
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of tunable peptide nucleic acid junctions†
Tanghui
Duan
a,
Liu
He
a,
Yu
Tokura
b,
Xin
Liu
c,
Yuzhou
Wu
 *ab and
Zhengshuang
Shi
*a
*ab and
Zhengshuang
Shi
*a
aHubei Key Laboratory of Bioinorganic Chemistry and Materia Medica, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Luoyu Road 1037, 430074 Hongshan, Wuhan, P. R. China. E-mail: wuyuzhou@hust.edu.cn; kevinshi@gmail.com
bMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wuyuzhou@mpip-mainz.mpg.de
cDepartment of Biomedical Engineering, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China
First published on 19th January 2018
Abstract
We report here the construction of 3-way and 4-way peptide nucleic acid (PNA) junctions as basic structural units for PNA nanostructuring. The incorporation of amino acid residues into PNA chains makes PNA nanostructures with more structural complexity and architectural flexibility possible, as exemplified by building 3-way PNA junctions with tunable nanopores. Given that PNA nanostructures have good thermal and enzymatic stabilities, they are expected to have broad potential applications in biosensing, drug delivery and bioengineering.
DNA/RNA and peptide/protein molecules possess many unique physicochemical properties that are selected during the natural evolution over millions of years.1 They are perfect building blocks to form sophisticated nanomachineries with a variety of biological functions in living organisms.2,3 Inspired by nature, both DNA/RNA and peptide/protein based nanotechnologies have attracted mounting attention during the past 35 years.4–6 DNA/RNA nanostructures can be designed with arbitrary shapes and complicated geometries; they were demonstrated to have great potential for application in molecular computation, macromolecular scaffolding and drug delivery.7,8 While DNA nanostructures from computer-assisted designs could be obtained with defined shape, size, and stoichiometry,9,10 their stabilities are highly dependent on buffer conditions;11 more importantly, their functions and applications are limited compared to those from proteins. Peptides/proteins can be manipulated to fabricate diverse nanostructures such as nanotubes, nanovesicles and nanofibers, and they are widely used in drug release, gene delivery and tissue engineering.12,13 While it is difficult to design peptide/protein nanostructures in a similar precise fashion to DNA nanostructure designs, peptide/protein nanostructures can incorporate motifs with intrinsic structural robustness and complexity, architectural flexibility and chemical versatility.14 From this aspect, an ideal material would take advantage of both DNA and protein building blocks. Chemically joining DNA and peptides/proteins together was successful but with many limitations, and joining DNA and peptides together block-by-block remains a challenge due to their distinct physicochemical properties.15–17 Using PNAs instead of DNAs might overcome the issue since both PNA and peptide building blocks are compatible with standard solid-phase synthesis protocols,18,19 while the desired features of DNA and peptides are retained in the nanostructures constructed.
PNA is a synthetic analogue of DNA/RNA, which was first designed by Nielsen and co-workers in 1991.20 They reported the replacement of sugar phosphate backbones in DNA/RNA by neutral N-(2-aminoethyl)glycine units, which possess the same base-pair recognition properties as those of DNA/RNA and similar properties characteristic of peptides.19,21 The stability of PNA double chains is higher than the corresponding DNA/RNA structures since PNAs are neutral.22 Also, lacking related proteases or nucleases makes PNAs digestion-resistant in cells.23 PNAs were used in numerous biological and chemical applications over the past two decades.24,25
Herein, we designed PNA 4-way (Fig. 1A) and 3-way (Fig. 1B) junctions (named as P4J and P3J respectively) to demonstrate the possibility of using PNA to design nanostructures similar to those from DNA. Junctions could act as rigid and functional motifs for more complicated 2D and 3D nanostructures;26 both junctions were studied quite extensively in the literature.26–29 The sequences (Table 1) are designed according to the principles proposed by Seeman30 and were originally inspired by the DNA Holliday junction.31 Cavities in DNA 3-way junctions are capable of accommodating guest molecules;26,28 here we have designed PNA 3-way junctions with variable sizes of cavities by inserting one, three or five alanine residues in the middle of the PNA chains (chains 7–15; they are named P3J1a, P3J3a and P3J5a as shown in Fig. 1C–E, respectively). For each chain, one or two lysine residues were incorporated into the C-terminus or N-terminus of PNA sequences to increase solubility (Table 1). For chains 5–15, the N-terminus was capped with 4-pentynoic acid and the C-terminus was modified with lysine-N3; efficient click reactions are expected to occur at the vertices of the nanostructures after correct hybridization.
