
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiels–Alder reactions between hexafluoro-2-butyne and bis-furyl dienes: kinetic versus thermodynamic control†
Kseniya K.
Borisova
 a,
Eugeniya V.
Nikitina
a,
Roman A.
Novikov
b,
Victor N.
Khrustalev
a,
Pavel V.
Dorovatovskii
c,
Yan V.
Zubavichus
c,
Maxim L.
Kuznetsov
d,
Vladimir P.
Zaytsev
a,
Alexey V.
Varlamov
a and
Fedor I.
Zubkov
*a
a,
Eugeniya V.
Nikitina
a,
Roman A.
Novikov
b,
Victor N.
Khrustalev
a,
Pavel V.
Dorovatovskii
c,
Yan V.
Zubavichus
c,
Maxim L.
Kuznetsov
d,
Vladimir P.
Zaytsev
a,
Alexey V.
Varlamov
a and
Fedor I.
Zubkov
*a
aOrganic Chemistry Department, Faculty of Science, Peoples’ Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow, 117198, Russian Federation. E-mail: fzubkov@sci.pfu.edu.ru
bV. A. Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 32 Vavilov St., Moscow, 119991, Russian Federation. E-mail: novikovfff@bk.ru
cNational Research Center “Kurchatov Institute”, 1 Acad. Kurchatov Sq., Moscow, 123182, Russia. E-mail: yzubav@gmail.com
dCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, Lisbon 1049-001, Portugal. E-mail: max@mail.ist.utl.pt
First published on 2nd February 2018
Abstract
The tandem [4+2] cycloaddition between hexafluoro-2-butyne and bis-furyl dienes, like difurfuryl ester, at room temperature leads to the kinetically controlled “pincer”-adducts – annulated 4a,8a-bis(trifluoromethyl)hexahydro-1,4:5,8-diepoxynaphthalenes. On the other hand, if these reactions proceed at 140 °C, only the thermodynamically controlled “domino”-adducts – annulated 2,3-bis(trifluoromethyl)hexahydro-1,4:5,8-diepoxynaphthalenes – are formed. Therefore, a very rare and unexpected example of full kinetic and thermodynamic control in the Diels–Alder reaction is reported in this paper.
Domino and tandem intramolecular Diels–Alder reactions of furans (IMDAF reaction) are widely used in organic synthesis for different purposes due to their simplicity, reliability and controlled stereochemistry.1 In particular, the products of the IMDAF reaction are often applied as starting materials for the synthesis of functionally substituted naphthalenes, indoles, isoindoles, quinolines and isoquinolines.1c
The products of the reaction between furans and alkynes have a 7-oxabicyclo[2.2.1]heptene scaffold. The structural motif has great potential as a tool for the design and synthesis of a broad diversity of substances with various practically useful properties. For example, recently these cycloadducts have been used for the construction of polycyclic aromatic hydrocarbons – fragments of graphene, which can serve as a model for new carbon based electronic materials.2 The annulated 7-oxabicyclo[2.2.1]heptane moiety acts as a framework for molecular tweezers,3 various supramolecular systems,4 donor–bridge–acceptor molecules,5 a wide range of bioactive and natural compounds,6 high-molecular weight materials,6e,7etc.
Taking into account such widespread actual or potential applications of the IMDAF reaction products, design of new efficient molecular systems for these processes and the understanding of intimate mechanistic details and driving forces underlying these reactions are of obvious general importance.
Various alkynes were used in the tandem intramolecular [4+2] cycloaddition, but only limited information concerning the reactivity of fluorinated acetylenes is available.8 In 1985, this journal published pioneering work by Visnick and Battiste,9 who reported an unprecedented example of the total kinetic and thermodynamic control in the course of the domino [4+2] cycloaddition between hexafluorobut-2-yne and N,N′-dipyrrolylmethane (1, Scheme 1). When the reaction was carried out under kinetically controlled conditions (room temperature), only the pincer-type polycycle 3 was formed through the open intermediate 2. Upon heating, however, the pincer adduct 3 underwent an intramolecular rearrangement into the domino-type adduct 4. Both reactions proceeded in quantitative yields, unlike other known cycloadditions of this type,10 which usually lead to a mixture of products of kinetic and thermodynamic control, regardless of the reaction temperature. For a long time, the transformation discussed in that paper had been attractive in synthetic11 and theoretical12 chemistry research, but it remained unique due to the limited accessibility of the suitable starting bis-dienes.
