
Kinome chemoproteomics characterization of pyrrolo[3,4-c]pyrazoles as potent and selective inhibitors of glycogen synthase kinase 3†
Martin Golkowski *a,
Gayani K. Pererab,
Venkata Narayana Vidadalab,
Kayode K. Ojoc,
Wesley C. Van Voorhisc,
Dustin J. Maly*b and
Shao-En Ong*a
*a,
Gayani K. Pererab,
Venkata Narayana Vidadalab,
Kayode K. Ojoc,
Wesley C. Van Voorhisc,
Dustin J. Maly*b and
Shao-En Ong*a
aSchool of Medicine, Department of Pharmacology, University of Washington, Seattle, WA 98195, USA. E-mail: golkom@uw.edu; shaoen@uw.edu
bDepartment of Chemistry, University of Washington, Seattle, WA 98195, USA. E-mail: djmaly@uw.edu
cSchool of Medicine, Division of Allergy and Infectious Disease, Center for Emerging and Reemerging Infectious Disease (CERID), University of Washington, Seattle, WA 98109, USA
First published on 20th December 2017
Abstract
Glycogen synthase kinase 3 has evolutionarily conserved roles in cell signaling and metabolism and is a recognized drug target in neurological pathologies, most prominently bipolar disorder. More recently it has been suggested that GSK3 may be a target for the treatment of trypanosomatid parasite infections, e.g. with T. brucei, due to the lethal phenotype observed in parasite GSK3 short RNAi knockdown experiments. Here we investigated the kinome selectivity of a library of pyrrolo[3,4-c]pyrazol inhibitors that were developed against T. brucei GSK3 but that also interact with the human orthologue and other protein kinases. We applied label-free MS-based kinome chemoproteomics profiling with kinobeads to obtain the selectivity profiles of all 39 library members against 217 human protein and lipid kinases. This allowed us to study the structure–activity relationship of the library members as well as the chemical genetic relationships between kinase targets. As a result, we identified a novel and highly selective HsGSK3 inhibitor containing a 2-chloroaniline-substituted squaric acid amide pharmacophore that confers low nanomolar (IC50 = 2.8 nM) and sub-micromolar potency against purified and cellular HsGSK3. The inhibitor will be useful as a new lead for GSK3 inhibitor development and as a chemical genetic probe to study roles of GSK3 in cell signaling.
Introduction
The human glycogen synthase kinase 3 isoforms alpha and beta (HsGSK3α and β) are central regulators of numerous cellular processes such as Wnt-β-catenin signaling, insulin receptor signaling and glycogen metabolism as well as transcriptional regulation (JUN, NFAT, etc.) and microtubule dynamics.1–3 Lithium, the first line treatment of bipolar disorder is thought to act through inhibition of GSK3 and the enzyme is intensively studied as an effector of this disease and other neurological disorders such as depression, Fragile X syndrome, and Alzheimer's disease. Further, GSK3 has recognized roles in the development of cancer, diabetes mellitus and cardiac hypertrophy.4–9 More recently, the trypanosomatid parasite orthologues of mammalian GSK3 were found to be essential for survival of e.g. Trypanosoma brucei (T. brucei) and it has been proposed that the inhibition of parasite GSK3 may present an avenue for the development of targeted chemotherapies against African sleeping sickness and Chagas disease (trypanosomiasis) as well as Leishmaniosis.10–12Consequently, much effort has been directed toward the development of small molecule inhibitors that target human GSK3 or their parasite orthologues. A number of potent ATP-competitive HsGSK3 inhibitors that show low nanomolar affinity and varying degrees of kinome selectivity have been reported in the literature.13–15 Many of these compounds also potently inhibit TbGSK3 as well as other members of the human and parasite kinome.12,16 Among the main challenges that remain for the development of novel HsGSK3 inhibitors are kinome selectivity, especially over structurally related cyclin-dependent kinases (CDKs) and the optimization of in vivo efficacy, i.e. drug delivery to the target organ (brain). Ideally, parasite GSK3 inhibitors should possess high potency and selectivity over HsGSK3 and other human kinases to avoid systemic toxicity; substantial progress has been made towards this goal.17
In our parasite drug discovery platform, we have pursued the discovery of novel inhibitor scaffolds with superior affinity and selectivity for TbGSK3. Leveraging combinatorial medicinal chemistry strategies, we generated a pyrrolo[3,4-c]-pyrazol kinase inhibitor library. Our efforts yielded inhibitors with low nanomolar affinity against TbGSK3 but unfortunately these compounds failed to inhibit the growth of T. brucei. Instead, some of the library members showed an up to tenfold higher affinity to HsGSK3 over TbGSK3, which led us to investigate if our compound library contained molecules that could serve as novel leads for HsGSK3α and β drug discovery. Here we report the use of state-of-the-art mass spectrometry (MS)-based chemoproteomics and native kinases derived from human cellular extracts to characterize all library members’ selectivity profiles and target kinase structural properties on a kinome scale.
Materials and methods
Cell culture and harvest
C3A, HEK293T and HCT-116 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in the recommended medium. Base media, Dulbecco's minimum essential medium (DMEM) and Eagle's minimum essential medium (MEM), were from Gibco supplemented with penicillin–streptomycin–glutamine 100× (Thermo Fisher Scientific, Waltham, MA) and 10% Seradigm fetal bovine serum (FBS, VWR, Radnor, PA). Cells for proteomics experiments were harvested as described previously and cells for western blot analysis were harvested the same way.18Kinobead chemoproteomics experiments
The kinobead reagents were synthesized in house and the pull downs were performed as described previously with a few minor modifications.18 300 μg of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HCT-116 and HEK293T cell lysate at 2 mg mL−1 (150 μL) was used as the source for native kinases for each pulldown experiment. After washing of the beads after the 3 h pull down step the beads were resuspended in 25 μL 6 M aq. guanidine hydrochloride (Gdn*HCl) containing 100 mM tris (pH = 8.5), 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 10 mM 2-chloroacetamide (CAM). The samples were heated to 95 °C for 5 min and then processed further as described. All label-free kinase inhibitor competition experiments were done in biological duplicates and all DMSO control experiments were performed in biological quadruplicates on the same days on which inhibitor competition experiments were performed. Competitors were applied at 50 μM or 1 μM for single dose experiments and at 50 μM to 2.5 nM (3-fold dilution steps) for titration experiments. One set of DMSO control experiments was prepared and analyzed each month to guarantee consistent label-free quantification (LFQ) of kinase ratios. The structures of the kinobead capture reagents can be found in SI-PDF file (ESI,† Fig. S1).
:1 mixture of HCT-116 and HEK293T cell lysate at 2 mg mL−1 (150 μL) was used as the source for native kinases for each pulldown experiment. After washing of the beads after the 3 h pull down step the beads were resuspended in 25 μL 6 M aq. guanidine hydrochloride (Gdn*HCl) containing 100 mM tris (pH = 8.5), 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 10 mM 2-chloroacetamide (CAM). The samples were heated to 95 °C for 5 min and then processed further as described. All label-free kinase inhibitor competition experiments were done in biological duplicates and all DMSO control experiments were performed in biological quadruplicates on the same days on which inhibitor competition experiments were performed. Competitors were applied at 50 μM or 1 μM for single dose experiments and at 50 μM to 2.5 nM (3-fold dilution steps) for titration experiments. One set of DMSO control experiments was prepared and analyzed each month to guarantee consistent label-free quantification (LFQ) of kinase ratios. The structures of the kinobead capture reagents can be found in SI-PDF file (ESI,† Fig. S1).
