DOI:
10.1039/C7NJ02877F
(Paper)
New J. Chem., 2018,
42, 336-354
Cyclometallated ruthenium(II) complexes with 3-acetyl-2[H]-chromene-2-one derived CNS chelating ligand systems: synthesis, X-ray characterization and biological evaluation†
Received
4th August 2017
, Accepted 14th November 2017
First published on 30th November 2017
Abstract
Four new bivalent organoruthenium complexes incorporated with 3-acetylcoumarin-4(N)-substituted thiosemicarbazones were synthesized. Comprehensive spectral and analytical techniques such as elemental analyses, IR, UV-vis, 1H NMR and 13C NMR provided proof of the formation of the complexes (1–4). Single crystals obtained for two ligands and the four complexes were characterized by X-ray crystallographic studies, which indicated CNS tridentate coordination of the ligands through C–H activation at the C(4) carbon of the pyrone ring of coumarin. The ligands and complexes intercalated with calf thymus DNA (CT DNA), supported by EB displacement studies and DNA viscosity measurements. The compounds bound well with two serum albumins (bovine serum albumin (BSA) and human serum albumin (HSA)) and the quenching mechanism was found to be static. Antioxidant studies revealed the radical scavenging proficiency of the compounds. The compounds showed better antimicrobial activity against two Gram-positive bacteria (Staphylococcus aureus and Streptococcus pneumonie), two Gram-negative bacteria (Pseudomonas aeruginosa and Salmonella paratyphi) and five fungi (Candida albicans, Trichophyton rubrum, Aspergillus niger, Aspergillus fumigates and Candida tropicalis). The in vitro cytotoxic abilities of the compounds were analysed using the MTT assay on MCF-7 (human breast cancer) and A549 (human lung carcinoma) cell lines and compared with standard cisplatin. Activities greater than cisplatin were observed for the ligands as well as the complexes. Assays on the human normal keratinocyte cell line HaCaT showed that the compounds were non-toxic to those cells. In all the biological studies done, the complexes displayed better activity than the ligands and among them, complex 3 stood out as the best, owing to the greater electron donating ability of the N-terminal ethyl group on the thiosemicarbazone. The results implied that N-terminal substitution played a significant role in bringing out the bioactivity of the complexes.
Introduction
Thiosemicarbazones are among the widely studied ligands due to their versatile nature and interesting profile in medicinal chemistry.1 They themselves are biologically active and generally act as chelating ligands for a number of metal ions by bonding through their sulfur and N1/N2 hydrazinic nitrogen atoms. Thiosemicarbazones functionalized with an additional donor site are now becoming important ligands and exhibit improved activities in pharmacological2 and electrochemical fields.3 They display anticancer,4–8 antiparasitic,2 antitubercular,9 antiviral, antimicrobial, antitumour,10 antiamoebic11 and antimalarial activities.12 Coumarin is a phytochemical made of fused benzene and an α-pyrone ring, essentially found in green plants of the family Orchidaceae, Leguminaceae, Rutaceae, Umbelifferae and Labiatae. Many compounds containing coumarins have been reported to exhibit anti-coagulant, antibacterial, antifungal and anticancer properties and they are employed as biological inhibitors, chemotherapeutics and bio-analytical reagents.13 Some coumarin derivatives such as novobiocin, clorobiocin and coumermycin A1 are commercialized as antibiotics.14,15 Various coumarins are able to scavenge reactive oxygen species (ROS) and reactive nitrogen species (RNS) and display potential tissue-protective antioxidant properties.16 Incorporation of a coumarin into these thiosemicarbazones usually results in enhancement of their biological activities. There is an emerging interest in these systems since their pharmacological response enhances upon complexation. Among the transition metal complexes, ruthenium complexes are found to be the best alternatives for platinum-based drugs since they offer diverse coordination modes,17 stable oxidation states, and a slow ligand exchange rate similar to Pt(II) and Pd(II), and mimic iron in their ability to bind transferrin and albumin.18,19 Their application in cancer therapy has now become a broad area of research after the synthesis of NAMI-A and KP1019 (successfully completed phase I and II clinical trials).20–22 Sufficient articles are available evidencing the cytotoxic nature of ruthenium complexes and include those reported by Garza-Ortiz et al.,23 Juinn Chow et al.24 and Chang et al.25 Having said the above facts and in the quest for new treatments against cancer, 3-acetylcoumarin containing thiosemicarbazone derivatives and their ruthenium(II) complexes have been synthesized, extensively investigated in biological studies, like DNA binding, protein binding, and antioxidant and antimicrobial activity studies, and developed as prospective anticancer agents against MCF-7 (human breast cancer) and A549 (human lung carcinoma) cell lines. A study of these compounds on human normal keratinocyte cell line HaCaT revealed their nontoxic nature to normal cells.
Results and discussion
Synthesis and characterisation
The functionalised ligands ((1E)-1-(1-(2-oxo-2H-chromen-3-yl)ethylidene)thiosemicarbazide) H2L1–4 were prepared by the condensation reactions of 3-acetyl-2H-chromen-2-one with appropriate 4(N)-substituted thiosemicarbazides (Scheme 1). The synthesized 3-acetylcoumarin-4(N)-substituted thiosemicarbazone ligands H2L1–4 were treated with the Ru(II) precursor [RuHCl(CO)(PPh3)3] (Scheme 1) to give the corresponding Ru(II) complexes 1–4. All the compounds were fully characterized by using various spectroscopic techniques and the structure of the ligands H2L2 and H2L3 and complexes 1–4 were confirmed by single-crystal X-ray diffraction analysis. The compounds were obtained in excellent yields. They are stable to air and moisture and are coloured. They were soluble in almost all organic solvents. The stability of the complexes in 1% aqueous DMSO, phosphate buffer–DMSO (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and Tris–HCl–DMSO (99:1) was confirmed using UV-visible spectroscopic techniques (Fig. S1 in the ESI†). The spectra which were recorded immediately and after 24 h, did not show any appreciable change in either the intensity or the position of the bands indicating the stability of the complexes in these solutions.
:1) and Tris–HCl–DMSO (99:1) was confirmed using UV-visible spectroscopic techniques (Fig. S1 in the ESI†). The spectra which were recorded immediately and after 24 h, did not show any appreciable change in either the intensity or the position of the bands indicating the stability of the complexes in these solutions.
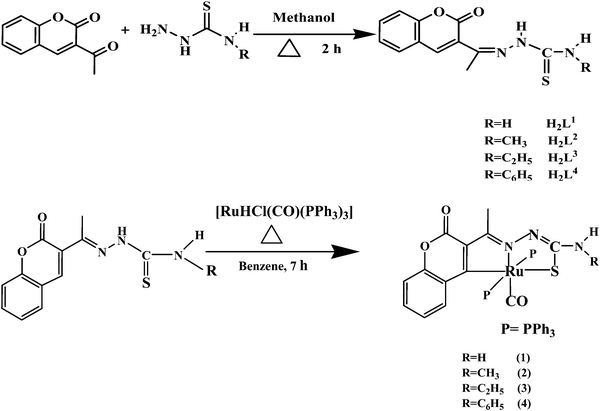 |
| | Scheme 1 Synthesis of new ligands H2L1–4 and their ruthenium(II) complexes. | |
Important spectral bands and their assignments for the newly synthesized compounds are given in the ESI† (Table S1). The IR spectra of the free ligands showed a very strong band around ∼1606 to 1617 cm−1 due to the stretching frequency of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group26 and this was shifted to lower frequencies (∼10 to 25 cm−1) upon complexation with the Ru(II) ion in the complexes, representing the involvement of the azomethine nitrogen atom in coordination.27 The bands observed at 3250–3265 cm−1 and 761–770 cm−1 in the ligands were due to the presence of hydrazinic N–H and thione CS groups respectively. In the spectra of the complexes (1–4), these bands disappeared and a new band appeared around 721–746 cm−1 corresponding to a possible ν(C–S) vibration indicating the coordination of a thiolate sulphur atom after enolisation prior to deprotonation.27 The complexation was further confirmed by the bands found at ∼694, 1084–1090 and ∼1402–1433 cm−1 representing the presence of triphenylphosphine. The strong band that appeared around 1917–1923 cm−1 was assigned to the terminally coordinated carbonyl group of the complexes 1–4.20,27,28
N group26 and this was shifted to lower frequencies (∼10 to 25 cm−1) upon complexation with the Ru(II) ion in the complexes, representing the involvement of the azomethine nitrogen atom in coordination.27 The bands observed at 3250–3265 cm−1 and 761–770 cm−1 in the ligands were due to the presence of hydrazinic N–H and thione CS groups respectively. In the spectra of the complexes (1–4), these bands disappeared and a new band appeared around 721–746 cm−1 corresponding to a possible ν(C–S) vibration indicating the coordination of a thiolate sulphur atom after enolisation prior to deprotonation.27 The complexation was further confirmed by the bands found at ∼694, 1084–1090 and ∼1402–1433 cm−1 representing the presence of triphenylphosphine. The strong band that appeared around 1917–1923 cm−1 was assigned to the terminally coordinated carbonyl group of the complexes 1–4.20,27,28
The electronic spectra of the complexes showed intense absorption bands at λ = 265–279 nm and 327–333 nm. The higher energy absorption band at 265–279 nm is ascribed to the intraligand (IL) transition (π → π*/n → π*) whereas the absorption band at 327–333 nm is assigned to the ligand to metal charge transfer (LMCT) transition. Similar data for the related ruthenium(II) compounds have also been reported in the literature.27,291H NMR spectra of the ligands H2L1–4 and complexes (1–4) are given in the ESI† (Fig. S2–S9). An examination of the NMR data showed that the ligands (H2L1–4) behaved as binegative tridentate chelates coordinated through the C(4) carbon atom of the pyrone ring, thiolate sulphur and imine nitrogen in all the complexes. The peaks at δ 10.37–10.86 ppm due to the N(2)–H proton in the free ligands disappeared after complexation, indicating the coordination of the thiolate sulfur of the ligands after deprotonation.27 The signal at δ 2.32–2.34 ppm due to the NC–CH3 proton30 of the free ligand underwent an upfield shift in the spectra of the complexes at δ 1.90–2.03 ppm, indicating that the coordination occured via the azomethine nitrogen atom.10 A singlet observed at δ 7.94–8.49 ppm for the ligands H2L1–4 due to the presence of a hydrogen atom at the C(4) carbon, disappeared in the spectra of the complexes, signifying the coordination of the ligands in the anionic form after deprotonation at the C(4) carbon of the pyrone ring. Resonances due to all the aromatic protons were observed in the range of δ 6.74–7.79 ppm.20,27,30 Two broad singlets appeared at δ 8.40 and δ 8.46 ppm corresponding to NH2 protons for H2L1. However in the spectrum of complex 1, this signal was observed as a broad singlet at δ 5.75 ppm. A quatret observed at δ 8.49–8.50 ppm corresponding to the presence of terminal –NH protons in H2L2, was shifted to δ 6.10–6.11 ppm in complex 2. In H2L3 and complex 3, a triplet was observed at δ 8.50–8.53 and δ 6.18–6.19 ppm respectively for the presence of terminal –NH protons. The methyl group protons appeared as a doublet at δ 2.10–3.01 ppm for H2L2 and complex 2, whereas the same appeared as a triplet around δ 0.737–1.14 ppm for H2L3 and complex 3. In addition, in the spectra of H2L3 and 3, a multiplet corresponding to the methylene group of the protons was observed at δ 2.21–3.60 ppm.31 A sharp singlet corresponding to the terminal –NH proton in H2L4 was found at δ 10.14 ppm and this signal underwent a negative shift in complex 4 and was found as a sharp singlet at δ 8.50 ppm.
X-ray crystallography
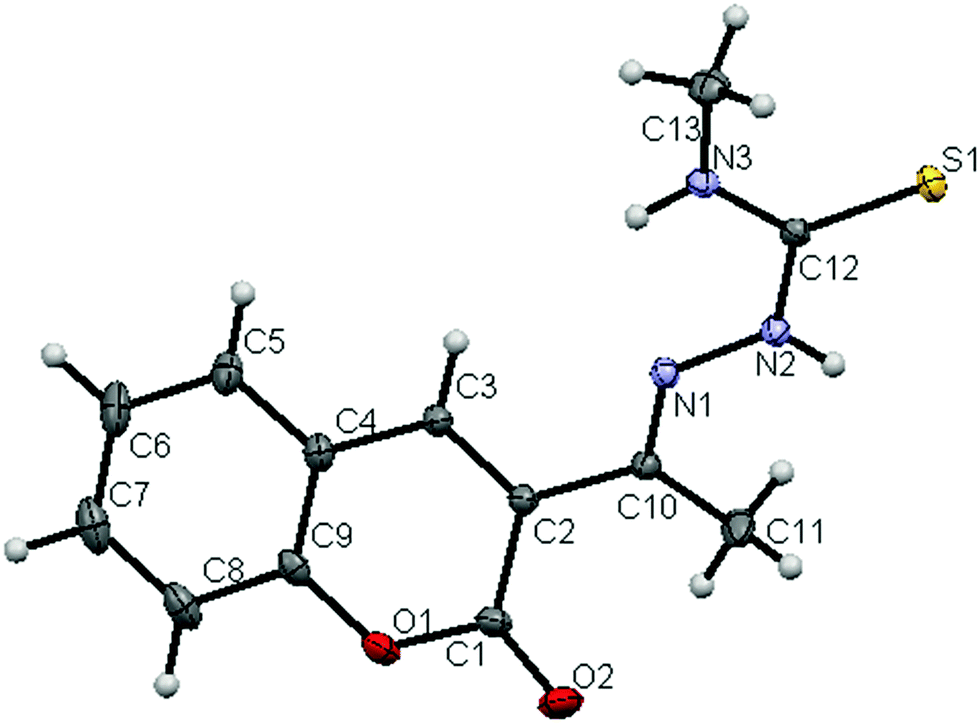
The molecular structures of the ligands (H2L2 and H2L3) and new ruthenium(II) complexes (1–4) have been determined by single crystal X-ray diffraction studies and their ORTEP drawings with the atom labeling are shown in Fig. 1–6. A summary of the structure refinement of the ligands and complexes is listed in Table S2 (ESI†) and Table 1 and selected inter-atomic distances and bond angles of the ligands (H2L2 and H2L3) and complexes (1–4) are summarized in Table S3 (ESI†) and Table 2.
