2−. Probe BNPT (10 μM) was incubated with 50 μM S2− in HEPES buffer at 37 °C. Upon addition of increasing concentrations of S2−, a dramatic increase of emission intensity at 503 nm was observed within 1 min, and the emission signal essentially reached a plateau within 5 min and showed a fairly fast reaction rate (Fig. 4).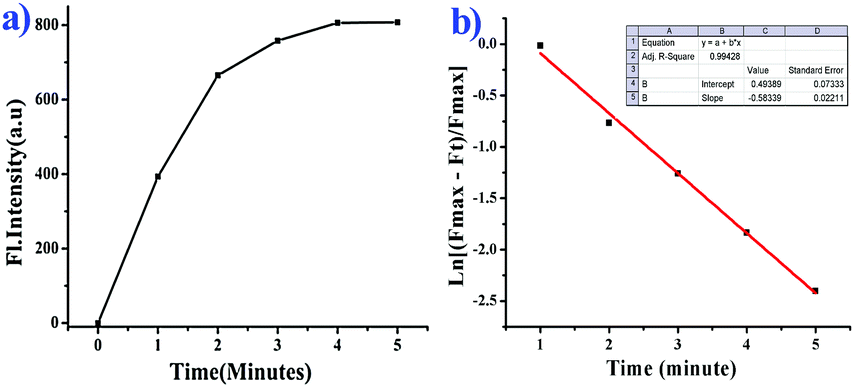 | ||
| Fig. 4 (a) Time course experiment of the probe BNPT in the presence of 5 equiv. S2− in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) at room temperature. (b) Pseudo-first-order kinetic plot. | ||
Through monitoring the fluorescence emission at 503 nm, the S2− mediated reaction was found to obey a pseudo-first-order law of rate constant K′ = 0.58 min−1 under S2− excess conditions. The absence of S2− in the tested solutions of probe BNPT meant that no changes in fluorescence response were detected; this result suggests that probe BNPT is a “fast-response” probe for S2− and may be suitable for real-time sensing of S2−.
Secondary recognition of Zn2+
The release of phenolic compound 5 from BNPT by S2− has been used for the secondary sensing property toward metal ions. To the solution (BNPT + S2−), an aqueous solution of Zinc perchlorate (5 mM) was added – a complete quenching was observed at 503 nm with concomitant increase of fluorescence at 426 nm (Fig. S5, ESI†). Fluorescence color changed from intense green to non-fluorescent. Interestingly, subsequent addition of S2− (5 mM) to the above solution retrieves the fluorescence (Fig. S6, ESI†). The color of the solution gradually changed from non-fluorescent to intense green. The secondary recognition capability of the compound 5 has been carried out by titrating it with different cations, viz., Na+, K+, Mg2+, Ca2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Cd2+, Zn2+, and Hg2+.Compound 5 does not show any significant fluorescence change toward alkali and alkaline earth metal cations; however, it shows quenching of fluorescence intensity in the rest of the cations due to inhibition of ESIPT on chelation. Thus, in the case of zinc ions, it shows maximum quenching and the sigmoidal plot exhibits saturation at ∼3.5 equiv., as compared with all other transition-metal ions studied, which show saturation at much higher equivalents, suggesting the strong chelating affinity of Zn2+ toward 5 (Fig. 5). The fast and selective response of compound 5 toward Zn2+ as compared with many cations, including other transition-metal ions, allows it to be a selective sensing molecular system for Zn2+.
 | ||
| Fig. 5 Fluorescence spectra of BNPT (10 μM) as a function of cations/compound 5 mole ratio obtained during the titration of BNPT with different cations in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4). | ||
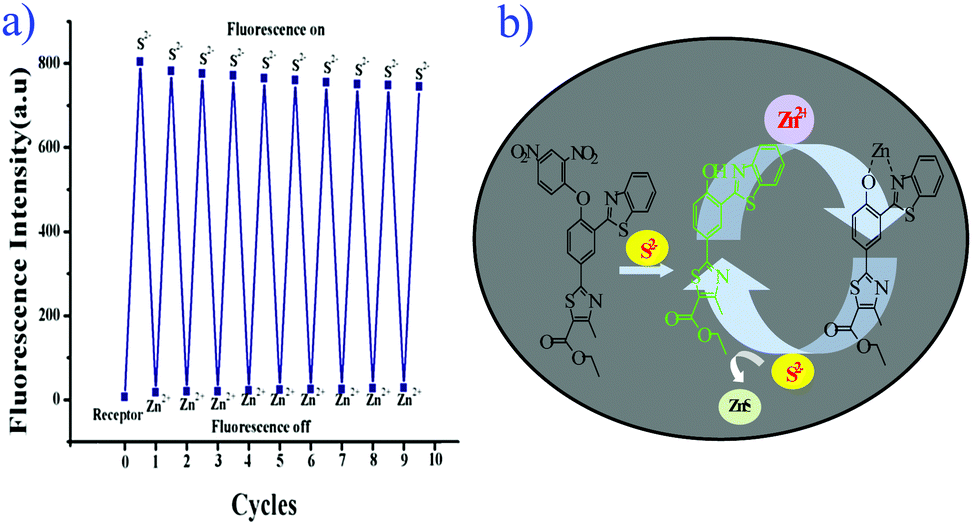 | ||
| Fig. 6 (a) Reversible switching cycles of fluorescence intensity (λex = 347 nm) by alternate addition of S2− and Zn2+ in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4). (b) Schematic representation of reversible nature of compound 5. | ||
The chemical reversibility behavior of the compound 5 with Zn2+, followed by the removal of Zn2+ by S2−, was studied in aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4). Hence, compound 5 is a reversible and reusable sensor for Zn2+ and its zinc complex [Zn-5] as a secondary sensor for S2−.