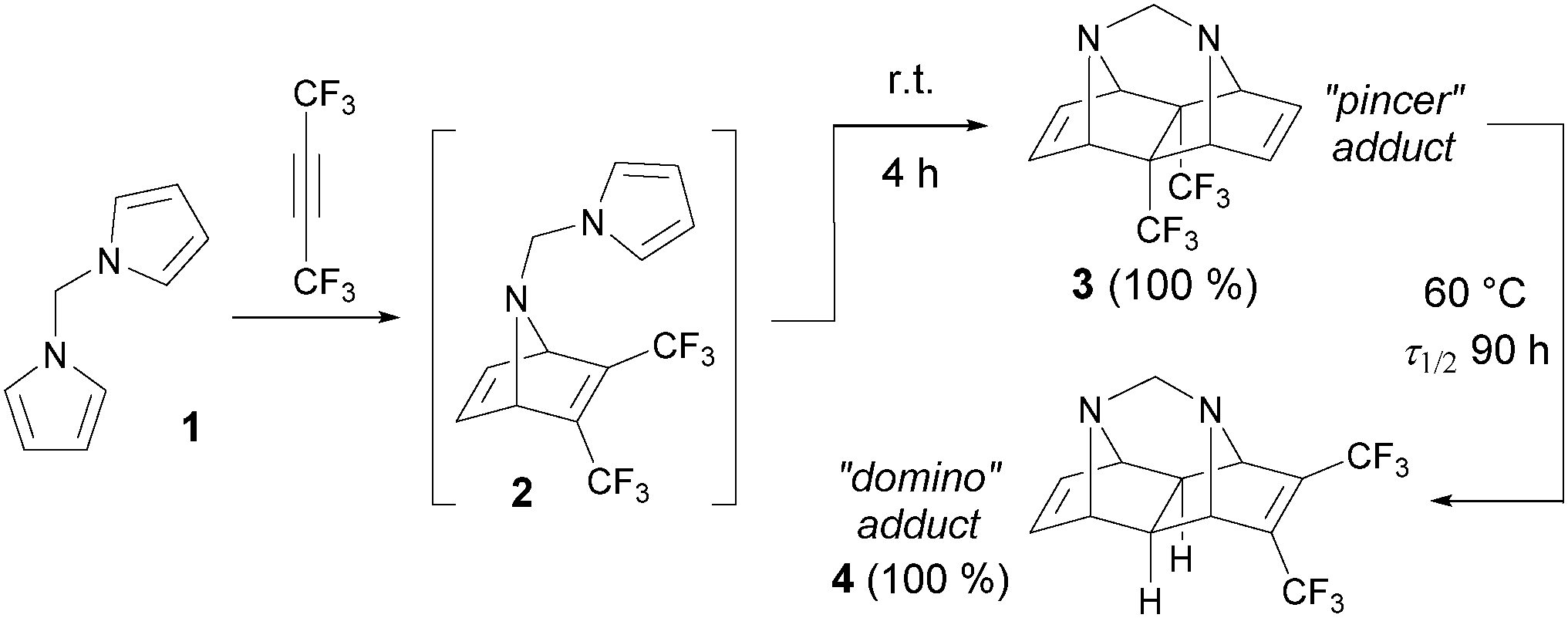 | ||
| Scheme 1 Example of the total temperature control in the tandem intramolecular Diels–Alder reaction.9a | ||
In 1997, the second example similar to system 1 appeared in a report by Lautens and Fillion who demonstrated that difuryl bis-dienes in a reaction with alkynes are able to give pincer-cycloadducts analogous to 3 under kinetically controlled conditions.13
Having obtained a significant insight from previous reports about the importance of kinetic and thermodynamic control in intra/intermolecular [4+2]-cycloaddition reactions, our main goal in this study was to expand the available classes of dienes and dienophiles for the implementation of the tandem Diels–Alder reaction under kinetic or thermodynamic control conditions. In this work, we focused on bis-furyldienes 5 that can be easily obtained in a two-step process,10a,13a and on hexafluorobutyne as a dienophile. The latter, apparently, is the most active known dienophile and usually ensures excellent yields of the cycloaddition products at low temperatures. However, the experimental treatment of this alkyne has some peculiarities due to its gaseous state under normal conditions.