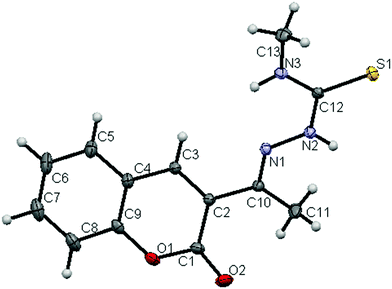 |
| | Fig. 1 ORTEP diagram of the ligand [H2-3AC-mtsc] (H2L2) (thermal ellipsoids at the 30% probability level). | |
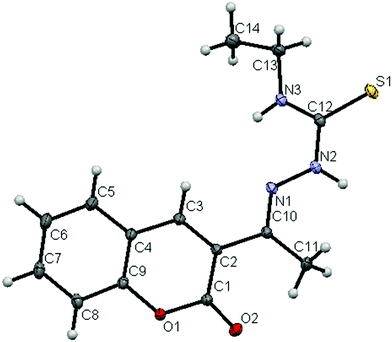 |
| | Fig. 2 ORTEP diagram of the ligand [H2-3AC-etsc] (H2L3) (thermal ellipsoids at the 30% probability level). | |
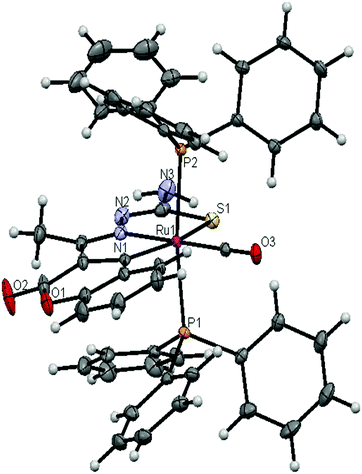 |
| | Fig. 3 ORTEP diagram of the complex [Ru(3AC-tsc)(CO)(PPh3)2] (1) (thermal ellipsoids at the 30% probability level). | |
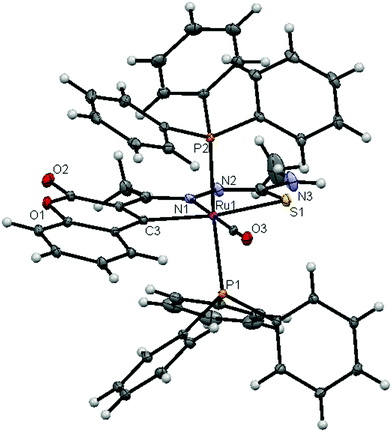 |
| | Fig. 4 ORTEP diagram of the complex [Ru(3AC-mtsc)(CO)(PPh3)2] (2) (thermal ellipsoids at the 30% probability level). | |
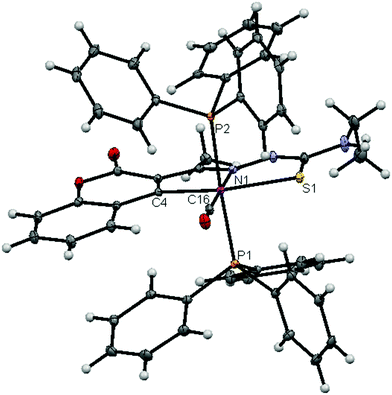 |
| | Fig. 5 ORTEP diagram of the complex [Ru(3AC-etsc)(CO)(PPh3)2] (3) (thermal ellipsoids at the 30% probability level). | |
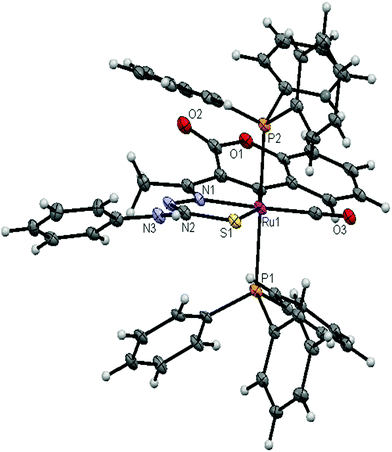 |
| | Fig. 6 ORTEP diagram of the complex [Ru(3AC-ptsc)(CO)(PPh3)2] (4) (thermal ellipsoids at the 30% probability level). | |
Table 1 Crystallographic data of the new Ru(II) complexes (1–4)
| Identification code |
[Ru(3AC-tsc)CO(PPh3)2] |
[Ru(3AC-mtsc)CO(PPh3)2] |
[Ru(3AC-etsc)CO(PPh3)2] |
[Ru(3AC-ptsc)CO(PPh3)2] |
| Empirical formula |
C49H39N3O3P2RuS·(0.46)H2O |
C50H41N3O3P2RuS |
C51H43N3O3P2RuS·(0.5)C6H6 |
C55H43N3O3P2RuS |
| Formula weight |
921.19 |
985.52 |
980.01 |
988.99 |
| Temperature |
100.0(5) K |
90.0(5) K |
90.0(5) K |
100.0(5) K |
| Wavelength |
0.71073 Å |
0.71073 Å |
0.71073 Å |
1.5418 Å |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
| Unit cell dimensions |
|
a
|
11.7445(4) Å |
11.8754(4) Å |
13.0418(3) Å |
12.8090(8) Å |
|
b
|
13.1594(5) Å |
12.4147(4) Å |
13.4769(3) Å |
34.6852(19) Å |
|
c
|
15.1515(6) Å |
17.2352(6) Å |
13.6076(3) Å |
10.9190(5) Å |
|
α
|
86.303(2)° |
80.4346(18)° |
79.4942(12)° |
90° |
|
β
|
81.895(2)° |
78.9857(15)° |
80.9076(11)° |
108.019(4)° |
|
γ
|
65.417(2)° |
63.9950(15)° |
71.6378(11)° |
90° |
| Volume |
2108.11 Å3 |
2231.85 Å3 |
2218.88 Å3 |
4613.19 Å3 |
|
Z
|
2 |
2 |
2 |
4 |
| Density |
1.451 mg m−3 |
1.466 mg m−3 |
1.467 mg m−3 |
1.424 mg m−3 |
| Absorption coefficient |
0.546 mm−1 |
0.600 mm−1 |
0.523 mm−1 |
4.217 mm−1 |
|
F(000) |
945 |
1010 |
1010 |
2032 |
| Crystal size |
0.25 × 0.16 × 0.05 mm |
0.26 × 0.25 × 0.04 mm |
0.19× 0.15 × 0.05 |
0.34 × 0.14 × 0.01 mm |
| Crystal shape |
Plate |
Plate |
Plate |
Lath |
|
θ range for data collection |
1.702° to 30.539° |
1.925° to 33.167° |
1.531° to 31.045° |
2.548° to 68.312° |
| Limiting indices |
−16 ≤ h ≤ 15, −18 ≤ k ≤ 18, −21 ≤ l ≤ 21 |
−17 ≤ h ≤ 18, −19 ≤ k ≤ 18, −26 ≤ l ≤ 26 |
−18 ≤ h ≤ 18, −19 ≤ k ≤ 19, −19 ≤ l ≤ 19 |
−15 ≤ h ≤ 15, −41 ≤ k ≤ 41, −13 ≤ l ≤ 13 |
| Independent reflections |
12859 (R(int) = 0.0426) |
16826 (R(int) = 0.0286) |
14189 (R(int) = 0.0346) |
8369 (R(int) = 0.0713) |
| Absorption correction |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
12859/1/549 |
16826/0/548 |
14189/1/582 |
8369/1/590 |
| Goodness-of-fit on F2 |
1.049 |
1.072 |
1.016 |
1.024 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0479, wR2 = 0.1067 |
R
1 = 0.0337, wR2 = 0.0847 |
R
1 = 0.0261, wR2 = 0.0608 |
R
1 = 0.1113, wR2 = 0.2881 |
|
R indices (all data) |
R
1 = 0.0773, wR2 = 0.1193 |
R
1 = 0.0418, wR2 = 0.0881 |
R
1 = 0.0348, wR2 = 0.0644 |
R
1 = 0.1647, wR2 = 0.3223 |
Table 2 Selected bond lengths (Å) and bond angles (°) of the new Ru(II) complexes (1–4)
| Complex 1 |
Complex 2 |
Complex 3 |
Complex 4 |
| Bond lengths |
| Ru1–C3 |
2.078(2) |
Ru1–C3 |
2.066(1) |
Ru1–C4 |
2.064(1) |
Ru1–C1 |
2.05(1) |
| Ru1–N1 |
2.083(2) |
Ru1–N1 |
2.090(1) |
Ru1–N1 |
2.084(1) |
Ru1–N1 |
2.070(9) |
| Ru1–P1 |
2.3718(7) |
Ru1–P1 |
2.3671(5) |
Ru1–P1 |
2.3746(5) |
Ru1–P1 |
2.377(3) |
| Ru1–P2 |
2.3641(7) |
Ru1–P2 |
2.3868(5) |
Ru1–P2 |
2.3675(5) |
Ru1–P2 |
2.375(3) |
| Ru1–S1 |
2.4470(7) |
Ru1–S1 |
2.4542(4) |
Ru1–S1 |
2.4491(4) |
Ru1–S1 |
2.430(4) |
| Ru1–C13 |
1.851(4) |
Ru1–C14 |
1.853(1) |
Ru1–C16 |
1.855(1) |
Ru1–C19 |
1.87(1) |
|
|
| Bond angles |
| S1–Ru1–P1 |
87.57(2) |
S1–Ru1–P1 |
87.76(2) |
S1–Ru1–P1 |
89.12(1) |
S1–Ru1–P1 |
91.9(1) |
| S1–Ru1–P2 |
85.92(2) |
S1–Ru1–P2 |
89.98(2) |
S1–Ru1–P2 |
87.60(1) |
S1–Ru1–P2 |
88.0(1) |
| S1–Ru1–N1 |
78.81(7) |
S1–Ru1–N1 |
78.97(4) |
S1–Ru1–N1 |
79.51(3) |
S1–Ru1–N1 |
79.7(3) |
| S1–Ru1–C3 |
156.91(7) |
S1–Ru1–C3 |
157.38(4) |
S1–Ru1–C4 |
158.12(4) |
S1–Ru1–C1 |
157.8(3) |
| S1–Ru1–C13 |
103.25(8) |
S1–Ru1–C14 |
101.88(5) |
S1–Ru1–C16 |
100.83(4) |
S1–Ru1–C19 |
101.3(4) |
| P1–Ru1–P2 |
171.15(3) |
P1–Ru1–P2 |
175.02(2) |
P1–Ru1–P2 |
176.41(2) |
P1–Ru1–P2 |
179.0(1) |
| P1–Ru1–N1 |
92.07(7) |
P1–Ru1–N1 |
91.87(4) |
P1–Ru1–N1 |
90.96(3) |
P1–Ru1–N1 |
91.2(3) |
| P1–Ru1–C3 |
90.69(7) |
P1–Ru1–C3 |
94.73(4) |
P1–Ru1–C4 |
92.30(4) |
P1–Ru1–C1 |
92.9(3) |
| P1–Ru1–C13 |
88.41(8) |
P1–Ru1–C14 |
88.70(5) |
P1–Ru1–C16 |
88.25(4) |
P1–Ru1–C19 |
90.6(4) |
| P2–Ru1–N1 |
92.54(7) |
P2–Ru1–N1 |
92.05(4) |
P2–Ru1–N1 |
89.88(3) |
P2–Ru1–N1 |
88.6(3) |
| P2–Ru1–C3 |
97.65(7) |
P2–Ru1–C3 |
89.06(4) |
P2–Ru1–C4 |
91.30(4) |
P2–Ru1–C1 |
88.1(3) |
| P2–Ru1–C13 |
87.26(8) |
P2–Ru1–C14 |
87.42(5) |
P2–Ru1–C16 |
90.92(4) |
P2–Ru1–C19 |
89.5(4) |
| N1–Ru1–C3 |
78.24(9) |
N1–Ru1–C3 |
78.49(6) |
N1–Ru1–C4 |
78.64(5) |
N1–Ru1–C1 |
78.3(4) |
| N1–Ru1–C13 |
177.9(1) |
N1–Ru1–C14 |
179.0(6) |
N1–Ru1–C16 |
179.14(5) |
N1–Ru1–C19 |
177.9(4) |
| C3–Ru1–C13 |
99.7(1) |
C3–Ru1–C14 |
100.65(6) |
C4–Ru1–C16 |
101.03(6) |
C1–Ru1–C19 |
100.5(5) |
| Ru1–S1–C12 |
95.88(9) |
Ru1–S1–C12 |
95.57(6) |
Ru1–S1–C13 |
95.22(5) |
Ru1–S1–C12 |
95.4(4) |
| Ru1–P1–C14 |
113.85(9) |
Ru1–P1–C15 |
115.89(5) |
Ru1–P1–C17 |
113.37(4) |
Ru1–P1–C20 |
112.3(4) |
| Ru1–P1–C20 |
113.67(9) |
Ru1–P1–C21 |
117.49(5) |
Ru1–P1–C23 |
113.82(4) |
Ru1–P1–C26 |
116.2(4) |
| Ru1–P1–C26 |
116.37(8) |
Ru1–P1–C27 |
113.84(6) |
Ru1–P1–C29 |
116.41(5) |
Ru1–P1–C32 |
118.4(4) |
| Ru1–P2–C32 |
115.44(8) |
Ru1–P2–C33 |
117.11(5) |
Ru1–P2–C35 |
117.25(5) |
Ru1–P2–C38 |
119.1(4) |
| Ru1–P2–C38 |
114.0(1) |
Ru1–P2–C39 |
116.38(5) |
Ru1–P2–C41 |
118.20(4) |
Ru1–P2–C44 |
113.8(4) |
| Ru1–P2–C44 |
114.64(9) |
Ru1–P2–C45 |
112.92(5) |
Ru1–P2–C47 |
112.35(4) |
Ru1–P2–C50 |
112.6(4) |
| Ru1–N1–N2 |
125.1(2) |
Ru1–N1–N2 |
125.3(1) |
Ru1–N1–N2 |
124.94(8) |
Ru1–N1–N2 |
125.1(7) |
| Ru1–C3–C2 |
112.0(2) |
Ru1–C3–C2 |
112.2(1) |
Ru1–C4–C3 |
112.38(9) |
Ru1–C1–C2 |
131.1(9) |
| Ru1–C3–C4 |
132.2(2) |
Ru1–C3–C4 |
132.1(1) |
Ru1–C4–C5 |
131.9(1) |
Ru1–C1–C9 |
113.0(8) |
| Ru1–N1–C10 |
118.3(3) |
Ru1–N1–C10 |
118.0(1) |
Ru1–N1–C11 |
117.89(9) |
Ru1–N1–C10 |
118.1(8) |
| Ru1–C13–O3 |
178.2(2) |
Ru1–C14–O3 |
177.7(1) |
Ru1–C16–O3 |
176.9(1) |
Ru1–C19–O3 |
177.0(1) |
Crystal structure of the ligands H2L2 and H2L3
The ligands crystallized with monoclinic symmetry in the P21/c (H2L2) and C2/c (H2L3) space group respectively. The crystal structure of the ligands [H2-3AC-mtsc] (H2L2) and [H2-3AC-etsc] (H2L3) showed that the thiocarbonyl sulphur (S1) and the imine nitrogen N(1) atom in the thiosemicarbazone are trans to each other across the C(12)–N(2) bond. The structural arrangement corresponds to the E-isomer. The bond distances in the thiosemicarbazones agree well with the values observed for other thiosemicarbazones,22,26 where the C(12)S(1) bond distance was found as the thione form with 1.6793(7) Å and 1.6821(8) Å for H2L2 and H2L3 respectively. Furthermore, no large charge delocalization was detected, as shown by the clean double bond character of the C(10)–N(1) distance of 1.290(1) Å and 1.294(1) Å for the ligands H2L2 and H2L3 respectively. In ligand H2L2, two intermolecular hydrogen bondings were observed between the hydrogen atom of N(3)–H(3) of one molecule and the O(2) oxygen atom of another molecule and vice versa (Fig. S10; Table S4, ESI†).