LC-MS/MS analysis
The LC-MS/MS analyses were performed as described previously with the following minor modifications.18 Peptide samples were separated on 20 cm long fused silica capillary columns (100 μM ID) packed with 3 μm 120 Å reversed phase C18 beads (Dr Maisch, Ammerbuch, DE). The LC gradient was 120 min long with 10–35% B at 300 nL min−1. LC solvent A was 0.1% aq. acetic acid and LC solvent B was 0.1% acetic acid, 99.9% acetonitrile.Data analyses
MS raw files were computed using the MaxQuant software package (V1.5.2.8) with the Uniprot human FASTA file (published on 07.22.2015). All other parameters were kept as previously described.18 LFQ ratios were calculated from the MaxQuant protein intensity values (log2 transformed and normalized by subtracting the mean value from each intensity data column) of DMSO control experiments and compound competition experiments. All MS raw files and MaxQuant output files were deposited in the MassIVE online repository (https://massive.ucsd.edu/) under the accession number MSV000081471.
Unsupervised hierarchical clustering was performed using the Perseus software package (V1.5.6.0)19 using the following settings. Distance: euclidean; linkage: average; process with k-means enabled; number of clusters: 300; maximal number of iterations: 10; number of restarts: 1. For correlation analysis p-values were corrected for multiple comparisons according to Bonferroni as α/m where m is the number of comparisons and α is the significance level. Only kinases that were quantified in at least two out of four replicates in the DMSO control experiments (MaxQuant protein intensity calculated, n = 170) and that interacted with ≥3 of the 39 compounds (log2 LFQ ratio ≥ 2) were used for these analyses (total of 32 kinases). Further, to account for kinase-compound interactions among these 32 kinases that may have led to quantitative competition (kinase not detected in presence of competitor), data imputation in Perseus (V1.5.6.0) was performed. Missing intensity values in the compound competition experiments were sampled from a hypothetical distribution downshifted by 1.5 and having a width of 0.2. For DMSO control experiments no data imputation was performed and missing LFQ ratios were calculated from the imputed dataset (see ESI,† Excel file S1).
Graphs of the titration curves for calculation of the EC50 values (competition) and IC50 values (GSK3 inhibition) were plotted and analyzed using the GraphPad Prism software package (V5.0a) with a least-squares nonlinear regression model for curve fitting (one site – Fit logIC50 function).
Western blot analysis
C3A cells were seeded on 12 well plates in the ATCC-recommended medium at 10000 cells per well and grown until reaching 90% confluency. Treatment was either DMSO alone (control, 0.1% final) or inhibitor dissolved in DMSO (0.1% final) at concentrations ranging from 10 μM to 0.5 nM (3-fold dilution steps, i.e. 10 μM, 3.3 μM, 1.1 μM, 0.37 μM, 0.12 μM, 41 nM, 14 nM, 4.6 nM, 1.5 nM, 0.5 nM) for 2 h. Cells were then washed twice with ice cold PBS and harvested in 50 μL modified RIPA buffer with a cell scraper.18 After pelleting the insoluble fraction at 4 °C and 21000 × g, cell lysate supernatants were mixed with 4× NuPAGE LDS sample buffer (Life Technologies, Carlsbad, CA) containing 50 mM dithiothreitol (DTT), vortexed briefly and then heated to 95 °C for 5 min. Proteins were separated using Bolt 4–12% Bis–Tris Plus pre-cast gels (Invitrogen, Carlsbad, CA) and proteins were electro transferred onto Amersham Protran 0.2 μm nitrocellulose (GE Healthcare, Chicago, IL). Membranes were blocked with 5% non-fat milk powder dissolved in tris-bufferd saline containing 0.1% Tween-20 (TBST) for 1 h at RT. For probing of (phospho)proteins the following primary antibodies were used (Cell Signaling Technology, Danvers, MA): GAPDH (D16H11) XP® Rabbit mAb (HRP Conjugate) #8884; Phospho-Glycogen Synthase (Ser641) Antibody #3891. Primary antibodies were probed using goat anti-rabbit IgG–peroxidase antibody (Sigma-Aldrich, St. Louis, MO) and visualized with the Clarity western ECL substrate (Bio-Rad, Hercules, CA) using a FluorChem E gel scanner (Protein Simple, San Jose, CA). Western blotting bands (pGYS1-S641, GAPDH) were quantified using the AlphaView (V3.3.0) software and the pGYS1 signal was normalized to the GAPDH signal.