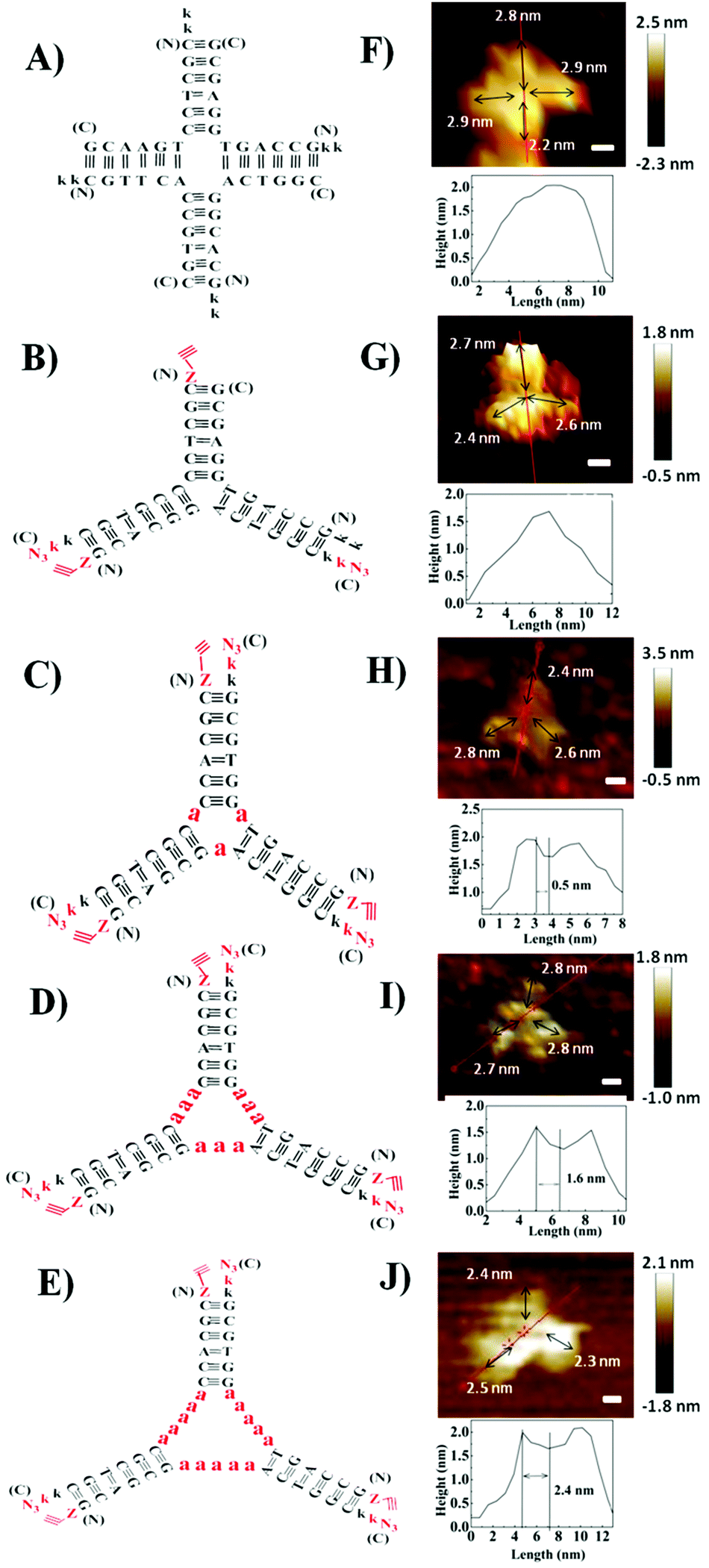 | ||
| Fig. 1 Schemes of PNA and PNA–peptide nanostructures for P4J (A), P3J (B), P3Ja1 (C), P3Ja3 (D) and P3Ja5 (E) are on the left; AFM images of the P4J (F), P3J (G), P3Ja1 (H), P3Ja3 (I) and P3Ja5 (J) nanostructures are on the right. Images are shown with a scale bar at of 2 nm. The height cross section is indicated by the red line. | ||
| Nanostructure | Chain number | Sequence of PNA chains | Expected (m/z) | Found (m/z) |
|---|---|---|---|---|
| P4J (chains 1–4); and P3J (chains 4–6) | Chain 1 | kkCGTTCACCGTGC–NH2 | 3475.47 | 3475.39 |
| Chain 2 | kkGCACGGACTGGC–NH2 | 3549.49 | 3549.55 | |
| Chain 3 | kkCGCTCCTGAACG–NH2 | 3484.48 | 3484.11 | |
| Chain 4 | kkGCCAGTGGAGCG–NH2 | 3589.50 | 3589.17 | |
| Chain 5 | Z-GCACGGACTGGCk–k(N3) | 3656.51 | 3656.63 | |
| Chain 6 | Z-CGCTCCCCGTGCk–k(N3) | 3543.48 | 3543.26 | |
| P3Ja1 (chains 7–9) | Chain 7 | Z-GCCAGTaGGTGCGk–k(N3) | 3758.64 | 3758.81 |
| Chain 8 | Z-GCAGCGaACTGGCk–k(N3) | 3727.55 | 3727.43 | |
| Chain 9 | Z-CGCACCaCGCTGCk–k(N3) | 3623.53 | 3623.67 | |
| P3Ja3 (chains 10–12) | Chain 10 | Z-GCCAGTaaaGGTGCGk–k(N3) | 3900.62 | 3900.60 |
| Chain 11 | Z-GCAGCGaaaACTGGCk–k(N3) | 3869.62 | 3869.77 | |
| Chain 12 | Z-CGCACCaaaCGCTGCk–k(N3) | 3765.60 | 3765.51 | |
| P3Ja5 (chains 13–15) | Chain 13 | Z-GCCAGTaaaaaGGTGCGk–k(N3) | 4042.69 | 4042.50 |
| Chain 14 | Z-GCAGCGaaaaaACTGGCk–k(N3) | 4011.70 | 4011.63 | |
| Chain 15 | Z-CGCACCaaaaaCGCTGCk–k(N3) | 3907.67 | 3907.79 | |