Crystal structure description of the new Ru(II) complexes (1–4)
The crystal structure of the complexes 1–4 consists of a six-coordinated ruthenium ion with a C2N1S1P2 core, where the equatorial plane was constituted by the C(4) carbon of the pyrone ring, N1 hydrazinic nitrogen and thiolate sulfur atom of thiosemicarbazone. The fourth equatorial position is occupied by a carbonyl carbon. The phosphorous atoms (P1 and P2) from the two triphenylphosphine molecules have taken the two apical vertices. The crystals of the complexes (1–3) belong to the triclinic system with the P space group and the crystals of complex 4 belong to the monoclinic system with the P21/c space group. The coordination environment around the Ru(II) ion can be described as a distorted octahedron due to the deviation of the bond angles from the required 90° and 180° (Table 2). The five member chelate rings have a N(1)–Ru(1)–S(1) bite angle of 78.81(7)° for complex (1), 78.97(4)° for complex (2), 79.51(3)° for complex (3) and 79.70(3)° for complex (4), and a C–Ru(1)–N(1) bite angle of 78.24(9)° for complex (1), 78.49(6)° for complex (2), 78.64(5)° for complex (3) and 78.30(4)° for complex (4). The presence of a strong π-acidic CO ligand restricts the triphenylphosphine ligands from mutually occupying the cis position, which is often preferred for better π-interaction. Hence the PPh3 groups occupied trans positions with respect to each other. Furthermore, the bulkier thiosemicarbazone ligand forced the phosphorous ligand not to lie in the equatorial position. The carbonyl group occupied the site trans to the N1 nitrogen, which is confirmed from the bond angle of N(1)–Ru(1)–C(13) = 177.9(1)° for complex 1, N(1)–Ru(1)–C(14) = 179.0(6)° for complex 2, N(1)–Ru(1)–C(16) = 179.14(5)° for complex 3 and N(1)–Ru(1)–C(19) = 177.9(4)° for complex 4.27,32 The bond length of Ru(1)–P(1) and Ru(1)–P(2) distances was found as 2.3718(7) Å and 2.3641(7) Å for complex 1, 2.3671(5) Å and 2.3868(5) Å for complex 2, 2.3746(5) Å and 2.3675(5) Å for complex 3, and 2.378(3) Å and 2.375(3) Å for complex 4. The bond distances of Ru(1)–P(1) and Ru(1)–P(2) are comparatively longer than those observed for basal planar bonds, such as Ru(1)–N(1) [2.070–2.090 Å], Ru(1)–Cpyrone [2.05–2.078 Å] and Ru(1)–CCO [1.851–1.87 Å]. The lengthening of the Ru–P bonds is due to the higher trans influence of bulky triphenylphosphine.
The selected bond distances of complexes 1–4 such as Ru–P, Ru–O, Ru–S, Ru–N and Ru–C and bond angles agree very well with the similar reported ruthenium(II) complexes.20,22,27,28,33–35 In the free ligands, the S1 and N1 atoms are in an E conformational arrangement with respect to the C12–N2 bond and upon coordination to the metal, a rotation of 180° about the C12–N2 bond occured which placed the S1 and N1 atoms in the Z conformation.
DNA binding properties
Electronic spectra of the ligands H2L1–4 and complexes 1–4 recorded in the presence and absence of CT-DNA are given in Fig. S11 in the ESI.† Upon incremental addition of DNA, the absorption bands of the ligands exhibited hypochromism of about 18.43–34.60% with a red shift of 1–4 nm at around 330 nm. Whereas, the LMCT absorption band of the complexes (1–4) exhibited the same phenomenon of hypochromism about 33.15–56.68% with a red shift of about 3 nm. The binding constant Kbin values are calculated as the ratio of the slope to the y intercept in plots of [DNA]/[εa − εf] versus [DNA] (Fig. 7) and are given in Table 3. These values revealed that the ligands and the organometallic Ru(II) complexes behaved as better binders to DNA. Chelation of the ligands with the metal ion enhanced the binding ability to CT-DNA compared with free ligands. From the results obtained, the order of binding strength of the compounds with CT-DNA is 3 > 2 > 1 > 4 > H2L3 > H2L2 > H2L1 > H2L4 and their binding ability increased with increase in the electron-donating ability of the substituent on the terminal nitrogen of the coordinated thiosemicarbazones. Complex 3 has higher magnitude of binding with CT-DNA, which revealed that the binding ability to DNA increases by increasing the aliphatic chain on the terminal nitrogen. These results are comparable with the earlier reports describing the intercalative mode of various metallo intercalators.20,27,29 The intercalative mode of binding of the compounds with DNA has been proven by additional experiments such as an ethidium bromide displacement assay and viscosity measurements.
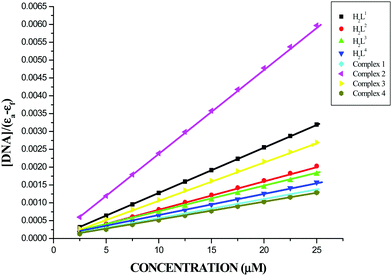 |
| | Fig. 7 Binding isotherms of the ligands H2L1–4 and complexes 1–4 with CT-DNA. | |
Table 3 The binding constant (Kbin) and quenching constant (KSV) values for the interactions of the ligands H2L1–4 and complexes (1–4) with CT-DNA
| Compounds |
Binding constant Kbin (M−1) |
Quenching constant Kq (M−1) |
| (H2L1) |
2.0308 ± 0.286 × 105 |
1.52 ± 0.002 × 103 |
| (H2L2) |
2.1693 ± 0.275 × 105 |
2.10 ± 0.002 × 103 |
| (H2L3) |
3.8299 ± 0.275 × 105 |
2.35 ± 0.004 × 103 |
| (H2L4) |
1.7449 ± 0.324 × 105 |
1.50 ± 0.002 × 103 |
| Complex 1 |
7.9023 ± 0.320 × 105 |
2.90 ± 0.007 × 103 |
| Complex 2 |
11.9388 ± 0.332 × 105 |
3.04 ± 0.006 × 103 |
| Complex 3 |
16.1480 ± 0.320 × 105 |
3.87 ± 0.006 × 103 |
| Complex 4 |
7.7066 ± 0.334 × 105 |
2.68 ± 0.007 × 103 |
Ethidium bromide-displacement assay
To further elucidate the binding mode of the ligands H2L1–4 and complexes (1–4) with CT-DNA, a competitive binding assay with EB-bound DNA was performed. Binding of a second molecule to DNA by stacking interactions between adjacent DNA base pairs, displaces the ethidium bromide from the EB bound DNA model and decreases the fluorescence intensity.36,37 The fluorescence quenching extent of the EB-bound DNA model can be used to determine the relative DNA binding abilities of the second molecule. The emission spectra of the EB-bound CT DNA in the absence and presence of each compound have been recorded for [EB] = 10 μM, and [DNA] = 10 μM for increasing concentrations of each compound. Addition of compounds of different concentrations (10–100 μM) to CT-DNA pretreated with EB ([DNA]/[EB] = 1) caused a reduction in the emission intensity of about 13.42, 19.59, 18.98, 14.07, 22.26, 22.83, 27.88 and 20.22% together with a red shift of 2–3 nm for H2L1, H2L2, H2L3, H2L4, and complexes 1, 2, 3 and 4 respectively (Fig. S12, ESI†). The observed quenching of DNA–EB fluorescence for ligands H2L1–4 and complexes 1–4 suggested that they displace EB from the DNA–EB complex and they can probably interact with CT DNA by the intercalative mode. The quenching constant (KSV) values are calculated as the slope from the plot of I0/Icorrversus [Q] (Fig. 8) and are given in Table 3. The values increased in the order H2L4 < H2L1 < H2L2 < H2L3 < 4 < 1 < 2 < 3. The results are consistent with the absorption spectral observations. Furthermore, the calculated KSV values of the compounds are significant when compared to the reported values.27,38
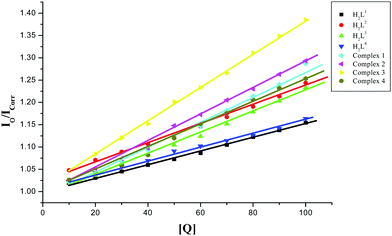 |
| | Fig. 8 Stern–Volmer plot of the fluorescence titration of the ligands H2L1–4 and complexes (1–4) (10–100 μM) with DNA–EB (10 μM). | |
Viscosity measurement
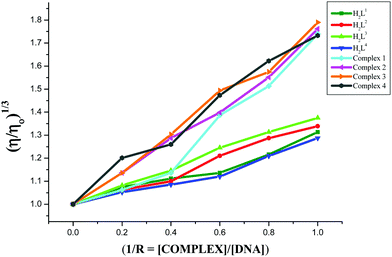
In the presence of DNA intercalators, the viscosity of CT-DNA is known to be significantly altered. Viscosity experiments were carried out on CT-DNA in Tris–HCl/NaCl buffer (100 μM) by varying the concentrations of the compounds (0–100 μM) and their data are given in Fig. 9. Upon increasing the concentration of the compounds, the relative DNA viscosity was increased. The plots of (η/η0)1/3vs. [compound]/[DNA] ratio showed the intercalative binding mode of the compounds to CT-DNA. The viscosity of the CT-DNA in the presence of the complexes increased to a greater extent when compared with their parent ligands. This may be explained by the chelation of the ligands with Ru(II) ions, and it strengthens the interaction between the complex and CT-DNA, leading to an increase in the DNA length, and thus increases the viscosity of CT-DNA. In addition, the plot revealed that the ability of the compounds to increase the viscosity of CT-DNA depends upon the substitution on the N-terminal nitrogen of the ligand and the increasing order of viscosity of CT-DNA by the compounds is 3 (NH-ethyl) > 2 (NH-methyl) > 1 (NH-hydrogen) > 4 (NH-phenyl) > H2L3 > H2L2 > H2L1 > H2L4 which is consistent with their DNA binding results.
 |
| | Fig. 9 Effect of the ligands H2L1–4 and complexes (1–4) on the viscosity of CT-DNA. | |
DNA cleavage studies
The cleavage of supercoiled plasmid DNA pBR322 by the ligands H2L1–4, [RuHClCO(PPh3)3] and Ru(II) complexes (1–4) was carried out by using 1.5% agarose gel electrophoresis in Tris–acetic acid–ethylenediamine tetraacetic acid buffer. The ligands cleaved SC DNA into nicked circular NC DNA. However, complexes 1–4 cleaved SC DNA into nicked circular NC DNA and linear DNA (Fig. 10). The study revealed that the complexes cleaved DNA more efficiently than the ligands. The mechanisms of DNA cleavage basically include three types – photo-induced cleavage, oxidative cleavage and hydrolytic cleavage. Photocleavage allows the use of light to trigger nuclease activity, while oxidative cleavage is mediated by the presence of additives such as H2O2. In contrast, DNA hydrolysis involves the cleavage of phospho diester bonds to generate fragments i.e. nucleophilic attack on the DNA phosphate backbone. In the present case, the compounds did not require addition of any external agents or light for effective DNA cleavage. In the complexes, the metal acts as a Lewis acid and activates the phosphate group towards nucleophilic attack, generating a five coordinate intermediate, which can be stabilized by a catalyst. Hence, the probable mechanism of cleavage may be due to DNA hydrolysis.39 The DNA cleavage strength of complexes follows the order 3 ≈ 2 > 1 > 4, this results in a pattern almost similar to the order of the binding strength of the complexes with CT-DNA. The given results confirm that our organometallic Ru(II) complexes have a great impact on the strength of DNA degradation.