In vitro kinase assay
The assay using 32P-labeled ATP and recombinant human and T. brucei GSK3B was performed as described previously.10Kinase inhibitor synthesis
All pyrrolo[3,4-c]-pyrazol inhibitors were synthesized in house and their purity was ≥95% as determined by LC analysis. Detailed synthetic procedures and compound characterization can be found in the electronic ESI† (PDF file S1).Results and discussion
Design of the pyrrolo[3,4-c]pyrazol inhibitor library
Based on previous reports indicating that amino pyrazol-based inhibitors can act as potent and selective TbGSK3-short inhibitors, we decided to build a compound library based on the pyrrolo[3,4-c]pyrazol scaffold (Fig. 1a).12,17 This scaffold was previously used in clinically relevant inhibitors of human aurora kinase A (HsAURKA) and the breakpoint cluster region – Abelson tyrosine kinase (BCR-ABL) fusion protein (danusertib, PHA-739358).20–22 The scaffold has the advantage that highly diverse compound libraries can be generated with only three synthetic chemical operations starting from a commercially available precursor (ESI,† PDF file S1).20 To find novel compounds with activity against GSK3, we focused on probing the effect of substitution at the N-5 position (R1). Ligands presented from this position project into the hydrophobic pocket II adjacent to the purine binding pocket of the kinase ATP-binding pocket.23 Variation of the amide ligands presented from the C-3 position (R2) that interact with the hydrophobic pocket I and the solvent interface was kept at a minimum (Fig. 1a and also Table 1). The crystal structure of the pyrrolo[3,4-c]pyrazol inhibitor PHA-680632 in complex with HsAURKA at 3 Å resolution is available and this structure served as a valuable resource for inhibitor design (Fig. 1b). Here, PHA-680632 adopts a type I binding mode and the aryl residue presented from N-5 position via a urea linkage was found to be positioned under the phosphate-binding loop (P-loop), rotated by 90° relative to the flat heterocyclic core.20 | ||
| Fig. 1 Binding mode of pyrrolo[3,4-c]pyrazol inhibitors. (a) Binding mode typical for type I kinase inhibitors, including the interactions with the hinge region and the hydrophobic pockets I and II. (b) Co-crystal structure of PHA-680632 and the HsAURKA catalytic domain. Ribbon color indicates the type of secondary structure; red = α-helix, yellow = β-sheet, white = disordered.24 PDBID: 2J50. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | No. | R1 | R2 | No. | R1 | R2 | No. |
 |
A | 1 |  |
A C |
13 14 |
 |
A | 28 |
 |
A D |
2 3 |
 |
A | 15 |  |
A C |
29 30 |
 |
A | 4 |  |
A | 16 |  |
A | 31 |
 |
A C |
5 6 |
 |
A B C D E |
17 18 19 20 21 |
 |
A | 32 |
 |
A | 7 |  |
A D |
22 23 |
 |
A C |
33 34 |
 |
A | 8 | 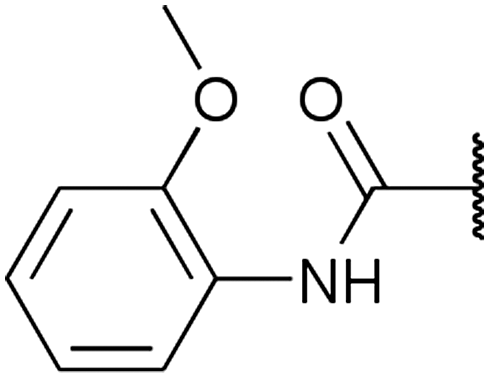 |
A | 24 | 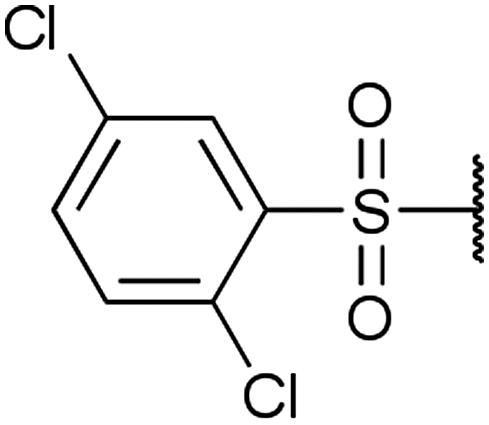 |
A | 35 |
 |
A | 9 |  |
D | 25 |  |
A C |
36 37 |
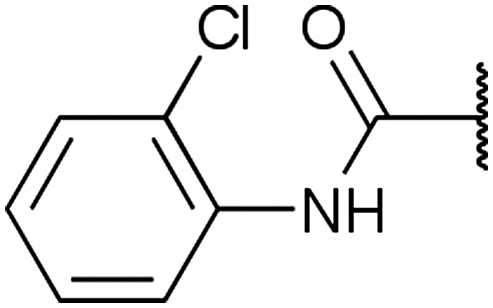 |
A B C |
10 11 12 |
 |
A C |
26 27 |
 |
A C |
38 39 |

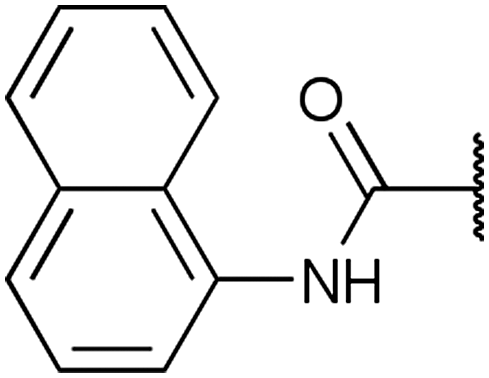
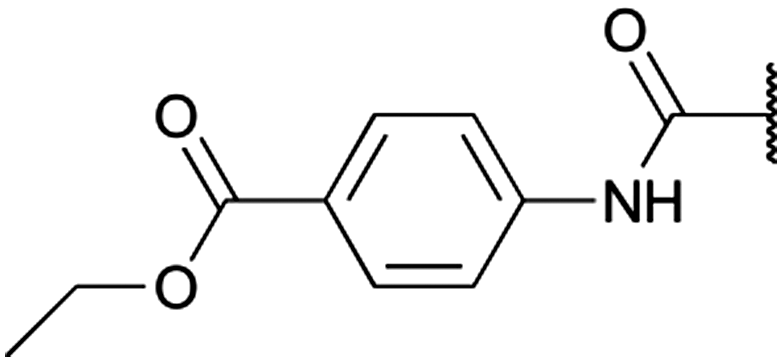
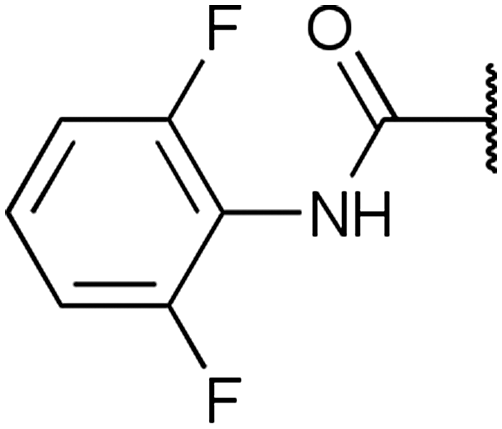
To diversify our inhibitor library we introduced four different lipophilic acyl groups of varying size at the C-3 position (R2); a butyric acid or cyclopropanecarboxylic acid amide served as small lipophilic substituents, a 4-methoxycyclohexanecarboxylic acid amide as a medium sized lipophilic substituent and a 2-naphtaleneacetic acid amide as a bulky lipophilic substituent (1–20, 22–39, Table 1). Compound 21 is unique in that it bears an N-methyl-4-piperidinecarboxylic acid amide residue at this position. From the N-5 position (R1) of the dihydropyrrole moiety, aryl-groups are displayed via a urea, amide, sulfonamide or squaric acid amide linkage. These aryl groups contain polar small (–F) and medium sized (–CF3, –Cl, –OMe, COOEt) substituents alone or in pairs positioned around the aromatic ring. Further, we included three compounds that contain the sterically less demanding n-butyl amine urea, butyric acid amide and aniline urea substituents as controls. This resulted in a total of 24 different substituents at the R1 position that served to probe the hydrophobic pocket II of kinase domains (Fig. 1 and Table 1).