The 1:1 stoichiometry of [Zn-5] was established from the Job's plot (Fig. S12, ESI†). The formation of a 1:1 complex was further confirmed by HR-MS studies. The molecular ion peak appears at m/z 558.8810, and corresponds to the mass of [BNPT + Zn2+ + ClO4]+ (Fig. S4, ESI†).
The binding constant for the formation of [Zn-5] complex was calculated on the basis of change of emission at 503 nm by considering 1:1 binding stoichiometry. The binding constant (K) determined by the Benesi–Hildbrand plot experiments was found to be 4.0 × 105 M−1 (Fig. S14, ESI†).
Theoretical calculations
Computational calculations were carried out using the Gaussian 09 program to explain the electronic structural properties of probe BNPT and its corresponding phenolic product 5, as well as to establish the complexation features of product 5 by Zn2+. To do that, initially the structures of BNPT and its corresponding phenolic product 5 were optimized in DFT. The optimized geometries and spatial distributions and orbital energies of HOMO and LUMO of BNPT and resultant product 5 are given in Fig. 7. The optimized 5 was subjected to the interaction with Zn2+ that was further optimized by Lanl2dz with the same ECP to obtain the complex [Zn-5]. The optimized geometries and the calculated electron distributions in the frontier molecular orbitals HOMO and LUMO of the phenolic product 5 – Zn-complex are shown in ESI† (Fig. S16 and Table S1). The major chromophore exists in a nearly planar conformation, extending from the phenoxide moieties to the acceptors thiazole and benzthiazole moieties of BNPT. Such a planar conformation enables an efficient π-conjugation and favors the efficient ICT transition and ESIPT process between the donor and the acceptor groups. | ||
| Fig. 7 The optimized configuration of (a) BNPT and (b) compound 5. HOMO–LUMO energy gaps for respective compounds and interfacial plots of the orbitals: (c) BNPT and (d) compound 5. | ||
In particular, for the probe BNPT, the HOMOs are majorly localized on the benzothiazole and thiazole parts whereas LUMOs are widely spread out over the dinitrobenzene (DNB) part that results in PET process ON and it effects the initial weak fluorescent of the probe. The charge separation in the excited state is much more pronounced in the phenolic species that corresponds to the n → π* transition of the phenolic group to the thiazole moiety. Furthermore, the energy gap between the HOMO and LUMO of the probe BNPT was 2.87 eV higher than that of their corresponding phenolic compound (1.86 eV) (Table S2, ESI†), in good agreement with the red shift in the absorption observed upon the treatment of probes with S2−.
The TDDFT calculations were performed at the B3LYP/6-31+G(d,p) level to explain the optical properties of the ground and excited states behavior of BNPT and compound 5. The vertical transitions i.e., the calculated λmax, main orbital transition, and oscillator strength (f) are listed in Table S1, (ESI†). The vertical transitions calculated by TDDFT of BNPT and compound 5 were found to have good agreement with the experimental data. The studies suggest that the vertical transitions observed at ∼319 and ∼366 nm are comparable to those of the experimentally observed spectra at ∼312 and ∼347 nm for BNPT and compound 5, respectively. The main contributing transitions for BNPT and compound 5 are S0 → S14 (∼56.60%) and S0 → S1 (∼95.60%), respectively, which correspond to energy states arising from HOMO−1 → LUMO and HOMO → LUMO/HOMO → LUMO+1, respectively. The calculated red shift is in good agreement with experimental results due to the n → π* transition. The TDDFT calculations have been carried out for zinc complex also and the corresponding electronic transition details are included in the ESI† (Fig. S16 and Tables S1, S2).