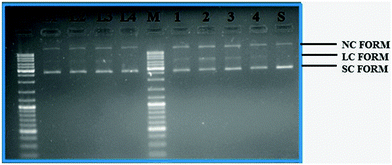 |
| | Fig. 10 Gel electrophoresis diagram showing the cleavage of supercoiled pBR322 DNA by ligands H2L1–4 and complexes 1–4 in 5% DMSO and 95% 5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2 and 37 °C with an incubation time of 2 h. Lanes M: marker; lane L1: ligand 1 (50 μM). Lane L2: ligand 2 (50 μM); lane L3: ligand 3 (50 μM); lane L4: ligand 4 (50 μM); lane 1: complex 1 (50 μM). Lane 2: complex 2 (50 μM); lane 3: complex 3 (50 μM); lane 4: complex 4 (50 μM); lane S: metal precursor (50 μM); forms SC, NC, and LC are supercoiled, nicked circular, and linear circular DNA, respectively. | |
Protein binding studies
The protein binding affinity of the ligands H2L1–4 and new ruthenium(II) complexes 1–4 was studied by using serum albumins such as BSA and HSA. The emission spectra of BSA and HSA solutions exhibit an intense fluorescence emission maximum at 346 nm and 345 nm, respectively, due to the tryptophan residues, when excited at 280 and 290 nm respectively.40,41 The quenching and changes observed in the fluorescence emission spectra of the serum albumins (BSA and HSA) in the presence of 3-acetyl coumarin Schiff base ligands H2L1–4 and ruthenium complexes (1–4) are shown in Fig. S13 and S14 in the ESI.† Addition of the above test compounds to BSA solution resulted in a significant decrease in the fluorescence intensity of BSA at 346 nm with 43.12%, 56.16%, 66.90% and 50.29% of the initial fluorescence intensity of BSA with a blue shift of 2–4 nm for H2L1–4 and 73.54%, 61.47%, 74.83% and 69.79% of the initial fluorescence intensity of BSA with a 2–3 nm blue shift for complexes 1, 2, 3 and 4 respectively. Addition of the compounds to a human serum albumin HSA solution resulted in a quenching of the HSA emission band in the range of 44.64–53.37% by ligands H2L1–4 and more pronounced quenching by the complexes (1–4) in the range of 54.37–55.36% (Fig. 11). These results revealed a definite interaction of the ligands and new complexes with the BSA and HSA proteins. The absorption spectra of the serum albumins in the absence and presence of the ligands and the complexes are given in Fig. S15 in the ESI.† On adding the ligands and complexes to the albumins, the absorbance intensity of the serum albumins was quenched with a red shift of 2 nm. The observed changes indicated a static quenching mechanism of serum albumins such as BSA and HSA by the ligands and complexes.
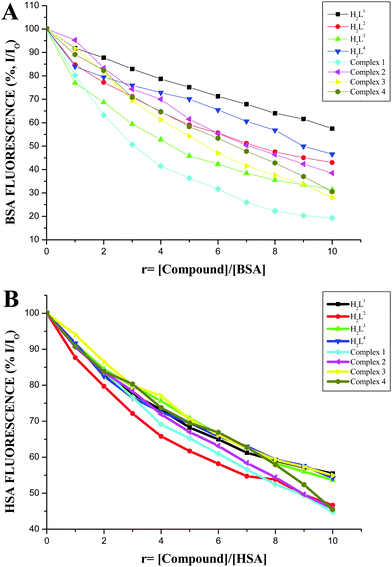 |
| | Fig. 11 (A) Plot of % relative fluorescence intensity (% I/I0) vs. r (r = [compound]/[BSA]). (B) Plot of % relative fluorescence intensity (% I/I0) vs. r (r = [compound]/[HSA]). | |
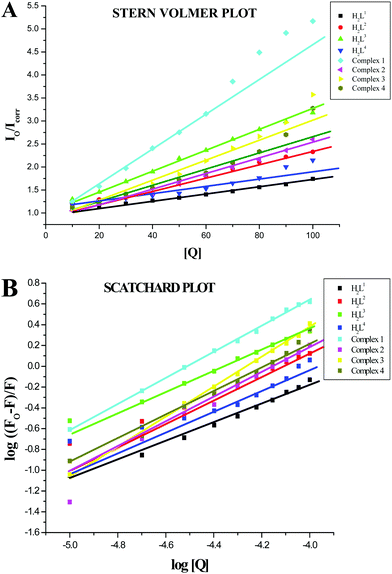
The values of the Stern–Volmer quenching constant (KSV) and the quenching constant (kq) for compounds interacting with serum albumins (BSA or HSA) were calculated by using the Stern–Volmer quenching equation (eqn (S2), ESI†).42 The Stern–Volmer quenching constant can be obtained as a slope from the plot of [Q] verses the ratio of the fluorescence intensity in the absence of (I0) and in the presence of (I) the quencher (Fig. 12A and 13A; Table 4). The observed linearity in the plots (Fig. 12A and 13A) indicates the ability of the compounds to quench the emission intensity of serum albumins. The quenching constant values for the quenching of serum albumins by the compounds were calculated and cited in Table 4. These values (kq ≈ 1012 M−1 s−1) suggested the good binding propensity of the compounds with serum albumins through a static quenching mechanism.42 These results agree well with the results obtained from the absorption spectral studies.
 |
| | Fig. 12 (A) Stern–Volmer plot of the fluorescence titration of the ligands H2L1–4 and complexes (1–4) (10–100 μM) with BSA (10 μM). (B) Scatchard plot of the fluorescence titration of the ligands H2L1–4 and complexes (1–4) (10–100 μM) with BSA (10 μM). | |
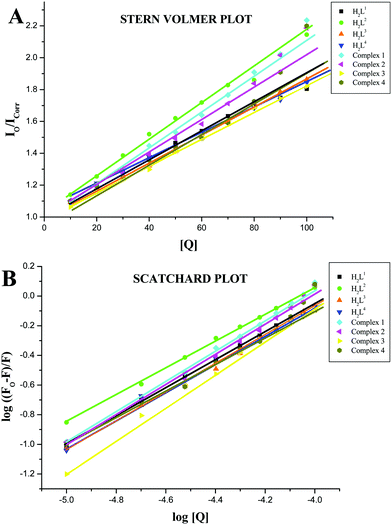 |
| | Fig. 13 (A) Stern–Volmer plot of the fluorescence titration of the ligands H2L1–4 and complexes (1–4) (10–100 μM) with HSA (10 μM). (B) Scatchard plot of the fluorescence titration of the ligands H2L1–4 and complexes (1–4) (10–100 μM) with HSA (10 μM). | |
Table 4 Quenching constant (KSV), binding constant (Kbin) and number of binding sites (n) for the interactions of the ligands H2L1–4 and complexes (1–4) with BSA and HSA
| Compounds |
Stern–Volmer quenching constant (KSV) (M−1) |
Quenching constant (kq) (M−1 s−1) |
Binding constant (Kbin) (M−1) |
Number of binding sites (n) |
| BSA |
| (H2L1) |
0.711 ± 0.019 × 104 |
0.711 ± 0.019 × 1012 |
3.479 ± 0.0147 × 103 |
0.8948 ± 0.024 |
| (H2L2) |
1.311 ± 0.022 × 104 |
1.311 ± 0.022 × 1012 |
4.924 ± 0.016 × 103 |
0.8967 ± 0.024 |
| (H2L3) |
2.160 ± 0.033 × 104 |
2.160 ± 0.033 × 1012 |
9.009 ± 0.026 × 103 |
0.9050 ± 0.019 |
| (H2L4) |
1.028 ± 0.083 × 104 |
1.028 ± 0.083 × 1012 |
1.046 ± 0.077 × 103 |
0.7671 ± 0.083 |
| Complex 1 |
4.603 ± 0.022 × 104 |
4.603 ± 0.022 × 1012 |
4.269 ± 0.079 × 105 |
1.2482 ± 0.018 |
| Complex 2 |
1.695 ± 0.022 × 104 |
1.695 ± 0.022 × 1012 |
9.348 ± 0.237 × 105 |
1.2375 ± 0.054 |
| Complex 3 |
2.624 ± 0.014 × 104 |
2.624 ± 0.014 × 1012 |
5.224 ± 0.260 × 106 |
1.2440 ± 0.090 |
| Complex 4 |
2.194 ± 0.019 × 104 |
2.194 ± 0.019 × 1012 |
1.722 ± 0.247 × 105 |
1.2442 ± 0.056 |
|
|
| HSA |
| (H2L1) |
0.955 ± 0.030 × 104 |
0.955 ± 0.030 × 1012 |
4.130 ± 0.019 × 103 |
0.9213 ± 0.020 |
| (H2L2) |
1.077 ± 0.029 × 104 |
1.077 ± 0.029 × 1012 |
4.607 ± 0.015 × 103 |
0.9026 ± 0.016 |
| (H2L3) |
0.871 ± 0.029 × 104 |
0.871 ± 0.029 × 1012 |
6.395 ± 0.010 × 103 |
0.9699 ± 0.020 |
| (H2L4) |
0.788 ± 0.018 × 104 |
0.788 ± 0.018 × 1012 |
3.940 ± 0.013 × 103 |
0.9175 ± 0.030 |
| Complex 1 |
1.173 ± 0.046 × 104 |
1.173 ± 0.046 × 1012 |
1.711 ± 0.033 × 104 |
1.0516 ± 0.031 |
| Complex 2 |
0.855 ± 0.035 × 104 |
0.855 ± 0.035 × 1012 |
1.215 ± 0.035 × 104 |
1.0693 ± 0.021 |
| Complex 3 |
1.215 ± 0.016 × 104 |
1.215 ± 0.016 × 1012 |
2.281 ± 0.026 × 104 |
1.1046 ± 0.027 |
| Complex 4 |
1.090 ± 0.028 × 104 |
1.090 ± 0.028 × 1012 |
1.081 ± 0.034 × 104 |
1.0158 ± 0.036 |
The fluorescence quenching data obtained from the interaction of the compounds with serum albumins were further analyzed to obtain the binding constant Kbin and number of binding sites per albumin (n) values, which can be calculated from the Scatchard equation (eqn (S3), ESI†). The plots of log[(F° − F)/F] against log[Q] showed a linear relationship with the slope equal to n (Fig. 12B and 13B) and the obtained values are given in Table 4. The binding site for the compounds is almost equal to 1 for serum albumins and the binding between the serum albumins and the compounds is in a 1:1 stoichiometric ratio. From the binding constant values, it is clear that the ligands H2L1–4 and complexes (1–4) showed strong binding affinity with BSA and HSA.