Theoretical DFT calculations (IEFPCM-M06-2X/6-311++G**) of the reaction between F3CC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCF3 and three dienes (5a, 5c, and 5g) were carried out (for computational details, see the ESI†). The reaction starts with the cycloaddition of F3CCCCF3 at one of the furan moieties which occurs in a concerted fashion viaTS1 (Fig. 1) and represents the rate-limiting step of the whole process with the activation barrier of 23.1–26.8 kcal mol−1 (in terms of ΔG≠).
CCF3 and three dienes (5a, 5c, and 5g) were carried out (for computational details, see the ESI†). The reaction starts with the cycloaddition of F3CCCCF3 at one of the furan moieties which occurs in a concerted fashion viaTS1 (Fig. 1) and represents the rate-limiting step of the whole process with the activation barrier of 23.1–26.8 kcal mol−1 (in terms of ΔG≠).
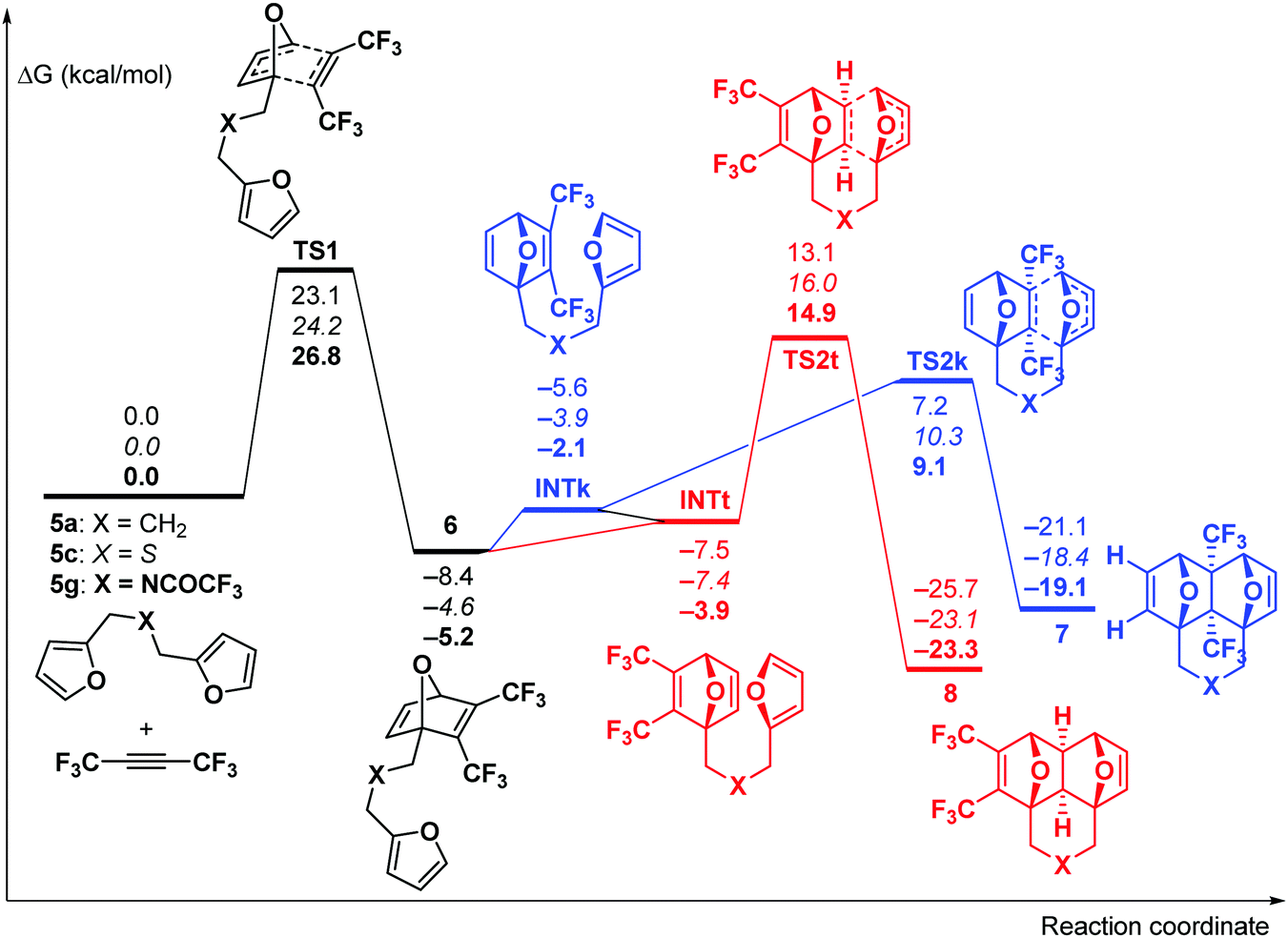 | ||
| Fig. 1 Gibbs free energy profile for the reaction between bis-dienes 5a, c, and g and F3CCCCF3. The relative energies are given in kcal mol−1 for X = CH2 (plain text), S (italic) and NC(O)CF3 (bold). | ||
Further, the reaction could proceed via two competing channels, i.e. either leading to the pincer-type products 7viaTS2k or resulting in the formation of the domino product 8viaTS2t. The calculations showed that the first channel is more kinetically favourable (by 5.7–5.9 kcal mol−1), which is in agreement with the published experimental observations9,13 on the exclusive formation of the pincer products at room temperature. Meanwhile, the domino products 8 are more thermodynamically stable than 7 by 4.2–4.7 kcal mol−1, and this fact may cause the isomerization of 7 into 8 at elevated temperature. Indeed, the calculated activation barriers for the 7 → 8 isomerization via the retro-Diels–Alder reaction of 7 followed by the intramolecular [4+2]-cycloaddition in the chain intermediate 6 to give 8 are 34.0–34.4 kcal mol−1. These values are reasonable to permit such a process at a sufficiently high temperature. Taking into account these predictions, the following experiments were performed.