Detection of S2− in environmental water samples
The potential utilities of BNPT were also checked by proof-of concept experiments through the analysis of S2− in aqueous solution for practical applications. Because S2− has carcinogenic properties and has been widely used in a variety of industrial processes, the detection of S2− in aqueous samples is of interest.Tap and river paper water were collected from the laboratory and Ganges river, respectively, and were spiked with S2− (0–15 μM) into these samples. The concentration of S2− in these samples was determined by fluorescence titration using BNPT. The fluorescence intensities were proportional to the concentrations of S2− in the range of 0–15 μM in the two collected water samples (Fig. 8). Therefore, the presented probe BNPT could be used for trace S2− analysis in these environmental samples.
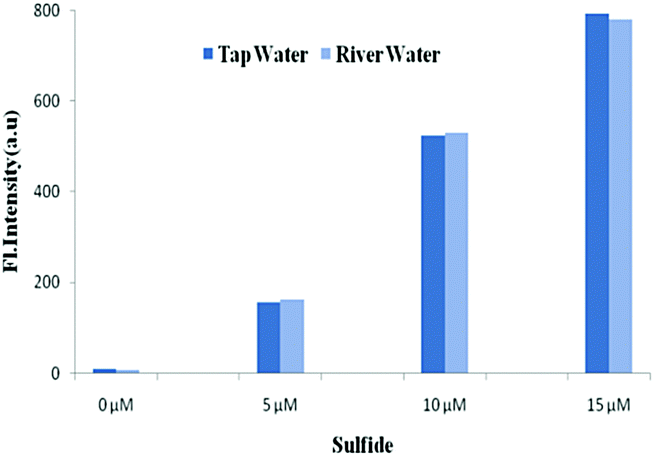 | ||
| Fig. 8 Fluorescence detection of S2− in tap water and river water with BNPT. | ||
To find the practical application of BNPT, the test strips were prepared by soaking the TLC plates into an aqueous acetonitrile (CH3CN:H2O = 3:2 v/v, 10 mM HEPES buffer, pH = 7.4) solution of BNPT (10 μM) and then drying in air. The test strips were utilized to sense S2− (0.1 mM) in aqueous solution. A significant change in color (non-fluorescent to intense green fluorescence) was observed with S2− under a 365 nm UV lamp. Therefore, the test strips based on BNPT can be used to detect the presence of trace amounts of S2− in an aqueous medium quickly and conveniently (Fig. 9).
 | ||
| Fig. 9 Sensing of S2− using a BNPT-coated TLC plate under a UV lamp. | ||
Application in intracellular S2− imaging
To demonstrate the applicability of BNPT in living cells, the suitability of the probe for visualization of S2− was investigated. First, cytotoxicity of BNPT in living cells was evaluated by MTT assay (Fig. S15, ESI†). The application of BNPT in cell biology showed excellent uptake of the receptor by HADF cells. The untreated HADF cells emitted much less green auto-fluorescence. The HADF cells treated with BNPT gave green fluorescence in lower intensity. This indicated that BNPT was well taken up by the cells. Therefore, a strong green fluorescence was observed in those cells treated with BNPT and followed by 1 h sulfide treatment (Fig. 10). These results revealed that non-fluorescent BNPT is cell compatible and gives strong fluorescence. The exact spectral behavior of the BNPT was also found in fluorescence titration. | ||
| Fig. 10 Fluorescence microscopic observation of HADF cells under green filter. Fluorescence with low intensity was observed in BNPT-treated cells compared to control. After addition of S2−, a strong green fluorescence was emitted by BNPT, scale bar 100 μm. | ||
Conclusion
In conclusion, we have designed and synthesized a new fluorescent chemoreactant probe BNPT based on the benzothiazole–thiazole arms, and explored its sulfide ion recognition property extensively using spectroscopic techniques. The proposed chemosensors exhibit good selectivity and sensitivity toward sulfide over other tested allied anions with ratiometric emission. The fluorescence enhancement mechanism is based on the cleavage of DNB from the fluorophore by S2−, which switches the weak emissive state to a strong emissive excited state. The ratiometric emission by the probe with S2− was well justified in terms of ICT and ESIPT due to deprotection of the DNB group. Probe BNPT displays a color change from colorless to greenish yellow upon addition of S2−, and thus can serve as a “naked-eye” probe for S2−. Therefore, BNPT