Furthermore, the larger Stern–Volmer quenching constant KSV values, quenching constant (kq) values and binding constant Kbin indicate a strong interaction between the albumins and the complexes over the ligands due to the chelation of the ligand with the metal ion.43 The higher binding affinity of the complexes over the ligands may be due to the efficient intercalation of base pairs of the protein moiety with the complexed metal ions. The quenching constant and binding constant values indicate that the ruthenium(II) complexes bind to both the albumins with the following order: complex 3 > complex 2 > complex 1 > complex 4. This observed trend can be explained on the basis of the electron donating ability and hydrophobicity of the compounds. It is obvious from the results that increase in the electron-donating ability of the substituent on the terminal nitrogen of the ligand increases the binding ability to albumins. This might be due to the increase in the electron density on the electron deficient metal centers. In addition, the observed blue shift can be correlated with their binding with protein which makes it more hydrophobic. The complex 3 possessing an ethyl group on the terminal nitrogen, exhibited better protein binding ability with enhanced hydrophobicity. Similar observations were reported in the literature.31,44 The quenching constants and binding constants found in these organometallic ruthenium(II) complexes agree well with those reported for other ruthenium(II) complexes.20,29,45,46
The synchronous fluorescence spectra of both the albumins with various concentrations (10–100 μM) of test compounds were recorded at Δλ = 15 nm, which is a characteristic of tyrosine, and Δλ = 60 nm, which is a characteristic of tryptophan residues (Fig. S16–S19, ESI†). In the synchronous fluorescence spectra of the tyrosine residues of BSA, the addition of the ligands to the solution of BSA showed a hypochromic effect and a hyperchromic effect was observed on addition of the complexes to BSA (Fig. S16 in the ESI†) with no shift in their emission wavelength maxima. The emission intensity of tyrosine in HSA has a hypochromism with negligible shift in the emission wavelength (Fig. S17, ESI†). On adding the test compounds to the BSA solution, the fluorescence intensity of tryptophan residues at 340 nm decreased up to 19.54–37.09% for ligands H2L1–4 and 60.50–75.57% for the complexes (1–4) with a bathochromic shift (Fig. S18 in the ESI†). In the synchronous fluorescence spectra of the tryptophan residues of HSA, the addition of the compounds to the solution of HSA showed hypochromism up to 22.52–43.63% and 48.05–63.71% for the ligands and complexes (1–4) respectively (Fig. S19, ESI†). Thus, synchronous fluorescence spectral studies showed that the fluorescence intensity of both tyrosine and tryptophan residues was affected by increasing the concentration of the compounds, but a significant decrease along with a blue shift of the fluorescence intensity of tryptophan has been observed. These results confirmed that the interaction of the ligands and the complexes with BSA/HSA affects the conformation of tryptophan much more than the tyrosine micro region. The strong interaction between the test compounds and serum albumins suggested that these compounds can easily be stored in protein and can be released to the desired targets.47
Antioxidative activity
Since the ligands H2L1–4 and complexes act as better binders to biomolecules such as CT-DNA and BSA, other biological studies such as of antioxidant activity were performed. Free radicals can induce DNA damage in humans which leads to aging and various diseases such as cancer, hypertension, cardiac infarction, atherosclerosis, rheumatism, cataracts and others.48 The free radical scavenging activity of the coumarin derivatives and Ru(II) complexes has been investigated, mainly in in vitro systems.13,16,28,49 The IC50 values of the ligands (48.20 ± 0.89–88.41 ± 1.45) and complexes (5.25 ± 0.010–8.41 ± 0.013) from a DPPH assay, showed that the compounds display appreciable radical scavenging activity, which is better than that of the standard vitamin C (98.72 ± 1.50) and is in the order of 3 > 2 > 1 > 4 (Fig. 14 and Table 5). Complex 3 exhibited potent radical scavenging ability over complexes 1, 2 and 4. This may be due to the more electron donating ethyl substitution in the terminal nitrogen of the ligand. In a phosphomolybdenum assay, the antioxidant activity of the compounds was measured from their ability to reduce Mo(VI) to Mo(V) with subsequent formation of a green phosphate/Mo(V) complex at acidic pH. This method is quantitative, since the antioxidant activity is expressed as the number of equivalents of ascorbic acid (Table 6). The total antioxidant activity of the compounds is in the following order: Ru(II) complexes > H2L1–4 > ascorbic acid > [RuHClCO(PPh3)3]. The four new Ru(II) complexes exhibited nearly 5–10 times higher activity than their corresponding ligands and standards, which clearly revealed that chelation of the ligand with metal ions plays an important role in the measured antioxidative properties. The radical scavenging ability of the new complexes is greater than that of a few other reported ruthenium complexes, containing Schiff base ligands.49
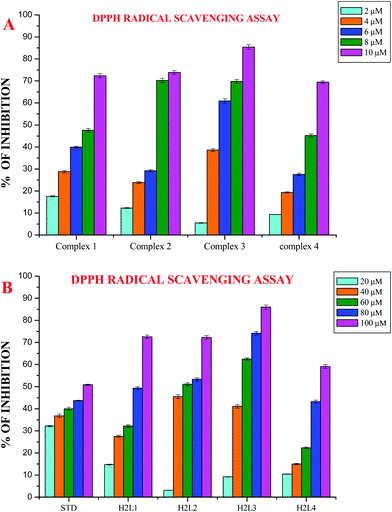 |
| | Fig. 14 (A) DPPH scavenging activity of the new Ru(II) complexes. (B) DPPH scavenging activity of ligands H2L1–4. Error bars represent the standard deviation of the mean (n = 3). | |
Table 5 The DPPH radical scavenging activity of the ligands, RuHClCO(PPh3)3 and new Ru(II) complexes
| Compounds |
IC50 value (μM) |
| Ascorbic acid |
98.72 ± 1.50 |
|
H2L1
|
80.58 ± 1.18 |
|
H2L2
|
57.11 ± 0.76 |
|
H2L3
|
48.20 ± 0.89 |
|
H2L4
|
88.41 ± 1.45 |
| Complex 1 |
8.18 ± 0.011 |
| Complex 2 |
7.01 ± 0.013 |
| Complex 3 |
5.25 ± 0.010 |
| Complex 4 |
8.41 ± 0.013 |
| [RuHClCO(PPh3)3] |
>100 |
Table 6 Estimation of the total antioxidant capacity of the ligands, RuHClCO(PPh3)3 and new Ru(II) complexes
| Compounds |
Ascorbic acid equivalents/μg mL |
|
H2L1
|
27.22 ± 0.78 |
|
H2L2
|
32.45 ± 0.65 |
|
H2L3
|
34.56 ± 0.69 |
|
H2L4
|
23.59 ± 0.47 |
| [RuHClCO(PPh3)3] |
07.02 ± 0.08 |
| Complex 1 |
70.09 ± 0.98 |
| Complex 2 |
74.16 ± 0.84 |
| Complex 3 |
78.09 ± 0.75 |
| Complex 4 |
68.74 ± 0.81 |
Antimicrobial activity
The participation of transition metal complexes in antimicrobial treatment is of special interest since bacteria achieve resistance to antibiotics through morphological and biochemical modifications. Ideal antimicrobial compounds affect only the prokaryotic cells while being inert for eukaryotic cells (human tissues). Bacterial species like Escherichia coli, Pseudomonas aeruginosa and Staphylococcus aureus cause food-borne diseases and nosocomial infections, whereas the fungus Candida albicans is responsible for respiratory infections and gastro-intestinal tract infections.50 Therefore it is necessary to develop new metal-based antimicrobial agents for effective inhibition of microbial growth. The antimicrobial activity of the synthesized ligands H2L1–4, metal precursor and Ru(II) complexes along with standard drugs (Gentamicin for bacteria and Ketaconazole for fungus) was determined against two Gram positive bacteria, Staphylococcus aureus andStreptococcus pneumonie, two Gram negative bacteria, Pseudomonas aeruginosa and Salmonella paratyphi, and five fungi (Candida albicans, Trichophyton rubrum, Aspergillus niger, Aspergillus fumigates and Candida tropicalis). The results are expressed as the zone of inhibition and minimum inhibitory concentration (MIC) and are given in Tables S5–S8 in the ESI† and Fig. 15 and 16. The effect of the compounds was susceptible to the concentration of the compound used for inhibition. From the results, we know that the compounds showed significant activity as compared to the standards Gentamicin and Ketoconazole. The results revealed that the acetyl coumarin Schiff base ligands showed moderate activity against all organisms, while the new organometallic Ru(II) complexes were found to be more toxic against all pathogens taken for investigation. Chelation increased the toxicity of the metal complexes better than the parent Schiff bases and this may be explained by Tweedy's chelation theory.51 Coordination reduces the polarity of the metal ion essentially because of the partial sharing of its positive charge with the donor groups within the chelate ring system formed during coordination and leads to increase in the lipophilic nature of the central metal atom, which favors its permeation more effectively through the lipid layer of the microorganism, thus destroying them more aggressively.51,52 Among the four complexes, complex 3 exhibited good activity against Staphylococcus aureus, Streptococcus pneumonie, and Pseudomonas aeruginosa followed by 2, 1 and 4 and in the case of Salmonella paratyphi the activity of the complexes follows the order of 3 > 2 > 4 > 1. On comparing the antifungal activity of the complexes, complex 3 was more active against four fungi, namely Aspergillus niger, Aspergillus fumigates, Trichophyton rubrum and Candida albicans, followed by complex 2, complex 1 and complex 4. When tested against Candida tropicalis, the activity of the complexes was in the order of 2 > 3 > 1 > 4. The variation in the antimicrobial activity of different metal complexes against different pathogens depends on the impermeability of the cell or the differences in ribosomes in microbial cells.53 In addition, the antimicrobial activity of the complexes was compared with already reported ruthenium complexes and it was found that the new Ru(II) complexes exhibited better activity.54
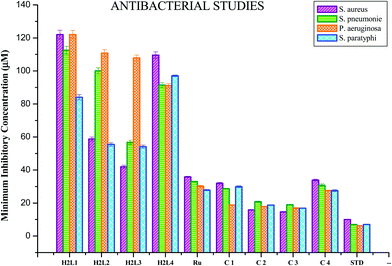 |
| | Fig. 15 Anti-bacterial activity of ligands H2L1–4, [RuHClCO(PPh3)3] and the new Ru(II) complexes (1–4). Error bars represent the standard deviation of the mean (n = 3). | |
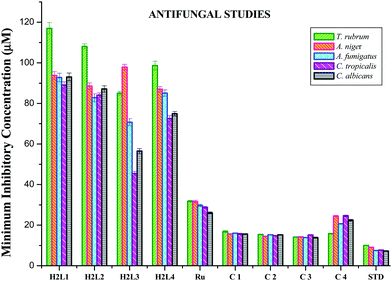 |
| | Fig. 16 Anti-fungal activity of ligands H2L1–4, [RuHClCO(PPh3)3] and the new Ru(II) complexes (1–4). Error bars represent the standard deviation of the mean (n = 3). | |
Antiproliferative studies
Cancer cell growth inhibition.
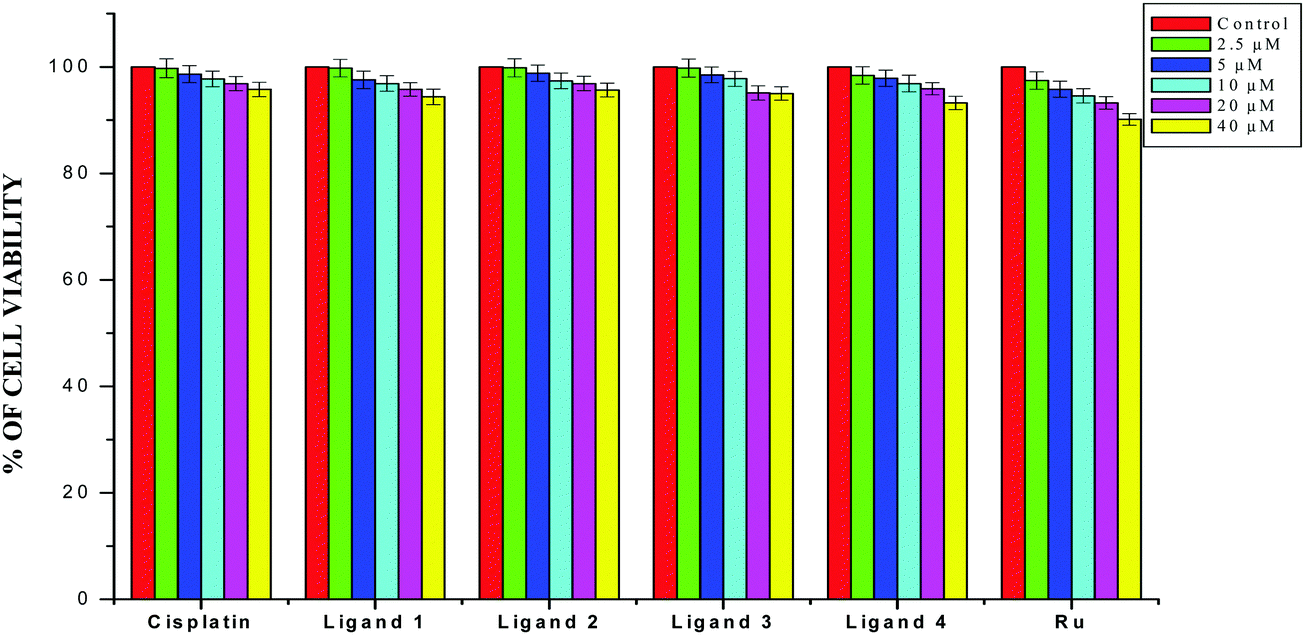
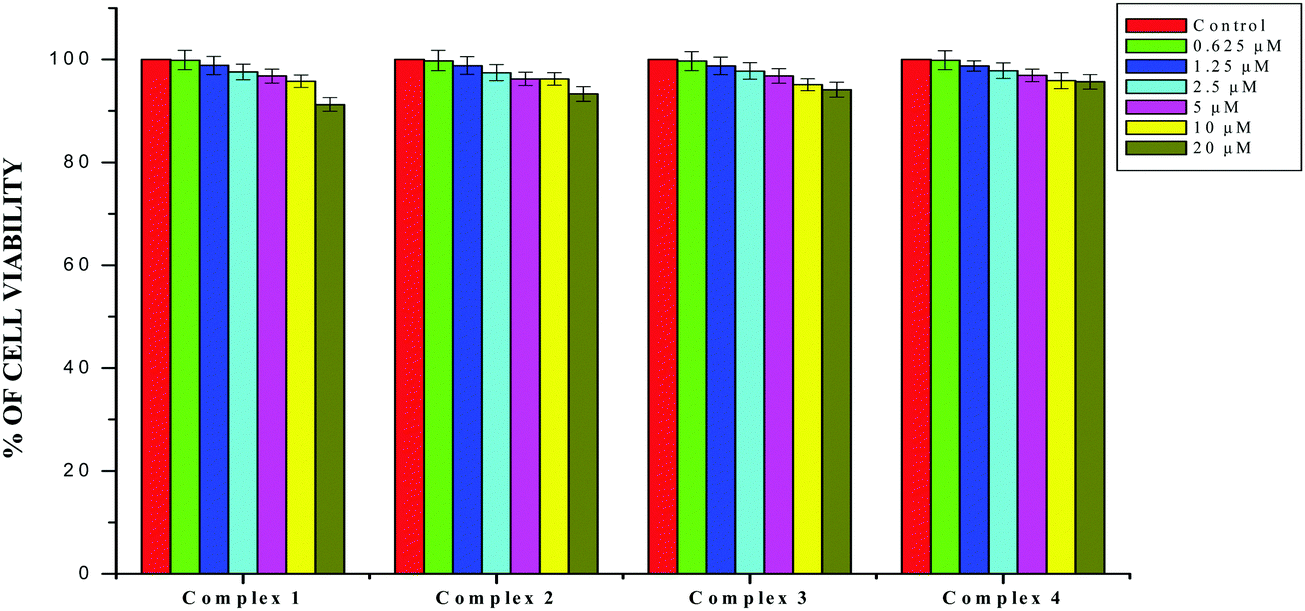