Under kinetically controlled conditions, the reaction of bis-dienes 5 with hexafluorobutyne was carried out in sealed glass ampules at −70 °C, with subsequent increase of temperature to ∼21–24 °C during 2–3 h. Toluene was chosen as a solvent due to its inertness, relatively low volatility and low melting point. Under these conditions, the reaction can occur within five days, but in order to reach a true equilibrium, all reactions were run for 10 days at r.t.
The 1H NMR data of the crude reaction mixtures obtained after gentle evaporation of the solvent are given in Table 1. In most cases, the cycloaddition occurs chemospecifically leading to the kinetically controlled adducts 7a, d, g, and h only in quantitative yields or proceeds with a high level of chemoselectivity; the ratio of pincer/domino adducts is 7/8 > 95/5 (entries e, f, and i). The ratio of diene/alkyne varied from 1/1 to 1/1.4, and in most cases did not significantly affect both the yield of the products and the composition of the reaction mixtures.
| Entry | X | Molar ratio of 5/C4F6a | Ratiob of 7/8 | 7 (%) | 8 , (%) |
|---|---|---|---|---|---|
| a All reactions were carried out in sealed ampules at r.t. for 10 days. b Ratio of 7/8 according to the 1H NMR analysis of the reaction mixtures obtained after solvent evaporation. c Isolated yields after recrystallization. d Domino-adducts 8 were obtained by heating of pincer-adducts 7 in o-xylene for 2 h. e See Table 2. | |||||
| a | CH2 | 1/1.1 | 100/0 | 69 | 62 |
| b | O | 1/1.1 | —e | 60 | 67 |
| c | S | 1/1.1 | —e | 76 | 76 |
| d | N-Bn | 1/1.4 | 100/0 | 71 | 66 |
| e | N-Ac | 1/1.1 | >95/5 | 62 | 68 |
| f | N-Bz | 1/1.1 | >96/4 | 70 | 76 |
| g | N-COCF3 | 1/1.4 | 100/0 | 56 | 77 |
| h | N-COCCl3 | 1/1 | 100/0 | 62 | 74 |
| i | N-CO2Me | 1/1 | >95/5 | 79 | 71 |
However, two examples in Table 1 (rows b: X = O and c: X = S) drop out of the series and gave rise to a lower chemoselectivity (Scheme 2).