can be used as a sensitive and selective ratiometric switch on fluorescence chemoreactant for S2−on the basis of fluorescence, absorption, and 1H NMR spectroscopy, ESI mass spectrometry, and visual fluorescent color changes. The in situ generated [Zn-5] complex was further explored for its secondary sensing properties for the detection of cations resulting in a selective turn-off fluorescence response toward zinc among the allied cations. The structural, electronic, and emission properties of the probe BNPT and their reaction product have been demonstrated using the DFT and TDDFT computational calculations. Furthermore, we have also demonstrated the proof-of-concept experiments with some natural water samples, wherein the probe can be used for an evaluation of sulfide ion in real water samples. Cell imaging studies were carried out using BNPT for visualization of S2− in living cells. Low cytotoxicity and excellent uptake by the cells make it a good chemoreactant for visualization of intracellular S2− in living cells.Experimental section
Materials and methods
Unless otherwise mentioned, materials were obtained from commercial suppliers and were used without further purification. 1H and 13C NMR spectra were recorded on a Brucker 400 MHz instrument. For NMR spectra, DMSO-d6 and CDCl3 were used as solvent using TMS as an internal standard. Chemical shifts are expressed in δ ppm units and 1H–1H and 1H–C coupling constants in Hz. Mass spectra were carried out using a Waters QTOF Micro YA 263 mass spectrometer. Fluorescence spectra were recorded on a Perkin Elmer Model LS 55 spectrophotometer. UV spectra were recorded on a JASCO V-530 spectrophotometer.General method of UV-vis and fluorescence titration
For UV-vis and fluorescence titrations, the stock solution of probe BNPT was prepared (c = 10.0 μM) in aqueous solution HEPES buffer (pH 7.4, 10 mM) in CH3CN–H2O (3:2, v/v). The solution of the guest anions, cations and amines in the order of 1.0 × 10−4 ML−1 was also prepared in aqueous solution HEPES buffer (pH 7.4, 10 mM) in CH3CN–H2O (3:2, v/v). The spectra of these solutions were recorded by means of UV-vis and the fluorescence methods. All the solvents were purchased from local suppliers and were distilled by the standard procedure before use.
Kinetic study
In kinetic studies of the probe for S2− detection, the apparent rate constant kobs for the reaction of BNPT with S2− was determined by fitting the fluorescence intensities to the pseudo-first-order equation:| Ln[(Fmax − Ft)/Fmax] = −kobst |
Cell imaging study
Human adult dermal fibroblast (HADF, Himedia Laboratories, India) cells were cultured in Dulbecco's modified eagle medium (DMEM, Himedia Laboratories, India) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% antibiotic–antimycotic solution (Himedia Laboratories, India) and seeded on a clean sterile cover slip placed in 24-well plates (Thermo Scientific, USA) with a serum-free medium and incubated for 24 h at 37 °C in a humidified CO2 incubator. Two sets of cells were treated with 10−4 M receptor (BNPT) for 1 h. After washing with phosphate buffered saline (PBS), one set of treated cells were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 minutes at room temperature, and another set of cells were treated with 10−2 M sulfide in serum-free DMEM followed by 1 h incubation. The sulfide-treated cells were washed with PBS and fixed using 4% paraformaldehyde. Fluorescence microscopy was carried out under a Nikon Inverted microscope (Nikon eclipse TíU, Japan) equipped with a 20× (S Plan Fluor) objective. 465–495 nm excitation filters were used and images were captured through a 515–555 nm emission filter.Synthesis of BNPT