The results obtained from the DNA/protein binding, antioxidant and antimicrobial studies provoked us to further investigate the biological activities of the newly synthesized Schiff base ligands and organometallic ruthenium(II) complexes. The in vitro antiproliferative effects of the ligands [H2L]1–4, metal precursor [RuHCl(CO)(PPh3)3] and new Ru(II) complexes (1–4) were measured by MTT assay in the human breast cancer cells MCF-7, human lung carcinoma cells A549 and human normal keratinocyte cells (HaCaT). For comparison purposes, the anticancer activity of cisplatin was evaluated under the same experimental conditions. The results were analyzed by means of a graph between the percentage of cell viability and concentration and expressed as IC50 values (Fig. 17–20; Table 7). All the compounds showed higher cytotoxic activity to both human lung carcinoma (A549) and human breast cancer cells lines (MCF-7) than the standard drug cisplatin under the same experimental conditions. The anticancer activity of the compounds follows the order ruthenium precursor (15.96 ± 0.21) < cisplatin (15.10 ± 0.05) < ligands H2L1–4 (11.58 ± 0.14–13.28 ± 0.16) < new ruthenium(II) complexes 1–4 (2.56 ± 0.10–3.06 ± 0.08) against human lung cancer cell lines (A549) and activity follows the order Ru precursor (20.10 ± 0.18) < ligands H2L1–4 (17.88 ± 0.15–19.18 ± 0.23) < cisplatin (16.79 ± 0.08) < new Ru(II) complexes 1–4 (2.29 ± 0.11–3.08 ± 0.08) against human breast cancer cell lines (MCF-7) respectively. All the ligands exhibited good cytotoxicity against the two cancer cell lines. Notably, under the same experimental conditions the IC50 values of complexes 1–4 ranged from 2.29 ± 0.11–3.08 ± 0.08 μM against MCF-7 cell lines and 2.56 ± 0.10–3.06 ± 0.08 against A549 cell lines compared to the IC50 of 16.79 ± 0.08 μM and 15.10 ± 0.05 for cisplatin against MCF-7 and A549 cells respectively. The chelation of the ligands with metal ions is responsible for high cytotoxicity of the complexes over the ligands. The enhancement in antitumor activity of the complexes can be related to the electron donating nature of the N-terminal substitution of the coordinated ligands. It should be noted that higher antiproliferative activity was observed in MCF-7 and A549 cells treated with ruthenium(II) complex 3 containing 3-acetylcoumarin-4(N)-ethylthiosemicarbazone than in those treated with ruthenium(II) complexes containing other ligands. On the basis of these results, the antiproliferative activity of these complexes has been arranged in the order 3 > 2 > 1 > 4. Interestingly, this observation is in agreement with the previous biological studies such as DNA binding, protein binding and antioxidant activity, suggesting that the anticancer activities of the tested compounds against cancer cell lines may be related to their ability to intercalate the base pairs of the DNA and/or their free radical scavenging activity. In order to investigate the selectivity of the compounds for cancer cells rather than normal cell lines, the compounds were also screened against human normal keratinocyte cells (HaCaT). The IC50 value of the compounds against HaCaT implies that the ligands and complexes are nontoxic to normal cells. From the literature, it is observed that some other Ru(II) complexes showed anticancer activity against human breast cancer cells MCF-7 and human lung carcinoma cell lines A549 with IC50 values in the range of 2.93–500 μM.1,17,20,24,27,29,55–58 However, the cytotoxic effectiveness of our organometallic Ru(II) complexes containing 3-acetylcoumarin-4(N)-substituted-thiosemicarbazone ligands against these cell lines was found to be higher than that of already reported Ru(II) complexes containing Schiff bases.1,17,20,24,27,29,55–58
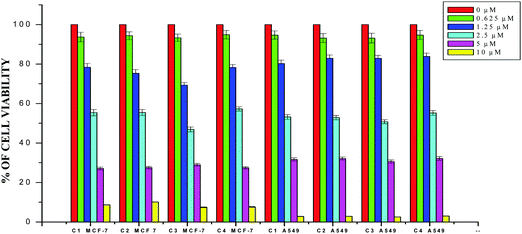 |
| | Fig. 17 The newly synthesized ruthenium(II) complexes (1–4) inhibit MCF-7 and A549 cell proliferation in a dose-dependent manner. MCF-7 and A549 cells were treated with different concentrations of the complexes for 48 h, the cell viability was determined and the results were expressed as percentage cell viability relative to the control. The results shown are means, from three separate experiments performed in triplicate. | |
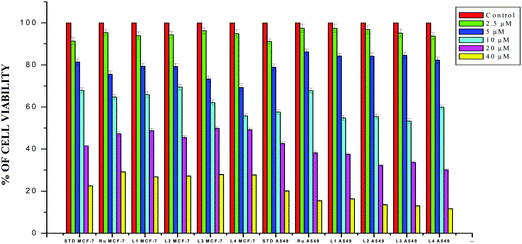 |
| | Fig. 18 The newly synthesized ligands H2L1–4, [RuHClCO(PPh3)3] and cisplatin inhibit MCF-7 and A549 cell proliferation in a dose-dependent manner. MCF-7 and A549 cells were treated with different concentrations of compounds for 48 h, the cell viability was determined and the results were expressed as percentage cell viability relative to the control. The results shown are means, from three separate experiments performed in triplicate. | |
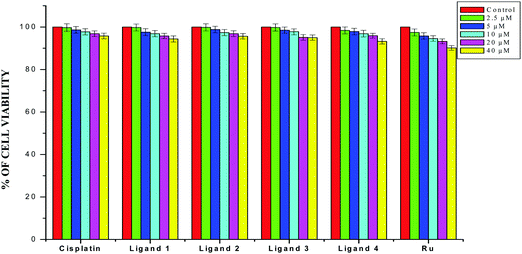 |
| | Fig. 19 The newly synthesized ligands H2L1–4, [RuHClCO(PPh3)3] and cisplatin inhibit HaCaT cell proliferation in a dose-dependent manner. HaCaT cells were treated with different concentrations of compounds for 48 h, the cell viability was determined and the results were expressed as percentage cell viability relative to the control. The results shown are means, from three separate experiments performed in triplicate. | |
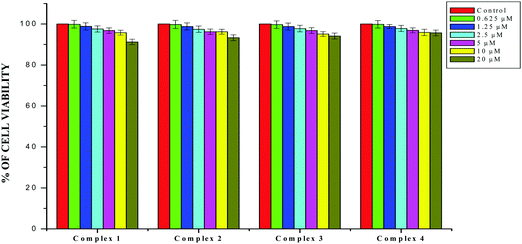 |
| | Fig. 20 The newly synthesized complexes (1–4) inhibit HaCaT cell proliferation in a dose-dependent manner. HaCaT cells were treated with different concentrations of compounds for 48 h, the cell viability was determined and the results were expressed as percentage cell viability relative to the control. The results shown are means, from three separate experiments performed in triplicate. | |
Table 7 The IC50 values for the human breast cancer cell line MCF-7, human lung carcinoma cancer cell line A549 and human normal keratinocyte cells (HaCaT) with the ligands H2L1–4, [RuHClCO(PPh3)3] and the new organometallic Ru(II) complexes for 48 h
| Compounds |
IC50 values (μM) |
| MCF-7 |
A549 |
HaCaT |
|
H2L1
|
19.18 ± 0.23 |
12.67 ± 0.14 |
>40 |
|
H2L2
|
18.17 ± 0.29 |
12.11 ± 0.19 |
>40 |
|
H2L3
|
17.88 ± 0.15 |
11.58 ± 0.14 |
>40 |
|
H2L4
|
18.47 ± 0.20 |
13.28 ± 0.16 |
>40 |
| [RuHClCO(PPh3)3] |
20.10 ± 0.18 |
15.96 ± 0.21 |
>40 |
| Complex 1 |
2.97 ± 0.11 |
2.89 ± 0.11 |
>20 |
| Complex 2 |
2.94 ± 0.09 |
2.81 ± 0.13 |
>20 |
| Complex 3 |
2.29 ± 0.11 |
2.56 ± 0.10 |
>20 |
| Complex 4 |
3.08 ± 0.08 |
3.06 ± 0.08 |
>20 |
| Cisplatin |
16.79 ± 0.08 |
15.10 ± 0.05 |
>40 |
Conclusion
Reaction of [RuHCl(CO)(PPh3)3] with 3-acetylcoumarin-4N-substituted thiosemicarbazones afforded four new organoruthenium(II) complexes. The products were characterized by various analytical and spectral techniques. The exact structures of the ligands and complexes have been determined by X-ray crystallography and the ligands were found to coordinate in a tridentate CNS donor manner. Preliminary biological studies like CT-DNA/protein binding were carried out. The compounds bound well with DNA and binding was through intercalation. The binding affinity of the ruthenium complexes with BSA and HSA was found to be potent and the synchronous spectral studies of the compounds showed that the complexes bound with BSA/HSA affecting the tryptophan residues through a static quenching mechanism. The results obtained from the antioxidant assays showed their significant efficacy in scavenging free radicals with the order of 3 > 2 > 1 > 4. The in vitro anticancer activity of all the compounds has been evaluated against the human breast cancer cell line MCF-7, human lung cancer cell line A549 and the human normal keratinocyte HaCaT cell line, and compared to that of cisplatin. The ligands exhibited significant antiproliferative activities with IC50 cytotoxicity coefficients similar to those of cisplatin, but the organometallic Ru(II) complexes were found to exhibit potent activities. An in vitro cell proliferation assay supported the non-cytotoxic nature of the compounds with high IC50 values against the HaCaT noncancerous cell lines. Chelation plays an important role in the enhancement of the biological activity of the complexes over the ligands. In particular, out of the complexes, complex 3 containing the more electron donating N-terminal ethyl group on thiosemicarbazone was shown to be the best. The outcome from the various biological studies namely DNA/protein binding, viscosity experiments, antioxidant and antimicrobial studies and evaluation of antiproliferative ability indicated that the biological activity of the complexes followed the pattern 3 > 2 > 1 > 4, showing the importance of N-terminal substitution in aiding their activity.
Experimental section
General procedures and measurements
All the reagents used were of analytical or chemically pure grade and used as received without further purification. Solvents were purified and dried according to the reported procedures.59 3-Acetyl-chromene-2-one (3-acetyl coumarin)60 and the metal precursor [RuHCl(CO)(PPh3)3] were prepared according to the literature procedure.61 Ethidium bromide (EB), bovine serum albumin (BSA), human serum albumin (HSA), calf thymus DNA (CT-DNA) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from HiMedia. The melting points of the compounds were determined by using Lab India apparatus. Infrared (IR) spectra were measured using a JASCO FT-IR 4100 instrument as KBr pellets between 400 and 4000 cm−1. Elemental analyses were measured by using a Vario EL III CHNS analyzer. The electronic spectra of the compounds were recorded using a JASCO V-630 spectrophotometer using DMSO as the solvent in the 800–200 nm range. Emission spectra were recorded by using a JASCO FP 6600 Spectrofluorimeter. 1H NMR spectra were recorded using a Bruker instrument at 400 MHz in DMSO at room temperature, with the chemical shift measured relative to tetramethylsilane (TMS).
X-ray crystallography
Suitable single crystals of the ligands H2L2 and H2L3 and complexes 1–4 were obtained from methanol, dichloromethane/methanol and benzene/methanol medium respectively. The data sets for X-ray diffraction studies of the ligands H2L2 and H2L3 and new complexes (1–3) were collected with Mo Kα (λ = 0.71073 Å) radiation using a Bruker kappa APEX-II DUO 1000 CCD diffractometer. The structure of complex 2 was a dichloromethane solvate with disordered solvent which was difficult to model with discrete, partially occupied molecules. The solvent contribution was removed using SQUEEZE, and amounted to 1.38 molecules of DCM per unit cell, or 0.69 per Ru complex. In the case of complex 4, only very thin, weakly scattering crystals of this compound were available. Data were collected from a two-component twin using Cu radiation (λ = 1.5418 Å) from a microfocus source. The quality of the results is thus much lower than for the other structures. All the calculations were performed by using the SHELXS-9762 and SHELXL-2014/7 programs.63
Synthesis of ((1E)-1-(1-(2-oxo-2H-chromen-3-yl)ethylidene)thiosemicarbazide) [H2-3AC-tsc] (H2L1)26.
Thiosemicarbazide (0.484 g, 5.314 mmol) was dissolved in 30 cm3 of methanol with continuous stirring and gently heated for a period of 30 min. This was added to a methanolic solution (20 cm3) of 3-acetyl-2H-chromen-2-one (1.000 g, 5.314 mmol). To this, a few drops of glacial acetic acid was added and the mixture was refluxed for 2 h with continuous stirring. The mixture was then cooled to room temperature whereby a yellow crystalline compound precipitated. This was collected by filtration, washed well with cold methanol and dried under vacuum. The compound was recrystallized from DMF–methanol (1:9 v/v). Yield: 83%. M.p: 192 °C. Anal. calcd for C12H11N3O2S: C, 55.15; H, 4.25; N, 16.08; S, 12.26. Found: C, 55.11; H, 4.23; N, 16.04; S, 12.23%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1718, ν(CN) 1606, ν(–NH2) 3388, ν(–NH) 3154, ν(CS) 761. UV-vis (DMSO), λmax(ε): 271 (11624) nm (dm3 mol−1 cm−1); 340 (12565) nm (dm3 mol−1 cm−1). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 7.946 (s, 1H, C4-H), δ 7.745–7.763 (dd, 1H, J = 1.2, 6, C5-H), δ 7.619–7.654 (m, 1H, C6-H), δ 7.369–7.435 (m, 2H, C7 and C8-H), δ 2.242 (s, 3H, –CH3), δ 10.443 (s, 1H, NH–CS), δ 8.402 and 8.469 (2s, 2H, –NH2).
A very similar method was followed to synthesize the following compounds.
Synthesis of ((1E)-1-(1-(2-oxo-2H-chromen-3-yl)ethylidene) 4(N)-methyl thiosemicarbazide) [H2-3AC-mtsc] (H2L2).
The ligand [H2-3AC-mtsc] was prepared from 4-(N)-methylthiosemicarbazide (0.559 g, 5.314 mmol) and 3-acetyl-2H-chromen-2-one (1.000 g, 5.314 mmol) in the presence of glacial acetic acid. Yield: 71%. M.p: 189 °C. Anal. calcd for C13H13N3O2S: C, 56.70; H, 4.76; N, 15.26; S, 11.64. Found: C, 56.67; H, 4.73; N, 15.22; S, 11.62%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1710, ν(CN) 1617, ν (terminal –NH) 3316, ν(–NH) 3365, ν(CS) 762. UV-vis (DMSO), λmax(ε): 268 (22090) nm (dm3 mol−1 cm−1); 344 (21834) nm (dm3 mol−1 cm−1). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 8.359 (s, 1H, C4-H), δ 7.772–7.790 (dd, 1H, J = 1.2, 6, C5-H), δ 7.626–7.661 (m, 1H, C6-H), δ 7.377–7.445 (m, 2H, C7 and C8-H), δ 2.239 (s, 3H, –CH3), δ 10.454 (s, 1H, NH–CS), δ 8.490–8.501 (q, 1H, terminal –NH), δ 3.004–3.013 (d, J = 3.6, 1H, terminal –NH–CH3).
Synthesis of ((1E)-1-(1-(2-oxo-2H-chromen-3-yl)ethylidene) 4(N)-ethyl thiosemicarbazide) [H2-3AC-etsc] (H2L3).