PNA and PNA–peptide nanostructures were self-assembled by thermal annealing. For P3J, P3J1a, P3J3a and P3J5a, each set of three individual single chains were mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratios, while for P4J, a set of four individual single chains (chains 1–4) were mixed in a 2:1:1:1 molar ratio. The PNA nanostructures were examined by cation exchange fast protein liquid chromatography (FPLC) and MALDI-TOF mass spectroscopy. FPLC traces (Fig. 2A) showed that there were two peaks (peaks 1 & 2) from P4J. Observing the mass of chain 1 only in peak 1 together with the retention time information signifies that peak 1 corresponds to chain 1, which was in excess within the reaction mixture; whereas the masses of all four chains (chains 1–4; Fig. S4, ESI†) that were found in peak 2 indicate the successful assembly of P4J, which showed higher affinity to the column compared to the single strand. For P3J, there is one major peak with a longer retention time compared to the single strand (Fig. 2B). As a control if only two chains were mixed, the corresponding assembly showed a slightly shorter retention time than that of P3J (Fig. 2B). Masses corresponding to all three chains (chains 4–6) were observed in the major peak of the P3J sample, while two chains (chains 5 & 6) were found in the major peak of the control sample (mixtures of chains 5 & 6; see Fig. S5 (ESI†) for details of mass data used for peak assignments).
:1:1 molar ratios, while for P4J, a set of four individual single chains (chains 1–4) were mixed in a 2:1:1:1 molar ratio. The PNA nanostructures were examined by cation exchange fast protein liquid chromatography (FPLC) and MALDI-TOF mass spectroscopy. FPLC traces (Fig. 2A) showed that there were two peaks (peaks 1 & 2) from P4J. Observing the mass of chain 1 only in peak 1 together with the retention time information signifies that peak 1 corresponds to chain 1, which was in excess within the reaction mixture; whereas the masses of all four chains (chains 1–4; Fig. S4, ESI†) that were found in peak 2 indicate the successful assembly of P4J, which showed higher affinity to the column compared to the single strand. For P3J, there is one major peak with a longer retention time compared to the single strand (Fig. 2B). As a control if only two chains were mixed, the corresponding assembly showed a slightly shorter retention time than that of P3J (Fig. 2B). Masses corresponding to all three chains (chains 4–6) were observed in the major peak of the P3J sample, while two chains (chains 5 & 6) were found in the major peak of the control sample (mixtures of chains 5 & 6; see Fig. S5 (ESI†) for details of mass data used for peak assignments).
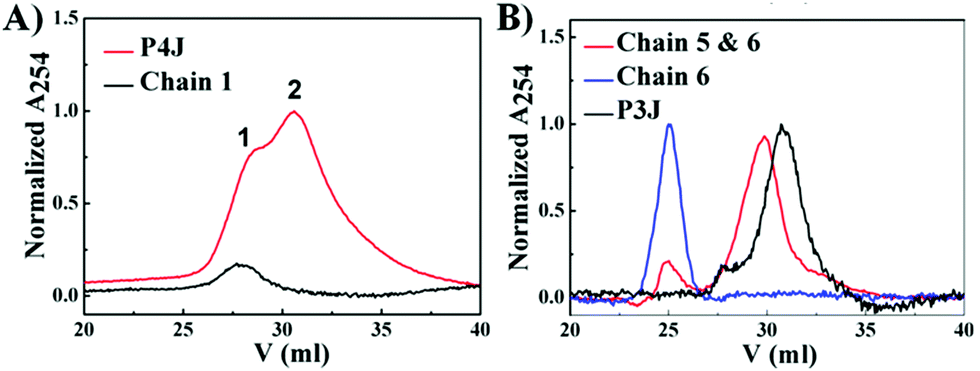 | ||
| Fig. 2 FPLC profiles of PNA P4J (A) and P3J (B). | ||
To visualize the self-assembled structures of P4J and P3J, atomic force microscopy (AFM) images were acquired. The images were taken in pure water using the liquid tapping mode. As shown in Fig. 1F, G and Fig. S6A, B (ESI†), the formations of the 4-way and 3-way junction structures were clearly distinguishable; we noted the resolutions of the images were not very high since the objects were fragile and small. Given that the distance between adjacent bases in PNA is 0.36 nm (the same as in DNA),32 the calculated length of each arm comprised of 6 bases is approximately 2.2 nm. The measured average length of each arm from the AFM images is 2.6 ± 0.2 nm (n = 70, see the ESI† for more images and measurement details),33 consistent with the theoretical calculations. The measured height of each structure is between 1.5 and 2 nm, consistent with the theoretical width of a PNA double helix.34 From all the evidence, we can conclude that desired nanostructures with defined size and shape are successfully constructed from our designed PNA single chains.
To characterize the PNA–peptide nanostructures (P3Ja1, P3Ja3 and P3Ja5) after self-assemblies, the hybridized PNA chains were locked together from the end of each arm using copper catalyzed azide–alkyne cycloaddition (CuAAC) reactions (see Fig. 1C and E). Then, one could find out which chains are connected together in the nanostructures by analyzing the MALDI-TOF mass spectra. Click chemistry was proven to be an efficient method to identify DNA and DNA–RNA G-quadruplex structures.35 Compared to FRET and NMR spectroscopy, which provide only averaged results, snapshots of samples after varying reaction times and different species trapped by the click reaction can be purified and analyzed.36 Details of the click reactions for the three PNA–peptide nanostructures are described in the ESI.† All products from the reactions were purified by HPLC (see Fig. S7, S8A, S9A and S10A, ESI†) and characterized by MALDI-TOF mass spectroscopy (see Fig. S8B–O, S9B–O and S10B–N, ESI†). MALDI-TOF mass spectroscopy (see Tables S1–S3, ESI†) revealed that there were three click reaction intermediate products corresponding to three possible reactions between two out of three different single chains, and no reaction products were observed between any two of the same sequences. We found products from reactions among three different single chains, and we observed no reaction products from three same sequences, or from two same sequences plus another single chain. If the nanostructures had not been formed, the probability of a single chain reacting to itself would be the same as the probability to other chains. However, in our self-assembled nanostructures, click reactions were observed between different single chains only, and not between the same sequences; the data indicate that the nanostructures were formed by complementary strands hybridized as expected. This is indeed verified by the control experiment using three random sequences that were not complementary. We observed click reaction intermediates between different sequences and between the same sequences with the same probability. The reaction efficiency was low and a large amount of single chains remained unreacted (see the ESI† for details).
The AFM images of the PNA–peptide nanostructures are shown in Fig. 1H–J and Fig. S6C–E (ESI†). The pores in the middle of the structure can be clearly observed on the cross section of the height profile. The theoretical length of each alanine is about 0.5 nm; thus the sizes of the nanopores for P3Ja1, P3Ja3 and P3Ja5 are expected to be about 0.5, 1.5 and 2.5 nm respectively. The measured average sizes of the nanopores for P3Ja1, P3Ja3 and P3Ja5 are 0.6 ± 0.1, 1.6 ± 0.1, and 2.5 ± 0.2 nm, respectively (n = 10, see the ESI† for more images and calculation details),33 which are consistent with theoretical sizes. The arm lengths of the PNA–peptide nanostructures are expected to be the same as that for P3J, whereas the measured average length of each arm is 2.3 ± 0.3 nm (n = 90).33 The conclusions from AFM corroborate with those from the click reactions.
The UV melting curves of all PNA nanostructures were monitored at 260 nm37 in pure water (see Fig. 3). The Tm values are calculated as described previously (see Fig. S3A–E, ESI†).38,39 UV measurements showed the PNA and PNA–peptide nanostructures are stable in water at room temperature. The Tm of P3J is 45.7 ± 0.3 °C; for P4J, a biphasic melting curve was observed, consistent with a report for the corresponding DNA Holliday junctions30 that was explained by the fact that there are more A–T base pairs in the horizontal arms than in the vertical arms. The melting temperatures from either phase are higher for the PNA nanostructure than those for the DNA Holliday junction. With P4J, Tm1 (1st phase) is 30.3 ± 0.5 °C and Tm2 (2nd phase) is 47.6 ± 0.4 °C; correspondingly, Tm1 = 10.2 ± 0.3 °C and Tm2 = 30.6 ± 0.5 °C were observed for the DNA Holliday junction30 with the same sequences. The results show that PNA nanostructures have higher thermal stability than the corresponding DNA nanostructures. This is expected due to the neutral backbone in PNAs. It is worthwhile to highlight that one critical limitation of using DNA to form larger nanostructures, such as in a DNA origami technique, is the requirement of a high concentration of bivalent cations to neutralize the negatively charged phosphate backbone. This limitation could be overcome easily by using PNAs since the neutral peptide backbone avoids the charge repulsion among helices. In addition, the Tm values of P3Ja1, P3Ja3 and P3Ja5 were found to be 43.8 ± 0.4 °C, 42.2 ± 0.4 °C, and 39.2 ± 0.2 °C, respectively. It is noteworthy that the elongation of the peptide chains reduces the stability of the nanostructures. It is understandable since a 12 mer DNA double helix is expected to be more stable than two 6 mer DNA double helixes linked by a flexible chain.
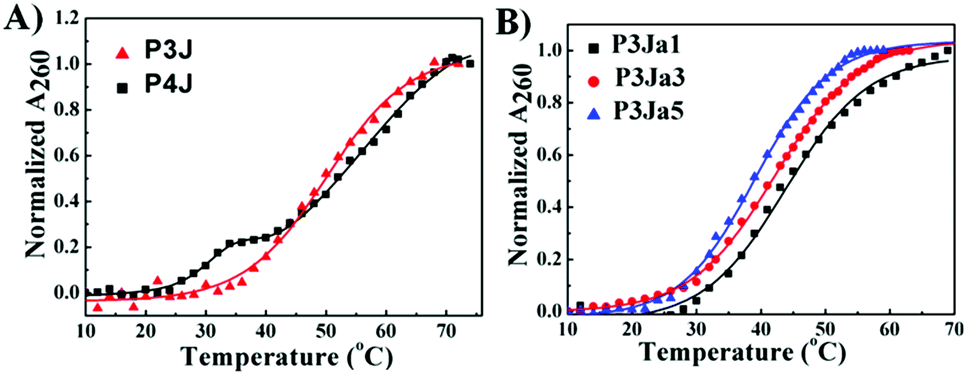 | ||
| Fig. 3 UV melting curves for PNA (A) and PNA–peptide nanostructures (B), and the corresponding lines are from fitting. | ||
In summary, defined nanostructures were constructed from PNA strands and PNA–peptide hybrid chains. The structures were characterized by FPLC, MALDI-TOF mass spectroscopy and AFM. UV melting curves reveal that PNA nanostructures have high thermostability even in pure water, which gives a significant advantage in comparison to DNA nanostructures. This indicates that PNAs could be promising substitutes for DNAs to overcome the stability limitations of DNA nanostructures. In addition, the peptide backbone allows easy incorporation of amino acids into PNA nanostructures, which provides attractive flexibility and expands the structural and functional versatilities into PNA nanostructures. This has been demonstrated by inserting amino acids in the middle of the PNA chains to create defined nanopores with different sizes within the designed 3-way PNA junction. It is expected that decorations of other functional groups using further reactions (such as the click chemistry as demonstrated in this study) would be straightforward; as a result, connecting PNAs further with other materials would provide PNA nanostructures with other advantages. Therefore, in view of the powerful DNA-based nanofabrication techniques and their broad applications in sensing, medicine and photonics, the development of PNA based nanotechnology would have more potential benefits and broader applications. The challenge in preparing larger PNA nanostructures is mainly the large scale and high throughput synthesis of PNA, which will certainly be overcome with the development of more efficient PNA synthesis techniques in the future.
This work was supported by the National Natural Science Foundation of China (21272081, 51703073) and the 1000 Young Talent Program of China. We thank the AFM facility in MPIP, the Analytical and Testing Centre of HUST, the Analytical and Testing Centre of the School of Chemistry and Chemical Engineering (HUST), and Research Core Facilities for Life Science (HUST) for instrument support. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Follmann and C. Brownson, Nat. Sci., 2009, 96, 1265–1292 CrossRef CAS PubMed.
- O. Berger, L. Adler-Abramovich and M. Levy-Sakin, et al. , Nat. Nanotechnol., 2015, 10, 353–360 CrossRef CAS PubMed.
- W. C. W. Chan, Biol. Blood Marrow Transplant., 2006, 12, 87–91 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- H. M. Meng, H. Liu and H. Kuai, et al. , Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- J. Li, C. Fan and H. Pei, et al. , Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- J. S. Shin and N. A. Pierce, J. Am. Chem. Soc., 2004, 126, 10834–10835 CrossRef CAS PubMed.
- C. J. Delebecque, A. B. Lindner and P. A. Silver, et al. , Science, 2011, 333, 470–474 CrossRef CAS PubMed.
- H. Zuo, S. Wu and M. Li, et al. , Angew. Chem., Int. Ed., 2015, 127, 15333–15336 CrossRef.
- T. C. Marsh, J. Vesenka and E. Henderson, Nucleic Acids Res., 1995, 23, 696–700 CrossRef CAS PubMed.
- H. A. Behanna, J. J. J. M. Donners and A. C. Gordon, et al. , J. Am. Chem. Soc., 2005, 127, 1193–1200 CrossRef CAS PubMed.
- M. E. Davis, P. C. H. Hsieh and T. Takahashi, et al. , Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS PubMed.
- Q. Luo, C. Hou and Y. Bai, et al. , Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- R. Liu, J. E. Barrick and J. W. Szostak, et al. , Methods Enzymol., 2000, 318, 268–293 CAS.
- T. Wang, A. Pfisterer and S. L. Kuan, et al. , Chem. Sci., 2013, 4, 1889–1894 RSC.
- S. Kovacic, L. Samii and G. Lamour, et al. , Biomacromolecules, 2014, 15, 4065–4072 CrossRef CAS PubMed.
- A. Gupta, R. Bahal, M. Gupta, P. M. Glazer and W. M. Saltzman, J. Controlled Release, 2016, 240, 302–311 CrossRef CAS PubMed.
- P. E. Nielsen, Curr. Opin. Biotechnol., 1999, 10, 71–75 CrossRef CAS PubMed.
- P. E. Nielsen and R. H. Berg, Science, 1991, 254, 1497 CAS.
- T. Ratilainen, A. Holmén, E. Tuite, P. E. Nielsen and B. Nordén, Biochemistry, 2000, 39, 7781–7791 CrossRef CAS PubMed.
- V. V. Demidov, V. N. Potaman and M. D. Frank-Kamenetskil, et al. , Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS PubMed.
- P. Lundberg, K. Kilk and Ü. Langel, Cell-Penetrating Peptide-Mediated Delivery of Peptide Nucleic Acid (PNA) Oligomers, Cold Spring Harbor Protocols, 2008, pdb. prot4889 Search PubMed.
- F. Mussbach, M. Franke and A. Zoch, et al. , J. Cell. Biochem., 2011, 112, 3824–3833 CrossRef CAS PubMed.
- J. D. Flory, S. Shinde and S. Lin, et al. , J. Am. Chem. Soc., 2013, 135, 6985–6993 CrossRef CAS PubMed; J. L. H. A. Duprey, Y. Takezawa and M. Shionoya., Angew. Chem., 2013, 125, 1250–1254 CrossRef.
- J. L. H. A. Duprey, Y. Takezawa and M. Shionoya, Angew. Chem., 2013, 125, 1250–1254 CrossRef.
- M. Probst, D. Wenger, S. M. Biner and R. Häner, Org. Biomol. Chem., 2012, 10, 755–759 CAS.
- S. Phongtongpasuk, S. Paulus, J. Schnabl, R. K. Sigel, B. Spingler, M. J. Hannon and E. Freisinger, Angew. Chem., Int. Ed., 2013, 52, 11513–11516 CrossRef CAS PubMed.
- H. Yang, F. Altvater and A. D. de Bruijn, et al. , Angew. Chem., Int. Ed., 2011, 50, 4620–4623 CrossRef CAS PubMed.
- N. C. Seeman and N. R. Kallenbach, Biophys. J., 1983, 44, 201–209 CrossRef CAS PubMed.
- R. Holliday, Genet. Res., 1964, 5, 282–304 CrossRef.
- P. S. Lukeman, A. C. Mittal and N. C. Seeman, Chem. Commun., 2004, 1694–1695 RSC.
- For the length of arms, n is the number of arms measured in all pictures. For the size of nanopores, n is the number of pictures measured.
- H. Rasmussen, J. S. Kastrup, J. N. Nielsen, J. M. Nielsen and P. E. Nielsen, Nat. Struct. Biol., 1997, 4, 98–101 CrossRef CAS PubMed.
- Y. Xu, Y. Suzuki and T. Ishizuka, et al. , Bioorg. Med. Chem., 2014, 22, 4419–4421 CrossRef CAS PubMed.
- Y. Xu, Y. Suzuki and M. Komiyama, Angew. Chem., Int. Ed., 2009, 48, 3281–3284 CrossRef CAS PubMed.
- E. A. Lesnik, L. M. Risen and D. A. Driver, et al. , Nucleic Acids Res., 1997, 25, 568–574 CrossRef CAS PubMed.
- D. Iverson, C. Serrano and A. M. Brahan, et al. , Arch. Biochem. Biophys., 2015, 587, 1–11 CrossRef CAS PubMed.
- J. L. Mergny and L. Lacroix, Oligonucleotides, 2003, 13, 515–537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures. See DOI: 10.1039/c8cc00108a |
| This journal is © The Royal Society of Chemistry 2018 |