
A quantitative and temporal map of proteostasis during heat shock in Saccharomyces cerevisiae†
Andrew F.
Jarnuczak
 ab,
Manuel Garcia
Albornoz
a,
Claire E.
Eyers
c,
Christopher M.
Grant
a and
Simon J.
Hubbard
*a
ab,
Manuel Garcia
Albornoz
a,
Claire E.
Eyers
c,
Christopher M.
Grant
a and
Simon J.
Hubbard
*a
aSchool of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester Academic Health Science Centre, Oxford Road, Manchester M13 9PT, UK. E-mail: simon.hubbard@manchester.ac.uk
bEuropean Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, UK
cDepartment of Biochemistry, Centre for Proteome Research, Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, UK
First published on 16th January 2018
Abstract
Temperature fluctuation is a common environmental stress that elicits a molecular response in order to maintain intracellular protein levels. Here, for the first time, we report a comprehensive temporal and quantitative study of the proteome during a 240 minute heat stress, using label-free mass spectrometry. We report temporal expression changes of the hallmark heat stress proteins, including many molecular chaperones, tightly coupled to their protein clients. A notable lag of 30 to 120 minutes was evident between transcriptome and proteome levels for differentially expressed genes. This targeted molecular response buffers the global proteome; fewer than 15% of proteins display significant abundance change. Additionally, a parallel study in a Hsp70 chaperone mutant (ssb1Δ) demonstrated a significantly attenuated response, at odds with the modest phenotypic effects that are observed on growth rate. We cast the global changes in temporal protein expression into protein interaction and functional networks, to afford a unique, time-resolved and quantitative description of the heat shock response in an important model organism.
Introduction
All organisms are potentially exposed to adverse environmental conditions at some point during their development. Much effort has therefore been devoted to elucidating mechanisms of stress response in both unicellular and multicellular eukaryotes.1–4 The budding yeast S. cerevisiae has provided an important model organism contributing to our understanding of environmental stress responses at a molecular level for many decades.5–9 Among different types of stresses, heat stress is generally very well understood.10–12 Caused by sudden temperature shifts of as little as a few degrees, it can have many damaging effects and results in pronounced physiological and metabolic changes.10,11 For example: increased protein unfolding and aggregation, destabilisation of cell wall or membranes, metabolic reprogramming, or cell cycle arrest.10,11 In response, a widespread reorganisation of gene expression has been observed. In cases of general cellular stress (proteotoxic, osmotic, starvation, etc.) it is termed the environmental stress response (ESR)8,9,13 and in the specific case of heat stress, it is simply called the heat shock response (HSR).10,11The key protein players in HSR are transcription factors, Hsf1p and the partially redundant Msn2p/Msn4p pair, in addition to various downstream heat-shock effector proteins which mainly act as molecular chaperones. Hsf1 binds to heat shock elements (HSE) in the promoter region of its target genes and activates their transcription.12,14,15 Hsf1 targets include many chaperones, genes of the protein transport and degradation machinery, cell signalling or transcription.16,17 Msn2/4 on the other hand binds to a stress responsive element (STRE) promoter motif and is also a regulator of multiple stresses.18 As noted, the best known heat-induced proteins are molecular chaperones, initially discovered and characterised as heat shock proteins (Hsps). Chaperones are necessary for protein biogenesis in normal conditions by facilitating and/or assisting de novo folding or protein translocation, and in stress conditions (as well as in normal conditions) they prevent aggregation and assist in the refolding of misfolded proteins.12 Hsp70s (for example, the constitutive Ssa1p/Ssa2p and stress-inducible Ssa3p/Ssa4p) and Hsp90 chaperones (Hsp82p redundant in function with its paralogue Hsc82p) with its cofactors, e.g. Sti1p, are all well documented to be induced in heat stress.10,13 Incidentally, it has been proposed that Hsp70 and Hsp90 chaperones are also involved in heat stress sensing by being titrated away from Hsf1p upon accumulation of misfolded proteins.10 Other proteins are also known to be upregulated in heat stress, including the disaggregase Hsp104p, antiaggregases Hsp26p and Hsp42p, and membrane stabilising Hsp12p.10 In addition to induction of heat shock genes, some gene expression is also transcriptionally repressed. These include growth-related and protein biosynthetic genes like ribosomal components and metabolic enzymes.9
Interestingly the mRNAs encoding two ribosomally-associated Hsp70 family members, Ssb1/2, were also reported to be rapidly reduced in abundance upon shift from 23 °C to 37 °C.19 Ssb1p and Ssb2p are chaperones associated with actively translating ribosomes and, together with Ssz1p and co-chaperone J-domain partner Zuo1p, form the ribosome-associated complex (RAC). Ssb1/2p bind and assist the folding of newly synthesised polypeptide chains, possibly by assisting its transport through the ribosome tunnel or by protecting it from aggregating until other chaperones actively take over the folding.20–22 Ssb1/2p have also been found to be necessary to maintain translational fidelity in yeast.23 Moreover, despite being 99% identical, differential function of Ssb1p and Ssb2p has been proposed in the literature.24 For example, only Ssb1p was able to rescue a phenotype observed in Tim18p mutant cells which are unable to live without mitochondrial DNA. Additionally, in the context of this study, the lack of Ssb1p would be expected to exacerbate the problem of thermal protein misfolding and deleteriously affect translation during heat stress. This presents Ssb1p mutant cells in particular, as an interesting target for deeper investigations of their unique heat stress response.
Evidently, HSR is a complex process that requires an integrative system-wide view in order to be fully understood. However, while HSR has been very well characterised on the global transcriptome level,8,9,25 perhaps surprisingly, no large-scale temporal proteome studies have so far been reported. Importantly, although no HSR proteomics surveys with temporal resolution have been performed, two relevant studies are available: a study by Nagaraj & Kulak et al.,26 and another one by Shui & Xiong et al.27 Nagaraj & Kulak et al.26 used a quadrupole Orbitrap mass spectrometer and a spike-in SILAC approach to quantify protein abundance changes in a wild type laboratory S288C yeast strain grown at 24 °C and transferred to 37 °C for half an hour. They achieved an impressive coverage of just over 3100 proteins and found many heat shock proteins to be up-regulated and ribosome biogenesis proteins down-regulated in response to elevated temperature. Additionally, evidence of regulation of translation through down-regulation of proteins implicated in translation initiation and elongation was highlighted in this study. Interestingly, the proteins found to be induced in heat shock were often highly up-regulated; for example Ssa4p had a log2 fold change of almost 8 and Hsp12p close to 4. In contrast, the down-regulated proteins had much smaller fold changes, often below 2 fold.
In a more recent study, Shui & Xiong et al.27 used iTRAQ and 2D reversed phase fractionation to investigate the proteome response to prolonged heat exposure in two thermotolerant industrial strains: ScY01 and its parental strain ScY. Comparisons with the Nagaraj et al.26 study supported the differentiation of the thermotolerant response (TR) from the heat shock response (HSR). Central carbon metabolism and lipid metabolism were decreased in thermotolerance but increased in heat shock response; for example Cyb2p, Gph1p, Faa1p, Hor2p, Cct1p, Aro10p were up-regulated in HSR but down-regulated in TR.
Those studies however examined only a single time point during HSR. Here we use a label-free quantitative proteomics approach to determine temporal changes in the yeast proteome at different times during heat stress induced by a mild temperature shift, from 30 °C to 37 °C; a first such study. We measured proteome-level expression for the haploid, a wild type (WT) strain and an isogenic strain lacking the Ssb1p chaperone (ssb1Δ). The resulting temporal profiles confirmed many of the HSR hallmark characteristics and were in excellent agreement with the literature. They also revealed additional insights not available from earlier microarray-based efforts or proteomics studies lacking temporal resolution. Of particular interest is that while heat stress response on the mRNA level appears to be almost immediate (minutes) and with large amplitude or distinct temporal patterns, a steady and uniform response (0.5–1 hour) on the protein level is visible, at least during the mild heat stress considered here. Additionally, we analyse the temporal profile of an ssb1Δ mutant in the same heat stress conditions and characterise at the molecular level how the deletion produces a markedly attenuated response compared to the WT cells. We interpret both data sets in conjunction with known chaperone-target protein interactions, to reveal a global picture of the entire temporal heat shock response, which demonstrates a systems-wide view of proteostasis in action.
Experimental
Yeast growth and proteomics sample preparation
The wild-type BY4742 (Matα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) and ssb1 mutant (ssb1::kanMX4) yeast strains used in this study were purchased from the Thermo Fisher Scientific Yeast Knockout (YKO) Collection. Batch cultures were grown in Yeast Nitrogen Base medium (supplemented with 10 g L−1 of glucose, 10 mL L−1 Arg, 20 mL L−1 Leu, 10 mL L−1 Ura, 3 mL L−1 His and 10 mL L−1 Lys) in biological quadruplicates. For the heat stress experiment, WT and ssb1Δ cells were grown at 30 °C in glass conical flasks until they reached mid-exponential growth phase (OD600 ∼ 1.9) and were then moved to a large rotating water bath pre-heated to 37 °C. In order to ensure the temperature equilibration inside the flasks was rapid, the water level in the bath was maintained above the culture level and the bath lid was closed. A 5 mL sample of unstressed cells was removed at time 0 min (T0) and then at 10 min (T10), 30 min (T30), 60 min (T60), 120 min (T120) and 240 min (T240) after transfer to 37 °C. For each time point four biological replicates were collected and cells were counted using a Cellometer cell counter (Cellometer AUTOM10 by Nexcelom. http://www.nexcelom.com). Cell pellets were collected by centrifugation at 4000 rpm, for 10 minutes at 4 °C and stored at −80 °C until use.Cell lysis was performed according to an earlier described protocol28 with minor modifications. Briefly, the cell pellet was re-suspended in ice-cold 50 mM NH4HCO3 with added ROCHE Complete mini protease and phosphatase inhibitor cocktails (Roche Diagnostics Ltd, West Sussex, UK), 1 tablet of each/10 mL of buffer, and washed twice by centrifugation (at 4000 rpm for 5 min at 4 °C) to remove any contaminants. Cell lysis was achieved by automated glass bead-beating using the MINILYS® homogenizer (Precellys, UK), 10× 1 minute cycles at 4 °C with 2 min break between each cycle, when lysates were cooled on ice. The lysate was centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 4 °C for 10 minutes) and protein collected from supernatant. The remaining cell debris was re-suspended in lysis buffer, collected by centrifugation (10 min, 16000 rpm, 4 °C) and combined with the protein supernatant.
000 rpm, 4 °C for 10 minutes) and protein collected from supernatant. The remaining cell debris was re-suspended in lysis buffer, collected by centrifugation (10 min, 16000 rpm, 4 °C) and combined with the protein supernatant.
For protein digestion, a volume equivalent to 25 million cells (∼100–150 μg protein) was diluted to 160 μL with 25 mM ammonium bicarbonate. Proteins were denatured with RapiGest™ detergent (10 μL of 1% (w/v), 80 °C for 10 minutes), reduced using 60 mM dithiothreitol (10 μL, 60 °C, 400 rpm shaking for 10 minutes), cooled on ice and alkylated 180 mM iodoacetamide (10 μL, incubation at room temperature in the dark for 30 minutes). Digestion was performed by addition of sequencing grade porcine trypsin (Promega, UK) in a 1:50 enzyme to protein ratio (10 μL of 0.2 μg μL−1 trypsin), incubation for 4.5 h followed by further trypsin addition (10 μL of 0.2 μg μL−1 trypsin), and overnight incubation at 37 °C. Trifluoroacetic acid (TFA) was added to the reaction mixture (1% (v/v) final concentration) to stop the digestion and hydrolyse RapiGest™ detergent. To aid peptide solubilisation, 2.5 μL acetonitrile:water (2:1) was also added and samples were incubated at 4 °C for at least 2 hours.