In vitro inhibition of GSK3 kinase activity
To investigate the capability of our compounds to inhibit the kinase activity of GSK3, we performed a series of in vitro titration experiments with recombinant human and T. brucei GSK3β against a subset of compounds from our library. The results indicated that the inhibitors 18, 19 and 39 have double-digit nanomolar IC50 values against TbGSK3β. At the same time, we observed a general trend that the compounds show lower relative IC50s against HsGSK3β activity. This suggested a preference of our compounds for the human orthologue and the possibility that inhibition of human or animal kinases may lead to systemic side effects in preclinical and clinical stages of drug development. Further, when tested in a growth assay against T. brucei, none of our compounds achieved a significant inhibitory effect. To our surprise, the squaramides 38 and 39 showed extremely low IC50 values against HsGSK3β with 4.7 and 2.8 nM, respectively (Table 2). This equals the IC50 values of some of the most potent HsGSK3α and β inhibitors developed to date, e.g. BRD1652,13 SB216763,25 AR-A014418,26 and CHIR9902127 that all have been tested in a kinome-wide screen for their selectivity (see Fig. 2).13 Accordingly, we hypothesized that our compound library, originally developed against TbGSK3 contains potent inhibitors of the human orthologue that may serve as leads for drug discovery and as chemical genetic probes against these enzymes.| Compound | HsGSK3β IC50 (S.D.) in nM | TbGSK3β IC50 (S.D.) in nM |
|---|---|---|
| a Mean of two replicate experiments. b Mean of three replicate experiments. c Mean of ≥4 replicate experiments. | ||
| 10 | 77 (14)b | 360 (100)b |
| 16 | 66 (32)a | 5700 (1300)b |
| 19 | 77 (18)c | 62 (12)c |
| 29 | 170 (1.6)a | 510 (78)a |
| 18 | 120 (35)b | 47 (7.7)b |
| 11 | 65 (10)b | 200 (34)b |
| 39 | 2.8 (0.6)a | 47 (19)c |
| 38 | 4.7 (0.8)c | 160 (16)c |
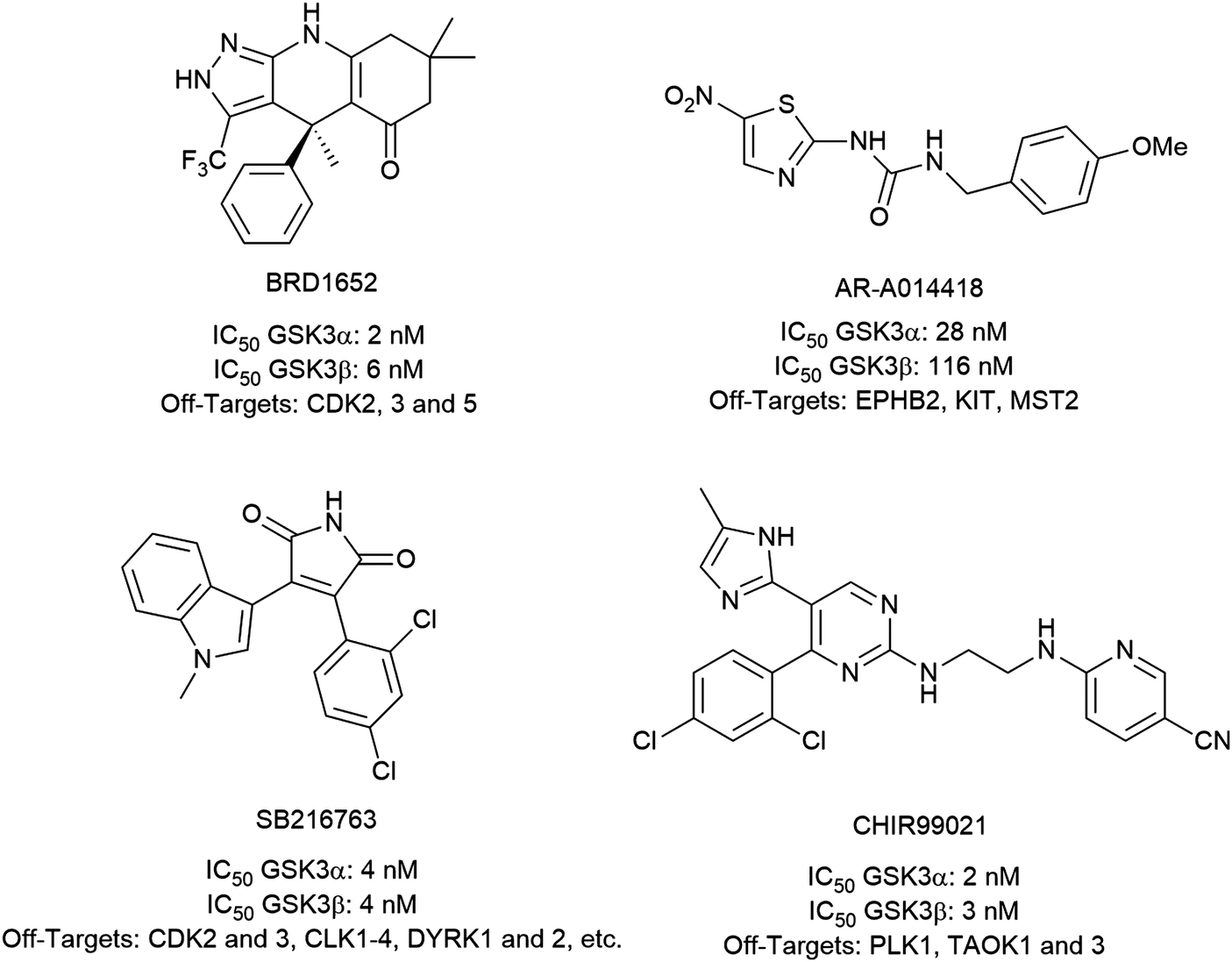 | ||
| Fig. 2 Structures, IC50 values and example off-targets of some potent HsGSK3α and β inhibitors described previously. | ||
Chemoproteomics characterization in human cell lysates
To gain further insights into the interaction of our compounds with human kinases we assessed the kinome selectivity of all members of the 39-member compound library. We applied our newly developed streamlined mass spectrometry (MS) chemoproteomics workflow based on the kinobead platform.18,28 Chemoproteomics profiling has been shown very recently to be highly valuable for guiding medicinal chemistry optimization of kinase inhibitors.29 For the competition-binding experiments, a 1:1 mixture of HEK293T and HCT-116 cell lysates served as the source for native human kinases. Protein quantification was performed in a label-free fashion,30 which led to the robust measurement of the abundance of 217 protein and lipid kinases. In the initial round of selectivity profiling, each compound was added to the lysate at a high (50 μM) concentration to facilitate the identification of even low affinity interactions. This yielded a comprehensive interaction map of all library members against a large fraction of the human kinome (Fig. 3a and ESI,† Excel file S1). In a second round of selectivity profiling, compounds that showed a high relative affinity to HsGSK3α and β and several other kinases were re-probed at a lower concentration (1 μM) to assess if a larger number of high affinity kinase interactors are present in the inhibitor library (Fig. 3c).