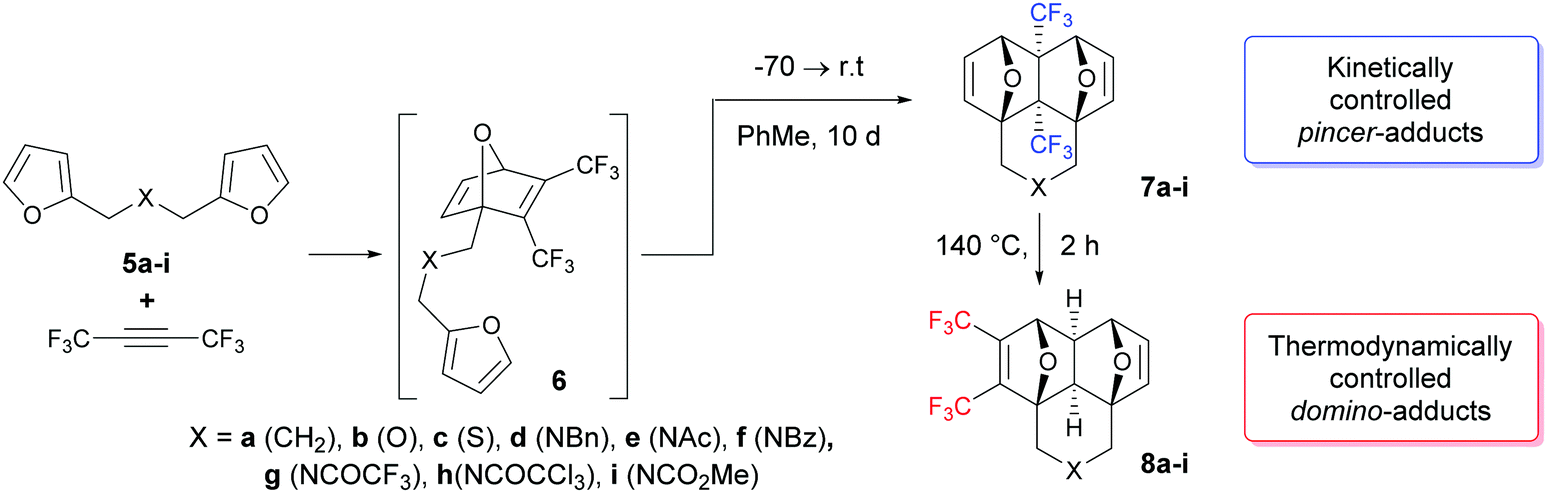 | ||
| Scheme 2 Synthesis of pincer and domino adducts 7 and 8. | ||
In these cases, in addition to the side domino-adducts 8b and c, the products of the double cycloaddition 9b and c and 10b and c were formed as inseparable diastereomeric pairs with a practically equal ratio of components (Scheme 3 and Table 2).
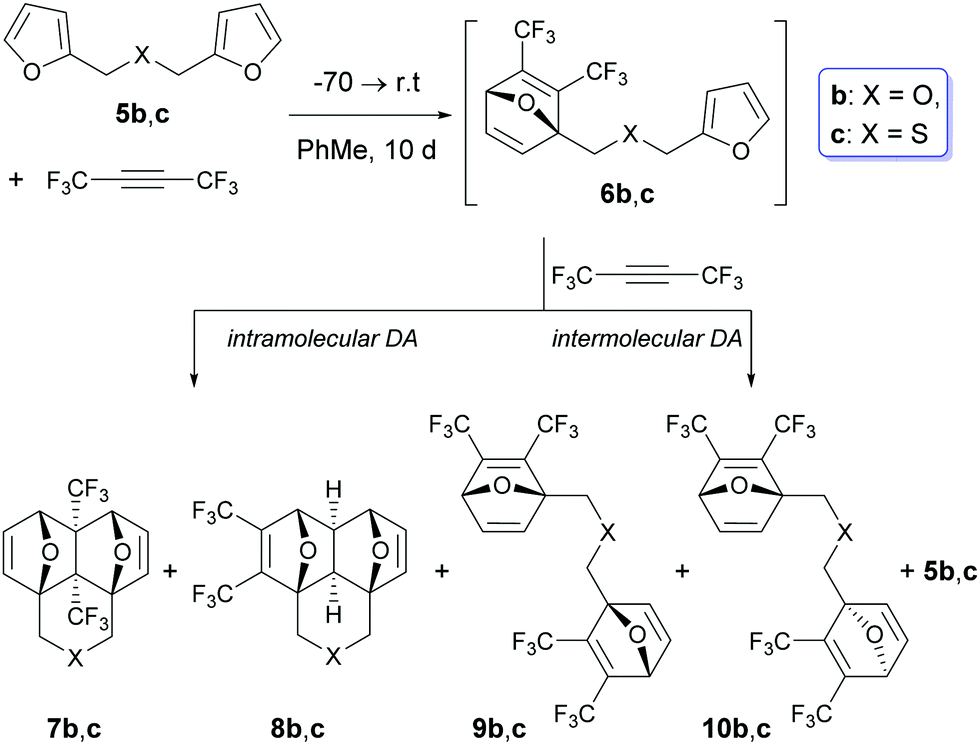 | ||
| Scheme 3 Compositions of the reaction mixtures in the case of the [4+2] cycloaddition between F3CCCCF3 and bis-furans 5b and c. | ||
CCF3 according to 1H NMR analysis
The formation of bis-adducts 9 and 10 suggests that in these examples, the intermolecular Diels–Alder reaction involving a subsequent interaction of the second molecule of hexafluorobutyne with intermediates 6b and c takes place. In other words, the intermolecular [4+2] cycloaddition between C4F6 and intermediate 6 starts to compete with the intramolecular Diels–Alder reaction in the chain intermediate.
Initially, the reactions of 5b and c were carried out with 1.0 mol equivalent of C4F6 under similar conditions: 10 days at r.t. (rows b and c in Table 2). At the equimolar ratio of 5/C4F6, the reaction mixtures contained 8–15% of unreacted starting bis-furans 5b and c, therefore the molar ratio of 5/C4F6 was changed by 1/1.1. Under these conditions (rows b′ and c′), the percentage of the initial 5b and c in the crude reaction mixtures was reduced to 4–5%. Simultaneously, a sharp increase (up to 28%) in the content of the double-adducts 9 and 10 was observed. Due to this, we did not perform further experiments with a larger excess of hexafluorobutyne. The data shown in Table 2 allow us to conclude that the excess of alkyne exerts a significant influence on the fraction of the double cycloaddition adducts 9 and 10, but at the same time, the ratio of 7/8 remains virtually constant (it changes only from 96/4 to 93/7).