Finally, RapiGest™ precipitate and any remaining particulate in the sample was removed by centrifugation (30 min, 13000 rpm at 4 °C). Additionally, two QC pools were prepared containing an equal mix of all WT digests (WT-QC) and another one of all ssb1Δ digests (ssb1Δ-QC).
LC-MS data acquisition
The digests were analysed on a nanoACQUITY UPLC™ system (WATERS) coupled to LTQ Orbitrap Velos (linear trap quadrupole with an Orbitrap mass analyser; Thermo). Peptides equivalent to 306818 cells, and 397727 cells for the QC runs were loaded onto a C-18 pre-column (Symmetry C18 Column, 100 Å pore size, 5 μm particle size, 180 μm × 20 mm, Waters, Manchester, UK) and trapped for 5 minutes at a flow rate of 5 μL min−1 in 99.9% solution A (water, 0.1% (v/v) formic acid). Peptides were then resolved on an analytical C18 column (nanoACQUITY UPLC™ HSS T3 C18 75 μm × 150 mm, 1.7 μm particle size, Waters, Manchester, UK) with a linear gradient of solution B (acetonitrile, 0.1% (v/v) formic acid) from 3% to 35% over 210 minutes at a flow rate of 300 nL min−1, followed by a ramp to 90% solution B, column wash and 15 minute re-equilibration. The total run time was 240 minutes. A 30 min blank injection was also performed between each tryptic sample to wash the columns and minimise carry-over between samples.
The LTQ Orbitrap was operated in data-dependent mode, with the twenty most intense precursor ions (with charge state ≥2) in the survey scan selected for collision induced dissociation (CID) fragmentation. The survey scan was acquired over the range m/z 350–2000 at a resolution of 30000 (m/z 400) in the Orbitrap analyser and the fragmentation was performed in the LTQ ion trap with the normalised collision energy of 35.
Label-free protein quantification
Mass spectrometry data was acquired in two blocks, WT and ssb1Δ samples separately. Acquisition order within each block was randomised and a QC sample was injected at the beginning, intermediately and at the end of each MS run, a total of 5 QC injections. The raw instrument data was then processed using MaxQuant (MQ) software.29 Protein identification was performed with the built-in Andromeda search engine.30 MS/MS spectra were searched against the S. cerevisiae canonical and isoform protein database (UniProt, 6721 entries, accessed April 2015) appended with 124 common laboratory contaminants. The parameters were as follows; digest reagent: trypsin, max missed cleavages: 2, max protein mass: 250 kDa, fixed modifications: cysteine carboxymethylation, variable modifications: protein N-terminal acetylation and methionine oxidation and serine, threonine and tyrosine (STY) phosphorylation. The mass tolerance was set to 6 ppm for MS1 and MS/MS to 0.5 Da. The initial MS1 mass calibration was performed within MQ with a 20 ppm tolerance. The false discovery rate (FDR) for accepted peptide spectrum matches and protein matches was set to 1%.For protein quantification, ‘re-quantify’ and ‘match between runs’ options were selected; match time window was set to 1 min after manual inspection of average chromatographic peak widths in each dataset. Label free quantification was performed with the MaxLFQ algorithm31 within MaxQuant. Minimum ratio count was set to 1 and quantification was based on razor and unique peptides only. Razor peptides are determined based the Occam's razor principle where a minimum set of proteins accounting for all identified peptides is used.32,33 All other MQ parameters were left in default. The raw mass spectrometry data was deposited to the ProteomeXchange Consortium34 with the identifier PXD006262.
Proteomics data handling procedure
The final quantification results from MSstats are available in Supplementary File S1 (ESI†). The original, unprocessed MQ output proteinGroups tables are available in Supplementary File 2 (WT) and 3 (ssb1Δ) (ESI†). The MQ evidence.txt files are uploaded to PRIDE with the id PXD006262.
Gene ontology enrichment analysis was performed with the online application Panther,38,39 directly on the Gene Ontology Consortium webpage (http://pantherdb.org/). The background set consisted of all proteins identified in a given MS experiment and the target set was the significantly changing proteins, or, in case of c-means fuzzy clustering, the proteins within each cluster and cluster membership >0.5. Only terms with a q-value below 0.05 were considered.
From the list of differentially abundant proteins, all molecular chaperones were linked to their attendant protein clients based on the filtered protein interaction dataset used previously.40,41 Based on this information, a bipartite network was created linking proteins by their chaperone-target interactions and a protein–protein interaction network analysis was created using Cytoscape42 with an organic layout. Additionally, all the differentially abundant proteins were linked to their Slim GO biological process terms and a bipartite network was created linking proteins by their shared GO terms from which a PPI network analysis was done as before.
Results
Generation of MS-based temporal protein profiles
Our experimental strategy followed the S. cerevisiae protein level response to heat stress induced by shift from 30 °C to 37 °C, the classic ‘heat shock’.10 This temperature results in a robust response but is low enough for cells to sustain growth and is physiologically relevant. As expected based on previous literature,43 the two strains under investigation; WT (wild type, BY4742, Mat ALPHA) and chaperone mutant ssb1Δ (BY4742, Mat ALPHA), had very similar growth rates in glucose-rich, batch cultures at normal temperature 30 °C, with estimated doubling times of 2.8 h and 2.9 h, respectively (Fig. S1, ESI†). Upon heat stress, at 37 °C, the growth rate was slowed in both strains, with estimated doubling times during the first 240 minutes increased to 3.8 h in WT and 3.6 h in ssb1Δ. The cultures were sampled in quadruplicate at 10 min (T10), 30 min (T30), 60 min (T60), 120 min (T120) and 240 min (T240) after transfer to 37 °C. The control (mid-exponential cells, 30 °C) was obtained just before the shift, at time T0 (Fig. 1A). After cell lysis, total protein extracts were digested with trypsin and mass spectrometry data was obtained in 48 single-shot LC-MS whole proteome analyses (Fig. 1A), using a randomised block design.44 Relative protein quantification was achieved using a label-free approach using MaxQuant,29,31 achieving excellent quantification repeatability in both the WT and ssb1Δ blocks of analysis (Fig. S2, ESI†). Altogether, we quantified 2141 proteins, at 1% PSM and protein-level FDR from all WT and ssb1Δ runs, representing around 50% of the expressed yeast proteome.45 After robust statistical analysis (see Material and methods), 1780 proteins in WT and 1970 in ssb1Δ were consistently quantified in each of the time points with 1609 proteins overlapping between the two datasets (Fig. 1B, Venn diagram). This provides good protein coverage and isoform resolution; ∼80% proteins in both strains were identified with 3 or more unique peptides across the whole dataset, with an average of 6 unique peptides per protein (boxplots, Fig. 1B). This supported the resolution and quantification of potentially interesting protein isoforms/paralogues. For example, all of the SSA subfamily members of the ubiquitously expressed HSP70 heat shock protein family could be quantified in the ssb1Δ dataset (3 out of 4; Ssa1p, Ssa2p and Ssa4p, could be quantified in the WT). However, because of high sequence similarity (99%) between Ssb isoforms, the two proteins Ssb1p and Ssb2p can not be reliably distinguished by any untargeted MS-based proteomic technique.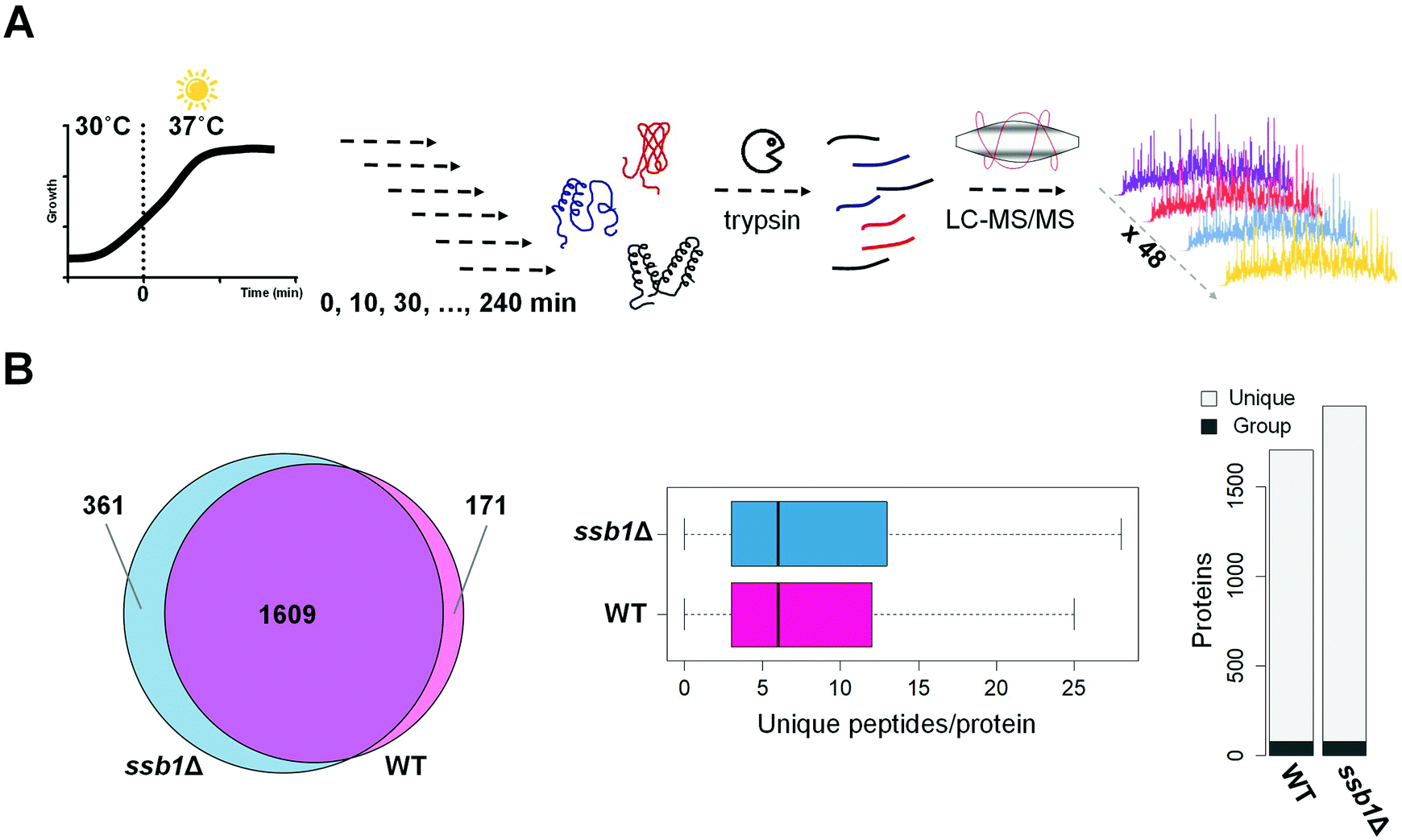 | ||
| Fig. 1 Overview of proteomics strategy and label free quantification results. (A) Yeast cultures (WT and ssb1Δ) were grown at 30 °C to mid exponential phase before transfer to 37 °C, and subsequent sampling at 0, 10, 30, 60, 120 and 240 minutes; the entire experiment was performed in four biological replicates. Cell extracts for each strain were prepared and protein digests analysed by LC-MS/MS using an LTQ-Orbitrap Velos. Mass spectrometry data was acquired for 24 WT and 24 ssb1Δ samples in two separate experimental blocks. (B) Left panel: Venn diagram shows the overlap of the number of quantified proteins in WT and ssb1Δ strains. Middle panel: Two boxplots display the number of unique peptides by which each protein was identified. Right panel: Barplots indicate the number of unique protein versus protein group identifications in the two datasets. | ||
Temporal profiles of relative protein abundance changes upon heat stress were calculated from the protein ratios at each time point with respect to time 0, represented by a heatmap for each strain shown side-by-side in Fig. 2. Each cell represents the model-based estimate of fold change from MSstats at a given time (expressed as log2 of the ratio) after transfer to 37 °C. The protein profiles for the WT and mutant cells were additionally clustered row-wise using Euclidean distance. This overview shows the global proteomic response to heat stress in the WT and mutant cells, with many proteins increasing (red) and decreasing (blue) in relative abundance post-stress; however, most show a change of less than 1.5 fold (corresponding to log2 = 0.58). Notably, many proteins display co-ordinated but significantly different behaviour in the mutant cells compared to wild type. For example, clusters 4 and 5 highlight a number of proteins that show increasing abundance post-stress in the wild type cells, but are depleted in the mutant. These highlight several biological processes that the mutant cells is no longer able to sustain in the absence of one of the RAC Hsp70s, including much of nucleotide and amino acid biosynthesis and metabolism.