 | ||
| Fig. 3 Kinome selectivity profiling and structural evolution of our pyrrolo[3,4-c]pyrazol compound library. (a) Heatmap of the log2 LFQ ratios for kinase competition in the kinobead MS assay at 50 μM compound concentration. Kinase competition profiles are clustered on the X-axis (unsupervised hierarchical clustering, see Materials and methods) with the phylogenetic relationship of the kinase catalytic domains conserved on the Y-axis. Only kinases that show a strong interaction with at least three out of 39 compounds (log2 LFQ ratio ≥2.0) and that were quantified in at least 2 out of 4 replicate experiments of all DMSO control experiments were used for clustering (n = 32). (b) Chemical structures illustrate the evolution of inhibitors from small substituents occupying the hydrophobic pocket II of protein kinases to sterically more demanding and diverse substituents (i.e., compounds A progressing to F); the impact on kinome binding selectivity for each compound is shown in (a). Bold: key structural feature; red highlight: newly added structural feature; grey highlight: retained structural feature. (c) Heatmap of log2 LFQ ratios for kinase competition in the kinobead assay of kinase-inhibitor interactions still detectable at 1 μM compound concentration. | ||
Application of a log2 fold-change threshold (FCT) of 2 for kinase abundance changes indicated the interaction of our compounds with 62 kinases at 50 μM concentration (ESI,† Excel file S1). To visualize the shift in kinome selectivity with the changing structural features present in the inhibitors (Fig. 3b), we performed unsupervised hierarchical clustering of LFQ ratios for kinase targets that bound to at least 3 out of 39 compound library members and that were quantified robustly across all DMSO control experiments (32 kinases). We did not consider kinases with fewer than three quantified ratios indicating significant competition as these scattered target interactions do not explain general trends in the structure–activity relationship (SAR) analysis and may increase the chance to identify false-positives in the co-competition–correlation analysis (see Chemical genetics analysis of kinases with chemoproteomics data below). Clustering was performed for the compounds (X-axis) and kinases were sorted according to the phylogenetic relationship between the catalytic domains on the Y-axis (Fig. 3a). The analysis revealed that at least five distinct groups (clusters) of compounds were present in the library that differ significantly in the number of kinase targets as well as the preference for families of related kinases. Compounds with the same or similar residue at the N-5 position (R1) typically clustered together in the same subgroup, which suggested that kinase selectivity was mainly influenced by the substituent projecting into the hydrophobic pocket II. The exceptions were compounds 3, 20 and 23 in which the presence of a bulky naphtyl group presented from the 3-position (R2) almost completely abrogated protein kinase binding.
Interestingly, besides the 62 protein and lipid kinases we also identified 241 non-kinase proteins that showed a log2 LFQ ratio of ≥2. More stringent selection for non-kinases that were competed by at least three compounds left 141 proteins of which 28 are associated with the gene ontology molecular function (GOMF) term “ATP binding” (see ESI,† Excel file S1). These proteins may either represent direct binders of our compounds (ATP-binding pocket present) or may have been competed as part of a kinase complex (secondary interactors).
SAR and structural evolution of the compound library
Chemoproteomics selectivity profiling with kinobeads granted deep insights into the interaction patterns of our compound library members with 217 native human kinases. This allowed us to track the changes in kinome selectivity accompanied by the evolution of structural features presented form the pyrrolo[3,4-c]pyrazol core scaffold (Fig. 3b). Nonetheless, we want to point out that single, high concentration competition data does not reflect absolute affinity values of competed kinases to the compounds but describes relative trends. We observed that the sterically less demanding compounds 1 and 2 showed a robust interaction with GSK3α and β as well as members of the cyclin-dependent kinase family (CDK1, 2 and 7) and the tyrosine kinase-like family (TKL), e.g. the bone morphogenic protein receptor type-1A (BMPR1A) and others (Fig. 3a, for the full interaction map see ESI,† Excel file S1). Substitution of the n-butyl/butyryl moiety with an aniline urea (4) or difluoroaniline urea substituent (7–9) led to a decrease in binding of GSK3 and the CDKs but interaction with the TKL kinases was retained; these compounds also formed a distinct sub-cluster which indicated that the fluorine substituents on the aniline portion did not contribute to kinome selectivity. Strikingly, when the aniline urea (as in 4) was modified with a chlorine atom at the 2-position (10–12), high relative affinity to GSK3 was restored and a significant gain in binding to TKL kinases and the casein kinase 2 catalytic subunit isoforms CSNK2A1-3 was observed. In contrast, interaction of 10–12 with CDKs was still decreased compared to 1 and 2. Substitution of the 2-chlorine with a methoxy group (24) or addition of another chlorine at the 3-position (16) had no impact on the relative affinity for GSK3 but strongly suppressed interactions with other protein kinases (with exception of the CK2 isoforms). The finding that 16 and 24 show exceptionally high selectivity for GSK3 may be valuable for future development of inhibitors with improved kinome selectivity. Addition of a chlorine atom at the 6-position of the 2-chloroaniline urea drastically altered the kinome selectivity (17–19, 21). Inhibitors with a 2,6-dichlorophenyl group at the R1 position show a gain in interaction with TKL kinases, in particular the LIM domain kinase 1 (LIMK1), the receptor tyrosine kinases (RTKs) epithelial discoidin domain-containing receptor 1 and 2 (DDR1/2) and the ephrin receptor isoforms A1, A7 and B4 and cyclin dependent kinases (especially CDK10); this was accompanied by a relative loss in binding of GSK3 and CK2. Compound 21, which displays the N-methyl piperidine moiety from the 3-position (R2) of the core scaffold, stood out of this sub-cluster of inhibitors in that it did not bind to RTKs. Exchange of the urea linkage in 10 and 12 (2-chloro anilines) with a squaramide moiety yielded compounds 38 and 39 that almost completely competed GSK3 from the kinobeads (log2 LFQ ratios ≥9), indicating a very high relative affinity of the squaramides for GSK3α and β among the compound library members. This is in agreement with our results from the in vitro GSK3β kinase activity assay (Table 2). All other compounds in the library show only moderate relative affinity to GSK3 and a varying degree of kinome selectivity.
To identify the presence of selective high affinity interactors, we re-tested compounds 11, 12, 16–18, 25, 38 and 39 in a second kinobead competition experiment at 1 μM concentration. The compounds 28, 33 and 36 were also re-tested because they efficiently competed the clinically relevant kinases ABL1, AURKA and PIP4K2 (ESI,† Excel file S1). Gratifyingly, the squaric acid amides 38 and 39 showed very high LFQ ratios (log2 > 7) exclusively for HsGSK3α and β competition at 1 μM competitor concentration whereas the 2-chloro aniline ureas showed much lower ratios (Fig. 3c). This indicated that the squaric acid amides have an exceptionally high affinity and selectivity for human GSK3. Three compounds, i.e.11, 17 and 18, still showed interaction with activin receptor type-1 (ACVR1 or ALK-2) at 1 μM concentration (11 also still bound GSK3) which indicated that these structures may represent leads for ALK-2 inhibitor development. Of note, 17 also interacted with LIMK1 at 1 μM. Compound 33 interacted with microtubule-associated serine/threonine protein kinase 3 (MAST3) at high and low competitor concentration which warrants investigation of 33 as a tool compound to probe the activity of this poorly characterized kinase. In contrast, no competition of ABL1, AURKA and PIP4K2 with 28, 33 and 36 could be detected at 1 μM concentration.