The ligand [H2-3AC-etsc] was prepared from 4-(N)-ethylthiosemicarbazide (0.637 g, 5.314 mmol) and 3-acetyl-2H-chromen-2-one (1.000 g, 5.314 mmol) in the presence of glacial acetic acid. Yield: 72%. M.p: 193 °C. Anal. calcd for C14H15N3O2S: C, 58.10; H, 5.23; N, 14.52; S, 11.07. Found: C, 58.07; H, 5.20; N, 14.49; S, 11.05%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1722, ν(CN) 1615, ν (terminal –NH) 3350, ν(–NH) 3315, ν(CS) 760. UV-vis (DMSO), λmax(ε): 286 (28230) nm (dm3 mol−1 cm−1); 344 (28942) nm (dm3 mol−1 cm−1). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 8.338 (s, 1H, C4-H), δ 7.786–7.804 (dd, 1H, J = 1.2, 6, C5-H), δ 7.627–7.662 (m, 1H, C6-H), δ 7.376–7.444 (m, 2H, C7 and C8-H), δ 2.237 (s, 3H, –CH3), δ 10.379 (s, 1H, NH–CS), δ 8.507–8.530 (t, J = 4.4, 1H, terminal –NH), δ 3.553–3.608 (p, 2H, terminal –NH–CH2), δ 1.114–1.142 (t, J = 5.6, 3H, –CH3).
Synthesis of ((1E)-1-(1-(2-oxo-2H-chromen-3-yl)ethylidene) 4(N)-phenyl thiosemicarbazide) [H2-3AC-ptsc] (H2L4).
The ligand [H2-3AC-ptsc] was prepared from 4-(N)-phenylthiosemicarbazide (887 g, 5.314 mmol) and 3-acetyl-2H-chromen-2-one (1.000 g, 5.314 mmol) in the presence of glacial acetic acid. Yield: 81%. M.p: 180 °C. Anal. calcd for C18H15N3O2S: C, 64.06; H, 4.48; N, 12.45; S, 9.50. Found: C, 64.02; H, 4.46; N, 12.42; S, 9.48%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1711, ν(CN) 1610, ν (terminal –NH) 3224, ν(–NH) 3205, ν(CS) 770. UV-vis (DMSO), λmax(ε): 274 (16323) nm (dm3 mol−1 cm−1); 341 (12808) nm (dm3 mol−1 cm−1). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 8.496 (s, 1H, C4-H), δ 7.778–7.797 (dd, 1H, J = 1.2, 6, C5-H), δ 7.632–7.681 (m, 1H, C6-H), δ 7.558–7.592 (m, 2H, C7 and C8-H), δ 7.181–7.215 and δ 7.321–7.462 (m, 5H, phenyl group protons), δ 2.320 (s, 3H, –CH3), δ 10.866 (s, 1H, NH–CS), δ 10.143 (s, 1H, terminal –NH).
Synthesis of the new ruthenium(II) complexes
Synthesis of [Ru(3AC-tsc)(CO)(PPh3)2] (1).
A solution of [H2-3AC-tsc] (0.027 g; 0.105 mmol) in 10 cm3 of benzene was added dropwise to a boiling solution of [RuHCl(CO)(PPh3)3] (0.100 g, 0.105 mmol) in benzene, refluxed for 7 h and allowed to stand for 4 days at room temperature. The reddish orange solid formed was filtered, washed with petroleum ether (60–80 °C) and crystallized from benzene and methanol mixture (1:1 v/v) to yield transparent, needle-like red crystals suitable for X-ray analysis. Yield: 67%. M.p. 187 °C. Anal. calcd for C49H39N3O3P2RuS: C, 64.45; H, 4.31; N, 4.60; S, 3.51. Found: C, 64.43; H, 4.28; N, 4.56; S, 3.47%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1683, ν(CN) 1596, ν(C–S) 721, ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) 1923, 1433, 1090, 694 (for PPh3). UV-vis (DMSO), λmax(ε): 265 (21021) nm (dm3 mol−1 cm−1) (intraligand transition); 327 (16624) nm (dm3 mol−1 cm−1) (LMCT s → d). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 7.085–7.681 (m, 32H, Ar–H), δ 6.671–6.708 (t, 1H, J = 7.8, C7-H), δ 6.860–6.880 (d, 1H, J = 8, C8-H), δ 2.075 (s, 3H, –CH3), δ 5.753 (br s, 2H, –NH2).
O) 1923, 1433, 1090, 694 (for PPh3). UV-vis (DMSO), λmax(ε): 265 (21021) nm (dm3 mol−1 cm−1) (intraligand transition); 327 (16624) nm (dm3 mol−1 cm−1) (LMCT s → d). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 7.085–7.681 (m, 32H, Ar–H), δ 6.671–6.708 (t, 1H, J = 7.8, C7-H), δ 6.860–6.880 (d, 1H, J = 8, C8-H), δ 2.075 (s, 3H, –CH3), δ 5.753 (br s, 2H, –NH2).
A similar method was followed to synthesize the other ruthenium(II) complexes.
Synthesis of [Ru(3AC-mtsc)(CO)(PPh3)2] (2).
Complex 2 was prepared by the same procedure as described for 1, with H2L2 (0.028 g; 0.105 mmol) and [RuHCl(CO)(PPh3)3] (0.100 g; 0.105 mmol). Needle shaped, transparent pink colour crystals were obtained on slow evaporation of the reaction mixture. Yield: 69%. M.p. 190 °C. Anal. calcd for C50H41N3O3P2RuS: C, 64.77; H, 4.46; N, 4.53; S, 3.45. Found: C, 64.73; H, 4.43; N, 4.51; S, 3.42%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1687, ν(CN) 1594, ν(C–S) 743, ν(CO) 1919, 1402, 1090, 695 (for PPh3). UV-vis (DMSO), λmax(ε): 269 (34758) nm (dm3 mol−1 cm−1) (intraligand transition); 328 (19890) nm (dm3 mol−1 cm−1) (LMCT s → d). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 7.130–7.302 (m, 31H, Ar–H), δ 7.600–7.618 (d, 1H, J = 7.2, C5-H), δ 6.742–6.780 (t, 1H, J = 7.6, C6-H), δ 6.873–6.855 (d, 1H, J = 7.2, C8-H), δ 1.90 (s, 3H, –CH3), δ 6.106–6.118 (q, 1H, terminal –NH), δ 2.106–2.118 (d, 3H, J = 4.8, –NH–CH3).
Synthesis of [Ru(3AC-etsc)(CO)(PPh3)2] (3).
Complex 3 was prepared by the same procedure as described for 1, with H2L3 (0.030 g; 0.105 mmol) and [RuHCl(CO)(PPh3)3] (0.100 g; 0.105 mmol). The red solid formed was filtered, washed with petroleum ether (60–80 °C) and crystallized from dichloromethane and methanol to yield orange crystals. Yield: 65%. M.p. 221 °C. Anal. calcd for C51H43N3O3P2RuS: C, 65.08; H, 4.61; N, 4.46; S, 3.40. Found: C, 65.06; H, 4.59; N, 4.43; S, 3.36%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1679, ν(CN) 1590, ν(C–S) 744, ν(CO) 1917, 1431, 1089, 695 (for PPh3). UV-vis (DMSO), λmax(ε): 268 (31127) nm (dm3 mol−1 cm−1) (intraligand transition); 333 (17453) nm (dm3 mol−1 cm−1) (LMCT s → d). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 7.132–7.550 (m, 31H, Ar–H), δ 7.568–7.571 (d, 1H, J = 1.2, C5-H), δ 6.729–6.750 (t, 1H, J = 7.6, C6-H), δ 6.846–6.866 (d, 1H, J = 8, C8-H), δ 1.901 (s, 3H, –CH3), δ 6.186 (t, 1H, terminal –NH), δ 2.210–2.360 (m, 2H, J = 4.8, –NH–CH2), δ 0.701–0.737 (t, 3H, J = 7.2, –CH2–CH3).
Synthesis of [Ru(3AC-ptsc)(CO)(PPh3)2] (4).
Complex 4 was prepared by the same procedure as described for 1, with H2L4 (0.035 g; 0.105 mmol) and [RuHCl(CO)(PPh3)3] (0.100 g; 0.105 mmol). The red solid formed was filtered, washed with petroleum ether (60–80 °C) and crystallized from dichloromethane and methanol to yield pink crystals. Yield: 68%. Mp 187 °C. Anal. calcd for C55H43N3O3P2RuS: C, 66.78; H, 4.39; N, 4.24; S, 3.24. Found: C, 66.76; H, 4.36; N, 4.21; S, 3.22%. FT-IR (ν, cm−1) in KBr: ν(CO lactone) 1671, ν(CN) 1595, ν(C–S) 746, ν(CO) 1917, 1428, 1084, 694 (for PPh3). UV-vis (DMSO), λmax(ε): 279 (33167) nm (dm3 mol−1 cm−1) (intraligand transition); 331 (28237) nm (dm3 mol−1 cm−1) (LMCT s → d). 1H NMR (400 MHz, DMSO-d6, δ ppm, J Hz): δ 6.919–7.566 (m, 35H, Ar–H), δ 7.569–7.587 (d, 1H, J = 7.2, C5-H), δ 6.723–6.779 (q, 2H, C6 and C7-H), δ 6.780–6.900 (d, 1H, J = 8, C8-H), δ 1.980 (s, 3H, –CH3), δ 8.501 (s, 1H, terminal –NH).
Stability of the compounds in solution
The biological activity of any metallodrug depends on its stability in solution. In this respect, it is necessary to evaluate the stability of the complexes in aqueous media. All the compounds used in this study were insoluble in water. But, they were very soluble in DMSO and 1% DMSO was sufficient for their dissolution in aqueous media, Tris–HCl buffer or phosphate buffer. In order to analyse the stability of the complexes, the electronic spectra of the compounds in 1% aqueous DMSO, phosphate buffer–DMSO (99:1) and Tris–HCl–DMSO (99:1) were monitored over 24 h at room temperature and their absorbance spectra were recorded.
DNA binding study
DNA binding studies, DNA viscosity studies and DNA cleavage experiments have been carried out according to the method described in earlier reports.20,29,64 The detailed procedures for these experiments are provided in the ESI.†
Serum albumin binding study
Protein binding (HSA and BSA) studies were done according to the earlier reported methods.20,29,65 The detailed procedures for the serum albumin (HSA and BSA) binding experiments are given in the ESI.†
DPPH˙ scavenging assay
The DPPH radical scavenging activity of the compounds was measured according to the literature method.66 100 μL of methanolic solutions of various concentrations (20–100 μM) of the experimental standard ascorbic acid, [RuHCl(CO)(PPh)3], and the ligands and various concentrations (2–10 μM) of the complexes were taken. About 4 mL of a 0.1 mM methanolic solution of DPPH was added to the 100 μL of the methanolic solution of the samples and standard (vitamin C) and shaken vigorously. A negative control was prepared by adding 100 μL of methanol in 4 mL of 0.1 mM methanolic DPPH solution. The tubes were allowed to stand for 20 min at 27 °C. The absorbance of the sample was measured at 517 nm against the blank (methanol).
Total antioxidant capacity by the phosphomolybdenum method
The total antioxidant capacity of the compounds was evaluated by the reported method.67 100 μL of a methanolic solution of the compounds (500 μg mL−1) was mixed with 2 mL of reagent solution (0.6 M sulfuric acid, 28 mM sodium phosphate and 4 mM ammonium molybdate) and the mixture was incubated at 95 °C for 90 min, and after the mixture had cooled to room temperature, the absorbance of the each solution was measured at 695 nm against the blank.
In vitro antimicrobial assay
The antimicrobial activity of the [RuHClCO(PPh3)3], ligands (H2L)1–4 and newly synthesized Ru(II) complexes was evaluated by the agar well diffusion method.68 The test organisms, which included two Gram-positive bacteria (Staphylococcus aureus and Streptococcus pneumonie), two Gram-negative bacteria (Pseudomonas aeruginos and Salmonella paratyphi) and five fungi (Candida albicans, Trichophyton rubrum, Aspergillus niger, Aspergillus fumigatu and Candida tropicalis), were obtained from the MTCC, Chandigarh, India and Microbiological laboratory, Coimbatore, Tamil Nadu, India. The required nutrient broth and potato dextrose broth were prepared and sterilized at 121 °C. The bacteria were grown in the nutrient broth and fungus were grown in the potato dextrose broth at 37 °C and maintained on slants at 4 °C. For all the bacterial strains, overnight cultures grown in broth were adjusted to an inoculum size of 106 CFU per mL for inoculation of the agar plates. The antimicrobial activity of the test compounds was checked with various concentrations (25 μg mL−1, 50 μg mL−1 and 100 μg mL−1) against all the test pathogens. Gentamicin and Ketoconazole were used as positive controls for the antibacterial and antifungal activities, respectively. Following an incubation period of 24 h at 37 °C, the anti-microbial activity was evaluated by the zone of inhibition. DMSO was used both as a solvent and negative control, and no zone of inhibition was observed for the negative control (i.e. DMSO). Each experiment was performed in triplicate and the results were represented as average zone of inhibition and minimum inhibitory concentration of all the test pathogens.
Cell cytotoxicity assay69
The effects of the compounds on the viability of human breast cancer cells (MCF-7), human lung cancer cells (A549) and human normal keratinocytes (HaCaT) were assayed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay.69 The quantity of formazan formed gave a measure of the number of viable cells. The cells were seeded at a density of 10000 cells per well in 200 μL of Dulbecco's Modified Eagle's Medium (DMEM) and were allowed to attach overnight in a CO2 incubator, and then the complexes dissolved in DMSO were added and diluted in cell culture media to a final concentration. After 48 h, the wells were treated with 20 μL MTT (5 mg mL−1 PBS (phosphate buffered saline)) and incubated at 37 °C for 4 h. The purple formazan crystals that formed were dissolved in 200 μL of DMSO and read at 570 nm in a micro plate reader. Data were obtained as the average of three independent sets of experiments all performed in triplicate for each concentration and the medium containing no test compound served as the control. The graph was plotted between the percentage of cell viability and concentration of the compounds. The IC50 values were calculated from the percentage of cell viability versus concentration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author G. K. greatly acknowledges DST, New Delhi, India for the INSPIRE fellowship (IF140225 dated 23.01.2014). The author S. D. greatly acknowledges UGC, New Delhi, India for the UGC-BSR fellowship (F.25-1/2014-15(BSR)/7-26/2007(BSR) dated 05.11.2015).