Probe BNPT was synthesized by stirring 2-(3-benzothiazol-2-yl-4-hydroxy-phenyl)-4-methyl-thiazole-5-carboxylic acid ethyl ester (5) and 2,4-dinitrofluorobenzene in the presence of K2CO3, in dry DMF at 80 °C, to give probe BNPT in 72% yield (Scheme 2). | ||
| Scheme 2 Synthesis of BNPT. | ||
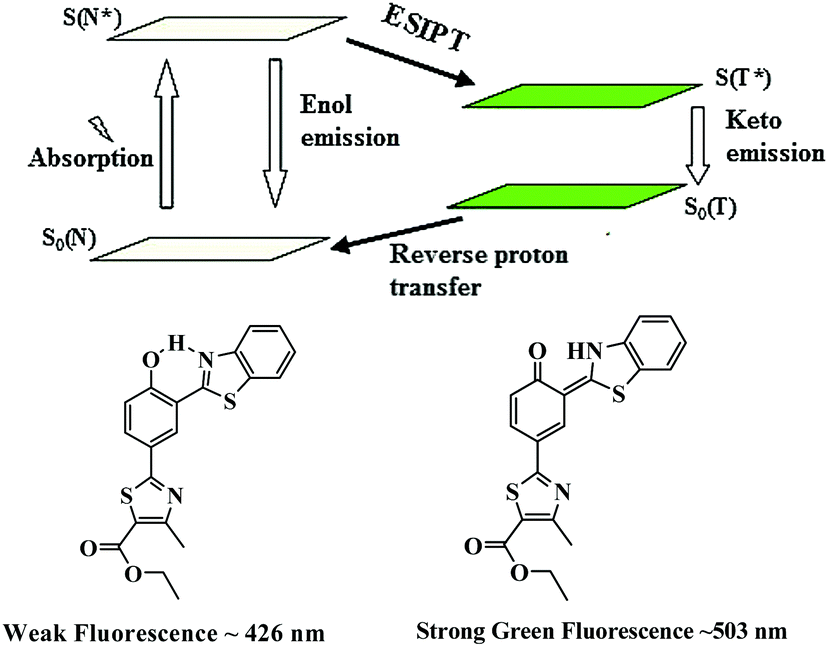 | ||
| Scheme 3 Schematic representation of ESIPT process of BNPT. | ||
Experimental procedure
To a stirred solution of compound 5 [2-(3-benzothiazol-2-yl-4 hydroxyphenyl)-4-methylthiazole-5-carboxylic acid ethyl ester] (200 mg, 0.50 mmol) in DMF (5 mL) was added K2CO3 (698 mg, 5.05 mmol) followed by the addition of 1-fluoro-2,4-dinitrobenzene (282 mg, 1.51 mmol) at rt. Then the reaction mixture was heated at 80 °C for 5 h. TLC showed SM was consumed and formed a polar spot. The reaction mixture was cooled at rt and then diluted with ethyl acetate (50 mL). The combined organic solution was washed with cold water (10 mL × 3), dried over sodium sulfate and concentrated. The crude residue was purified by column chromatography over silica to afford desired compound BNPT (120 mg, 42%) [2-[3-benzothiazol-2-yl-4-(2,4-dinitrophenoxy)phenyl]-4-methylthiazole-5-carboxylic ethyl ester] as a yellow solid. 1H NMR (400 MHz, d6-DMSO) δ (ppm): 9.14 (s, 1H), 9.01 (s, 1H), 8.49 (d, J = 9.48 Hz, 1H), 8.26 (d, J = 8.76 Hz, 1H), 8.19–8.14 (m, 2H), 7.6–7.49 (m, 4H), 4.36 (q, J = 7.08 Hz, 2H), 2.71 (s, 3H), 1.33 (t, J = 6.96 Hz, 3H). 13C NMR (100 MHz, CDCl3): 166.92, 161.90, 161.15, 159.62, 154.66, 152.41, 152.27, 142.21, 139.74, 135.51, 131.97, 130.04, 129.34, 128.97, 126.98, 126.57, 125.83, 123.52, 122.85, 122.20, 122.13, 121.41, 118.39, 61.37, 17.41, 14.23. MS (ESI): m/z calc. for C26H18N4O7S2: 562.58; found: 563.3 [M + H]+.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge DST-SERB [File No. EMR/2016/006640] for financial support. SSA, SM, AKP, and RS thank the UGC-MANF, UGC, SERB-NPDF, and UGC-RGNF, New Delhi, for the financial and research support. Special thanks are given to C. Bodhak and R. Bag from Calcutta University for characterization support.Notes and references
- (a) J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC; (b) M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583–7601 RSC; (c) Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC; (d) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed; (e) Q. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC; (f) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- N. S. Lawrence, J. Davis and R. G. Compton, Talanta, 2000, 52, 771–784 CrossRef CAS PubMed.
- Hydrogen Sulfide, World Health Organization, Geneva, 1981 (Environmental Health Criteria, No. 19).
- (a) X. Cao, W. Lin and L. He, Org. Lett., 2011, 13, 4716 CrossRef CAS PubMed; (b) S. A. Patwardhan and S. M. Abhyankar, Colourage, 1988, 35, 15 CAS.
- (a) R. F. Huang, X. W. Zheng and Y. J. Qu, Anal. Chim. Acta, 2007, 582, 267 CrossRef CAS PubMed; (b) N. S. Lawrence, J. Davis and R. G. Compton, Talanta, 2000, 52, 771–784 CrossRef CAS PubMed.
- Z. Pawlak and A. S. Pawlak, Talanta, 1999, 48, 347–353 CrossRef CAS PubMed.
- B. Vallejo, P. Richter, I. Toral, C. Tapia and M. D. L. Castro, Anal. Chim. Acta, 2001, 436, 301–307 CrossRef CAS.
- W. Puacz, W. Szahun and K. Linke, Analyst, 1995, 120, 939–941 RSC.
- B. Divjak and W. Goessler, J. Chromatogr. A, 1999, 844, 161–169 CrossRef CAS.
- (a) R. Huang, X. Zheng and Y. Qu, Anal. Chim. Acta, 2007, 582, 267–274 CrossRef CAS PubMed; (b) F. Maya, J. M. Estela and V. Cerda, Anal. Chim. Acta, 2007, 601, 87–94 CrossRef CAS PubMed.