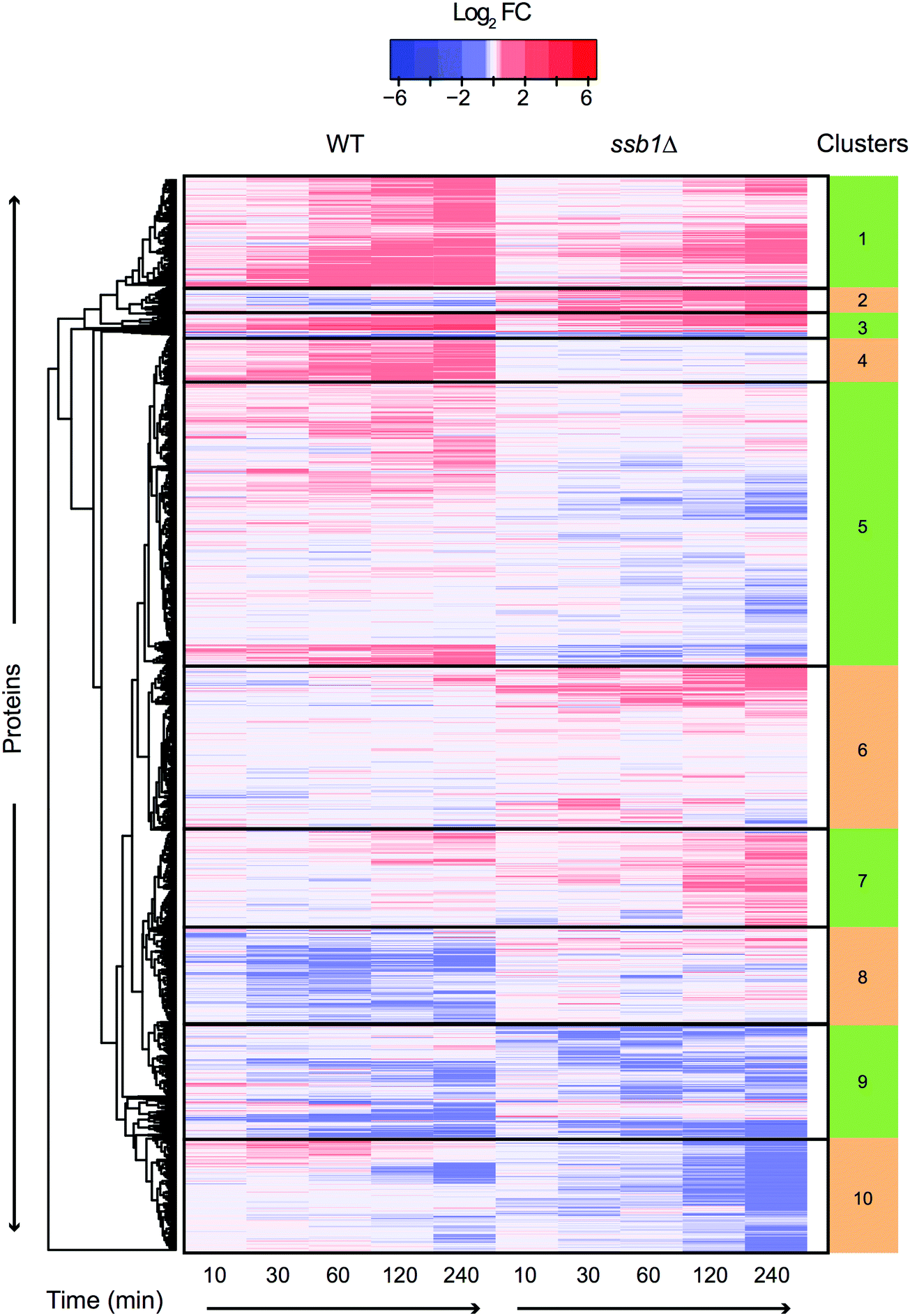 | ||
| Fig. 2 Heatmap and functional enrichment of temporal protein profiles during HSR. Heat map visualisation of the entire quantitative proteomic profiles under the heat shock response in WT and ssb1Δ. At each time point (column) log2 ratio to time 0 is plotted. Each row represents a protein and rows were clustered using Euclidean distance using all timepoints, so that similarities in gene profiles between the two strains can be observed. This is evident from the coherent clusters which display common Gene Ontology functional enrichments (see Supplementary File S4, ESI†) based on GO slim. Clusters display both common (e.g. cluster 1) and opposed (e.g. cluster 5) expression patterns between WT and ssb1Δ strains. | ||
Other functional enrichments are associated with the clusters, including the archetypal HSR such as clusters 1 and 3 which include up regulated proteins involved in protein folding and response to heat, and clusters 9 and 10 which are enriched for down regulated proteins involved in ribosomal processing and protein synthesis.
Identification of significantly changing protein profiles
Significant protein fold changes across the time points were determined using the MSstats package, adjusting p-values via the Benjamini & Hochberg false discovery rate method46 and also applying an empirical cutoff. Subsequently, 267 (WT) and 313 (ssb1Δ) proteins were determined to be significantly differentially abundant (see Methods) in at least one time point during the 240 min time course, corresponding to ∼15% of all quantified proteins. Only 78 proteins were found to be changing in both strains, a surprisingly small overlap. However, those common changes were highly correlated between the two strains in later time points. In particular, T0/T60 ratio in WT versus T0/T60 ratio in ssb1Δ correlated; Spearman = 0.67, p-value < 2.2 × 10−16 and T0/T120; Spearman = 0.74, p-value < 2.2 × 10−16. This suggests a confined ‘core’ response that is essentially unaffected by the deletion, supplementing a much greater strain-specific response likely to be linked to the molecular function of the Ssb1 protein, as discussed later. Next, we focused our analysis on the WT heat shock response.Hallmark proteins of heat stress response (HSR)
The power of the proteomics approach becomes evident from its ability to pinpoint proteins with well-known roles in heat stress. Among the proteins with most statistically significantly increases in relative abundance in the WT were the small heat shock proteins with chaperone activity, Hsp26p (adjusted p-value = 1.0 × 10−16), the membrane associated Hsp12p (adjusted p-value = 3.6 × 10−8) (Fig. 3A) as well as Hsp30p, all previously documented to be induced by heat stress.47–49 Interestingly, Hsp30p had no detectable peptides and therefore no reported intensity in the T0 time point, but was quantified with an average of 2 unique peptides in all other samples (Fig. S4, ESI†). We were therefore unable to calculate a fold change for Hsp30p, but based on the distribution of valid fold changes in the experiment we estimate it increases in abundance by at least 3-fold in response to heat.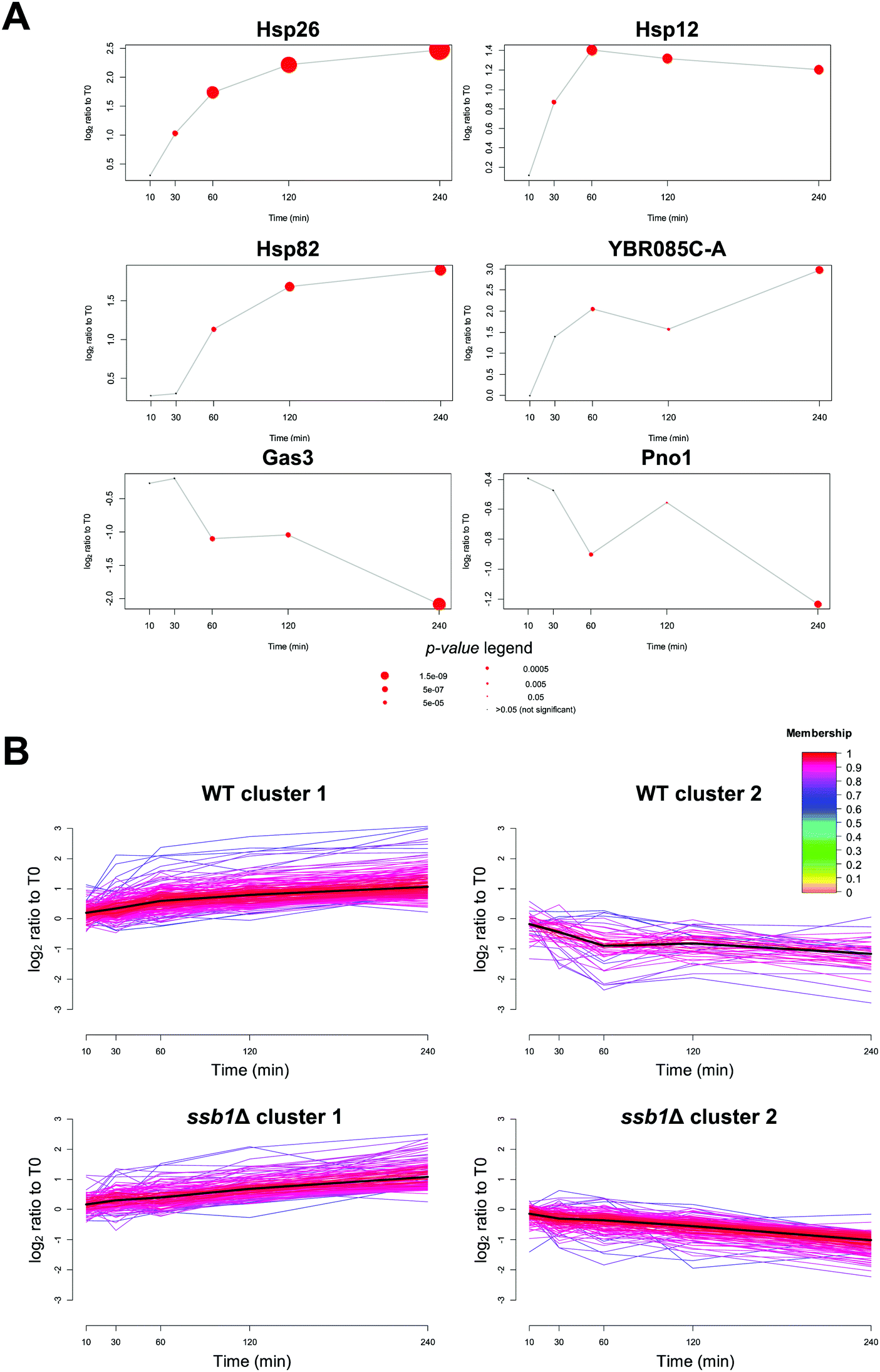 | ||
| Fig. 3 Temporal protein profiles after heat shock stress in WT and ssb1Δ cells. (A) Example protein profiles for well-established heat stress responders with most significant up-regulation (Hsp26, Hsp12, Hsp82), as well as YBR085C-A, the highest induced uncharacterized protein. Gas3 and Pno1p are examples of the most significantly repressed proteins. Red circles signify statistically significant expression change at any given time point, and the size of the circle represents significance (bigger is more significant). (B) Unsupervised clustering of temporal protein profiles. Two dominant clusters were obtained using fuzzy c-means algorithm for both WT and ssb1Δ cells, displaying proteins with membership greater than 0.7 (210 and 48 proteins in clusters 1 and 2 in WT, and 140 and 168 in the ssb1Δ mutant strain). Notably, more proteins display the cluster 2 “decreasing” abundance profile in the mutant strain. | ||
We also quantified both Hsp90 chaperone paralogs, Hsp82p and Hsc82p. Hsc82p was moderately (>2-fold, adjusted p-value 2.4 × 10−5) induced at T60, 120 and 240. Comparatively, the Hsp82p isoform increased in abundance by over 3-fold two hours after stress (adjusted p-value 5.5 × 10−9) (Fig. 3A). This data is in excellent agreement with the transcriptome literature; HSP82 is strongly transcriptionally induced via the Hsf1p factor, while HSC82 is expressed constitutively and also stimulated by Hsf1p, but increases only marginally upon heat stress.50,51
Amongst the 1773 quantified proteins in the WT cells (Supplementary File S1, ESI†), many that displayed significant change and have not previously been associated with the HSR. Most notably, an uncharacterised protein YBR085C-A was induced up to eight-fold over the time course (adjusted p-value 1.4 × 10−6) (Fig. 3A). YBR085C-A has been shown to increase in response to DNA damage in a high-throughput study but has not previously been associated with heat stress response.52 A PSI-BLAST search53 of YBR085C-A against the NCBI non-redundant database found no hits with annotated function. However, a literature search of known DNA-binding regulators revealed a number of transcription factors associated with the promoter region of this gene and thus likely regulating its expression. In particular, a study by Harbison et al.54 provided direct evidence of two heat shock transcription factors, Hsf1p and Msn2p, occupying the DNA regulatory sites of YBR085C-A. The other transcription factors with experimental evidence of YBR085C-A DNA binding are displayed in Table S1 (ESI†). More generally, we also note that up-regulated proteins at 240 minutes are significantly enriched for targets of several stress response transcription factors55 including Msn2p, Msn4p, Hsf1p (adjusted p-value < 1 × 10−12) in all cases.