The rearrangement of the pincer-adducts 7 to the domino-products 8 is observed at temperatures higher than 110 °C (1H NMR experiments with a temperature gradient were carried out) and proceeds rather quickly at 120 °C. According to NMR spectroscopic data in boiling o-xylene, the products of thermodynamic control 8 were formed in 100% yields at 1–2 h, but after recrystallization they were isolated in moderate yields (Table 1).
Since the last transformation 7 → 8 occurred in reasonable time without any by-products, the half-life and rate constant have been calculated using dynamic 1H NMR spectroscopy assuming first-order kinetics. The reaction involving the rearrangements of 7b, c, and g to 8b, c, and g was carried out in an NMR tube in CDCl2–CDCl2 (for details, see the ESI†). At 120 °C, the half-life (τ1/2) was 37.5 min, 80 min, and 42.5 min (for X = O, S, and N–COCF3, respectively), which corresponds to the first-order reaction rate constants of 0.0185 min−1, 0.0087 min−1, and 0.0163 min−1. The transformation of 7b to 8b (X = O) is the quickest one along the series. As expected, at 140 °C in the same solvent, the reactions proceed even faster. For example, for the synthesis of compound 8b, τ1/2 was ∼4.6 min with the first-order reaction rate constants of 0.151 min−1. The rearrangement is so quick at 140 °C that it is difficult to accurately measure the reaction rate by the NMR method.
Our attempts to carry out the analogical dynamic NMR experiment for the first step of the reaction under discussion (the cycloaddition of C4F6 to 5a) in a sealed NMR tube failed due to its insufficient strength against the elevated pressure.
The spatial structures of the regioisomer pairs 7c, d, and g/8c, d, and g have been determined by single-crystal X-ray diffraction using synchrotron radiation (see the ESI†).