- (a) X. Cao, W. Lin and L. He, Org. Lett., 2011, 13, 4716–4719 CrossRef CAS PubMed; (b) D. Jimenez, R. M. Mãnez, F. Sancenon, J. V. Ros-Lis, A. Benito and J. Soto, J. Am. Chem. Soc., 2003, 125, 9000–9001 CrossRef CAS PubMed; (c) X. Yang, L. Wang, H. Xu and M. Zhao, Anal. Chim. Acta, 2009, 631, 91–95 CrossRef CAS PubMed; (d) M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 7390–7392 RSC; (e) F. Hou, L. Huang, P. Xi, J. Cheng, X. Zhao, G. Xie, Y. Shi, F. Cheng, X. Yao and D. Bai, Inorg. Chem., 2012, 51, 2454–2460 CrossRef CAS PubMed; (f) X. Lou, H. Mu, R. Gong, E. Fu, J. Qin and Z. Li, Analyst, 2011, 136, 684–687 RSC; (g) L. Zhang, X. Lou, Y. Yu, J. Qin and Z. Li, Macromolecules, 2011, 44, 5186–5193 CrossRef CAS; (h) J. Liu, J. Chen, Z. Fang and L. Zeng, Analyst, 2012, 137, 5502–5505 RSC; (i) M.-Q. Wang, K. Li, J.-T. Hou, M.-Y. Wu, Z. Huang and X.-Q. Yu, J. Org. Chem., 2012, 77, 8350–8354 CrossRef CAS PubMed; (j) R. Zhang, X. Yu, Y. Yin, Z. Ye, G. Wang and J. Yuan, Anal. Chim. Acta, 2011, 691, 83–88 CrossRef CAS PubMed; (k) Y. Qian, J. Karpus, O. Kabil, S. Y. Zhang, H. L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed; (l) J. Zhang, X. Xu and X. Yang, Analyst, 2012, 137, 1556–1558 RSC; (m) G. Zhou, H. Wang, Y. Ma and X. Chen, Tetrahedron, 2013, 69, 867–870 CrossRef CAS; (n) L. Tang, M. Cai, Z. Huang, K. Zhong, S. Hou, Y. Bian and R. Nandhakumar, Sens. Actuators, B, 2013, 185, 188–194 CrossRef CAS.
- (a) T. Dudev, Chem. Rev., 2003, 103, 773 CrossRef CAS PubMed; (b) P. Jiang and Z. C. Guo, Chem. Rev., 2004, 248, 205 CAS.
- (a) G. Danscher and M. J. Stoltenberg, J. Histochem. Cytochem., 2005, 53, 141 CrossRef CAS PubMed; (b) C. J. Chang, J. Jaworski, E. M. Nolan, M. Sheng and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1129 CrossRef CAS PubMed.
- (a) P. D. Zalewski, S. H. Millard, I. J. Forbes, O. Kapaniris, A. Slavotinek, W. H. Betts, A. D. Ward, S. F. Lincoln and I. J. Mahadevan, J. Histochem. Cytochem., 1994, 42, 877 CrossRef CAS PubMed; (b) B. Lukowiak, B. Vandewalle, R. Riachy, J.-K. Conte, V. Gmyr, S. Belaich, J. Lefebvre and F. J. Pattou, J. Histochem. Cytochem., 2001, 49, 519 CrossRef CAS PubMed.
- (a) M. P. Cuajungco and G. J. Lees, Neurobiol. Dis., 1997, 4, 137 CrossRef CAS PubMed; (b) A. I. Bush and R. E. Tanzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7317 CrossRef CAS PubMed; (c) A. A. Larson and K. F. J. Kitto, J. Pharmacol. Exp. Ther., 1997, 282, 1319 CAS; (d) C. Hogstrand, P. Kille, R. I. Nicholson and K. M. Taylor, Trends Mol. Med., 2009, 15, 101 CrossRef CAS PubMed.
- (a) D. J. Eide, Biochim. Biophys. Acta, 2006, 1763, 711 CrossRef CAS PubMed; (b) T. Gunnlaugsson, M. Glynn, M. T. Gillian, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094 CrossRef CAS.
- (a) Z. Guo, G. Chen, G. Zeng, Z. Li, A. Chen, J. Wang and L. Jiang, Analyst, 2015, 140, 1772 RSC; (b) F. Yu, X. Han and L. Chen, Chem. Commun., 2014, 50, 12234 RSC; (c) J. Li, C. Yin and F. Huo, RSC Adv., 2015, 5, 2191 RSC.