Chemical genetics analysis of kinases with chemoproteomics data
Chemoproteomics datasets for a series of inhibitors, like the one presented here, are very useful for the comparison of relative affinities of kinase targets to small molecule compounds (X-axis, Fig. 3a). Despite the high kinome coverage afforded by MS-chemoproteomics, it is more difficult to draw conclusions about similarities of kinase structural properties from these datasets.31 Kinase binding to the kinobead matrix, and thus the MS signal intensity, is dependent on each kinase's abundance in the lysate as well as the kinase's affinity to the individual kinobead matrix components, making it difficult to compare between kinases. In contrast, it has been shown previously that absolute affinity values of kinase-compound interactions can serve to group kinases according to structural features of the ATP-binding pocket.32 A possible workaround to make chemoproteomics binding data comparable between different kinases is the calculation of apparent dissociation constants from kinobead titration experiments,33 but here we intended to explore if single-dose competition data can be used to describe shared chemical genetic traits of kinases.We hypothesized that the correlation of LFQ competition patterns across large compound panels (series of log2 LFQ competition ratios) could facilitate the comparison of structural properties of kinases by using the compounds of the chemical series as probes. To investigate this possibility, we calculated a Pearson's correlation coefficient value (r) for each possible kinase pair from the 32 kinases that bound at least 3 competitors (496 possible combinations). In this analysis, the significance threshold for a Bonferroni-corrected p-value for pairwise functional association was r ≥ 0.58 (α = 0.05). The resulting symmetrical 32 × 32 data matrix (in which each value represents an r value of a kinase pair) was subjected to unsupervised hierarchical clustering in both dimensions to identify clusters of kinases that share similar inhibitor binding patterns (i.e. high r values; Fig. 4a). The validity of this approach was supported by the clustering of highly similar kinases and additionally several unexpected kinase associations were observed (Fig. 4b). Examples include a cluster of RTKs containing DDR1/2 and the ephrin receptors A1, A7 and B4 (r ranging from 0.75 to 0.96); the binding profiles of members of this cluster also correlated closely with the profile of CDK10 (r ranging from 0.71 to 0.83). A distinct cluster of the TGFβ-receptor family members ACVR1 and ACVR1B, BMPR1A and the TGFBR1 also comprised LIMK1 and the CAMK family member BRSK2 (r ranging from 0.62 to 0.80). Interestingly, GSK3A and B together with the CK2 catalytic subunit isoforms CSNK2A1-3 formed an isolated cluster indicating a close relationship of these kinases (Pearson's r for these interactions ranged from 0.76 to 0.95).
 | ||
| Fig. 4 Chemical genetic relationship of kinases interacting with inhibitor library members. (a) Heatmap showing the Pearson's r values for correlation analysis of competitor–kinase interactions (n = 39; log2 LFQ ratios at 50 μM inhibitor concentration) among all possible kinase pairs (n = 496; see Materials and methods). The data matrix was subjected to unsupervised hierarchical clustering in both dimensions to uncover kinase clusters with a similar inhibitor binding profile (high Pearson's r). The significance threshold for a Bonferroni-corrected p-value (496 comparisons) of 0.05 was r ≥ 0.58. (b) Kinome dendrogram showing the phylogenetic relationship of the 32 protein kinases that interacted with at least 3 out of 39 compounds from our library. Colored bars connect the kinase sub-groups with a significant chemical genetic association that were selected for discussion (for a complete matrix of r values see ESI,† Excel file S1). | ||
These findings and the observation that the substituents at the N-5 position (R1) dominate kinase selectivity suggest that single-dose MS-chemoproteomics data may be used to compare the structural properties of the hydrophobic pocket II in those kinases that interact with members of our compound library. The data may aid the discovery of preferred ligands for individual kinases and help to predict commonly encountered off-targets. We foresee that similar unbiased analyses of kinome binding with inhibitor libraries can guide the design of high affinity ligands for other kinases.
Detailed in vitro and in vivo characterization of a high affinity HsGSK3 interactor
We used a kinobead competition-titration experiment to compare the relative affinity of the compound that shows the strongest interaction with the main target GSK3, i.e. squaramide 39, with other kinase off-targets (Fig. 5a). Compound 39 was applied at 10 different concentrations ranging from 50 μM to 2.5 nM and MS label-free quantification yielded robust titration curves against ≥170 kinases present in the cell extract. With this assay, we determined that the EC50s for competition of HsGSK3α and β by 39 were 99 ± 23 nM and 57 ± 10 nM, respectively. Gratifyingly, the closely related kinase CDK2 (a common off-target of GSK inhibitors) showed an approximately 100-fold higher EC50 (4.5 ± 1.2 μM) and even higher values were observed for the other potential off-target interactors CSNK2A1–3 (19 ± 4.6 μM). Interestingly, we could also obtain titration curves for two known interactors of GSK3 and three interactors of CK2 which have been competed as part of a kinase complex. GSK3-beta interaction protein (GSKIP, EC50 = 21 ± 3.8 nM)34 and axin-1 (AXIN1, EC50 ∼ 2.5 nM)35 were competed with EC50 values in a similar range as HsGSK3α and β (Fig. 5b, upper panel). Further, the EC50s of casein kinase II subunit beta (CSNK2B, 11 ± 2.9 μM),36 protein DEK (DEK, EC50 = 16 ± 3.2 μM)37 and NF-kappa-B-activating protein (NKAP, 9.6 ± 3.6 μM)37 are very similar to the EC50 of CK2 competition (Fig. 5b, lower panel). From these data and the results from in vitro kinase inhibition (Table 2), we concluded that 39 is a highly selective and tight binder of HsGSK3α and β, comparable with the most advanced GSK3 inhibitors published to date (Fig. 2). Thus, 39 may be potentially useful for specific in vitro/in vivo inhibition of HsGSK3. | ||
| Fig. 5 Kinome chemoproteomics titration experiment and cellular activity of pyrrolo[3,4-c]pyrazol GSK3 inhibitors. (a) 10-point titration curves for compound 39 (see structure) constructed from the log2 LFQ competition ratios that show the main targets HsGSK3α and β (upper panel) and the two off-targets with the highest relative affinity after the main targets (lower panel). Compound competition was performed in duplicate and concentrations ranged from 50 μM to 2.5 nM (3-fold dilution steps). Data points are the means and error bars are the S.D. (b) Titration curves for HsGSK3α and β (upper panel) and CSNK2A1/3 interactors (lower panel) that were competed as part of a protein complex. Plotting is the same as in (a). (c) Western blot analysis of phospho-GYS1 S641 in extracts of C3A cells pretreated for 2 h with either DMSO (vehicle) or compounds 10, 38 and 39 at ten different concentrations ranging from 10 μM to 0.5 nM (3-fold dilution steps, upper panel). Quantification of the pGYS1 bands (biological duplicate) yielded titration curves and IC50 values (data points are the means and error bars are the S.D., lower panel). | ||
Consequently, we evaluated the ability of 39 to inhibit HsGSK3 enzymatic activity in cells by means of monitoring glycogen synthase 1 (GYS1) phosphorylation on serine 641; this is a well characterized GSK3 target phosphorylation site with important roles in metabolism.38 By western blot profiling of several human hepatocellular carcinoma cell lines, we determined that C3A cells showed high basal phosphorylation of GYS1 S641 (Fig. 5c). We performed a titration experiment in which we treated C3A cells with 39 as well as 38 and the 2-chloroaniline urea 10 at ten different concentrations ranging from 10 μM to 0.5 nM. Western blot analysis and quantification of the pGYS1 signal showed that 39 could reduce GYS1 S641 phosphorylation by ca. 70% after 2 h of exposure to 10 μM drug; this effect persisted down to a concentration of 0.4 μM. Plotting of a titration curve revealed a cellular IC50 of 39 for suppression of GYS1 phosphorylation of 0.32 ± 0.12 μM (Fig. 5c, lower panel). In contrast, 38 and 10 showed only a reduction of ca. 50% and 20% at 10 μM compound concentration and IC50 values of 0.56 ± 0.17 μM and 1.6 ± 0.45 μM, respectively. From these data, we concluded that 39 can efficiently suppress HsGSK3 activity in cells and that this compound may serve as a chemical genetic tool to perturb kinase activity in a variety of cellular and perhaps in vivo models.