References
- B. Demoro, R. F. M. de Almeida, F. Marques, C. P. Matos, L. Otero, J. C. Pessoa, I. Santos, A. Rodríguez, V. Moreno, J. Lorenzo, D. Gambino and A. I. Tomaz, Dalton Trans., 2013, 42, 7131–7146 RSC.
- M. Adams, C. de Kock, P. J. Smith, K. M. Land, N. Liu, M. Hopper, A. Hsiao, A. R. Burgoyne, T. Stringer, M. Meyer, L. Wiesner, K. Chibalea and G. S. Smith, Dalton Trans., 2015, 44, 2456–2468 RSC.
- B. Ghosh, P. Adak, S. Naskar, B. Pakhira, P. Mitra, R. Dinda and S. K. Chattopadhyay, Inorg. Chim. Acta, 2017, 459, 1–14 CrossRef CAS.
- A. I. Matesanz, J. Perlesb and P. Souza, Dalton Trans., 2012, 41, 12538–12547 RSC.
- D. Palanimuthu, S. V. Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 56, 722–734 CrossRef CAS PubMed.
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, J. Biol. Inorg. Chem., 2012, 17, 409–423 CrossRef CAS PubMed.
- D. Palanimuthu and A. G. Samuelson, Inorg. Chim. Acta, 2013, 408, 152–161 CrossRef CAS.
- W. Hernandez, A. J. Vaisberg, M. Tobar, M. Alvarez, J. Manzur, Y. Echevarrıa and E. Spodine, New J. Chem., 2016, 40, 1853–1860 RSC.
- S. D. Khanye, B. Wan, S. G. Franzblau, J. Gut, P. J. Rosenthal, G. S. Smith and K. Chibale, J. Organomet. Chem., 2011, 696, 3392–3396 CrossRef CAS.
- A. A. Ali, H. Nimir, C. Aktas, V. Huch, U. Rauch, K. H. Schäfer and M. Veith, Organometallics, 2012, 31, 2256–2262 CrossRef CAS.
- G. L. Parrilha, J. G. Da Silva, L. F. Gouveia, A. K. Gasparoto, R. P. Dias, W. R. Rocha, D. A. Santos, N. L. Speziali and H. Beraldo, Eur. J. Med. Chem., 2011, 46, 1473–1482 CrossRef CAS PubMed.
- K. Alomar, V. Gaumet, M. Allain, G. Bouet and A. Landreau, J. Inorg. Biochem., 2012, 115, 36–43 CrossRef CAS PubMed.
- P. Datta, A. P. Mukhopadhyay, P. Manna, E. R. T. Tiekink, P. C. Sil and C. Sinha, J. Inorg. Biochem., 2011, 105, 577–588 CrossRef CAS PubMed.
- B. S. Creaven, M. Devereux, D. Karcz, A. Kellett, M. McCann, A. Noble and M. Walsh, J. Inorg. Biochem., 2009, 103, 1196–1203 CrossRef CAS PubMed.
- A. Kulkarni, S. A. Patil and P. S. Badami, Eur. J. Med. Chem., 2009, 44, 2904–2912 CrossRef CAS PubMed.
- M. B. Halli, R. B. Sumathi and M. Kinni, Spectrochim. Acta, Part A, 2012, 99, 46–56 CrossRef CAS PubMed.
- H. Huang, P. Zhang, Y. Chen, K. Qiu, C. Jin, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13135–13145 RSC.
- P. Kalaivani, R. Prabhakaran, F. Dallemer and K. Natarajan, RSC Adv., 2014, 4, 51850–51864 RSC.
- P. Kalaivani, R. Prabhakaran, E. Vaishnavi, T. Rueffer, H. Lang, P. Poornima, R. Renganathan, V. Vijaya Padma and K. Natarajan, Inorg. Chem. Front., 2014, 1, 311–324 RSC.
- E. Jayanthi, S. Kalaiselvi, V. Vijaya Padma, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2016, 45, 1693–1707 RSC.
- J. Hildebrandt, H. Gorls, N. Hafner, G. Ferraro, M. Durst, I. B. Runnebaum, W. Weigand and A. Merlino, Dalton Trans., 2016, 45, 12283–12287 RSC.
- W. Su, Q. Qian, P. Li, X. Lei, Q. Xiao, S. Huang, C. Huang and J. Cui, Inorg. Chem., 2013, 52, 12440–12449 CrossRef CAS PubMed.
- A. Garza-Ortiz, P. Uma Maheswari, M. Siegler, A. L. Spek and J. Reedijk, New J. Chem., 2013, 37, 3450–3460 RSC.
- M. J. Chow, C. Licona, D. Y. Q. Wong, G. Pastorin, C. Gaiddon and W. H. Ang, J. Med. Chem., 2014, 57, 6043–6059 CrossRef CAS PubMed.
- S. W. Chang, A. R. Lewis, K. E. Prosser, J. R. Thompson, M. Gladkikh, M. B. Bally, J. J. Warren and C. J. Walsby, Inorg. Chem., 2016, 55, 4850–4863 CrossRef CAS PubMed.
- D. S. Ranade, A. M. Bapat, S. N. Ramteke, B. N. Joshi, P. Roussel, A. Tomas, P. Deschamps and P. P. Kulkarni, Eur. J. Med. Chem., 2016, 121, 803–809 CrossRef CAS PubMed.
- P. Kalaivani, R. Prabhakaran, P. Poornima, F. Dallemer, K. Vijayalakshmi, V. Vijaya Padma and K. Natarajan, Organometallics, 2012, 31, 8323–8332 CrossRef CAS.
- T. Sathiya Kamatchi, P. Kalaivani, P. Poornima, V. Vijaya Padma, F. R. Fronczek and K. Natarajan, RSC Adv., 2014, 4, 2004–2022 RSC.
- K. Jeyalakshmi, J. Haribabu, N. S. P. Bhuvanesh and R. Karvembu, Dalton Trans., 2016, 45, 12518–12531 RSC.
- S. A. Patil, S. N. Unki, A. D. Kulkarni, V. H. Naik and P. S. Badami, Spectrochim. Acta, Part A, 2011, 79, 1128–1133 CrossRef CAS PubMed.
- G. Kalaiarasi, R. Jain, H. Puschman, S. Poorna Chandrika, K. Preethi and R. Prabhakaran, New J. Chem., 2017, 41, 2543–2560 RSC.
- S. Mandal, D. K. Seth and P. Gupta, Inorg. Chim. Acta, 2013, 397, 10–20 CrossRef CAS.
- F. Basuli, S. M. Peng and S. Bhattacharya, Inorg. Chem., 2001, 40, 1126–1133 CrossRef CAS PubMed.
- B. Demoro, C. Sarniguet, R. Sanchez-Delgado, M. Rossi, D. Liebowitz, F. Caruso, C. Olea-Azar, V. Moreno, A. Medeiros, M. A. Comini, L. Otero and D. Gambino, Dalton Trans., 2012, 41, 1534–1543 RSC.
- R. Ramachandran, G. Prakash, S. Selvamurugan, P. Viswanathamurthi, J. G. Malecki and V. Ramkumar, Dalton Trans., 2014, 43, 7889–7902 RSC.
- F. J. Meyeralmes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS.
- R. F. Pasternack, M. Caccam, B. Keogh, T. A. Stephenson, A. P. Williams and E. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS.
- M. Fernández, E. Rodríguez Arce, C. Sarniguet, T. S. Morais, A. I. Tomaz, C. O. Azar, R. Figueroa, J. D. Maya, A. Medeiros, M. Comini, M. Helena Garcia, L. Otero and D. Gambino, J. Inorg. Biochem., 2015, 153, 306–314 CrossRef PubMed.
- K. R. Sangeetha Gowda, B. B. Mathew, C. N. Sudhamani and H. S. Bhojya Naik, Biomed. Biotechnol., 2014, 2, 1–9 Search PubMed.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 2nd edn, 1999 Search PubMed.
- M. R. Eftink and C. A. Ghiron, Biochemistry, 1976, 15, 672–680 CrossRef CAS PubMed.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, US, 3rd edn, 2006 Search PubMed.
- M. Zampakou, M. Akrivou, E. G. Andreadou, C. P. Raptopoulou, V. Psycharis, A. A. Pantazaki and G. Psomas, J. Inorg. Biochem., 2013, 121, 88–99 CrossRef CAS PubMed.
- P. Kalaivani, R. Prabhakaran, F. Dallemer, P. Poornima, E. Vaishnavi, E. Ramachandran, V. Vijaya Padma, R. Renganathan and K. Natarajan, Metallomics, 2012, 4, 101–113 RSC.
- F. Beckford, J. Thessing, J. Woods, J. Didion, N. Gerasimchuk, A. Gonzalez-Sarrias and N. P. Seeram, Metallomics, 2011, 3, 491–502 RSC.
- T. S. Morais, F. C. Santos, L. Corte-Real and M. H. Garcia, J. Inorg. Biochem., 2013, 129, 94–101 CrossRef CAS PubMed.
- T. E. Kydonaki, E. Tsoukas, F. Mendes, A. G. Hatzidimitriou, A. Paulo, L. Papadopoulou, D. Papagiannopoulou and G. Psomas, J. Inorg. Biochem., 2016, 160, 94–105 CrossRef CAS PubMed.
- K. Tsai, T. G. Hsu, K. M. Hsu, H. Cheng, T. Y. Liu, C. F. Hsu and C. W. Kong, Free Radical Biol. Med., 2001, 31, 1465–1472 CrossRef CAS PubMed.
- K. Sampath, S. Sathiyaraj, G. Raja and C. Jayabalakrishnan, J. Mol. Struct., 2013, 1046, 82–91 CrossRef CAS.
-
(a) M. B. Ferrari, S. Capacchi, G. Pelosi, G. Reffo, P. Tarasconi, R. Al-bertini, S. Pinelli and P. L. Helicin, Inorg. Chim. Acta, 1999, 286, 134–141 CrossRef;
(b) L. R. Dinelli, A. A. Batista, K. Wohnrath, M. P. de Araujo, S. L. Queiroz, M. R. Bonfadini, G. Oliva, O. R. Nascimento, P. W. Cyr, K. S. MacFarlane and B. R. James, Inorg. Chem., 1999, 38, 5341–5345 CrossRef CAS;
(c) H. Nikaido, J. Bacteriol., 1996, 178, 5853–5859 CrossRef CAS PubMed.
- B. G. Tweedy, Phytopathology, 1964, 55, 910–914 Search PubMed.
- R. Prabhakaran, A. Geetha, M. Thilagavathi, R. Karvembu, V. Krishnan, H. Bertagnolli and K. Natarajan, J. Inorg. Biochem., 2004, 98, 2131–2140 CrossRef CAS PubMed.
- P. G. Lawrence, P. L. Harold and O. G. Francis, Antibiot. Chemother., 1957, 4, 1980 Search PubMed.
- K. Shanker, R. Rohini, V. Ravinder, P. Muralidhar Reddy and Y. Ho, Spectrochim. Acta, Part A, 2009, 73, 205–211 CrossRef PubMed.
- J. Hildebrandt, H. Görls, N. Häfner, G. Ferraro, M. Dürst, I. B. Runnebaum, W. Weigand and A. Merlino, Dalton Trans., 2016, 45, 12283–12287 RSC.
- S. Richter, S. Singh, D. Draca, A. Kate, A. Kumbhar, A. S. Kumbhar, D. Maksimovic-Ivanic, S. Mijatovic, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2016, 45, 13114–13125 RSC.
- L. Chen, F. Peng, G. Li, X. Jie, K. Cai, C. Cai, Y. Zhong, H. Zeng, W. Li, Z. Zhang and J. Chen, J. Inorg. Biochem., 2016, 156, 64–74 CrossRef CAS PubMed.
- M. Kubanik, W. Kandioller, K. Kim, R. F. Anderson, E. Klapproth, M. A. Jakupec, A. Roller, T. Söhnel, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2016, 45, 13091–13103 RSC.
-
A. I. Vogel, Textbook of Practical Organic Chemistry, Longman, London, 5th edn, 1989, p. 268 Search PubMed.
- A. Pangal, J. A. Shaikh, G. Muiz, V. Mane and K. Ahmed, Int. Res. J. Pharm., 2013, 4, 108–110 CrossRef.
- N. Ahmed, J. J. Levision, S. D. Robinson and M. F. Uttley, Inorg. Synth., 1974, 15, 45–64 Search PubMed.
-
G. M. Sheldrick, SHELXL-97, Program for Structure Refinement, University of Gottingen, Gottingen, Germany, 1997 Search PubMed.
-
G. M. Sheldrick, SHELXL-2014/7: Program for the Solution of Crystal Structures, University of Gottingen, Gottingen, Germany, 2014 Search PubMed.
- S. Sathiyaraj, K. Sampath, R. J. Butcher, R. Pallepogu and C. Jayabalakrishnan, Eur. J. Med. Chem., 2013, 64, 81–89 CrossRef CAS PubMed.
- W. J. Lian, X. T. Wang, C. Z. Xie, H. Tian, X. Q. Song, H. T. Pan, X. Qiao and J. Y. Xu, Dalton Trans., 2016, 45, 9073–9087 RSC.
- M. S. Blios, Nature, 1958, 29, 1199–1200 CrossRef.
- P. Prieto, M. Pineda and M. Aguilar, Anal. Biochem., 1999, 269, 337–341 CrossRef CAS PubMed.
- I. Weigand and K. Hilpert, Nat. Protoc., 2008, 3, 163–175 CrossRef PubMed.
- T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for the ligands H2L2–3 and complex 1–4. CCDC 1565376 (H2L2), 1565377 (H2L3), 1565378 (1), 1565379 (2), 1565380 (3) and 1565381 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj02877f |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a