- (a) D. Diamond and M. A. Mckervey, Chem. Soc. Rev., 1996, 25, 15–24 RSC; (b) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed; (c) F. Unob, Z. Asfari and J. Vicens, Tetrahedron Lett., 1998, 39, 295 CrossRef; (d) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 3, 205 Search PubMed; (e) I. A. Bagatin, E. S. de Souza, A. S. Ito and H. E. Toma, Inorg. Chem. Commun., 2003, 6, 288 CrossRef CAS; (f) G. Ozturk and E. U. Akkaya, Org. Lett., 2004, 6, 241 CrossRef CAS PubMed; (g) S. K. Kim, S. H. Lee, J. Y. Lee, R. A. Bartsch and J. S. Kim, J. Am. Chem. Soc., 2004, 126, 16499 CrossRef CAS PubMed; (h) J. Y. Lee, S. K. Kim, J. H. Jung and J. S. Kim, J. Org. Chem., 2005, 70, 1463 CrossRef CAS PubMed; (i) S. Khatua, S. H. Choi, J. Lee, K. Kim, Y. Do and D. G. Churchill, Inorg. Chem., 2009, 48, 2993 CrossRef CAS PubMed; (j) K. Kim, Y. Ha, L. Kaufman and D. G. Churchill, Inorg. Chem., 2012, 51, 928 CrossRef CAS PubMed.
- (a) Z. Guo, G. H. Kim, I. Shin and J. Yoon, Biomaterials, 2012, 33, 7818–7827 CrossRef CAS PubMed; (b) M. J. Kim, K. Kaur, N. Singh and D. O. Jang, Tetrahedron, 2012, 68, 5429–5543 CrossRef CAS; (c) L. Tang, M. Cai, P. Zhou, J. Zhao, K. Zhong, S. Hou and Y. Bian, RSC Adv., 2013, 3, 16802–16809 RSC; (d) M. M. Henary, Y. Wu and C. J. Fahrni, Chem. – Eur. J., 2004, 10, 3015–3025 CrossRef CAS PubMed.
- (a) M. Chen, X. Lv, Y. Liu, Y. Zhao, J. Liu, P. Wang and W. Guo, Org. Biomol. Chem., 2011, 9, 2345–2349 RSC; (b) M. Taki, J. L. Wolford and T. V. O’Halloran, J. Am. Chem. Soc., 2004, 126, 712–713 CrossRef CAS PubMed; (c) Y. Xu and Y. Pang, Dalton Trans., 2011, 40, 1503–1509 RSC.
- (a) L. Tang, X. Dai, K. Zhong, X. Wen and D. Wu, J. Fluoresc., 2014, 24, 1487–1493 CrossRef CAS PubMed; (b) S. Janakipriya, S. Tamilmani and S. Thennarasu, RSC Adv., 2016, 6, 71496–71500 RSC.
- (a) A. Helal and H.-S. Kim, Tetrahedron Lett., 2009, 50, 5510–5515 CrossRef CAS; (b) A. Helal and H. S. Kim, Spectrochim. Acta, Part A, 2013, 105, 273–279 CrossRef CAS PubMed.
- (a) M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 7390–7392 RSC; (b) X. Lou, H. Mu, R. Gong, E. Fu, J. Qin and Z. Li, Analyst, 2011, 136, 684–687 RSC; (c) L. Zhang, X. Lou, Y. Yu, J. Qin and Z. Li, Macromolecules, 2011, 44, 5186–5193 CrossRef CAS; (d) F. Hou, L. Huang, P. Xi, J. Cheng, X. Zhao, G. Xie, Y. Shi, F. Cheng, X. Yao and D. Bai, Inorg. Chem., 2012, 51, 2454–2460 CrossRef CAS PubMed; (e) J. Liu, J. Chen, Z. Fang and L. Zeng, Analyst, 2012, 137, 5502–5505 RSC; (f) M.-Q. Wang, K. Li, J.-T. Hou, M.-Y. Wu, Z. Huang and X.-Q. Yu, J. Org. Chem., 2012, 77, 8350–8354 CrossRef CAS PubMed; (g) Y. Qian, J. Karpus, O. Kabil, S.-Y. Zhang, H.-L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed.
- (a) E. J. Neil and B. D. Smith, Coord. Chem. Rev., 2006, 250, 3068–3080 CrossRef; (b) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS.