In conclusion, a rare example of full kinetic and thermodynamic reaction control in the process of the tandem IMDAF reaction of bis-furyl dienes with hexafluoro-2-butyne has been discovered. At room temperature, the reaction occurs chemoselectively leading to adducts of the pincer-[4+2] cycloaddition. The exclusive formation of domino-adducts is observed at elevated temperatures. This reaction could be useful for the construction of natural product and supramolecular frameworks and can serve as an outstanding example of reaction reversibility to be used in physical chemistry courses.
Funding for this research was provided by the Ministry of Education and Science of the Russian Federation (award No. 4.1154.2017/4.6) and by the Fundação para a Ciência e a Tecnologia (FCT, Portugal) (project UID/QUI/00100/2013).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews on tandem intramolecular Diels–Alder cycloaddition in organic synthesis, see: (a) J. E. Sears and D. L. Boger, Acc. Chem. Res., 2016, 49, 241–251 CrossRef CAS PubMed; (b) P. T. Parvatkar, H. K. Kadam and S. G. Tilve, Tetrahedron, 2014, 70, 2857–2888 CrossRef CAS; (c) A. Padwa and S. K. Bur, Tetrahedron, 2007, 63, 5341–5378 CrossRef CAS PubMed; (d) J. Wu, L. Sun and W.-M. Dai, Tetrahedron, 2006, 62, 8360–8372 CrossRef CAS; (e) K.-I. Takao, R. Munakata and K.-I. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed; (f) J. D. Winkler, Chem. Rev., 1996, 96, 167–176 CrossRef CAS PubMed.
- (a) S. Eda, F. Eguchi, H. Haneda and T. Hamura, Chem. Commun., 2015, 51, 5963–5966 RSC; (b) A. Criado, M. Vilas-Varela, A. Cobas, D. Peréz, D. Peña and E. Guitián, J. Org. Chem., 2013, 78, 12637–12649 CrossRef CAS PubMed; (c) F. Furrer, A. Linden and M. C. Stuparu, Chem. – Eur. J., 2013, 19, 13199–13206 CrossRef CAS PubMed.
- (a) R. B. Murphy, R. E. Norman, J. M. White, M. V. Perkins and M. R. Johnston, Org. Biomol. Chem., 2016, 14, 8707–8720 RSC; (b) R. N. Warrener, D. Margetic, A. S. Amarasekara, D. N. Butler, I. B. Mahadevan and R. A. Russell, Org. Lett., 1999, 1, 199–202 CrossRef CAS.
- (a) T.-C. Chou and Y.-J. Li, Tetrahedron, 2015, 71, 5620–5633 CrossRef CAS; (b) C. H. Oh, H. J. Yi and K. H. Lee, Bull. Korean Chem. Soc., 2010, 31, 683–688 CrossRef CAS; (c) M. Eckert-Maksić, D. Margetić, S. Kirin, D. Milić and D. Matković-Čalogović, Eur. J. Org. Chem., 2005, 4612–4620 CrossRef.
- S. Chakrabarti, M. Liu, D. H. Waldeck, A. M. Oliver and M. N. Paddon-Row, J. Am. Chem. Soc., 2007, 129, 3247–3256 CrossRef CAS PubMed.
- (a) S. Roscales and J. Plumet, Nat. Prod. Commun., 2017, 12, 713–732 Search PubMed; (b) V. S. Enev, W. Felzmann, A. Gromov, S. Marchart and J. Mulzer, Chem. – Eur. J., 2012, 18, 9651–9668 CrossRef CAS PubMed; (c) A. Gromov, V. Enev and J. Mulzer, Org. Lett., 2009, 11, 2884–2886 CrossRef CAS PubMed; (d) C. S. Schindler and E. M. Carreira, Chem. Soc. Rev., 2009, 38, 3222–3241 RSC; (e) P. Vogel, Tetrahedron, 1999, 55, 13521–13642 CrossRef CAS.
- (a) D. Margetić, M. Eckert-Maksić, P. Trošelj and Z. Marinić, J. Fluorine Chem., 2010, 131, 408–416 CrossRef; (b) R. N. Warrener, D. Margetić, P. J. Foley, D. N. Butler, A. Winling, K. A. Beales and R. A. Russell, Tetrahedron, 2001, 57, 571–582 CrossRef CAS.