Conclusion
We provide data demonstrating the value of kinobead-based chemoproteomics MS profiling in characterizing kinome binding for entire libraries of novel kinase inhibitors. Using unsupervised hierarchical clustering of quantitative MS competition data, we show that the method is sufficiently sensitive to group together structurally related compounds with similar target profiles. We thus characterized a 39-member library of pyrrolo[3,4-c]pyrazol inhibitors that were originally developed against TbGSK3 and obtained their SAR against 217 human protein and lipid kinases. We found that chemoproteomics data can be used to extract chemical genetics information about protein kinase structural features that could potentially be exploited to guide the development of novel, high affinity kinase inhibitors. Tantalizingly, we discovered a novel class of squaramide compounds that potently and selectively inhibited human GSK3α and β. These compounds, in particular 39, showed low nanomolar inhibitory concentrations (2.8 nM) in an in vitro kinase assay and 39 was able to suppress HsGSK3 catalytic activity in live cells with an IC50 of 0.32 μM. Compound 39 also showed an approximately >100 lower affinity for the structurally related kinase CDK2 and even lower affinity for the chemogenetically associated kinases CSNK2A1–3, the only two off-targets for which we could calculate EC50 values in the human kinome. We concluded that inhibitor 39 fulfills all criteria for being a high quality pharmacological probe39 for HsGSK3α and β and it will be exciting to explore its potential as a therapeutic for diseases caused by aberrant regulation of GSK3 and chemical genetic tool compound in future experiments.Conflicts of interest
The authors declare that there are no competing financial interests.Acknowledgements
We want to thank the members of the Ong and Maly Labs for comments on the manuscript and fruitful discussions. This work was supported by grants from the National Institutes of Health issued under the award numbers R01GM086858, R01GM083926, R01AI111341, R21EB018384, and R21CA177402, AI110743-01, AI123690 and AI080625 and by a postdoctoral research fellowship of the German Research Foundation (DFG) awarded to M. G. (GO 2358/1-1). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- E. Beurel, S. F. Grieco and R. S. Jope, Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases, Pharmacol. Ther., 2015, 148, 114–131 CrossRef CAS PubMed.
- E. M. Hur and F. Q. Zhou, GSK3 signalling in neural development, Nat. Rev. Neurosci., 2010, 11(8), 539–551 CrossRef CAS PubMed.
- D. Wu and W. Pan, GSK3: a multifaceted kinase in Wnt signaling, Trends Biochem. Sci., 2010, 35(3), 161–168 CrossRef CAS PubMed.
- P. Cohen and M. Goedert, GSK3 inhibitors: development and therapeutic potential, Nat. Rev. Drug Discovery, 2004, 3(6), 479–487 CrossRef CAS PubMed.
- H. Lal, F. Ahmad, J. Woodgett and T. Force, The GSK-3 family as therapeutic target for myocardial diseases, Circ. Res., 2015, 116(1), 138–149 CrossRef CAS PubMed.
- K. MacAulay, B. W. Doble, S. Patel, T. Hansotia, E. M. Sinclair, D. J. Drucker, A. Nagy and J. R. Woodgett, Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism, Cell Metab., 2007, 6(4), 329–337 CrossRef CAS PubMed.
- M. A. Mines and R. S. Jope, Glycogen synthase kinase-3: a promising therapeutic target for fragile x syndrome, Front. Mol. Neurosci., 2011, 4, 35 Search PubMed.
- L. K. Chico, L. J. Van Eldik and D. M. Watterson, Targeting protein kinases in central nervous system disorders, Nat. Rev. Drug Discovery, 2009, 8(11), 892–909 CrossRef CAS PubMed.
- C. Hooper, R. Killick and S. Lovestone, The GSK3 hypothesis of Alzheimer's disease, J. Neurochem., 2008, 104(6), 1433–1439 CrossRef CAS PubMed.
- K. K. Ojo, J. R. Gillespie, A. J. Riechers, A. J. Napuli, C. L. Verlinde, F. S. Buckner, M. H. Gelb, M. M. Domostoj, S. J. Wells, A. Scheer, T. N. Wells and W. C. Van Voorhis, Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy, Antimicrob. Agents Chemother., 2008, 52(10), 3710–3717 CrossRef CAS PubMed.
- N. G. Jones, E. B. Thomas, E. Brown, N. J. Dickens, T. C. Hammarton and J. C. Mottram, Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome-wide RNAi screen, PLoS Pathog., 2014, 10(1), e1003886 Search PubMed.
- R. O. Oduor, K. K. Ojo, G. P. Williams, F. Bertelli, J. Mills, L. Maes, D. C. Pryde, T. Parkinson, W. C. Van Voorhis and T. P. Holler, Trypanosoma brucei glycogen synthase kinase-3, a target for anti-trypanosomal drug development: a public–private partnership to identify novel leads, PLoS Neglected Trop. Dis., 2011, 5(4), e1017 CAS.
- F. F. Wagner, J. A. Bishop, J. P. Gale, X. Shi, M. Walk, J. Ketterman, D. Patnaik, D. Barker, D. Walpita, A. J. Campbell, S. Nguyen, M. Lewis, L. Ross, M. Weiwer, W. F. An, A. R. Germain, P. P. Nag, S. Metkar, T. Kaya, S. Dandapani, D. E. Olson, A. L. Barbe, F. Lazzaro, J. R. Sacher, J. H. Cheah, D. Fei, J. Perez, B. Munoz, M. Palmer, K. Stegmaier, S. L. Schreiber, E. Scolnick, Y. L. Zhang, S. J. Haggarty, E. B. Holson and J. Q. Pan, Inhibitors of Glycogen Synthase Kinase 3 with Exquisite Kinome-Wide Selectivity and Their Functional Effects, ACS Chem. Biol., 2016, 11(7), 1952–1963 CrossRef CAS PubMed.
- T. Kramer, B. Schmidt and F. Lo Monte, Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer's Disease Models, Int. J. Alzheimer's Dis., 2012, 2012, 381029 Search PubMed.
- L. Meijer, M. Flajolet and P. Greengard, Pharmacological inhibitors of glycogen synthase kinase 3, Trends Pharmacol. Sci., 2004, 25(9), 471–480 CrossRef CAS PubMed.
- M. D. Urbaniak, T. Mathieson, M. Bantscheff, D. Eberhard, R. Grimaldi, D. Miranda-Saavedra, P. Wyatt, M. A. Ferguson, J. Frearson and G. Drewes, Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei, ACS Chem. Biol., 2012, 7(11), 1858–1865 CrossRef CAS PubMed.