In contrast to widespread upregulation in the WT strain (211 out of the 267 differentially abundant proteins), only 56 proteins exhibited a significant decrease in relative abundance. For example, the most statistically significantly down-regulated proteins were Mup1p (adjusted p-value = 1.1 × 10−5), Gas3p (adjusted p-value = 3.5 × 10−10) and Pno1p (adjusted p-value = 3.1 × 10−6) (Fig. 3A). The general trends in protein regulation observed here were also recapitulated in other large-scale proteome studies; from the Mann group26 (30 min incubation at 37 °C), and a study by Tyagi and Pedrioli56 (minimum 4 doublings at 37 °C). When considering the proteins reported as significantly changing in at least one study, we observed positive, albeit weak pairwise correlation between the relevant time points (inter-study Spearman correlations 0.44–0.62, see Fig. S5, ESI†). This is typical of inter-lab correlations found in methodologically different quantitative proteomic studies.57 Full protein lists identified in this study are available in Files S2 and S3 (ESI†) for the WT and the ssb1Δ samples.
Slow and sustained proteome remodelling in response to moderate heat stress
A distinguishing feature of our experimental strategy compared to previously reported proteome studies is the temporal resolution of the dataset. The 10 and 30 minute time points represent the early HSR, and 60, 120 and 240 minutes the mid to late response, whereas other studies have considered longer times designed to reflect thermo-tolerance adaptations (e.g. 16 h growth at 40 °C by Shui & Xiong et al.27). We sought to capitalise on this temporal resolution and identify groups of proteins with different profiles across T0, T10, T30, T60, T120 and T240 time points by performing c-means fuzzy clustering.36,58Clustering analysis of both strains revealed only two major temporal profiles (Fig. 3B); proteins that were steadily increasing in abundance over time (cluster 1), and those that were slowly decreasing (cluster 2). No dramatic shifts or transitory regulation in protein expression was evident. This is in contrast to observations involving regulation of phosphorylation in response to heat, where up to 15 clusters showing monotonous and adaptation-like profiles could be identified.59 A GO-term enrichment analysis revealed relatively few significant terms within WT cluster 1 (Bonferroni-adjusted p-value < 0.05) but included expected terms such as protein refolding (p-value = 2.2 × 10−3, enrichment = 6.2), oligosaccharide metabolic processes (p-value = 4.4 × 10−2, enrichment = 5.3) and cellular response to heat (p-value = 2.2 × 10−3, enrichment = 4.4). In contrast, cluster 2 proteins displaying decreasing abundance (Fig. 3B), had no significantly enriched GO-process terms, consistent with non-specific degradation throughout the heat stress response. Full GO enrichment data are provided in Supplementary File S4 (ESI†).
A notable example of coordinated regulation in the heat stress response involved proteins annotated with carbohydrate metabolism GO terms involved in the trehalose-synthesis pathway. Trehalose is a storage disaccharide produced in yeast in a relatively simple metabolic pathway (Fig. 4). It is a general stress protectant and contributes to the suppression of denatured protein aggregation. Following heat stress, the trehalose-synthesis protein (TPS) complex constituents (TPS1p, TPS2p, TPS3p, and TSL1p) were all significantly up-regulated; roughly 2-fold after 120 minutes of heat, with the exception of TPS3p, a regulatory subunit of TPS, which showed much smaller changes (Fig. 4). Similarly, the most abundant and highest affinity hexose transporter Hxt7p is up-regulated in preference to the others. These examples highlight the ability of proteomics to track the temporal molecular response in a coordinated fashion at the protein level across this pathway. We next turned our focus to the differences observed in the mutant strain.
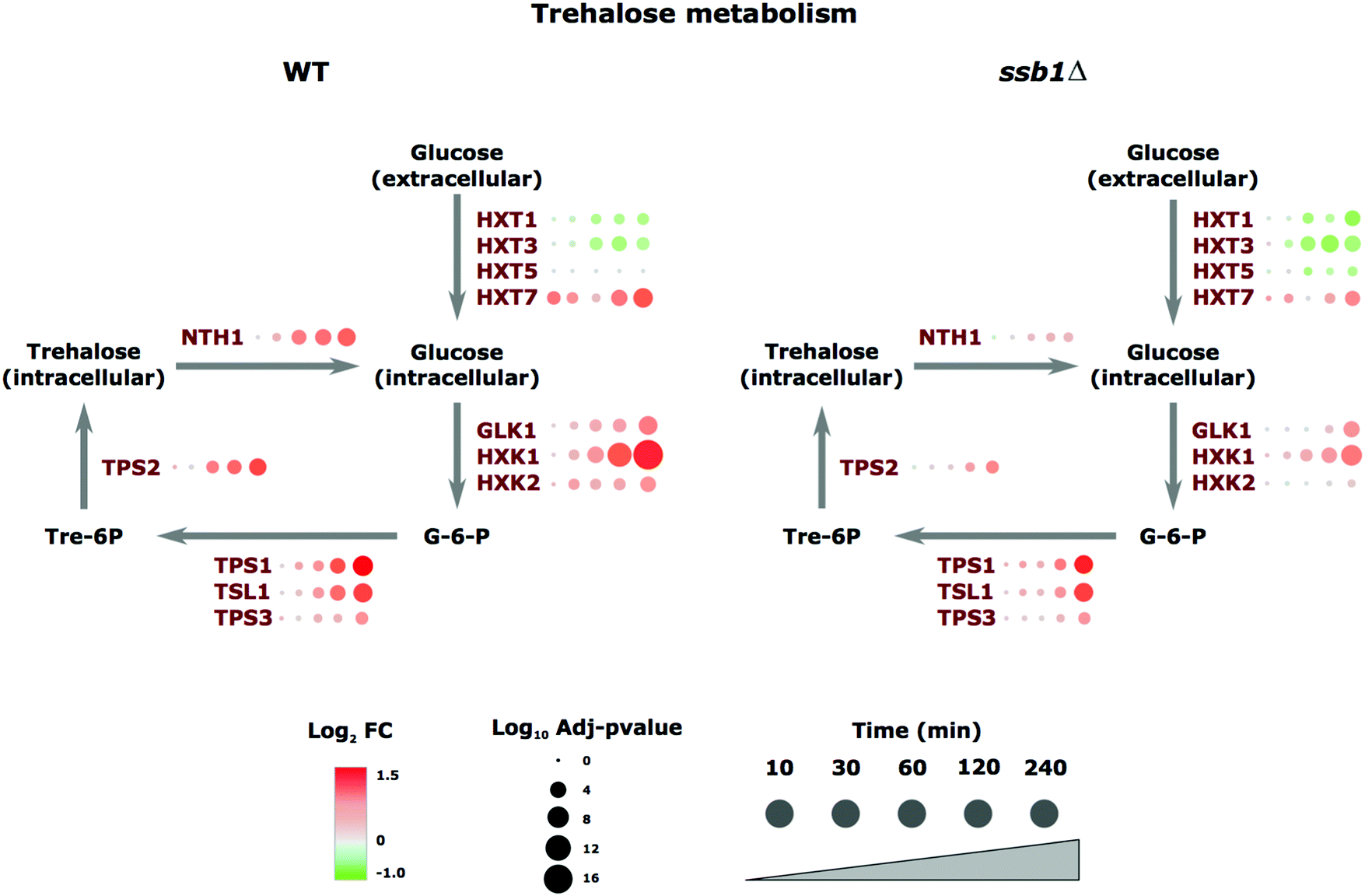 | ||
| Fig. 4 Heatshock induced proteome changes in the trehalose biosynthetic pathway. A bubble-plot overlaid on the trehalose pathway displaying relative protein abundance changes under heat stress of key enzymes. Red indicates abundance increases, green a decrease, with circle size corresponding to statistical significance of the changes. The increases are typically over 2-fold over unstressed cells after 240 minutes in WT cells, but this is attenuated in the ssb1Δ mutant. | ||
ssb1Δ HSR is different to the wild type response
In the case of the ssb1Δ mutant strain that lacks this key member of the HSP70 family we observed notable differences in the membership of the clusters shown in Fig. 3 and their associated gene ontology enrichments. Whilst clusters 1 and 2 retained the same overall trend in the two strains, a closer look at the dynamics of regulation and GO term analysis of the clusters reveals differences in their HSR response (Supplementary File S4, ESI†). Whilst ssb1Δ cluster 1 retains proteins with functional enrichments in GO categories ‘protein refolding’, ‘protein folding’, and ‘response to abiotic stimulus’, the corresponding increases in relative protein abundance were generally not observed until later time points (T60 at earliest) and showed relatively small increases, between roughly 1.4 and 2-fold change.GO term enrichment analysis of cluster 2 (down-regulated) showed a very different picture in the mutant; 168 proteins were present in this cluster (compared to 48 in WT) with significant enrichment for cytoplasmic translation (p-value = 1.8 × 10−16, enrichment = 4.4), peptide biosynthetic process (p-value = 6 × 10−10, enrichment = 2.8) and ribosome biogenesis/assembly (p-value = 9.4 × 10−8/1.1 × 10−2, enrichment = 2.6/3.7). This shows a more pronounced down-regulation of protein synthesis under stress. A direct comparison of the fold changes between WT and ssb1Δ at each time point highlights the differences in the global response as well as in individual proteins in each strain (Fig. S6, ESI†). This data confirms the trends captured by the clustering, we observed a general lack of response in the mutant strain in the early time points (T0–T60), and an inability to up-regulate key proteins at the later time points (T120–T240). These molecular details are informative when contrasting with the phenotypic response observed in the absence of heat shock,60 where deletion leads to only a modest decrease in fitness, consistent with our previous study.61 Here, we can see how loss of a key member of the RAC leads to loss of ribosome and proteins involved in translation in general after stress in comparison to the WT response.
Proteome, unlike transcriptome is not affected early in HSR
The vast majority of the proteome did not display statistically significant changes in the early time points (T10 and T30). Even proteins with large differential regulation later in the experiment remained mostly unchanged T10–T30. This is in stark contrast to transcriptome-driven gene expression studies such as the seminal microarray study by Gasch et al.,9 that reported large mRNA changes within minutes of introduction of heat stress, mirrored in more recent studies by Strassburg et al.62 There the authors measured mRNA changes in yeast cells exposed to heat stress in a time course experiment almost identical to ours. We decided to use this closely matched dataset to investigate the correlation between mRNA expression and protein abundance changes. To take full advantage of the time-resolved data, we performed global correlation analysis where a correlation coefficient is calculated based on all measurements at each time point. For the first analysis, we selected only the protein-transcript pairs for which a significant protein change was measured (resulting in 248 pairs).Despite a strong correlation within experimental sub-types (transcriptome or proteome) and between time points, with R between 0.7–0.9, we observed a poor global correlation between the transcriptome and proteome response (R < 0.3) during the early heat shock (Fig. 5A). However, correlation increased globally over time, with the highest correlations (R > 0.45) being observed between the early transcript (15 to 60 min) and late protein changes (120 to 240 min), (Fig. 5A). These findings are consistent with the immediate molecular response to elevated temperature, by regulating mRNA transcription via Hsf1p (but also potentially through post-translational modifications63 or modulating enzyme activities64). The attendant changes in protein level take longer to materialise due to the time it takes to synthesise the responding target proteins. This lag is further supported by the observation that expression of the majority of transcripts peaks between 30 and 60 minutes, while most protein expression peaks 240 minutes after stress (Fig. 5B). As visible in Fig. 5B, the average delay in proteome response is estimated to be between 1 and 3 hours. However, the lack of fine-grained resolution makes it only a rough estimate. Furthermore, although the imperfect correlations between the transcriptome and proteome can most obviously be attributed to post-translational regulation, at least some of the variance is likely caused by measurement noise and technical differences between the two datasets.
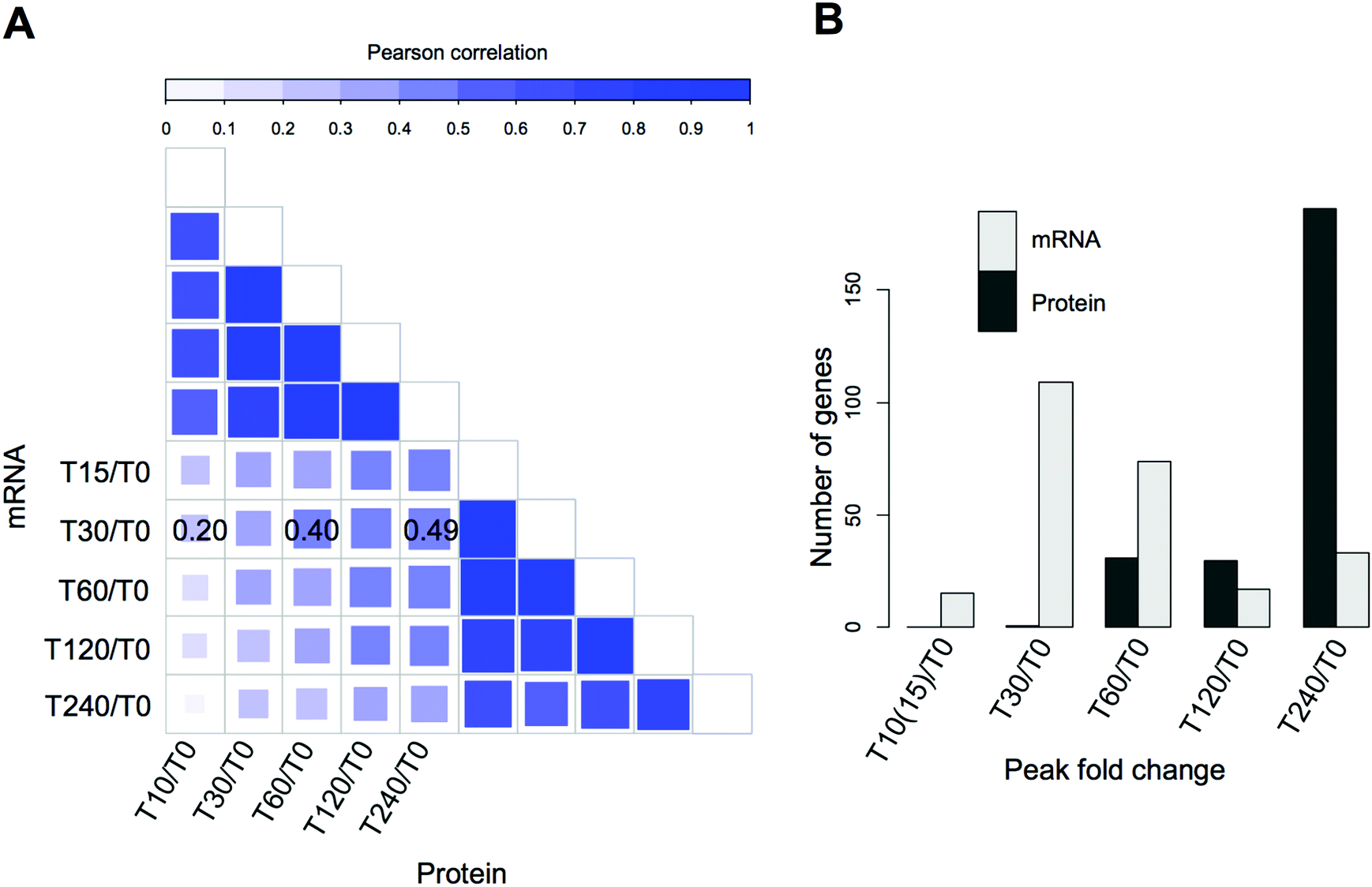 | ||
| Fig. 5 Integration of proteome and transcriptome changes in the heat stress response of the wild type strain. (A) Correlations between yeast transcriptome data taken from Strassburg et al.,25 compared to our quantitative proteomic profiles, comparing relative expression changes at individual time points. The upper and right-hand quadrants compare intra-technique expression profiles showing high correlations between transcriptomes, and between proteomes, and lower correlations when transcriptomes and proteomes are compared. The highest correlations between transcriptome and proteome are indicated on the plot; a time lag is also evident between early and late time points. (B) Histograms showing the number of differentially expressed transcripts and proteins whose expression peaks at each given time point with respect to unstressed cells at time zero. Protein expression is still rising in differentially expressed genes at 240 minutes, highlight the lag in the proteome with respect to the transcriptome. | ||
Global chaperone responses to the proteome under heat shock
As chaperones are the principal molecular responders to heat shock, we examined their relative protein abundance changes across the time course (Fig. 6), comparing the response in both the WT and mutant strain. This provides a unique picture of the global response for 60 quantified chaperones, in the context of their assigned classes following Gong et al.41 Here, the added value of the temporal experiment is apparent, as consistent step-changes in relative abundance are obvious over time, rather than a simple “before/after” two time-point experiment that could be more prone to false positives. Clear patterns are apparent, highlighting how selected members of the Small chaperone family (Hsp12p, Hsp26p), Hsp60 and Hsp90s (Hsc82p, Hsp82p) all increase over the 240 minutes post-exposure. In parallel, selected Hsp70s increase in relative abundance to help maintain proteostasis. Notably, only a restricted set of chaperones are significantly increased in response to heat shock and indeed a few, as has been described before, decrease; Erj5p being the most obvious. This is consistent with its function as an endoplasmic reticulum-based transmembrane protein, where decrease or loss of function leads to a constitutive increase in the unfolded protein response.65 A modest decrease in Ssb2p and also Ssb1p is also observed over 240 min, genes that are also known to be down-regulated under heat stress.11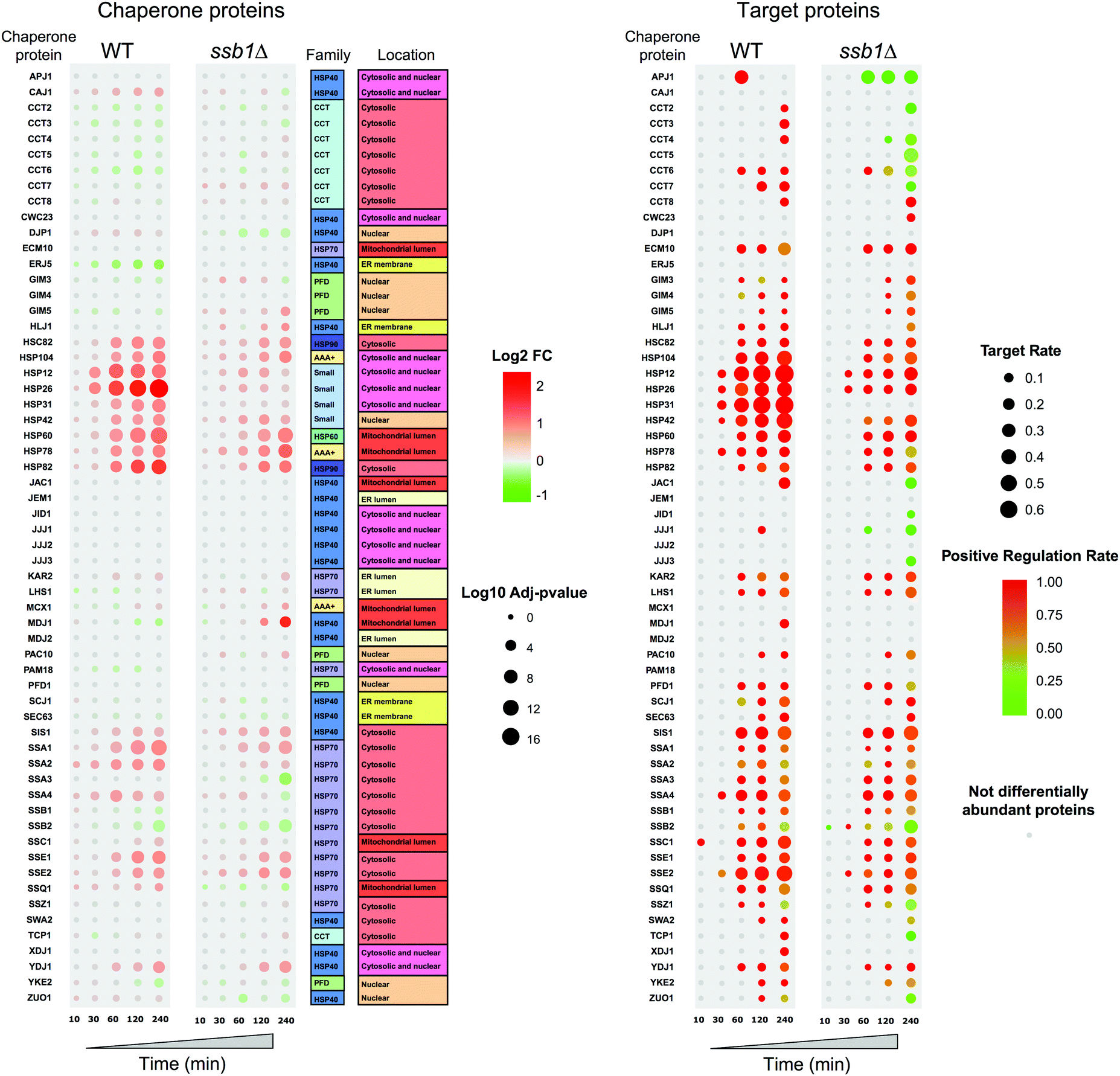 | ||
| Fig. 6 Global proteome changes in the yeast chaperome under heat stress. Left: The bubble-plot shows protein abundance for all chaperones, in both wild type and mutant cells, across the 240 minute time course. Red circles indicate increasing protein abundance changes according to the key, with green indicating decrease. The full set of 60 chaperones detected in the label-free mass spectrometry data are shown, indicating a localised response from a limited set of chaperones, chiefly in the small class. Right: The panel shows the corresponding changes in the annotated client proteins, showing the relative fraction of chaperone-specific targets detected in the differentially abundant protein set at that time point (adjusted p < 0.05), i.e. the “Target Rate”. Circles are coloured by the average abundance change (red increasing, green decreasing). Protein substrates follow the trends of their parent chaperones. The changes in both the chaperones and their targets are attenuated for the mutant strain. | ||
The global concerted heat shock response in wild type cells is not matched in the ssb1Δ mutant. Here, the global effects of Ssb1p absence are observed, leading to a marked attenuation of the main Hsp12p and Hsp26p increase and a generally weaker response across all the related heat shock factors. It is apparent that the loss of this key RAC associated Hsp70 prevents the cell from eliciting a full HSR, and it has knock-on effects on several areas of the chaperone network. For example, Mdj1 increases by 240 min; Mdj1 is a mitochondrial Hsp40 that is highly connected in the chaperone network and involved in many key functions including folding of nascent peptides and degradation of misfolded proteins66,67 – which are presumed to accrue at higher rates in the absence of Ssb1p under heat stress. Ssa3p also shows a marked decrease at 240 min and the Erj5p effect is lost.
In parallel to the chaperones, we also examined attendant changes in their target proteomes inferred from cognate interactions mapped by affinity-purification studies,41 shown on the right in Fig. 6. A key feature here is the high correspondence between chaperone abundance changes and their mapped target proteins, which display correlated changes during the heat stress. For each chaperone, we display the fraction of their target proteins present in the differentially abundant proteins (target rate, represented by circle size in Fig. 6), coloured by whether they largely increase (red) or decrease (green) in relative abundance. The target rate reaches as high as 60% in some cases. We would not expect to see 100% for several reasons. Firstly, chaperones are expected to attempt to maintain proteostasis (i.e. maintain protein substrate levels) rather than increase them. Therefore it is important to note we do not see a concerted decrease in relative abundance for those proteins that are targets of the chaperones that increase in abundance. Interestingly, chaperones also function by targeting misfolded clients for degradation pathways, a route to a nominal decrease in abundance. However, we do not observe this phenomenon directly in the quantitative proteomic data. Finally, shotgun proteomics remains a stochastic technique that under-samples the proteome, resulting in some undetected proteins from each chaperone target set. We also note that the data offers strong support for the validity of the protein interactome data, with a direct demonstration of how the specific targets of given chaperones are indeed protected from down-regulation under a heat shock stress.
Fig. 6 also shows how the targets of the chaperones in the mutant yeast strain are affected, with a matched attenuation of the response to heat. Indeed, in several instances the average level of some chaperone targets is notably reduced, such as those for Apj1p, Ssb2p and most of the CCT complex. This highlights the widespread effects on the cellular proteome of deleting a key member of the RAC complex. By 240 minutes, targets of all three members of the remaining RAC proteins (Ssb2p, Zuo1p and Ssz1p) are reduced in abundance, highlighting the ‘sick’ nature of the cells.
The nature of the global response can also be visualised by overlaying this quantitative data on the chaperone–substrate protein interaction network of differentially abundant proteins, as shown in Fig. 7. During the HSR, the chaperones and many of their attendant targets increase in the WT cells. For example, after 10 minutes, an increase in the highest-affinity glucose transporter Hxt7p is noted, followed by “small” group chaperones after 30 minutes; by 60 minutes, a clear grouping of Hsp12p, Hsp26p, Hsp26p and Hsp104p has formed, with attendant interconnected targets along with Hsp60p, both HSP90s and selected cytosolic HSP70s. Targets of these chaperones including trehalose biosynthetic enzymes Tps1p and Tsp2p are evident by 120 minutes along with the chaperones Sse2p and a growing interlinked network of targets by 240 minutes. This pattern is essentially replicated by the mutant strain but in a much-attenuated form (see the lower half of Fig. 7); an increase in Hsp26p levels is not detected at all and indeed by 240 minutes two key HSP70s (Ssa3p and the other RAC-associate Ssb2p) are down-regulated. This down-regulation of Ssb2p affects many of its protein clients including numerous ribosomal proteins.
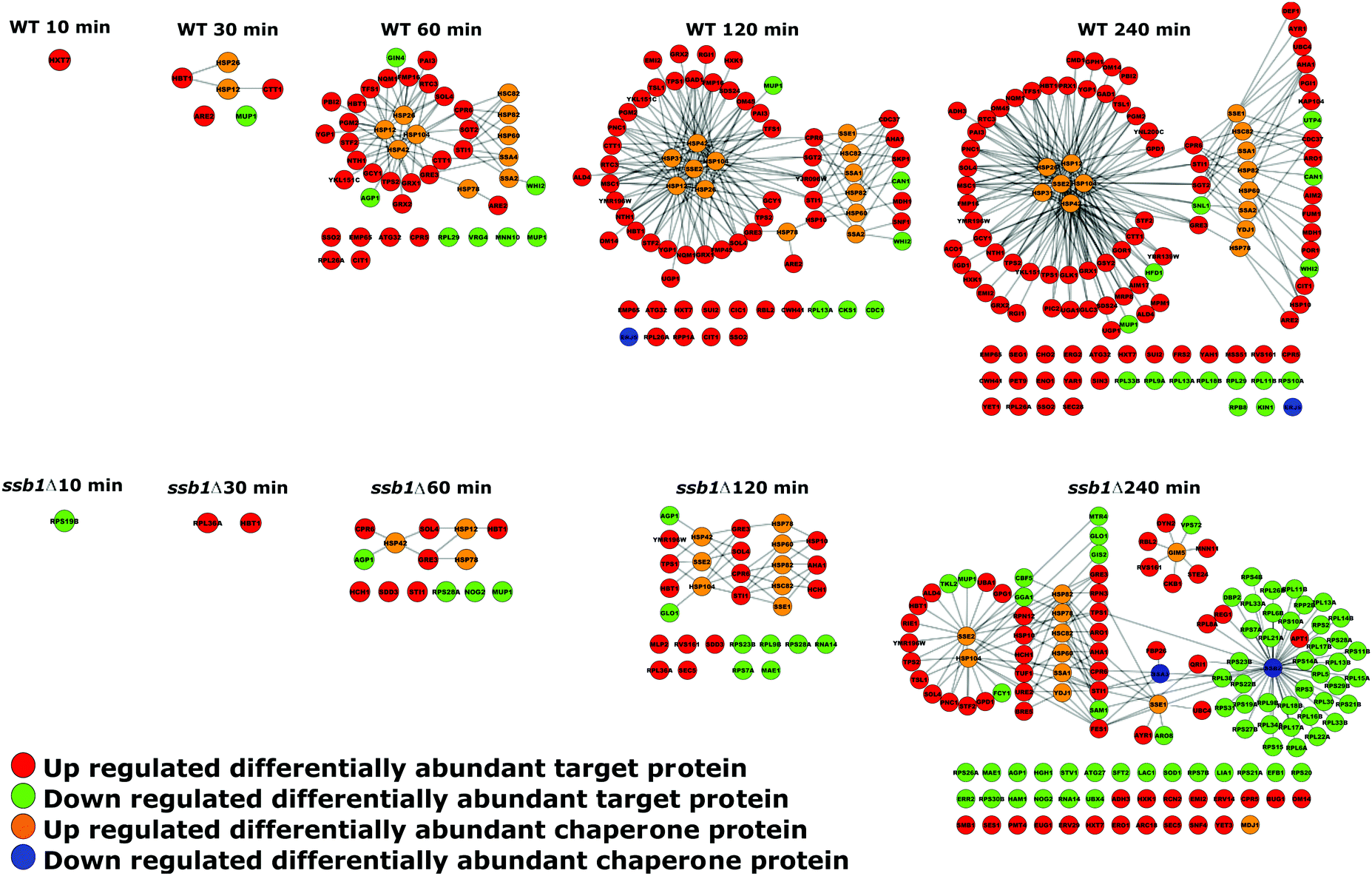 | ||
| Fig. 7 Temporal changes to the yeast chaperone-linked protein interaction network under heat shock. Proteins displaying differential abundance are shown as nodes in the graph, linked to cognate protein substrates via edges. Only chaperones or chaperone targets displaying significant abundance changes at each time point are shown, with WT cells in the upper panel and ssb1Δ mutant in the lower panel. | ||
The quantitative changes observed are also reflected in coherent common functional responses (Fig. 8, originally noted in Fig. 2). Here, we observe global changes to the entire functional network under heat stress, where the links between protein nodes are coloured by common Gene Ontology (biological process) functional categories. Several features are evident here over time, with expansion of the common protein clusters linked to protein folding, response to heat and carbohydrate metabolic processes. Again, the mutant strain exhibits an altered attenuated response pattern particularly in the earlier time points with fewer proteins up-regulated. In contrast, by 240 min a substantial number of proteins are reduced in abundance (green circles), most notably linked to cytoplasmic translation. This group is constituted by the majority of ribosomal proteins, which have depressed abundance levels compared to the cells at T0.
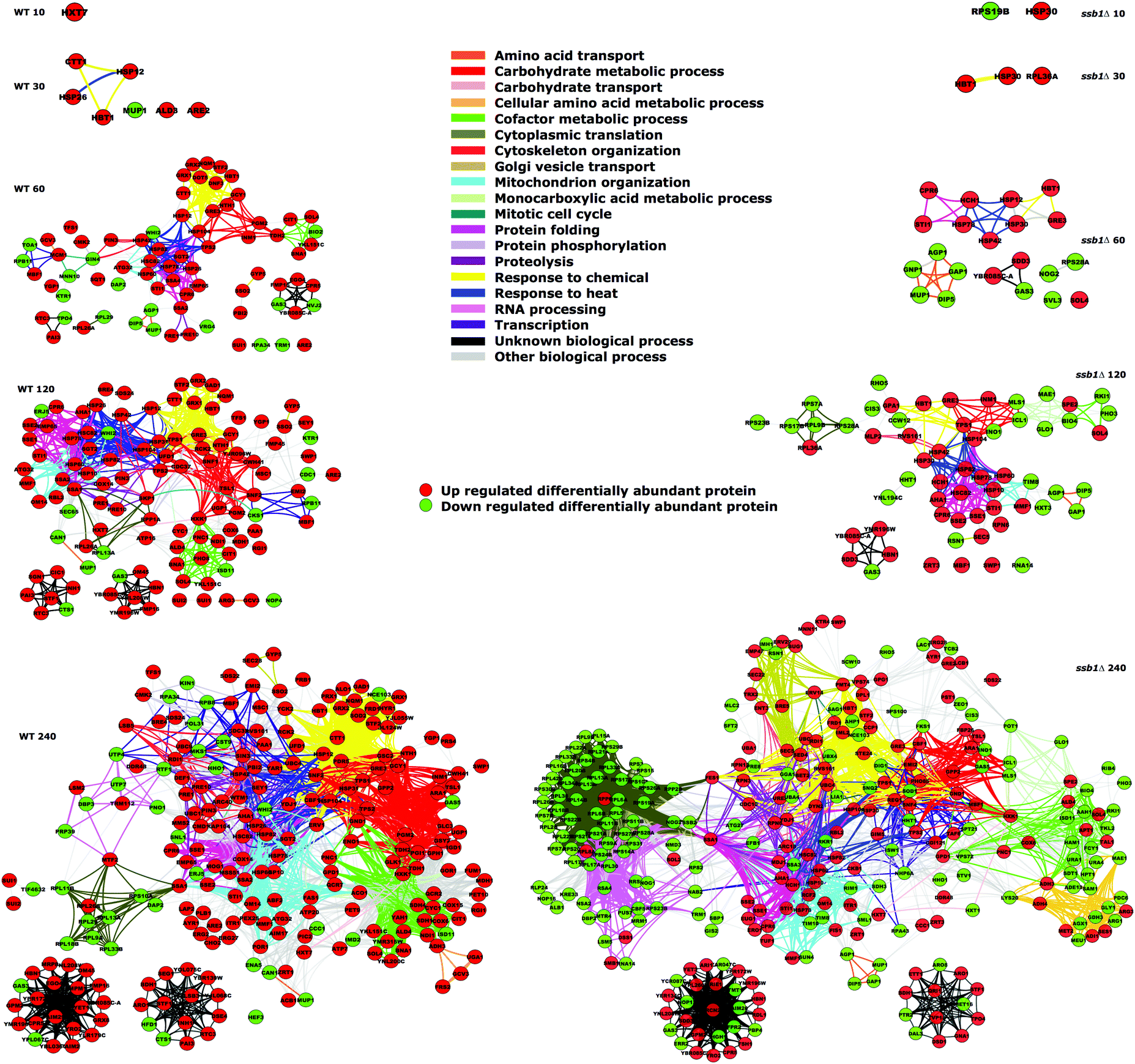 | ||
| Fig. 8 Functional network changes to the proteome during heat shock. Differentially abundant proteins during the heat shock response are shown as nodes in the two graphs (WT on the left, and ssb1Δ mutant on the right), with edges linking proteins that share common Gene Ontology biological process terms. The growth of the heat shock response is apparent as concerted blocks of common function expand over time, notably involved in carbohydrate metabolism, protein folding and response to heat. Equally, the ability of the mutant ssb1Δ strain to respond is evident, particularly in complete down-regulation of cytoplasmic translation capacity as almost all ribosomal proteins are decreased in abundance by 240 minutes. | ||
Discussion
Budding yeast has been a model organism for elucidating the molecular mechanisms of the heat shock response, most prominently, with global transcriptome profiling.8,9,25 However, comprehensive time-resolved proteomics studies are so far not available. Here, we addressed this gap and used single-shot LC-MS label-free quantitative proteomics to elucidate both the initial response, and longer term general adaptation to mild heat stress. In contrast to most previous heat shock studies, the shift from 30 °C to 37 °C investigated here is a smaller fluctuation in temperature. Accordingly, we observed subtle changes in protein expression, typically less than 1.5 fold, throughout the five time points sampled (T10, T30, T60, T120 and T240). This level of confident protein quantification was achieved through a robust normalisation strategy coupled to empirical cut offs derived from observed variance in WT:WT and ssb1Δ:ssb1Δ signal. Nevertheless, some limitations inherent to all “relative quantification” experiments remain. For example, since we do not directly measure absolute protein abundances, it is not possible to disentangle the contribution of individual versus global synthesis rates to explain changes in relative protein abundance. Effectively, we measure the percentage contribution of each protein to the total cellular proteome and how this changes over time. We also note that a small lag is expected before the cells truly start experiencing the elevated temperature in our experiments, due to the time it takes for the media inside the culture flasks to equilibrate to the new temperature. Consequently, it makes the precise determination of the early time point kinetics difficult to assess accurately. Nevertheless, the amplitude of protein fold changes across the time-course of the experiment should not be significantly affected.Our data confirm abundance changes in many hallmark HSR proteins, and for the first time, provides a detailed description of temporal patterns of expression for those proteins. In excellent agreement with the vast literature of heat shock response in S. cerevisiae,10,11 we detected a general down-regulation of proteins related to growth and protein synthesis, most notably ribosome biogenesis, and a widespread increase in abundance of stress-related proteins,47,68–70 including the Ssa family proteins whose modest up-regulation under mild heat stress is consistent with their role in preventing aggregation. However, there is a much greater up-regulation of small heat shock proteins, Hsp26p, Hsp12p, Hsp30p and Hsp42, coupled with a moderate induction of Hsp104p (1.5 fold 60 minutes after stress). This indicates that the cell puts greater emphasis on protein stabilisation by preventing aggregation and maintaining membrane and cytoskeletal organisation prior to kick-starting refolding and reactivation of the denatured proteins. We previously measured the absolute abundance changes in chaperone levels under heat shock using targeted mass spectrometry, with Hsp26p increasing to around ∼250000 copies per cell.71 Hsp26 and Hsp42 are cytosolic chaperones that form oligomers and bind unfolded target proteins in order to stop them from aggregating. While Hsp42p is active in both normal and stress conditions, Hsp26p functions only in stress conditions,70 even though both are regulated by Hsf1p and Msn2/Msn4p transcription factors.72 Interestingly, proteins displaying significant abundance increases between 60–240 minutes were enriched for known targets of several stress response transcription factors (Fig. S7, ESI†), including Msn2/4p, Hsf1p, Aft1p, Rpn4p. Rpn4p is the key regulator of proteasomal activity, which as expected, is up-regulated to deal with a higher burden of protein degradation associated with misfolded protein accumulation linked to heat shock. Rpn4p is also coupled to other stress response transcription factors such as Pdr1p and Pdr3p, two zinc-finger TFs that activate Rpn4 expression.73 Although high-throughput shotgun proteomics is not always sensitive enough to detect transcription factors, it is reassuring to be able to detect the quantitative effects on their targets in a consistent manner.
Since chaperones perform multiple proteostatic roles, including disaggregation and refolding, de novo folding of nascent polypeptides, and aiding degradation of misfolded proteins, the increased folding workload under stress inevitably leads to increases in abundance of the chaperone itself. In the wider context of established chaperone biology,74,75 and the known effects of heat on the proteome (for example, protein denaturation and aggregation) our results can be explained by two main scenarios leading to the overall chaperone-target changes observed here. Scenario one, where proteins prone to unfolding and aggregation in heat accumulate in the cell, but are protected by their cognate chaperones from a general down-regulation, they manifest as relative increases in protein abundance; scenario two, where proteins (other than chaperones) that are functionally important to counter the increase in temperature, such as trehalose biosynthesis pathway components, are actively upregulated. In both cases, there is a need to increase the amount of chaperone activity to either help refold the unfolded/misfolded proteins or to help with de novo folding.
Our clustering of quantitative protein profiles in Fig. 3 pointed to only two major expression patterns present during heat stress response and minimal transient behaviour was evident. More specifically, we did not detect many protein groups or biological processes displaying complex temporal patterns above simple up- or down-regulation; we did not observe a large set of proteins displaying any effective “recovery” step from the stress. This general behaviour is different to that observed at the mRNA level. Gasch et al.9 showed an almost symmetrical transient response of mRNA up- and down-regulation. Unsurprisingly, the time scale of protein response was also different. Gene expression peaks within minutes of stress and returns to a new steady state within hours. These dynamics were not observed for protein level changes, which required longer times to manifest and largely did not appear to return to basal levels. Although the stress and growth conditions differ slightly between our proteomics and the Gasch mRNA studies,9 the same expression change trends were also observed in another study which integrated matching yeast proteomics and transcriptomics results when adapting to high osmolarity.76 The authors there also reported relatively large in amplitude and transient mRNA changes and dampened protein levels for over a 1000 transcript-protein pairs.
In addition to the WT heat shock experiment, we performed a corresponding study with cells lacking the Ssb1 chaperone. Although this protein is in fact down-regulated in heat shock, it is an interesting target due to its key role in nascent polypeptide folding. Indeed, as expected, our analyses show how its deletion exacerbates the effects of stress. The level of protein response exhibited in WT cells was lower and the abundance of most proteins did not change until late in the time course. ssb1Δ cells do not appear to be capable of a strong heat stress response because the cells either do not sense the stimulus, are already pre-adapted to stress, or are unable to respond appropriately. The latter appears more likely, given the attenuated and delayed response we observe. For example, HSF1 targets continue to be over-represented in proteins up-regulated at 240 min in the ssb1Δ cells (adjusted p-value < 4 × 10−10) although only 16 are detected compared to 64 in the WT. Similarly, we note that Hsp12p and Hsp26p are themselves targets of Ssb1p (as well as many other chaperones), limiting the cells ability to deploy its full complement of stress response molecular repertoire. In the face of this inability to form an appropriate response, for example by producing more chaperones with de novo folding function, the cells have a limited RAC functionality. This strategy is consistent with the less protein production in general, and therefore less misfolded and aggregate-prone proteins in the cell. Curiously, less potential for aggregation would reduce the need for expressing stabilising proteins like Hsp12p, Hsp26p.
Previously, we have shown that cells deleted for Ssb1p are able to maintain protein homeostasis and do not undergo major proteome remodelling in normal conditions.28 We proposed that this is possible owing to other HSP70 chaperones taking over the folding duties of Ssb1p and spreading the workload across the network. Here, we suggest that under heat stress the system is pushed to breaking point leading to a collapse in the requisite chaperone and protein synthetic systems, epitomised by the systemic down-regulation of ribosomal proteins shown in Fig. 8 after 240 minutes.
More generally, our results demonstrate the power of quantitative proteomics coupled to integrated bioinformatics to inform on the molecular events underpinning a cellular response to stress in a temporal fashion. Additionally, by consideration of a deletant strain in parallel, the global consequences of the loss of that gene on the ability of the cell to respond to stress may be characterised and used to rationalise the molecular deficiencies that lead to phenotypes.
Overall, the results presented in this study provide a detailed description of the heat shock response in S. cerevisiae at the protein level, integrating this information with gene expression changes. This analysis allows us to examine expression patterns of individual proteins in fine detail, and to evaluate the more holistic global response, uncovering important trends on a system level. Finally, the data obtained here are in excellent agreement with the vast, mainly transcriptome-based literature of heat shock response, but emphasises that novel insights can still be gained from carefully controlled large-scale quantitative proteomics studies that cannot be deduced by examining the transcriptome alone.
Conflicts of interest
There are no competing or conflicting interests from any of the authors.Acknowledgements
The authors acknowledge support from the Biotechnology and Biological Sciences Research Council in the form of a studentship to AFJ as part of the Systems Biology Doctorial Training Centre, and grant support to SJH, CMG and CEE in the form of a LoLa grant (BB/G009112/1), and additional BBSRC support for MGA (BB/M025748/1).References
- D. J. Barshis, J. T. Ladner, T. A. Oliver, F. O. Seneca, N. Traylor-Knowles and S. R. Palumbi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1387–1392 CrossRef CAS PubMed.
- K. W. Kim and Y. Jin, FEBS Lett., 2015, 589, 1644–1652 CrossRef CAS PubMed.
- E. de Nadal, G. Ammerer and F. Posas, Nat. Rev. Genet., 2011, 12, 833–845 CAS.
- R. C. Taylor, K. M. Berendzen and A. Dillin, Nat. Rev. Mol. Cell Biol., 2014, 15, 211–217 CrossRef CAS PubMed.
- H. Karathia, E. Vilaprinyo, A. Sorribas and R. Alves, PLoS One, 2011, 6, 10 Search PubMed.
- S. Hohmann, Microbiol. Mol. Biol. Rev., 2002, 66, 300–372 CrossRef CAS PubMed.
- S. M. O'Rourke, I. Herskowitz and E. K. O'Shea, Trends Genet., 2002, 18, 405–412 CrossRef.
- H. C. Causton, B. Ren, S. S. Koh, C. T. Harbison, E. Kanin, E. G. Jennings, T. I. Lee, H. L. True, E. S. Lander and R. A. Young, Mol. Biol. Cell, 2001, 12, 323–337 CrossRef CAS PubMed.
- A. P. Gasch, P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein and P. O. Brown, Mol. Biol. Cell, 2000, 11, 4241–4257 CrossRef CAS PubMed.
- K. A. Morano, C. M. Grant and W. S. Moye-Rowley, Genetics, 2012, 190, 1157–1195 CrossRef CAS PubMed.
- J. Verghese, J. Abrams, Y. Y. Wang and K. A. Morano, Microbiol. Mol. Biol. Rev., 2012, 76, 115–158 CrossRef CAS PubMed.
- K. Richter, M. Haslbeck and J. Buchner, Mol. Cell, 2010, 40, 253–266 CrossRef CAS PubMed.
- M. B. Eisen, P. T. Spellman, P. O. Brown and D. Botstein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14863–14868 CrossRef CAS.
- P. K. Sorger and H. R. B. Pelham, Cell, 1988, 54, 855–864 CrossRef CAS PubMed.
- G. Wiederrecht, D. Seto and C. S. Parker, Cell, 1988, 54, 841–853 CrossRef CAS PubMed.
- J. S. Hahn, Z. Z. Hu, D. J. Thiele and V. R. Iyer, Mol. Cell. Biol., 2004, 24, 5249–5256 CrossRef CAS PubMed.
- E. J. Solis, J. P. Pandey, X. Zheng, D. X. Jin, P. B. Gupta, E. M. Airoldi, D. Pincus and V. Denic, Mol. Cell, 2016, 63, 60–71 CrossRef CAS PubMed.
- A. P. Schmitt and K. McEntee, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5777–5782 CrossRef CAS.
- N. Lopez, J. Halladay, W. Walter and E. A. Craig, J. Bacteriol., 1999, 181, 3136–3143 CAS.
- C. Pfund, N. Lopez-Hoyo, T. Ziegelhoffer, B. A. Schilke, P. Lopez-Buesa, W. A. Walter, M. Wiedmann and E. A. Craig, Embo J., 1998, 17, 3981–3989 CrossRef CAS PubMed.
- R. J. Nelson, T. Ziegelhoffer, C. Nicolet, M. Wernerwashburne and E. A. Craig, Cell, 1992, 71, 97–105 CrossRef CAS PubMed.
- M. Gautschi, H. Lilie, U. Funfschilling, A. Mun, S. Ross, T. Lithgow, P. Rucknagel and S. Rospert, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3762–3767 CrossRef CAS PubMed.
- M. Rakwalska and S. Rospert, Mol. Cell. Biol., 2004, 24, 9186–9197 CrossRef CAS PubMed.
- K. Peisker, M. Chiabudini and S. Rospert, Biochim. Biophys. Acta, Mol. Cell Res., 2010, 1803, 662–672 CrossRef CAS PubMed.
- K. Strassburg, D. Walther, H. Takahashi, S. Kanaya and J. Kopka, Omics, 2010, 14, 249–259 CrossRef CAS PubMed.
- N. Nagaraj, N. A. Kulak, J. Cox, N. Neuhauser, K. Mayr, O. Hoerning, O. Vorm and M. Mann, Mol. Cell. Proteomics, 2012, 11, 11 Search PubMed.
- W. Q. Shui, Y. Xiong, W. D. Xiao, X. N. Qi, Y. Zhang, Y. P. Lin, Y. F. Guo, Z. D. Zhang, Q. H. Wang and Y. H. Ma, Mol. Cell. Proteomics, 2015, 14, 1885–1897 CAS.
- A. F. Jarnuczak, C. E. Eyers, J. M. Schwartz, C. M. Grant and S. J. Hubbard, Proteomics, 2015, 15, 3126–3139 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- J. Cox, N. Neuhauser, A. Michalski, R. A. Scheltema, J. V. Olsen and M. Mann, J. Proteome Res., 2011, 10, 1794–1805 CrossRef CAS PubMed.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Mol. Cell. Proteomics, 2014, 13, 2513–2526 CAS.
- A. I. Nesvizhskii and R. Aebersold, Mol. Cell. Proteomics, 2005, 4, 1419–1440 CAS.
- O. Serang and W. Noble, Stat. Interface, 2012, 5, 3–20 CrossRef PubMed.
- J. A. Vizcaino, E. W. Deutsch, R. Wang, A. Csordas, F. Reisinger, D. Rios, J. A. Dianes, Z. Sun, T. Farrah, N. Bandeira, P. A. Binz, I. Xenarios, M. Eisenacher, G. Mayer, L. Gatto, A. Campos, R. J. Chalkley, H. J. Kraus, J. P. Albar, S. Martinez-Bartolome, R. Apweiler, G. S. Omenn, L. Martens, A. R. Jones and H. Hermjakob, Nat. Biotechnol., 2014, 32, 223–226 CrossRef CAS PubMed.
- M. Choi, C. Y. Chang, T. Clough, D. Broudy, T. Killeen, B. MacLean and O. Vitek, Bioinformatics, 2014, 30, 2524–2526 CrossRef CAS PubMed.
- L. Kumar and M. E. Futschik, Bioinformation, 2007, 2, 5–7 CrossRef PubMed.
- V. Schwammle and O. N. Jensen, Bioinformatics, 2010, 26, 2841–2848 CrossRef PubMed.
- H. Mi, A. Muruganujan, J. T. Casagrande and P. D. Thomas, Nat. Protoc., 2013, 8, 1551–1566 CrossRef PubMed.
- H. Mi, A. Muruganujan and P. D. Thomas, Nucleic Acids Res., 2013, 41, D377–D386 CrossRef CAS PubMed.
- P. Brownridge, C. Lawless, A. B. Payapilly, K. Lanthaler, S. W. Holman, V. M. Harman, C. M. Grant, R. J. Beynon and S. J. Hubbard, Proteomics, 2013, 13, 1276–1291 CrossRef CAS PubMed.
- Y. Gong, Y. Kakihara, N. Krogan, J. Greenblatt, A. Emili, Z. Zhang and W. A. Houry, Mol. Syst. Biol., 2009, 5, 275 CrossRef PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- E. A. Craig and K. Jacobsen, Mol. Cell. Biol., 1985, 5, 3517–3524 CrossRef CAS PubMed.
- A. L. Oberg and D. W. Mahoney, BMC Bioinf., 2012, 13, 18 CrossRef PubMed.
- L. M. F. de Godoy, J. V. Olsen, J. Cox, M. L. Nielsen, N. C. Hubner, F. Frohlich, T. C. Walther and M. Mann, Nature, 2008, 455, 1251–1254 CrossRef CAS PubMed.
- Y. Benjamini and Y. Hochberg, J. R. Stat. Soc.: Ser. B, Methodol., 1995, 57, 289–300 Search PubMed.
- M. Haslbeck, S. Walke, T. Stromer, M. Ehrnsperger, H. E. White, S. X. Chen, H. R. Saibil and J. Buchner, Embo J., 1999, 18, 6744–6751 CrossRef CAS PubMed.
- S. Welker, B. Rudolph, E. Frenzel, F. Hagn, G. Liebisch, G. Schmitz, J. Scheuring, A. Kerth, A. Blume, S. Weinkauf, M. Haslbeck, H. Kessler and J. Buchner, Mol. Cell, 2010, 39, 507–520 CrossRef CAS PubMed.
- J. M. Cherry, E. L. Hong, C. Amundsen, R. Balakrishnan, G. Binkley, E. T. Chan, K. R. Christie, M. C. Costanzo, S. S. Dwight, S. R. Engel, D. G. Fisk, J. E. Hirschman, B. C. Hitz, K. Karra, C. J. Krieger, S. R. Miyasato, R. S. Nash, J. Park, M. S. Skrzypek, M. Simison, S. Weng and E. D. Wong, Nucleic Acids Res., 2012, 40, D700–D705 CrossRef CAS PubMed.
- K. A. Borkovich, F. W. Farrelly, D. B. Finkelstein, J. Taulien and S. Lindquist, Mol. Cell. Biol., 1989, 9, 3919–3930 CrossRef CAS PubMed.
- D. McDaniel, A. J. Caplan, M. S. Lee, C. C. Adams, B. R. Fishel, D. S. Gross and W. T. Garrard, Mol. Cell. Biol., 1989, 9, 4789–4798 CrossRef CAS PubMed.
- J. M. Tkach, A. Yimit, A. Y. Lee, M. Riffle, M. Costanzo, D. Jaschob, J. A. Hendry, J. W. Ou, J. Moffat, C. Boone, T. N. Davis, C. Nislow and G. W. Brown, Nat. Cell Biol., 2012, 14, 966–976 CrossRef CAS PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed.
- C. T. Harbison, D. B. Gordon, T. I. Lee, N. J. Rinaldi, K. D. Macisaac, T. W. Danford, N. M. Hannett, J. B. Tagne, D. B. Reynolds, J. Yoo, E. G. Jennings, J. Zeitlinger, D. K. Pokholok, M. Kellis, P. A. Rolfe, K. T. Takusagawa, E. S. Lander, D. K. Gifford, E. Fraenkel and R. A. Young, Nature, 2004, 431, 99–104 CrossRef CAS PubMed.
- D. Tabas-Madrid, R. Nogales-Cadenas and A. Pascual-Montano, Nucleic Acids Res., 2012, 40, W478–W483 CrossRef CAS PubMed.
- K. Tyagi and P. G. Pedrioli, Nucleic Acids Res., 2015, 43, 4701–4712 CrossRef CAS PubMed.
- C. Lawless, S. W. Holman, P. Brownridge, K. Lanthaler, V. M. Harman, R. Watkins, D. E. Hammond, R. L. Miller, P. F. Sims, C. M. Grant, C. E. Eyers, R. J. Beynon and S. J. Hubbard, Mol. Cell. Proteomics, 2016, 15, 1309–1322 CAS.
- J. C. Bezdek, Pattern Recognition with Fuzzy Objective Function Algorithms, Kluwer Academic Publishers, 1981 Search PubMed.
- E. Kanshin, P. Kubiniok, Y. Thattikota, D. D'Amours and P. Thibault, Mol. Syst. Biol., 2015, 11, 813 CrossRef PubMed.
- E. A. Craig and K. Jacobsen, Mol. Cell. Biol., 1985, 5, 3517–3524 CrossRef CAS PubMed.
- A. F. Jarnuczak, C. E. Eyers, J. M. Schwartz, C. M. Grant and S. J. Hubbard, Proteomics, 2015, 15, 3126–3139 CrossRef CAS PubMed.
- K. Strassburg, D. Walther, H. Takahashi, S. Kanaya and J. Kopka, Omics, 2010, 14, 249–259 CrossRef CAS PubMed.
- E. Kanshin, P. Kubiniok, Y. Thattikota, D. D'Amours and P. Thibault, Mol. Syst. Biol., 2015, 11, 17 CrossRef PubMed.
- P. W. Chen, L. L. Fonseca, Y. A. Hannun and E. O. Voit, PLoS Comput. Biol., 2013, 9, 12 CrossRef.
- M. Carla Fama, D. Raden, N. Zacchi, D. R. Lemos, A. S. Robinson and S. Silberstein, Biochim. Biophys. Acta, 2007, 1773, 232–242 CrossRef CAS PubMed.
- I. Wagner, H. Arlt, L. van Dyck, T. Langer and W. Neupert, EMBO J., 1994, 13, 5135–5145 CAS.
- P. Walsh, D. Bursac, Y. C. Law, D. Cyr and T. Lithgow, EMBO Rep., 2004, 5, 567–571 CrossRef CAS PubMed.
- M. Wernerwashburne, J. Becker, J. Kosicsmithers and E. A. Craig, J. Bacteriol., 1989, 171, 2680–2688 CrossRef CAS.
- W. R. Boorstein and E. A. Craig, J. Biol. Chem., 1990, 265, 18912–18921 CAS.
- M. Haslbeck, N. Braun, T. Stromer, B. Richter, N. Model, S. Weinkauf and J. Buchner, Embo J., 2004, 23, 638–649 CrossRef CAS PubMed.
- R. J. Mackenzie, C. Lawless, S. W. Holman, K. Lanthaler, R. J. Beynon, C. M. Grant, S. J. Hubbard and C. E. Eyers, Proteomics, 2016, 16, 2128–2140 CrossRef CAS PubMed.
- M. Amoros and F. Estruch, Mol. Microbiol., 2001, 39, 1523–1532 CrossRef CAS PubMed.
- G. Owsianik, L. Balzi and M. Ghislain, Mol. Microbiol., 2002, 43, 1295–1308 CrossRef CAS PubMed.
- S. M. Hill, S. Hanzen and T. Nystrom, EMBO Rep., 2017, 18, 377–391 CrossRef CAS PubMed.
- A. L. Horwich, Cell, 2014, 157, 285–288 CrossRef CAS PubMed.
- M. V. Lee, S. E. Topper, S. L. Hubler, J. Hose, C. D. Wenger, J. J. Coon and A. P. Gasch, Mol. Syst. Biol., 2011, 7, 12 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mo00050b |
| This journal is © The Royal Society of Chemistry 2018 |