- For cycloaddition reactions between furans (pyrroles) and fluorinated alkynes, see: (a) D. Kozai, Y. Kabasawa, M. Ebert, S. Kiyonaka, Firman, Y. Otani, T. Numata, N. Takahashi, Y. Mori and T. Ohwada, Mol. Pharmacol., 2014, 85, 175–185 CrossRef PubMed; (b) A. A. Kislukhin, C. J. Higginson, V. P. Hong and M. G. Finn, J. Am. Chem. Soc., 2012, 134, 6491–6497 CrossRef CAS PubMed; (c) K. M. Guckian, E. Y.-S. Lin, L. Silvian, J. E. Friedman, D. Chin and D. M. Scott, Bioorg. Med. Chem. Lett., 2008, 18, 5249–5251 CrossRef CAS PubMed; (d) T. Yanagimoto, T. Toyota, N. Matsuki, Y. Makino, S. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2007, 129, 736–737 CrossRef CAS PubMed; (e) G.-D. Zhu, M. A. Staeger and S. A. Boyd, Chem. Lett., 2000, 3345–3348 CAS; (f) L. R. Domingo, M. T. Picher and M. J. Aurell, J. Phys. Chem. A, 1999, 103, 11425–11430 CrossRef CAS.
- (a) M. Visnick and M. A. Battiste, J. Chem. Soc., Chem. Commun., 1985, 1621–1622 RSC; (b) G. Sun, D. N. Butler, R. N. Warrener and D. Margetić, J. Heterocycl. Chem., 2015, 52, 1195–1200 CrossRef CAS.
- For selected reports on kinetic and thermodynamic control in the Diels–Alder reaction, see: (a) R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS PubMed; (b) C. Taffin, G. Kreutler, D. Bourgeois, E. Clot and C. Périgaud, New J. Chem., 2010, 34, 517–525 RSC; (c) J. D. White, F. W. J. Demnitz, H. Oda, C. Hassler and J. P. Snyder, Org. Lett., 2000, 2, 3313–3316 CrossRef CAS PubMed; (d) A. P. Marchand, B. Ganguly, W. H. Watson and S. G. Bodige, Tetrahedron, 1998, 37, 10967–10972 CrossRef; (e) M. Manoharan and P. Venuvanalingam, J. Chem. Soc., Perkin Trans. 2, 1997, 1799–1804 RSC; (f) S. G. Bott, A. P. Marchand and K. A. Kumar, J. Chem. Crystallogr., 1996, 26, 281–286 CrossRef CAS; (g) D. Suarez, T. L. Sordo and J. A. Sordo, J. Org. Chem., 1995, 60, 2848–2852 CrossRef CAS; (h) P. D. Bartlett and C. Wu, J. Org. Chem., 1985, 50, 4087–4092 CrossRef CAS; (i) L. A. Paquette, M. J. Wyvratt, H. C. Berk and R. E. Moerck, J. Am. Chem. Soc., 1978, 100, 5845–5855 CrossRef CAS.
- L. F. Tietze, G. Brasche and K. M. Gericke, in the book Domino reactions in organic synthesis, Wiley-VCH Verlag GmbH & Co. KgaA, 2006, pp. 280–336 Search PubMed.
- For the density functional theory study of the reaction between hexafluorobut-2-yne and 1, see: (a) L. R. Domingo, M. Arnó and J. Andrés, J. Am. Chem. Soc., 1998, 120, 1617–1618 CrossRef CAS; (b) L. R. Domingo, M. T. Picher, M. Arnó, J. Andrés and V. S. Safont, THEOCHEM, 1998, 426, 257–262 CrossRef CAS.
- (a) M. Lautens and E. Fillion, J. Org. Chem., 1997, 62, 4418–4427 CrossRef CAS PubMed; (b) L. R. Domingo, M. T. Picher and J. Andrés, J. Org. Chem., 2000, 65, 3473–3477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Single-crystal X-ray descriptions for 7c, d, and g and 8c, d, and g; computational details and tables with energies and atomic coordinates; detailed synthetic procedures and spectral data for compounds 5, 7, and 8. CCDC 1570123–1570128. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc09466c |
| This journal is © The Royal Society of Chemistry 2018 |