- R. Urich, R. Grimaldi, T. Luksch, J. A. Frearson, R. Brenk and P. G. Wyatt, The design and synthesis of potent and selective inhibitors of Trypanosoma brucei glycogen synthase kinase 3 for the treatment of human african trypanosomiasis, J. Med. Chem., 2014, 57(18), 7536–7549 CrossRef CAS PubMed.
- M. Golkowski, R. S. Vidadala, C. K. Lombard, H. W. Suh, D. J. Maly and S. E. Ong, Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors, J. Proteome Res., 2017, 16(3), 1216–1227 CrossRef CAS PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, The Perseus computational platform for comprehensive analysis of (prote)omics data, Nat. Methods, 2016, 13(9), 731–740 CrossRef CAS PubMed.
- D. Fancelli, D. Berta, S. Bindi, A. Cameron, P. Cappella, P. Carpinelli, C. Catana, B. Forte, P. Giordano, M. L. Giorgini, S. Mantegani, A. Marsiglio, M. Meroni, J. Moll, V. Pittala, F. Roletto, D. Severino, C. Soncini, P. Storici, R. Tonani, M. Varasi, A. Vulpetti and P. Vianello, Potent and selective Aurora inhibitors identified by the expansion of a novel scaffold for protein kinase inhibition, J. Med. Chem., 2005, 48(8), 3080–3084 CrossRef CAS PubMed.
- D. Fancelli, J. Moll, M. Varasi, R. Bravo, R. Artico, D. Berta, S. Bindi, A. Cameron, I. Candiani, P. Cappella, P. Carpinelli, W. Croci, B. Forte, M. L. Giorgini, J. Klapwijk, A. Marsiglio, E. Pesenti, M. Rocchetti, F. Roletto, D. Severino, C. Soncini, P. Storici, R. Tonani, P. Zugnoni and P. Vianello, 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazoles: identification of a potent Aurora kinase inhibitor with a favorable antitumor kinase inhibition profile, J. Med. Chem., 2006, 49(24), 7247–7251 CrossRef CAS PubMed.
- A. Gontarewicz, S. Balabanov, G. Keller, R. Colombo, A. Graziano, E. Pesenti, D. Benten, C. Bokemeyer, W. Fiedler, J. Moll and T. H. Brummendorf, Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I, Blood, 2008, 111(8), 4355–4364 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Targeting cancer with small molecule kinase inhibitors, Nat. Rev. Cancer, 2009, 9(1), 28–39 CrossRef CAS PubMed.
- A. S. Rose and P. W. Hildebrand, NGL Viewer: a web application for molecular visualization, Nucleic Acids Res., 2015, 43(W1), W576–W579 CrossRef CAS PubMed.
- M. P. Coghlan, A. A. Culbert, D. A. Cross, S. L. Corcoran, J. W. Yates, N. J. Pearce, O. L. Rausch, G. J. Murphy, P. S. Carter, L. Roxbee Cox, D. Mills, M. J. Brown, D. Haigh, R. W. Ward, D. G. Smith, K. J. Murray, A. D. Reith and J. C. Holder, Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription, Chem. Biol., 2000, 7(10), 793–803 CrossRef CAS PubMed.
- R. Bhat, Y. Xue, S. Berg, S. Hellberg, M. Ormo, Y. Nilsson, A. C. Radesater, E. Jerning, P. O. Markgren, T. Borgegard, M. Nylof, A. Gimenez-Cassina, F. Hernandez, J. J. Lucas, J. Diaz-Nido and J. Avila, Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418, J. Biol. Chem., 2003, 278(46), 45937–45945 CrossRef CAS PubMed.
- D. B. Ring, K. W. Johnson, E. J. Henriksen, J. M. Nuss, D. Goff, T. R. Kinnick, S. T. Ma, J. W. Reeder, I. Samuels, T. Slabiak, A. S. Wagman, M. E. Hammond and S. D. Harrison, Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo, Diabetes, 2003, 52(3), 588–595 CrossRef CAS PubMed.
- M. Golkowski, J. L. Brigham, G. K. Perera, G. E. Romano, D. J. Maly and S. E. Ong, Rapid profiling of protein kinase inhibitors by quantitative proteomics, MedChemComm, 2014, 5(3), 363–369 RSC.
- S. Heinzlmeir, J. Lohse, T. Treiber, D. Kudlinzki, V. Linhard, S. L. Gande, S. Sreeramulu, K. Saxena, X. Liu, M. Wilhelm, H. Schwalbe, B. Kuster and G. Medard, Chemoproteomics-Aided Medicinal Chemistry for the Discovery of EPHA2 Inhibitors, ChemMedChem, 2017, 12(12), 999–1011 CrossRef CAS PubMed.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ, Mol. Cell. Proteomics, 2014, 13(9), 2513–2526 CAS.
- J. T. Metz, E. F. Johnson, N. B. Soni, P. J. Merta, L. Kifle and P. J. Hajduk, Navigating the kinome, Nat. Chem. Biol., 2011, 7(4), 200–202 CrossRef CAS PubMed.
- D. Huang, T. Zhou, K. Lafleur, C. Nevado and A. Caflisch, Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis, Bioinformatics, 2010, 26(2), 198–204 CrossRef CAS PubMed.
- G. Medard, F. Pachl, B. Ruprecht, S. Klaeger, S. Heinzlmeir, D. Helm, H. Qiao, X. Ku, M. Wilhelm, T. Kuehne, Z. Wu, A. Dittmann, C. Hopf, K. Kramer and B. Kuster, Optimized chemical proteomics assay for kinase inhibitor profiling, J. Proteome Res., 2015, 14(3), 1574–1586 CrossRef CAS PubMed.
- M. Varjosalo, S. Keskitalo, A. Van Drogen, H. Nurkkala, A. Vichalkovski, R. Aebersold and M. Gstaiger, The protein interaction landscape of the human CMGC kinase group, Cell Rep., 2013, 3(4), 1306–1320 CrossRef CAS PubMed.
- T. Nakamura, F. Hamada, T. Ishidate, K. Anai, K. Kawahara, K. Toyoshima and T. Akiyama, Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level, Genes Cells, 1998, 3(6), 395–403 CrossRef CAS PubMed.
- O. Marin, F. Meggio, S. Sarno and L. A. Pinna, Physical dissection of the structural elements responsible for regulatory properties and intersubunit interactions of protein kinase CK2 beta-subunit, Biochemistry, 1997, 36(23), 7192–7198 CrossRef CAS PubMed.
- M. Varjosalo, R. Sacco, A. Stukalov, A. van Drogen, M. Planyavsky, S. Hauri, R. Aebersold, K. L. Bennett, J. Colinge, M. Gstaiger and G. Superti-Furga, Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS, Nat. Methods, 2013, 10(4), 307–314 CrossRef CAS PubMed.
- Y. Wang and P. J. Roach, Inactivation of rabbit muscle glycogen synthase by glycogen synthase kinase-3. Dominant role of the phosphorylation of Ser-640 (site-3a), J. Biol. Chem., 1993, 268(32), 23876–23880 CAS.
- P. Workman and I. Collins, Probing the probes: fitness factors for small molecule tools, Chem. Biol., 2010, 17(6), 561–577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mo00006e |
| This journal is © The Royal Society of Chemistry 2018 |