- (a) X. Lou, H. Mu, R. Gong, E. Fu, J. Qin and Z. Li, Analyst, 2011, 136, 684–687 RSC; (b) F. Hou, J. Cheng, P. Xi, F. Chen, L. Huang, G. Xie, Y. Shi, H. Liu, D. Bai and Z. Zeng, Dalton Trans., 2012, 41, 5799–5804 RSC; (c) D. Zhang and W. Jin, Spectrochim. Acta, Part A, 2012, 90, 35–39 CrossRef CAS PubMed; (d) X. Gu, C. Liu, Y. Zhu and Y. Zhu, Tetrahedron Lett., 2011, 52, 5000–5003 CrossRef CAS; (e) C. Kar, M. D. Adhikari, A. Ramesh and G. Das, Inorg. Chem., 2013, 52, 743–752 CrossRef CAS PubMed.
- (a) W. Lin, L. Yuan, Z. Cao, Y. Feng and L. Long, Chem. – Eur. J., 2009, 15, 5096 CrossRef CAS PubMed; (b) X. Chen, S. Ko, M. J. Kim, I. Shin and J. Yoon, Chem. Commun., 2010, 46, 2751 RSC; (c) S. Lim, J. O. Escobedo, M. Lowry, X. Xu and R. Strongin, Chem. Commun., 2010, 46, 5707 RSC; (d) Y.-Q. Sun, M. Chen, J. Liu, X. Lv, J.-F. Li and W. Guo, Chem. Commun., 2011, 47, 11029 RSC; (e) H. Zhang, P. Wang, Y. Yang and H. Sun, Chem. Commun., 2012, 48, 10672 RSC.
- (a) H. Zhang, C. Zhang, R. Liu, L. Yi and H. Sun, Chem. Commun., 2015, 51, 2029 RSC; (b) J. Bouffard, Y. Kim, T. M. Swager, R. Weissleder and S. A. Hilderbrand, Org. Lett., 2008, 10, 37 CrossRef CAS PubMed; (c) S. Wang, W. Deng, D. Sun, M. Yan, H. Zheng and J. Xu, Org. Biomol. Chem., 2009, 7, 4017 RSC; (d) X. Li, S. Qian, Q. He, B. Yang, J. Li and Y. Hu, Org. Biomol. Chem., 2010, 8, 3627 RSC; (e) S. Ji, H. Guo, X. Yuan, X. Li, H. Ding, P. Gao, C. Zhao, W. Wu, W. Wu and J. Zhao, Org. Lett., 2010, 12, 2876 CrossRef CAS PubMed.
- (a) L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928 CrossRef CAS PubMed; (b) X.-F. Yang, Q. Huang, Y. Zhong, Z. Li, H. Li, M. Lowry, J. O. Escobedo and R. M. Strongin, Chem. Sci., 2014, 5, 2177 RSC; (c) S.-Y. Lim, K.-H. Hong, D. I. Kim, H. Kwon and H.-J. Kim, J. Am. Chem. Soc., 2014, 136, 7018 CrossRef CAS PubMed; (d) Y.-S. Guan, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, RSC Adv., 2014, 4, 8360 RSC; (e) H. Lv, X.-F. Yang, Y. Zhong, Y. Guo, Z. Li and H. Li, Anal. Chem., 2014, 86, 1800 CrossRef CAS PubMed; (f) J. Liu, Y.-Q. Sun, H. Zhang, Y. Huo, Y. Shi and W. Guo, Chem. Sci., 2014, 5, 3183 RSC.
- (a) M. H. Lee, J. H. Han, P.-S. Kwon, S. Bhuniya, J. Y. Kim, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 1316 CrossRef CAS PubMed; (b) X. Cao, W. Lin and Q. Yu, J. Org. Chem., 2011, 76, 7423 CrossRef CAS PubMed; (c) D. Jung, S. Maiti, J. H. Lee, J. H. Lee and J. S. Kim, Chem. Commun., 2014, 50, 3044 RSC; (d) R. Wang, L. Chen, P. Liu, Q. Zhang and Y. Wang, Chem. – Eur. J., 2012, 18, 11343 CrossRef CAS PubMed; (e) H. S. Jung, J. H. Han, Y. Habata, C. Kang and J. S. Kim, Chem. Commun., 2011, 47, 5142 RSC; (f) Y. Bao, Q. Li, B. Liu, F. Du, J. Tian, H. Wang, Y. Wang and R. Bai, Chem. Commun., 2012, 48, 118 RSC; (g) P. Wang, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Org. Lett., 2012, 14, 520 CrossRef CAS PubMed; (h) S. R. Malwal, A. Labade, A. S. Andhalkar, K. Sengupta and H. Chakrapani, Chem. Commun., 2014, 50, 11533 RSC.
- A. K. Mahapatra, P. Karmakar, S. Manna, K. Maiti and D. Mandal, J. Photochem. Photobiol., A, 2017, 334, 1–12 CrossRef CAS.
- J. M. Sosa, M. Vinkovi and D. V. Topic, Croat. Chem. Acta, 2006, 79, 489–495 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj03207b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |