
Impact of anti-biofouling surface coatings on the properties of nanomaterials and their biomedical applications
Yuancheng
Li
a,
Yaolin
Xu
a,
Candace C.
Fleischer
 a,
Jing
Huang
b,
Run
Lin
c,
Lily
Yang
d and
Hui
Mao
*a
a,
Jing
Huang
b,
Run
Lin
c,
Lily
Yang
d and
Hui
Mao
*a
aDepartment of Radiology and Imaging Sciences, Emory University School of Medicine, Atlanta, GA 30322, USA. E-mail: hmao@emory.edu
bVascular Biology Program, Boston Children's Hospital, Boston, MA 02115, USA
cDepartment of Radiology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong 510080, People's Republic of China
dDepartment of Surgery, Emory University School of Medicine, Atlanta, GA 30322, USA
First published on 30th October 2017
Abstract
Understanding and subsequently controlling the non-specific interactions between engineered nanomaterials and the biological environment have become increasingly important for further developing and advancing nanotechnology for biomedical applications. Such non-specific interactions, also known as the biofouling effect, mainly associate with the adsorption of biomolecules (such as proteins, DNAs, RNAs, and peptides) onto the surface of nanomaterials and the adhesion or uptake of nanomaterials by various cells. By altering the surface properties of nanomaterials, the biofouling effect can lead to in situ changes of physicochemical properties, pharmacokinetics, functions, and toxicity of nanomaterials. This review provides discussions on the current understanding of the biofouling effect, the factors that affect the non-specific interactions associated with biofouling, and the impact of the biofouling effect on the performance and functions of nanomaterials. An overview of the development and applications of various anti-biofouling coating materials to preserve and improve the properties and functions of engineered nanomaterials for intended biomedical applications is also provided.
 Yuancheng Li | Dr Yuancheng Li is currently a postdoctoral researcher in Department of Radiology and Imaging Sciences at Emory University School of Medicine. He received his BS (2006) in applied chemistry from Beijing Technology and Business University in China, and PhD (2012) in organic chemistry from Auburn University in USA. Dr Li's research interests include the development of novel anti-biofouling nanomaterials for in vitro diagnosis and in vivo molecular imaging and therapy, as well as the metabolite detection and quantification by nuclear magnetic resonance spectroscopy. |
 Yaolin Xu | Dr Yaolin Xu received his BS in biological engineering from Shanxi University in 2009, and PhD in chemical engineering from the University of Alabama in 2014. He joined Dr Hui Mao's research group as a postdoctoral researcher in Department of Radiology and Imaging Sciences at Emory University in 2015. Dr Xu's current research is focused on the development of novel iron oxide nanoparticle based MRI contrast agents, and utilization of these magnetic-nanomaterials for targeted cancer imaging and therapy, and for the fast and reliable in vitro detection of biomarkers in biological samples. |
 Candace C. Fleischer | Dr Candace Fleischer received her PhD from the Georgia Institute of Technology in 2014. She completed post doctoral training in the Departments of Biomedical Engineering and Radiology and Imaging Sciences at Emory University. She is currently an Assistant Professor of Radiology and Imaging Sciences at Emory University School of Medicine. Her research is focused on the development of new spectroscopic methods for applications in cancer and neurological injury. |
 Jing Huang | Dr Jing Huang received her PhD from the College of Chemical and Molecular Engineering of Peking University in 2010. During her doctorate study, she worked on developing novel MRI contrast agents for biomedical imaging applications. She worked in the Department of Radiology and Imaging Sciences at Emory University in 2011–2015 as a postdoctoral researcher, focusing on magnetic nanomaterials-based nanotheranostics for targeted cancer imaging and therapy. She is currently working at Boston Children's Hospital as a research fellow. |
 Lily Yang | Dr Lily Yang is Nancy Panoz Chair Professor in Cancer Research in Department of Surgery at Emory University School of Medicine. Dr Yang received her medical training at West China University of Medical Sciences in China and then the Chinese Academy of Preventive Medicine. She received her PhD in Molecular and Cellular Biology at Brown University. Dr Yang's research interests focus on the development of novel targeted cancer nanotherapeutics and imaging methods using multifunctional drug carriers to address the major challenges in clinical oncology of cancer detection, targeted drug delivery, assessment of therapeutic response using non-invasive imaging, and image-guided surgery. |
 Hui Mao | Dr Hui Mao is Professor of Radiology in the Department of Radiology and Imaging Sciences at Emory University School of Medicine with joint appointment as Professor of Emory-Georgia Tech Joint Department of Biomedical Engineering. Dr Mao received bachelor and master degrees in chemistry from Nanjing University in China, and PhD in biophysical chemistry from Georgia State University, followed with post-doctoral training at Vanderbilt University. Research in his laboratory focuses on MRI-based molecular imaging, nanotechnology for imaging, drug delivery and in vitro diagnostics, NMR/MRS-based metabolomics for the discovery of biomarkers and imaging markers for disease detection, guiding intervention and monitoring treatments. |
1. Introduction
With a growing number of engineered nanomaterials developed for biomedical applications (e.g., imaging probes, drug delivery carriers, and liquid-biopsies), their non-specific interactions with the biological milieu have drawn increasing attention due to the effects of such interactions on in situ physicochemical properties, pharmacokinetics, functions, and toxicities of the nanomaterials.1 As illustrated in Fig. 1, these non-specific interactions, also known as the biofouling effect, often result in: (1) surface adsorption of biomolecules (e.g., proteins, DNA, RNA, and peptides) present in the biological media used for in vitro investigations or the circulating blood and organs in vivo, onto the surface of exogenous materials; and (2) adhesion or uptake of the materials by various cells, for example, macrophages from the mononuclear phagocyte system (MPS). These two types of interactions are closely and dynamically associated with one another, with variable contributions during the process by which a biological system encounters and interacts with engineered materials. It has been suggested that engineered materials with a “synthetic identity”, determined by its physicochemical properties after synthesis, are modified and given a distinct “biological identity” after interacting non-specifically with biomolecules.2 The biologically modified materials then function and interact with cells based on their new “biological identity” rather than the original design. The properties and intended functions of exogenous engineered materials may be altered in ways including, but not limited to, change of biodistribution and blood circulation half-life, loss of affinity of the targeting ligand, untimely release of payload drugs, or gain of immunogenicity leading to cytotoxicity.1b,3 As biofouling effects exist for most interfaces between the biological environment and engineered materials, a better understanding of the biofouling effect is important to further improve the design, function, and performance of engineered biomaterials, particularly various nanoparticles (NP) developed for biomedical applications, such as in vivo ligand-mediated targeted imaging and drug delivery,1c biosensors,4 and in vitro diagnostic devices (e.g., biomarker targeted detection of cells and analytes).5 | ||
| Fig. 1 Illustration of non-specific interactions of NPs with biomolecules, such as oligonucleotides, amino acids or peptides, and proteins, which can alter the physicochemical properties, pharmacokinetics, cytotoxicity, immune response, and biomarker targeting of the NPs. | ||
To reduce or even inhibit the biofouling effect, current efforts have been focused on the development of surface coating materials with anti-biofouling properties. A variety of coating materials, including synthetic polymers such as polyethylene glycol (PEG), poly(N-vinylpyrrolidone) (PVP), PEG-based copolymers, zwitterionic materials, and biomimetic materials such as polysaccharides and cell membranes, have been reported. Mechanistic investigations have also been carried out to understand the factors controlling interactions between artificial materials and biological systems such as the shape and size of the NPs, elasticity and rigidity of the NPs as well as the thickness and density of the coating, and the effects on delivery, biodistribution, and toxicity.6
In this review, we will provide an overview of the current understanding of interactions between engineered nanomaterials and the biological environment, including the factors that influence the interactions and the impact of biofouling on the performance and function of nanomaterials for biomedical applications. We will also discuss recent advances in the development of anti-biofouling surface coating materials. Given the scope and focus of this review, formulations/compositions of specific nanomaterials, which also possess significant impact on their biomedical applications, will not be discussed here. These topics have been covered by several excellent reviews and the references therein.4,7
2. Interactions between biological media and engineered nanoparticles
The high surface energy of engineered NPs results in rapid interactions with biomolecules and cells in highly dynamic and often irreversible manners, compromising the desired functionalities as the results of altered surface properties. Practically, engineered NPs designed for biomedical applications are tested, modified, and validated in vitro, either in solution or with cultured cells. However, when used for in vivo applications, the “optimized” NPs encounter a diverse and dynamic environment of cells, proteins, low molecular weight ions, and other biomolecules that can change the properties and functions of the NPs. The interactions of biomolecules (e.g., small molecules, proteins) and cells with engineered NPs are highly dependent on the physicochemical properties of the material including size, charge, and surface chemistry as well as the electronic and chemical properties of the biomolecules.2.1. NP Interactions with small molecules
Due to the negligible van der Waals forces of relatively small molecules (compared to proteins), electrostatic forces largely dominate the interactions between engineered NPs and small molecules in the biological environment. Anionic or cationic molecules, such as DNA, RNA, peptides, and amino acids, are highly attracted to NPs with charged surfaces. While small molecules with low molecular weight in the biological environment are abundant and diverse, this review will focus on nucleic acids and amino acids as representative examples.In addition to electrostatic forces, the conformation of DNA plays an important role in DNA–NP interactions. The double helical structure protects nitrogenous bases in double-stranded DNA (ds-DNA) from exposure to NPs, limiting the interactions of dsDNA with NPs.11 In comparison, single-stranded DNA (ss-DNA) is typically more flexible and can bind tightly to NPs by wrapping around the NP surface compared to relatively rigid ds-DNA.8 Sandstrom et al. demonstrated the role of ion-induced dipole dispersive interactions in the non-specific ds-DNA binding to gold NPs with a size of 13 nm. The size of the NP was crucial in this binding mechanism, as the polarizability was reduced significantly when the particle diameter decreased from 13 nm to 5 nm.12
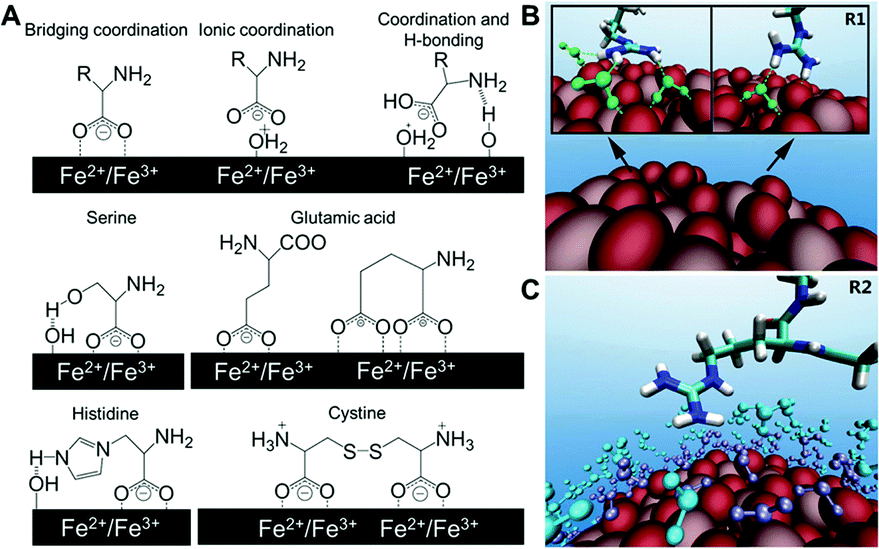 | ||
| Fig. 2 (A) Structures of different binding states of amino acids on an iron oxide NP surface and possible interactions of side chains with the surface in water. (B) Representative configurations of arginine forming hydrogen bonds with TiO2 NP (green dash lines), and (C) arginine with first (purple) and second (cyan) water layers on the NP surface. Adapted with permission.13,14 Copyright 2015 American Chemical Society and 2016 Nature Publishing Group. | ||
A more recent investigation on the interactions between TiO2 NPs and 20 standard amino acids by Liu et al. used all-atom molecular dynamics simulation to provide theoretical and analytical insights on previous experimental findings. It is found that the positively or negatively charged amino acids have substantially higher binding on TiO2 NPs than their non-charged counterparts.13 The simulations also revealed the role of water molecules in the interactions between NPs and amino acids. For example, the guanidinium group on arginine directly forms one or two hydrogen bonds with the TiO2 surface, resulting in stably adsorbed conformations. Arginine also forms hydrogen bonds with the first water layer in addition to the arginine–TiO2 interaction, further stabilizing its conformation. Moreover, extra stability can be provided by the hydrogen bonds between arginine and the second water layer (Fig. 2B and C). These results from simulation of molecular interactions provide unique molecular-level insights that might help interpret the experimental findings observed in complex biological systems, and improve our understanding of the molecular-level interactions between biomolecules and NPs. Although peptides and amino acids share similar electrostatic interactions with NPs, the conformation of peptides after binding NPs must also be considered. Berensmeier et al. constructed a short homo-peptide containing eight glutamic acid residues to study its interaction with IONPs.15 They observed that the conformation and coordination of peptides bound to IONPs was highly dependent on the pH. The peptide was bound with a monodentate coordination of carboxylate groups to the surface of IONPs as well as a randomly coiled peptide backbone at pH 5. However, a bidentate coordination or electrostatic interaction of the carboxylate groups with IONPs was preferred at pH 7 and 9, respectively. Additionally, α-helix and β-sheet conformations are more favourable for the bound peptides at higher pH.
A notable exception to the aforementioned non-specific interactions is the binding between the thiol group (cysteine and cysteine-containing peptides) and gold NPs, which does not involve electrostatic interactions yet forms a stable chemical bond. Although extensive efforts have focused on deciphering the structure and the chemical composition of the sulfur–gold interface, an atomic level understanding remains elusive.16 Nevertheless, this stable interaction has been widely used in the surface functionalization and applications of gold NPs.
2.2. NP interactions with proteins
Proteins, as with amino acids and peptides, interact with NP surfaces via electrostatic interactions, but their higher molecular weight and 3D structure leads to complex conformational and structural binding effects. It is well-established in the literature that upon exposure of NPs to plasma proteins in circulating blood (in vivo) or serum added to cell culture media (in vitro), these proteins rapidly adsorb to the NP surface forming a “protein corona”.1a,2,6a,17 The presence of a protein corona has been confirmed on NPs with varied diameters, chemical compositions, and surface functional groups.18 Despite the net negative charge of serum proteins, a protein corona has been observed on both cationic and anionic NP surfaces.18c,19 When NPs enter into bloodstream, the protein corona is initially composed primarily of albumin, the most abundant serum protein. However, the composition of the protein corona is highly dynamic and dependent upon the physical and chemical properties of the NP as well as the local biological environment.1a,18a,c Charge, size, and surface modifications of the NPs can all influence the corona composition.18b Initially, a “soft corona” is formed with weakly bound, low-affinity, abundant proteins and is eventually displaced by a long-lasting “hard corona” composed of high-affinity proteins.20 While an “adsorbome” of nearly 125 unique plasma proteins has been identified on the surface of NPs, two to six proteins are often detected on a single NP surface.6a Previous work by Fleischer and Payne demonstrated that on polystyrene NPs with a similar diameter but the opposite charge, the dominant corona protein was serum albumin for both cationic and anionic NPs.18c Schäffler et al. examined the protein corona on gold NPs with diameters of 5, 15, and 80 nm, and observed the adsorption of albumin, hemoglobin, fibrinogen, and apolipoprotein E on all of the gold NPs.21 However, myosin and apolipoproteins A2 and A4 were only detected on the 5 nm NPs, which also contained greater number of proteins per unit of surface area compared to the larger NPs. This NP size-dependent effect is likely due to the increased radius of curvature with decreasing NP diameter. Sakulkhu et al. characterized the protein corona on IONPs coated with polyvinyl alcohol (PVA) polymer or dextran with varied surface charges.18b They observed that PVA-coated IONPs had a greater number of bound corona proteins, along with a longer blood circulation time, than those with dextran coating for negatively and neutrally charged IONPs. Both soft and hard coronas on PVA- and dextran-coated IONPs were composed of albumin, serotransferrin, prothrombin, alpha-fetoprotein, and kininogen proteins. Interestingly, adsorbed bovine serum albumin (BSA) remains bound to the surface of polystyrene NPs during cellular binding, internalization, and active transport of the NP–protein complex.22 Characterization of the protein corona after NP transfer from plasma to cytosolic fluid confirmed that proteins from both solutions are present, suggesting that NPs retain a “memory” of the proteins encountered during biological transport.20aProtein–NP interactions are largely dominated by hydrophobic interactions, electrostatics, and van der Waals forces.20b,23 In the presence of cells, there is a continuous flux of ions in order to maintain cellular homeostasis and osmotic pressure.20b These ions can alter the local environment around the NP and protein, influencing the protein adsorption and corona profiles determined by long-range electrostatic interactions. Entropy-driven hydrophobic interactions are affected by the polarity of the surrounding solvent, the exposure of hydrophobic residues on the protein surface, and free water bound to both the protein and NPs.24 Local dipole moments on both NP and protein surfaces can alter weaker van der Waals interactions, particularly in the cases where the local environment is highly dynamic. While it is difficult to conclusively determine the corona composition in vivo at a particular time point due to the heterogeneity of the local biological environment, these studies establish the importance of considering the presence, influence, and dynamic nature of the protein corona when designing NPs for applications in biological environments both in vitro and in vivo.
2.3. NP interactions with cells
NPs can interact with cells via a number of pathways including non-specific interactions with phagocytic cells upon exposure to whole blood and receptor-mediated binding and internalization to endothelial cells. NP interactions with cells are driven by the NP properties including size, shape, chemical composition, and surface functionalization.25 Cytotoxicity, active cellular transport, and intracellular compartmentalization and distribution can all vary as a function of the NP. Internalization and cellular responses to NPs are cell-type dependent,25 with observed differences in NP uptake between phagocytic and non-phagocytic cells.26 Cellular binding and internalization of NPs can be mediated by NP surface ligands and targeting groups (e.g., proteins and antibodies), as well as by a number of cellular endocytic pathways (e.g., clathrin and caveolin). Additionally, scavenger receptors that are used to bind cellular waste and by-products can also bind to NPs and mediate internalization. Finally, non-specific interactions as a result of charged proteoglycans on the surface of most cells can mediate electrostatic interactions directly with charged NPs. A thorough review on cell–NP interactions as a function of NP properties can be found in ref. 27.Additionally, as most NPs will form a protein corona almost immediately after the exposure to serum or plasma proteins, the interactions of NPs with cells should often be considered by treating the NP as a protein–NP complex.17 It has been observed that during in vitro experiments, adsorbed BSA remains bound to the surface of polystyrene NPs during cellular binding, internalization, and active transport of the protein–NP complex, suggesting that the adsorbed protein can influence the entire cellular fate of a NP.22 While it is evident that the presence of a protein corona can affect NP interactions with cells, the dynamic and complex nature of the corona has also led to mixed conclusions as to the effect on cellular outcomes. Walkey et al. suggested that identification or “fingerprinting” of the protein corona could be used to predict the ways in which cells interact with gold and silver NPs,28 yet Dobrovolskaia et al. showed that the information on the corona composition was not sufficient to predict hemo-compatibility of colloidal gold NPs.29
While the presence of a protein corona can greatly influence NP interactions with cells, it is also important to recognize the dynamics of the cellular environment. Media conditioning, or cell secretion of biomolecules, can also affect NP aggregation, cellular binding, and NP retention.30 Dai, et al. identified that the cell-conditioned media can affect the biophysical properties of engineered particles as well as cytokine secretion and apoptosis.31 It is worth noting that the induction of an immune response and cytotoxicity are important consequences of direct NP interactions with cells, further described in Sections 3.3 and 3.4.
3. Impact of biofouling on engineered nanomaterials
As engineered NPs are transformed from their “synthetic identity” to a “biological identity” in the biological environment, biofouling can influence the physicochemical properties and functions of the NPs. Subsequently, there are notable changes in pharmacokinetics, cytotoxicity, immune response, and biomarker targeting, all of which are important factors to be taken into account when utilizing engineered nanomaterials for biomedical applications.3.1. Changes in physicochemical properties
The physicochemical properties of engineered materials are often altered from their original in vitro design following non-specific interactions with biomolecules in vivo. Changes in physico-chemical properties of artificial materials due to the biofouling effect primarily include: hydrodynamic diameter, zeta potential (i.e., effective surface charge), colloidal stability, imaging capability, and drug release.32 For instance, the formation of the protein corona on the surface enhanced the colloidal stability of layered double hydroxide NPs (LDH-NPs) by steric hindrance and neutralization of the positive zeta potential to protect them from disassembly at low pH.33 In comparison, Larson et al. reported the reduced stability and aggregation of methoxy-PEG-thiol coated gold NPs in the presence of cysteine and cystine at physiological concentrations due to the displacement of thiol groups on the NP by cysteine/cystine in biological media.34 In addition to the effect on the colloidal stability, the biofouling effect also impacts the performance of the NPs. In the case of IONPs used for magnetic resonance imaging (MRI) contrast enhancement, Amiri et al. demonstrated that the formation of a protein corona on IONPs with dextran surface coating resulted in slightly increased transverse relaxivity (r2) of the negatively charged IONPs and dramatically decreased r2 of the IONPs with positive surface charge, while neutral IONPs did not exhibit a change in the transverse relaxivity.35 MRI contrast stems from the signal difference between water molecules with different relaxation rates, which can be enhanced further by the presence of a contrast agent such as IONPs. Surface coating of IONPs may affect the MRI contrast enhancement by influencing the relaxation of water molecules via diffusion, hydration, and hydrogen bonding.36 Consequently, the relaxivity changes reported by Amiri et al. might not be observed for other coating materials.35 Consideration must be given to the fact that the adsorption of biomolecules of different thickness and hydration properties may alter the physical properties and performance of the NPs that were tested in vitro under relatively simple and controlled experimental conditions. Therefore, attempting to obtain quantitative measurements of NPs and NP performance should properly include characterization and consideration of the biofouling effect.3.2. Changes in pharmacokinetics
The pharmacokinetics of NPs is directly related to the efficiency and efficacy of NPs in sustaining their desired functions. The biofouling effect may trigger the non-specific uptake by the MPS in the liver, spleen, and bone marrow, greatly influencing the pharmacokinetics and biodistribution of the NPs. Since non-specific cellular uptake is size-, charge-, and surface chemistry-dependent, manipulation of these NP properties allows for the reduction of MPS uptake to prolong the blood circulation time and increase the chance of NPs reaching the target sites, thus may improve the blood half-life and delivery efficiency of NPs designed for non-invasive imaging and drug delivery.Upon intravenous injection, the most common in vivo delivery route, NPs will immediately interact with plasma proteins, phagocytic white blood cells (WBCs), and non-phagocytic platelets and red blood cells (RBCs) in the circulating blood. Albumins, the most abundant proteins in serum, make transient contacts non-specifically with NPs through ionic and hydrophobic interactions, and are then displaced easily by other proteins with less abundance yet higher affinities (Section 2.2).37 For example, serum lipoproteins have been demonstrated to decompose liposomal NPs through exchange of phospholipids.38 Adsorption of complement proteins (e.g., immunoglobulins) may increase the recognition of NPs by the MPS to trigger complement activation for the phagocytosis and clearance of NPs.39 Other constitutive proteins (e.g., fibronectin and fibrinogen) may also contribute to the clearance of NPs from blood through enhanced complement activation or receptor-mediated internalization.37 A recent study by Chen et al. indicated that surface opsonization can occur even after non-specific protein adsorption, and subsequent conformational changes of non-specifically adsorbed proteins may trigger antibody binding and/or complement activation.40
After being cleared from the blood, injected NPs are mostly trapped in reticuloendothelial system (RES) organs such as the liver, where Kupffer cells (the resident liver macrophages) play a predominant role in taking up opsonized NPs and aggregates.41 NPs that escape from the sequestration by the liver are sieved in the spleen due to the differences in blood flow and cell phenotype between the liver and spleen. The highly tortuous splenic parenchyma slows down the blood flow significantly (1.4% of total arterial blood from heart),37 thus maximizing the interaction of NPs with splenocytes and retention in the spleen. In addition, a stimulated biofouling trigged by the interactions of trapped NPs and B cells in the spleen can accelerate blood clearance of NPs, leading to a much shortened blood circulation time of NPs after the first injection.42
Finally, it is necessary to point out that the attenuation and inhibition of biofouling generally lengthens the blood circulation time of NPs, resulting in enhanced tumour accumulation1c,43 and sustained pharmacological effects.44 However, the prolonged residence of NPs in the bloodstream can also provoke undesired side effects.45 As the biofouling process is an inherent defensive mechanism of the immune system, complete inhibition or bypassing of biofouling may lead to unintended physiological responses and even damage to the host. The ideal scenario will be to achieve a balance between the optimal NP delivery efficiency and the minimal, tolerable immune response that can remove NPs from the body in a reasonable time frame.
3.3. Changes in cytotoxicity
Cytotoxicity is one of the most important parameters for the evaluation of engineered materials for biomedical applications. Adsorption of corona proteins has been shown to reduce cytotoxicity of NPs in the cellular environment.46 NP interactions with A549 lung epithelial cells in the absence of proteins caused cellular damage via NP-induced disruption of the cell membrane, while the presence of a corona mitigated subsequent cytotoxicity.47 The presence of a protein corona prevented hemolysis and aggregation of erythrocytes in vivo induced by silica NPs, suggesting that refined control of the protein corona may lead to better engineered NPs that are more biocompatible.48 On the other hand, although the protein corona can attenuate the cytotoxicity induced by NPs, the process of protein corona formation can disrupt normal physiological processes. Dose-dependent blood circulation times have been reported in which longer circulation can be achieved simply by increasing the injection dosage of NPs.49 Prolonged blood circulation following incremental dose increases is attributed to the depletion of plasma opsonins49 or saturation of macrophage phagocytic capacity,50 both of which can weaken the immunity of the host.3.4. Changes in immune response
Characterizing NPs in a realistic biological environment is important but often limited due to the practical challenges when considering that both engineered NP surfaces and adsorbed proteins can induce a separate immune response.46 Changes in serum albumin conformation on the surface of polymeric NPs may alter NP uptake by human monocytes and macrophages.51 More specifically, Deng et al. reported that gold NPs induced unfolding of bound fibrinogen (a plasma glycoprotein essential for blood clotting), which activated the Mac-1 receptor and the NF-κB pathway, a known mediator of inflammation that induced the downstream release of inflammatory cytokines.52 On the other hand, the protein corona was found to prevent cell death and inhibit the pro-inflammatory response induced by IONPs, suggesting that both pro- and anti-inflammatory responses may be induced depending on the NP properties and composition of the corona.53 Several existing mechanisms by which protein–NP interactions can induce an immune response, alter NP binding and uptake, and affect cytotoxicity have provided the guidance for carefully designing, preparing, and applying NPs for in vivo applications. However, fundamental studies with different models and systems that accurately mimic in vivo biological environments are still needed as the field of nanomedicine continues to develop and incorporate new engineered NPs for biomedical and clinical applications.3.5. Changes in biomarker targeting
A common strategy for developing NPs for targeted imaging and drug delivery is to functionalize the NP surface with targeting ligands, (e.g., small molecules, peptides, and antibodies). The consequence of the protein corona to this important capability is its subsequent effect on the interaction and interference of surface functionalized or ligand-decorated NPs with biomarker targets.6a,17 Adsorbed proteins can mask the conjugated ligands on the NP surface leading to the loss of targeting affinity,3,17 or induce changes in secondary structure of the adsorbed protein and redirect the targeting of protein corona–NP complexes to other unwanted sites.54 Importantly, the effect of the protein corona on NP targeting highly depends on both the protein corona composition and the physicochemical properties of NPs.17,28,55Work by the Dawson group demonstrated that transferrin (Tf)-functionalized silica NPs bind selectively to transferrin receptors (TFRC) in serum-free media, but the selective binding was lost in the presence of fetal bovine serum (FBS), a common supplement to cell culture media (Fig. 3A).3 However, Dai et al. observed that despite enhancements in cell membrane binding of layer-by-layer core–shell polymeric NPs after functionalization with a humanized A33 monoclonal antibody, the functionalized NP did not exhibit significant alteration in cell targeting in the presence of a protein corona (Fig. 3B).55 Due to the limited understanding of the protein corona and diverse factors from NPs that affect the composition of the protein corona (e.g., diameter and surface charge), understanding the roles of the protein corona formation and effect in targeting of NPs requires further investigation. For instance, a direct comparison between anti-biofouling and commercial (without anti-biofouling coating) IONPs was performed to examine the effect of the protein corona on cell targeting. The anti-biofouling IONPs conjugated with Tf (FITC-Tf-IONP) showed unaltered cell targeting to both targeted (D556) and non-targeted (A549) cells regardless of the protein corona, whereas the commercial IONPs (FITC-Tf-SHP) exhibited weakened targeting capability (i.e., decreased signal intensity difference between D556 and A549 cells) after the formation of the protein corona on the IONP surface (Fig. 3C and D).56 Interestingly, in a different study of IONPs conjugated with folic acid, Krais et al. found that the presence of serum proteins was required for folic acid-dependent uptake by ovarian cancer cells.57 These studies suggest that the protein corona can both inhibit and promote receptor-mediated binding, and targeting may still be possible even after the formation of a protein corona.
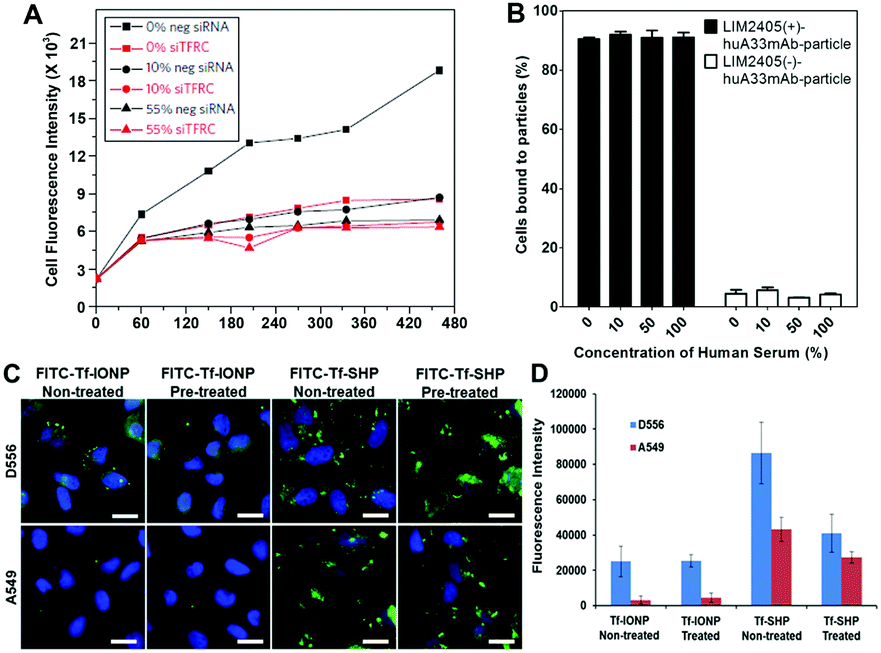 | ||
Fig. 3 (A) A549 cells are silenced for 72 h with a negative silencer control (neg siRNA) and for the transferrin receptor (siTFRC) before exposure to NPs. Median cell fluorescence intensity obtained by flow cytometry from A549 cells exposed to PEGylated human Tf particles in serum-free medium (0%), complete medium (10%) and medium supplemented with 55% serum (55%). The uptake is strongly reduced in cells silenced for TFRC. However, at increasing serum content the uptake decreases in cells with the TFRC and the effect of TFRC is also lost. (B) Cell membrane binding of polymeric NPs to LIM2405 ± cell mixtures (positive-to-negative cell ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of a protein corona. Cell mixtures were incubated with NPs at 4 °C for 1 h at a NP-to-positive cell ratio of 100:1. The percentage of cells bound to the NPs was determined by flow cytometry. (C) Fluorescent images of D556 medulloblastoma and A549 lung cancer cells incubated with FITC-Tf-IONP and FITC-Tf-SHP with or without FBS (100%) pre-treatment. Scale bar 20 μm. (D) Medium fluorescence intensities of FITC from FBS (100%) pre-treated or non-treated IONPs taken up by D556 medulloblastoma or A549 lung cancer cells. Adapted with permission.3,55,56 Copyright 2013 Nature Publishing Group, 2015 American Chemical Society, and 2017 Elsevier. :1) in the presence of a protein corona. Cell mixtures were incubated with NPs at 4 °C for 1 h at a NP-to-positive cell ratio of 100:1. The percentage of cells bound to the NPs was determined by flow cytometry. (C) Fluorescent images of D556 medulloblastoma and A549 lung cancer cells incubated with FITC-Tf-IONP and FITC-Tf-SHP with or without FBS (100%) pre-treatment. Scale bar 20 μm. (D) Medium fluorescence intensities of FITC from FBS (100%) pre-treated or non-treated IONPs taken up by D556 medulloblastoma or A549 lung cancer cells. Adapted with permission.3,55,56 Copyright 2013 Nature Publishing Group, 2015 American Chemical Society, and 2017 Elsevier. | ||
In addition to the potential for adsorbed proteins to mask targeting ligands or influence cell–NP interactions, the high energy NP surface can alter adsorbed protein structure and determine the cellular receptors for binding. Fleischer and Payne showed that while cationic and anionic polystyrene NPs both formed a protein corona composed primarily of serum albumin, only cationic NPs induce changes in the secondary structure of adsorbed albumin, directing these protein–NP complexes to bind to scavenger receptors on cell surfaces.54 Treuel et al. observed that chemical modification of adsorbed proteins alters the physical properties of the adsorbed protein layer as well as the cellular uptake of protein–NP complexes, suggesting that small changes to adsorbed protein structure can influence targeted delivery and cellular outcomes of NPs.58 For more detailed information on the effect of the corona on targeting, cytotoxicity, and immunotoxicity of bioconjugated NPs, we direct the reader to reviews by Caracciolo et al.,1a Kairdolf et al.,59 Corbo et al.,60 and the references therein.
4. Preventing biofouling with surface coating materials
One convenient and effective method to prevent biofouling on engineered materials is to coat the surface with a material that can reduce non-specific interactions with biological systems. Various synthetic and natural anti-biofouling materials have been developed for NPs including PEG, PVP, PEG- and PVP-related block copolymers, zwitterionic materials, polysaccharides, and cell membrane-derived coatings. Due to the simplicity of the chemical structures, PEG and PVP were developed as the first generation of anti-biofouling coatings for NPs. They both are generally easy to implement but less effective in providing anti-biofouling properties, in terms of the strength and duration, than the more recently developed PEG-derived copolymer and zwitterionic materials. Meanwhile, the newly reported cell membrane-derived coatings that render a natural camouflage for NPs to escape from the biofouling process have drawn broad attentions; although systematic investigations are of vast interest to fully understand this coating strategy.4.1. Poly(ethylene glycol) (PEG) for anti-biofouling
Coating surfaces with PEG, or PEGylation, has been widely applied for coating engineered materials in the field of biological research and medical device development, not only because of the non-toxic nature of PEG, but also its robust capability of reducing non-specific protein adsorption.61 A tightly hydrated layer formed via hydrogen-bonding between water molecules and ether groups of PEG chains is believed to provide the resistance against protein adhesion. Moreover, the flexible PEG chains provide steric hindrance which is associated with the dense hydrated layer, further reducing the adsorption of the protein molecules.62NP surfaces can be coated with PEG via covalent or non-covalent interactions. Non-covalent coating of PEG on NPs is generally obtained by hydrophobic/hydrophilic interactions. For instance, IONPs can be coated with PEG by drying the mixture of IONPs and PEG in chloroform and subsequently dispersing the residues in water.63 Compared with non-covalent coating, covalent PEGylation offers more stable coating on the NPs, consequently enhancing their colloidal stability. A variety of functional groups have been used to modify PEG molecules including thiols,64 dihydrolipoic acid (bidentate thiols),65 and silane66 to provide covalent and/or semi-covalent bonds with engineered materials. In addition, a biomimetic strategy has also been explored to covalently attach PEG molecules to NP surfaces. The most widely adapted linkers are dopamine67 and its derivatives,68 which employ a catechol segment for binding. The immobilization of loop- and brush-forms of PEG onto the substrate surface has been reported via the catechol linkage. Moreover, the loop-form of PEG exhibited better anti-biofouling performance than its brush-form analogue,68 indicating the importance of PEG orientation for anti-biofouling capability.
The mechanisms of the reduction of non-specific protein adsorption of NPs by PEGylation have also been investigated extensively with an excellent review by the Chan group.6a For example, Walkey et al. prepared a series of gold NPs with different PEG grafting densities. With increased PEG grafting density, the anchored PEG molecules gradually dehydrated, reducing space between neighbouring PEG chains, further limiting the possibility of protein diffusion and adsorption onto NPs. However, the high grafting density of PEG cannot completely block the serum-independent uptake pathway by macrophages against PEG-coated NPs.6b It is worth noting that PEG and poly(ethylene glycol phosphate) (PEEP) modification have been shown to affect the composition of the protein corona on the surface of polystyrene NPs in addition to the reduction of protein adsorption. The presence of adsorbed proteins, mainly clusterin proteins (also known as apolipoprotein J), is crucial for reducing non-specific cellular uptake.69
Although PEG is widely accepted as a NP surface coating for anti-biofouling, it has several disadvantages. PEG is found to be susceptible to oxidative damage and generation of reactive oxygen species.70 After coating with PEG, the hydrodynamic size of NPs can be significantly increased45 with attenuation of desired interactions between NPs and their biological targets.71 Moreover, a recent report showed that PEG may induce immunogenicity and subsequent production of anti-PEG antibodies in mice injected with NPs coated with PEG.42 However, due to the lack of specificity of most assays for anti-PEG antibodies, contradictory immune responses induced by anti-PEG antibodies have been reported. Standardized assays for anti-PEG antibodies and the development of reference sera are required to study and confirm the immunogenicity of PEG-coated NPs.72
4.2. Poly(N-vinylpyrrolidone) (PVP) for anti-biofouling
Due to the existence of both polar lactam groups and non-polar methylene groups, PVP can be dispersed into either aqueous solution or common organic solvents. Amphiphilic PVP exhibits excellent physiological inertness, complexing ability, chemical stability, and biocompatibility, making it a promising alternative to PEG as an anti-biofouling coating polymer.73 For example, the adsorption of BSA was decreased by 75% using PVP-coated silica compared to the unmodified material.74 Coating through hydrophobic/hydrophilic interaction is considered to be the easiest method to modify NP surfaces with PVP. However, several technologies have been developed for surface grafting of PVP to render better coating stability including photochemical grafting,75 γ-radiation-induced grafting,76 and plasma polymerization.77 While photochemical grafting78 and plasma polymerization79 cause little harm to the bulk properties of materials, their application is limited by the shape and composition of NPs.80In comparison to PEG, studies and applications of PVP for coating nanomaterials for biomedical applications are limited. The incorporation of PVP coatings onto various substrates (e.g., metals, ceramics, and polymers) has not been well established. Furthermore, in-depth understanding of the interactions between PVP coating and the biological system is still needed, especially for in vivo studies.
4.3. PEG-derivative copolymers for anti-biofouling
One approach to take advantage of the anti-biofouling properties of PEG while introducing more surface functions of NPs is to graft the PEG moiety to other polymers to produce copolymers for (1) enhancing anti-biofouling capability,81 and (2) constructing multifunctional materials.82 This approach may also mitigate the issue of competitive surface binding between biomolecules and PEG which causes displacement of the PEG coating on the NP surface and leads to changes in physicochemical properties and loss of stability.34Hydrophobic polymers have been grafted to PEG to assemble hydrophobic layers protecting the anchoring groups from contacting and being replaced by biomolecules, as well as inhibiting non-specific biomolecule interactions with the NP surface, which also contributes to the overall anti-biofouling effect. Various PEG based copolymers have been reported, such as PEG-b-PEI,83 PEG-block-polystyrene (PEG-b-PS),84 PEG-block-poly(ε-caprolactone) (PEG-b-PCL),85 PEG-block-poly(lactic acid) (PEG-b-PLA),86 poly(L-lysine)-g-PEG (PLL-g-PEG),87 and poly(oligo(ethylene glycol) methacrylate) (POEGMA).88 PEG-block-poly(γ-methacryloxypropyltrimethoxysilane) (PEG-b-PγMPS) prepared by Chen et al. represents a typical example of PEG-based copolymer coatings. The PEG-b-PγMPS coated NPs exhibited anti-biofouling properties in an in vitro macrophage uptake experiment and an in vivo biodistribution study in mice. After conjugating with tumour integrin αvβ3 targeting peptide (i.e., cyclo-RGD), specific cancer cell targeting by PEG-b-PγMPS coated IONPs to U87MG glioma cells with αvβ3 integrin over-expression was observed.81b
In addition to neutral PEG chains, an amine-functionalized PEG was utilized for the preparation of PEG-block-allyl glycidyl ether (PEG-b-AGE) to coat IONPs, which exhibited slightly positive surface charge (zeta potential = 1.85 ± 0.42 mV).81a Although charged PEG chains are believed to be less effective than non-charged chains for providing anti-biofouling properties,6a the slightly positively charged PEG-b-AGE coating demonstrated anti-biofouling capabilities by reducing serum protein adsorption, maintaining colloidal stability, and inhibiting non-specific cellular uptake. Unlike other coating polymers where introducing functional groups alters charge-free PEG and attenuates anti-biofouling properties,6a,81b the enhanced anti-biofouling property of PEG-b-AGE with positively charged PEG is possibly due to the weakened electrostatic interactions by PEG hydration. After conjugating a small molecule peptide (cyclo-RGD) or large protein (Tf) ligand to PEG-b-AGE coated IONPs using either thiol/maleimide click reaction or EDC/NHS coupling, specific cell targeting was observed for both RGD- and Tf-conjugated IONPs, while the anti-biofouling property was retained to prevent cellular uptake by off-target cells. Measurements of MRI relaxivity from IONPs bound to cells, semi-quantitatively indicating the degree of IONP binding and internalization, confirmed the anti-biofouling properties when comparing PEG-b-AGE coated IONPs to the same IONP cores but coated with the conventional amphiphilic copolymers without the anti-biofouling coating (Fig. 4A). Upon conjugation with Tf, both PEG-b-AGE coated IONPs and control IONPs exhibited MRI contrast changes (i.e. T2 weighted signal drop or darkening) between cells with surface targets and cells without targets. However, the signal-to-noise ratio for PEG-b-AGE coated IONPs was greatly improved compared to that from the control due to the reduction of background signal from non-specific uptake of targeted IONPs by cells without expression of the cell surface target. The observation of an enhanced signal-to-noise ratio implies the potential quantitative applications of the PEG-b-AGE coated NPs for biomarker targeted imaging. The application of PEG-b-AGE coated IONPs for improving biomarker targeted liquid biopsy, specifically for capturing circulating tumour cells via target-ligand interaction, has also been investigated in comparison with commercial IONPs or magnetic beads without anti-biofouling coating. After conjugating Tf as a targeting ligand, PEG-b-AGE coated IONPs demonstrated similarly high efficiency with commercial IONPs in capturing D556 medulloblastoma cells spiked in the blood samples, while exhibiting significantly higher specificity to targeted D556 cells with fewer non-targeted cells interfering with the detection (Fig. 4B and C). Isolation of 41 out of 100 spiked D556 cells from 1 mL of whole blood was achieved with minimal off-target blood cells.56
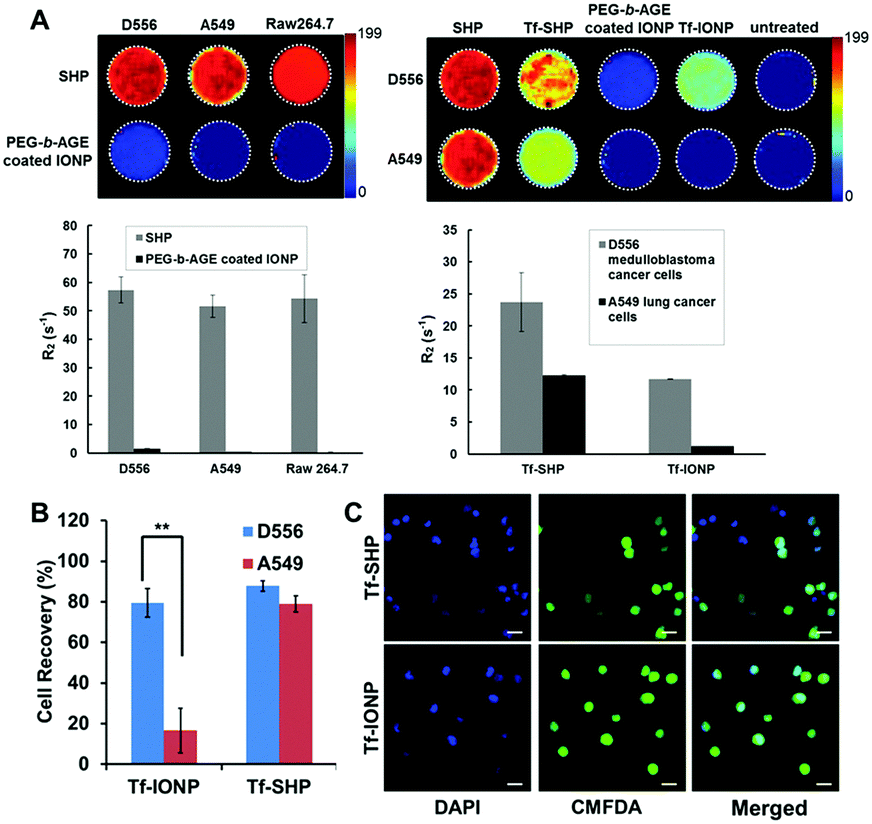 | ||
| Fig. 4 (A) R2 relaxometry map and R2 values of cell phantoms containing 8 × 106 D556 medulloblastoma cells, A549 lung cancer cells, and RAW264.7 macrophages after incubation with commercial SHP-20 and PEG-b-AGE coated IONPs with same core diameter (left panel); and R2 relaxometry map and R2 values of cell phantoms containing 8 × 106 D556 medulloblastoma cells and A549 lung cancer cells treated with SHP-20, Tf-conjugated SHP-20 (Tf-SHP), PEG-b-AGE coated IONP, and Tf-conjugated IONPs with PEG-b-AGE coating (Tf-IONP) (right panel). (B) D556 medulloblastoma cells (targeted) and A549 lung cancer cells (non-targeted) capture rate using Tf-conjugated anti-biofouling IONP (Tf-IONP) and commercial SHP (Tf-SHP) (**P ≤ 0.01). (C) Fluorescent images of D556 (pre-stained with CMFDA) and A549 cells captured by Tf-SHP and Tf-IONP. Scale bar 20 μm. Adapted with permission.56,81a Copyright 2015 Royal Society of Chemistry and 2017 Elsevier. | ||
4.4. Polysaccharides for anti-biofouling
The resistance of glycocalyx, a cell-membrane associated glycoprotein, to cellular adhesion and non-specific protein recognition, combined with the availability and simplicity of chemical modifications led to the exploration of using polysaccharides, a glycocalyx component, as anti-biofouling coatings.89 A number of techniques have been applied for the surface functionalization of NPs with polysaccharides,90 the most common utilizing (1) electrostatic interactions; (2) self-assembly of a hydrophilic polysaccharide with hydrophobic grafts; and (3) formation of a loop- or brush-structured layer through conjugation. Among the polysaccharides studied in recent work, hyaluronic acid (HA)91 and dextran92 are the most commonly employed polysaccharides. Park et al. conjugated PEI with HA for the delivery of small interfering RNA (siRNA). Compared to the siRNA–PEI complex that tends to aggregate due to non-specific serum protein adsorption, the HA functionalization shielded the siRNA/HA–PEI complex from aggregation in blood.91 Additionally, HA was shown to contribute to the extracellular stability of HA-coated structures, contributing to improved gene transfection. Xu et al. reported that the existence of a HA layer around a binary complex of DNA and disulfide cross-linked PEI strengthened the stability and transfection efficiency of the complex in a serum-containing environment. Their results also demonstrated the anti-biofouling property of HA in the protection of the DNA–PEI–HA complex from non-specific interactions with serum proteins.93In addition to the well-established properties of biocompatibility and biodegradability, dextran also exhibits anti-biofouling properties that have been used for protection of iron oxide90 and polymeric NPs.94 The surface density and conformation of dextran are considered to be important to the effectiveness of its anti-biofouling properties.92,95 For example, a brush-style, densely-packed dextran coating on polymeric NPs lengthened the distribution half-life and reduced clearance by the liver compared to NPs coated with a loosely-packed dextran layer.92 It has also been demonstrated that a covalently linked dextran layer is better than physically adsorbed dextran in protecting surfaces from non-specific interactions.96
Notwithstanding, several concerns remain when using polysaccharides as NP coatings for in vivo applications: (1) the properties of polysaccharides vary greatly as a function of molecular weight and structure, critically affecting their biological applications. (2) Reliable synthetic methods are needed to achieve consistent properties and sustainable results. Standardized procedures to maintain the purity of polysaccharide products should be developed due to the contaminants (such as pathogens and endotoxins) often associated with polysaccharides. (3) More detailed and rational experimental designs are required to investigate the effects, mechanisms, and fate of polysaccharides in biological systems, given that inconsistent results are obtained even when utilizing the “same” type of polysaccharide.
4.5. Zwitterionic materials for anti-biofouling
Electroneutral materials have greater anti-biofouling potential compared with their charged counterparts.1c Zwitterionic materials, bearing an equal amount of positive and negative charges on the same molecule, have an overall neutral charge and are considered a rational candidate for anti-biofouling applications. Upon attachment of zwitterionic materials to a surface, the charged moieties either fold intra-molecularly or interact with neighbouring molecules to neutralize the charges. The internal neutralization leads to a lack of exposed charged groups to bind non-specifically to charged adsorbents, resulting in a protective layer against biofouling.97 Zwitterionic coatings can be formed by low-molecular-weight molecules or polymers.Amino acids are natural candidates for zwitterionic materials since the amine groups (providing positive charge) and carboxyl groups (providing negative charge) are present in the same molecule. Among all natural amino acids containing thiols, cysteine and cystine have demonstrated the higher affinity towards NPs than thiol groups on coating polymers, leading to the displacement of coating polymers.34 Consequently, the application of amino acids, especially cysteine and its derivatives, may provide not only the zwitterionic feature but also better colloidal stability of coated NPs. When cysteine was used to assemble zwitterionic coating for quantum dots (QDs), desired anti-biofouling capability was achieved. However QD aggregation was also observed within a few hours of preparation due to the spontaneous oxidation of the cysteine groups.98 To enhance the stability, D-penicillamine was subsequently used for coating QDs, demonstrating comparable anti-biofouling capability yet better stability compared with cysteine coated QDs.99 The combination of cysteine and lysine as zwitterionic coating materials has also been reported for citrate-capped gold NPs. The outer zwitterionic layer was found to provide effective protection to the charged citrate layer to achieve zero protein adsorption.100 Moreover, glutathione, a cysteine derivative, was investigated as a zwitterionic coating material for gold NPs.101 After incorporating such zwitterionic gold NPs onto the surface of IONPs, the protein adsorption on the gold NP-covered-IONPs was 10-fold lower than the IONPs only coated with glutathione.102 In addition to natural amino acids, artificial structures with the zwitterionic feature have also drawn great attention. Among the developed structures, tertiary amines have been widely used to introduce the positive charge, while the negative charge can be provided by sulfonate (–SO3−),103 carboxylate (–CO2−),104 or phosphate (–PO3−).105
Compared with PEG coatings, which substantially increase the hydrodynamic diameters of NPs, zwitterionic coatings provided by low molecular weight molecules generally result in compact particles with little change on hydrodynamic diameter.45,97a Consequently, zwitterionic NPs coated by low molecular weight molecules are more suitable for deep tissue delivery as well as renal clearance.98 Moreover, the zwitterionic coating formed by low molecular weight molecules has little effect on the physical properties of NP cores. In the case of IONPs as MRI contrast agents, in vitro and in vivo MRI measurements of zwitterionic IONPs have shown retained superparamagnetism or saturation magnetization compared to as-synthesized hydrophobic IONPs.106
Evolved from zwitterionic phosphorylcholine self-assembled monolayers,107 the first zwitterionic polymer was developed by Chen et al. for a gold surface consisting of oligo phosphorylcholine.108 The linear polymer employed a tertiary amine as the cationic group and phosphate as the anionic group in each repeating unit. Whereas tertiary amines are still the most widely used cationic group, carboxylate and sulfonate emerge as anionic groups for the development of zwitterionic coating polymers such as phosphorylcholine,109 sulfobetaine,109a poly(carboxybetaine) (PCB),110 poly(sulfobetaine) (PSB),111 poly(carboxybetaine acrylamide) (PCBAA),112 poly(carboxybetaine methacrylate) (PCBMA),113 poly(sulfobetaine acrylamide) (PSBAA),114 and poly(sulfobetaine methacrylate) (PSBMA).115
Comparisons between zwitterionic polymers and PEG as well as among different zwitterionic polymers indicate that PCB and PSB have much lower hydration free energies than PEG, i.e., stronger hydration for PCB and PSB, as demonstrated by theoretical simulation and experimental analysis by Shao et al. Their analysis also suggested decreased water molecule mobility near the PCB and PSB coatings compared with PEG leading to greater repulsive forces against protein adsorption. PCB and PSB also interact with proteins differently from PEG. Amphiphilic PEG preferably covers the hydrophobic segment of proteins, while PCB and PSB have little interaction with the hydrophobic segment due to the local charges of zwitterionic materials. Their simulations to compare the differences between PCB and PSB in terms of hydration, ionic interaction, and self-association, led to the conclusion that PCB possessed stronger hydration than PSB and few self-associations. They found that PCB resisted non-specific protein adsorption even in complex media and was nearly inert to changes in environmental parameters. Meanwhile, PSB demonstrated strong hydration and moderate self-associations and were capable of resisting non-specific protein adsorption and responding to external stimuli.116
Another study by Han et al. performed a detailed comparison between QDs coated with sulfobetaine- and carboxybetaine-functionalized poly(imidazole) to investigate the influence of coating polymers on the cell binding. The carboxybetaine-bearing particles (CBPIL) exhibited non-specific binding to HeLa cells and vessels, whereas the sulfobetaine-bearing particles (SBPIL) showed minimal adsorption (Fig. 5). The differences in cell binding by CBPIL and SBPIL were associated with different spatial charge distributions. When comparing the better performing SBPIL with PEGylated control (PEGPIL), the zwitterionic SBPIL had a faster blood clearance time (an order of magnitude) compared to PEGPIL, but diffused into a tumour an order of magnitude more slowly than PEGPIL.117 Given that zwitterionic polymers are relatively new to the biomedical field, the behaviour of zwitterionic polymers in the biological environment and interactions with various biomolecules need to be better understood. This study suggests that even a small deviation in the polymer structure may affect the overall neutrality, subsequently leading to significant change in function. The structure-related charge distribution of zwitterionic polymers needs to be carefully considered in the design and optimization of zwitterionic polymers to gain or enhance the anti-fouling function.
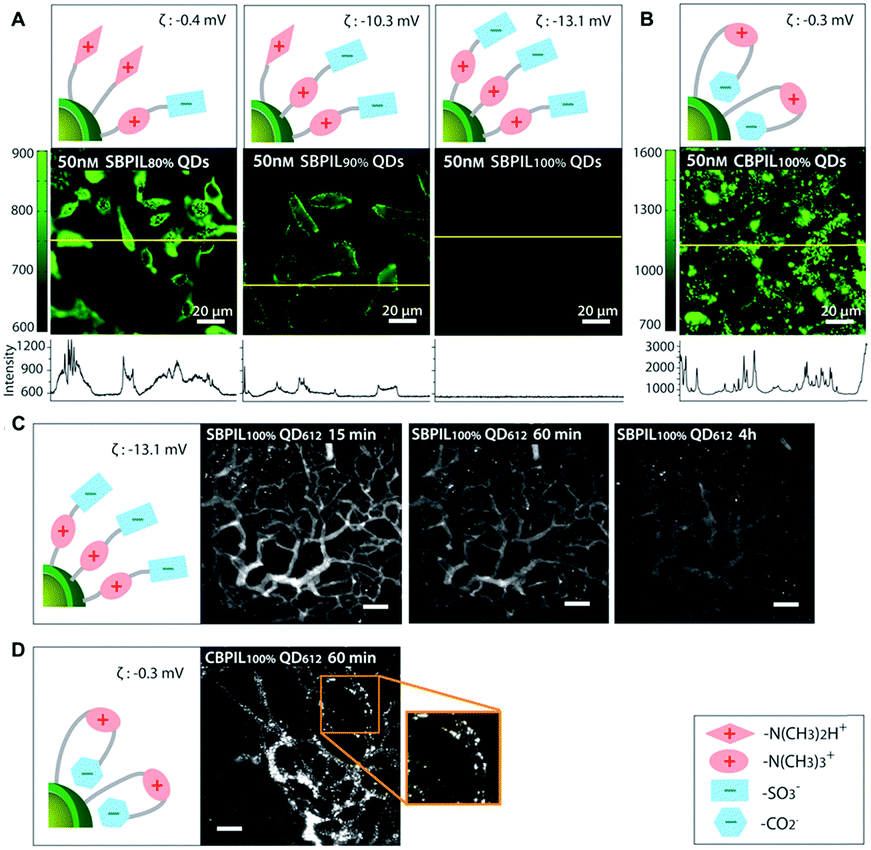 | ||
| Fig. 5 Influence of charge distribution on non-specific binding of QDs in vitro and in vivo. Charge distributions in the surface coatings were inferred from the different affinities of carboxylate groups and sulfonate groups towards QD surfaces. (A and B) Influence of surface charge distribution on non-specific binding to HeLa cells. Yellow lines indicate where the intensity profile was measured. (A) Significant decrease of the non-specific binding is observed when the betainization efficiencies increase from 80% to 100% for SBPIL QDs. The data show that exposed free amines (positively charged groups) trigger non-specific binding to cells. (B) CBPIL100% QDs exhibit a high level of non-specific binding owing to exposed amine groups. CBPIL100% QDs display significantly higher non-specific binding to cells than SBPIL QDs. (C and D) Influence of charge distribution on in vivo clearance. (C) Clearance of SBPIL QD612 from the vessels. The vessels of E0771 murine mammary adenocarcinoma grown in SCID mice (SCID = severe combined immunodeficiency) were imaged by multiphoton microscopy through a mammary fat pad window at 15 min, 60 min, and 4 h after intravenous injection. Note that SBPIL QD612 clears from the vessels without leaving any evidence of non-specific accumulation. Scale bar = 100 μm. (D) Clearance of CBPIL100% QDs from the vessels. An image taken at 60 min after the injection of CBPIL100% QDs illustrates that CBPIL100% QDs accumulate on the vessel walls non-specifically. Adapted with permission.117 Copyright 2013 Wiley. | ||
4.6. Cell membranes for anti-biofouling
Using a biomimetic approach to minimize the difference between endogenous materials and the biological environment to reduce the responses of biological systems at the interface is a potential solution to the biofouling problem, and has been increasingly applied for improving the biocompatibility, delivery, and pharmacokinetic profiles of NPs. A new strategy of adapting the plasma membrane of human platelets as a coating for polymeric NPs was reported by Hu et al.118 By using a natural cell membrane cloak that can be adoptively prepared to cover the surface, NPs are camouflaged with platelet-mimicking properties to reduce cellular uptake by macrophage-like cells (Fig. 6A–D). NP-induced complement activation was also attenuated. In addition, the NPs coated with the platelet membrane cloak demonstrated selective adhesion to damaged vasculatures and enhanced binding to platelet-adhering pathogens, indicating the disease-targeted delivery (Fig. 6E–H). In addition to using platelets, similar strategies using membranes from RBCs,119 WBCs,120 stem cells,121 cancer cells,122 and less commonly used bacteria123 have been explored.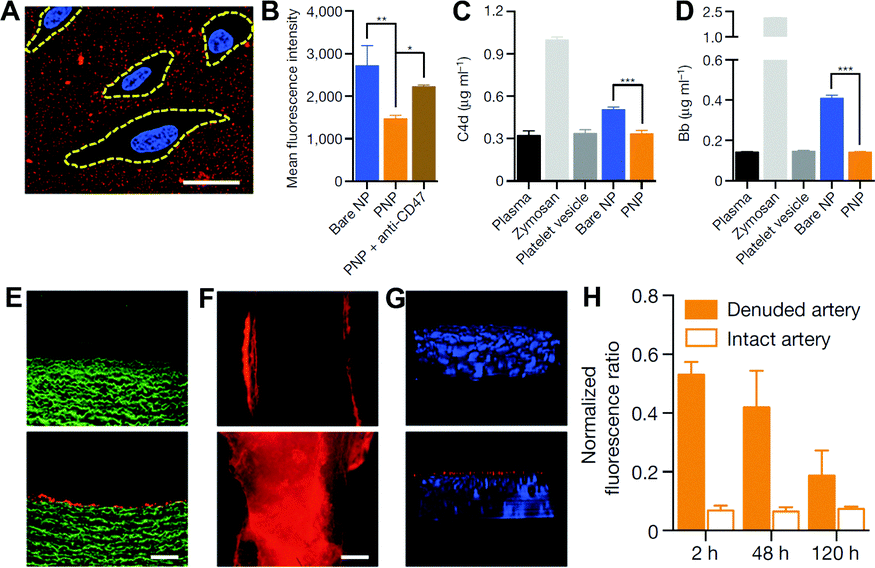 | ||
| Fig. 6 (A) Localization of platelet membrane coated NPs (stained in red) on collagen-coated tissue culture slides seeded with HUVECs (nuclei stained in blue). Cellular periphery is outlined based on cytosolic staining. Scale bar, 10 μm. (B) Flow cytometry analysis of NP uptake up human THP-1 macrophage-like cells (n = 3). Classical complement activation measured by C4d split product (C) and alternative complement activation measured by Bb split products (D) for bare NPs, platelet vesicles, and platelet membrane coated NPs in autologous human plasma (n = 4). Zymosan and untreated plasma are used as positive and negative controls, respectively. All bars represent means ± s.d. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Fluorescence images of (E) the cross-section (scale bar, 200 μm) and (F) the luminal side (scale bar, 500 μm) of undamaged (top) and damaged (bottom) arteries after platelet membrane coated NP incubation (tissue in green and NPs in red). (G) 3D reconstructed images of intact (top) and balloon-denuded (bottom) arterial walls from multi-sectional images after intravenous administration of platelet membrane coated NPs in rats (cell nuclei in blue and NPs in red). (H) Retention of platelet membrane coated NPs at the denuded and the intact arteries over 120 h after NP administration (n = 6). Adapted with permission.118 Copyright 2015 Nature Publishing Group. | ||
As a nascent technology, using cell membrane as NP coatings has many advantages by inheriting the selected cell membrane functions and properties of membrane proteins, glycans, and lipids from the source cells. Given the natural cell membrane coating, the engineered materials are generally identified as endogenous cells by the immune system, preventing NPs from non-specific protein adsorption and the uptake by the MPS.124 Moreover, by adapting different cell membranes, the coated materials impart biological properties and functions without complicated chemical modifications. For instance, NPs coated with RBC membranes can be recognized as RBCs, thus prolonging the blood circulation time.119 It has been shown that RBC membrane-coating increased the tumour accumulation of NPs by taking advantage of the enhanced retention and permeation (EPR) effect boosted by the prolonged blood circulation of NPs.119 Xuan et al. developed macrophage cell membrane-camouflaged gold nanoshells that not only exhibited good colloidal stability but also retained the original near-infrared adsorption of gold nanoshells. By recognizing tumour endothelia, the macrophage cell membrane provided better tumour accumulation of gold nanoshells than the RBC membrane coating approach.120 The membrane of mesenchymal stem cells has also been employed for gene delivery due to the unique surface-associated tumour targeting capability of mesenchymal stem cells. The NPs made of the mesenchymal stem cell membrane encapsulating plasmid DNA demonstrated the ability to inhibit the growth of metastatic orthotopic lung cancer and subcutaneous prostate cancer in mice.121 When membranes derived from 4T1 breast cancer cells were used for NP coating, highly cell-specific targeting of coated NPs to the primary tumour via homotypic binding was observed, which occurs when cancer cells adhere to one another to facilitate the growth of tumour mass.122
The current development of the cell membrane coating strategy is mostly focused on the exploration of different types of cell membranes. Many chemical modifications used in the traditional surface functionalization of NPs have yet to be incorporated. For example, the introduction of targeting ligands to the membrane coating for active targeting in addition to passive targeting enhanced by the inherent properties of the cell membrane has not yet been reported. As further investigations carried out, the cell membrane coating strategy may make significant contributions to the biomedical field.
5. Remarks and perspectives
With rapid advancements in the field of nanotechnology including nanomaterial development for biomedical applications, the non-specific interactions of engineered materials with the biological environment and the approaches to develop and use the anti-biofouling coating materials for controlling and improving function and properties of NPs will continue to draw attention and effort. As the “gold standard” for anti-biofouling coating materials, PEG has been intensively investigated as a model to understand the factors affecting anti-biofouling capabilities of coating materials. However, a number of coating materials designed and prepared according to the findings from PEG investigations, e.g., PVP polymers, PEG-based copolymers, zwitterionic materials, and polysaccharides, have demonstrated improved performance on different aspects of anti-biofouling as well as impacted the function and application of the NPs. Thorough and systematic studies to understand the anti-biofouling behaviours are needed for further development of these coating materials. Meanwhile, a novel artificial coating, i.e. a nascent platelet-membrane-derived coating, has been reported recently, closely followed by the development of different types of cell-membrane-derived coatings, such as RBCs, WBCs, stem cells, and cancer cells. The recognition of NPs by the host immune system can be greatly reduced by cloaking NPs with endogenous cell membranes. In addition, the cell-membrane-derived coating technology has only been employed to therapeutic delivery systems, while other artificial coating materials have been extensively utilized in a variety of applications including targeted drug delivery, in vivo imaging/biosensing, in vitro diagnostics, etc.Although tremendous effort has been put on the development of anti-biofouling coatings, there are only 14 examples approved for clinical trials.125 The long process of new drug approval by FDA, which typically takes 10 to 15 years, is the major obstacle for the translation of novel anti-biofouling coatings. Currently, most of the FDA approved anti-biofouling-related nanomedicines on clinical trials are PEG based, as they are the earliest and most widely used coating materials to provide anti-biofouling property. As the use of engineering nanomaterials for biomedical applications continues to expand, it is reasonable to expect that more and more nanomaterials with novel anti-biofouling coatings will be approved by FDA for clinical use in the future. Meanwhile, it is worth noting that the complete avoidance of biofouling may be harmful to the host as mentioned earlier. It is also unnecessary to compare the anti-biofouling capabilities of different materials directly, as materials with varied anti-biofouling capabilities may all find their applications in given diseases with different demand on anti-biofouling. All in all, there is a promising future for anti-biofouling materials, and the development of new materials remains an active area of research for nanomaterial translation and application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported in parts by grants (R01CA154846-05 and U01CA151810-04) from National Institutes of Health.References
- (a) G. Caracciolo, O. Farokhzad and M. Mahmoudi, Trends Biotechnol., 2017, 35, 257 CrossRef CAS PubMed; (b) S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS; (c) J. Huang, Y. Li, A. Orza, Q. Lu, P. Guo, L. Wang, L. Yang and H. Mao, Adv. Funct. Mater., 2016, 26, 3818 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901 CrossRef CAS PubMed.
- H. Cong, X. Xu, B. Yu, H. Liu and H. Yuan, Biomicrofluidics, 2016, 10, 044106 CrossRef PubMed.
- (a) C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780 RSC; (b) C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139 CrossRef CAS PubMed.
- (a) M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545 RSC; (b) B. Pelaz, C. Alexiou, R. A. Alvarez-Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grunweller, Z. Gu, N. J. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopecek, N. A. Kotov, H. F. Krug, D. S. Lee, C.-M. Lehr, K. W. Leong, X.-J. Liang, M. L. Lim, L. M. Liz-Marzan, X. Ma, P. Macchiarini, H. Meng, H. Mohwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjoqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H.-W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X.-E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313 CrossRef CAS PubMed.
- H. An and B. Jin, Biotechnol. Adv., 2012, 30, 1721 CrossRef CAS PubMed.
- H. H. P. Yiu, L. Bouffier, P. Boldrin, J. Long, J. B. Claridge and M. J. Rosseinsky, Langmuir, 2013, 29, 11354 CrossRef CAS PubMed.
- J. J. Storhoff, R. Elghanian, C. A. Mirkin and R. L. Letsinger, Langmuir, 2002, 18, 6666 CrossRef CAS.
- E. M. Boon, D. M. Ceres, T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2000, 18, 1096 CrossRef CAS PubMed.
- P. Sandstrom, M. Boncheva and B. Akerman, Langmuir, 2003, 19, 7537 CrossRef.
- S. P. Schwaminger, P. F. Garcia, G. K. Merck, F. A. Bodensteiner, S. Heissler, S. Gunther and S. Berensmeier, J. Phys. Chem. C, 2015, 119, 23032 CAS.
- S. Liu, X.-Y. Meng, J. M. Perez-Aguilar and R. Zhou, Sci. Rep., 2016, 6, 37761 CrossRef CAS PubMed.
- S. Schwaminger, S. A. Blank-Shim, I. Scheifele, P. Fraga-Garcia and S. Berensmeier, Faraday Discuss., 2017, 204, 233 RSC.
- E. Pensa, E. Cortes, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benitez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, Acc. Chem. Res., 2014, 47, 2651 CrossRef CAS PubMed.
- (a) C. C. Fleischer, U. Kumar and C. K. Payne, Biomater. Sci., 2013, 1, 975 RSC; (b) U. Sakulkhu, M. Mahmoudi, L. Maurizi, J. Salaklang and H. Hofmann, Sci. Rep., 2014, 4, 5020 CrossRef CAS PubMed; (c) C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2012, 116, 8901 CrossRef CAS PubMed; (d) S. H. De Paoli Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365 CrossRef CAS PubMed.
- D. Huhn, K. Kantner, C. Geidel, S. Brandholt, I. D. Cock, S. J. H. Soenen, P. Rivera_Gil, J.-M. Montenegro, K. Braeckmans, K. Mullen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253 Search PubMed.
- (a) M. Lundqvist, J. Stigler, T. Cedervall, T. Berggard, M. B. Flanagan, I. Lynch, G. Elia and K. A. Dawson, ACS Nano, 2011, 5, 7503 CrossRef CAS PubMed; (b) S.-T. Yang, Y. Liu, Y.-W. Wang and A. Cao, Small, 2013, 9, 1635 CrossRef CAS PubMed.
- M. Schaffler, M. Semmler-Behnke, H. Sarioglu, S. Takenaka, A. Wenk, C. Schleh, S. M. Hauck, B. D. Johnston and W. G. Kreyling, Nanotechnology, 2013, 24, 265103 CrossRef PubMed.
- (a) G. Doorley and C. K. Payne, Chem. Commun., 2011, 47, 466 RSC; (b) G. Doorley and C. K. Payne, Chem. Commun., 2012, 48, 2961 RSC.
- L. Treuel and G. U. Nienhaus, Biophys. Rev., 2012, 4, 137 CrossRef CAS PubMed.
- V. Hlady and J. Buijs, Curr. Opin. Biotechnol., 1996, 7, 72 CrossRef CAS PubMed.
- Z. Mao, X. Zhou and C. Gao, Biomater. Sci., 2013, 1, 896 RSC.
- X. Liu, N. Huang, H. Li, Q. Jin and J. Ji, Langmuir, 2013, 29, 9138 CrossRef CAS PubMed.
- (a) A. Verma and F. Stellacci, Small, 2010, 6, 12 CrossRef CAS PubMed; (b) L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 5 CrossRef PubMed.
- C. D. Walkey, J. B. Olsen, F. Song, R. Liu, H. Guo, D. W. H. Olsen, Y. Cohen, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 2439 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, B. W. Neun, S. Man, X. Ye, M. Hansen, A. K. Patri, R. M. Crist and S. E. McNeil, Nanomedicine, 2014, 10, 1453 CrossRef CAS PubMed.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 5515 CrossRef CAS PubMed.
- Q. Dai, J. Guo, Y. Yan, C.-S. Ang, N. Bertleff-Zieschang and F. Caruso, Biomacromolecules, 2017, 18, 431 CrossRef CAS PubMed.
- L. Treuel, K. A. Eslahian, D. Docter, T. Lang, R. Zellner, K. Nienhaus, G. U. Nienhaus, R. H. Stauber and M. Maskos, Phys. Chem. Chem. Phys., 2014, 16, 15053 RSC.
- C. Vasti, D. A. Bedoya, R. Rojas and C. E. Giacomelli, J. Mater. Chem. B, 2016, 4, 2008 RSC.
- T. A. Larson, P. P. Joshi and K. Sokolov, ACS Nano, 2012, 6, 9182 CrossRef CAS PubMed.
- H. Amiri, L. Bordonali, A. Lascialfari, S. Wan, M. P. Monopoli, I. Lynch, S. Laurent and M. Mahmoudi, Nanoscale, 2013, 5, 8656 RSC.
- J. Huang, X. Zhong, L. Wang, L. Yang and H. Mao, Theranostics, 2012, 2, 86 CrossRef CAS PubMed.
- N. Bertrand and J.-C. Leroux, J. Controlled Release, 2012, 161, 152 CrossRef CAS PubMed.
- K. M. Wasan, D. R. Brocks, S. D. Lee, K. Sachs-Barrable and S. J. Thornton, Nat. Rev. Drug Discovery, 2008, 7, 84 CrossRef CAS PubMed.
- D. Ricklin, G. Hajishengallis, K. Yang and J. D. Lambris, Nat. Immunol., 2010, 11, 785 CrossRef CAS PubMed.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2016, 12, 387 CrossRef PubMed.
- L. J. Dixon, M. Barnes, H. Tang, M. T. Pritchard and L. E. Nagy, Compr. Physiol., 2013, 3, 785 Search PubMed.
- A. S. Abu Lila, H. Kiwada and T. Ishida, J. Controlled Release, 2013, 172, 38 CrossRef CAS PubMed.
- H. Chen, L. Wang, Q. Yu, W. Qian, D. Tiwari, H. Yi, A. Y. Wang, J. Huang, L. Yang and H. Mao, Int. J. Nanomed., 2013, 8, 3781 Search PubMed.
- H. Arami, A. Khandhar, D. Liggitt and K. M. Krishnan, Chem. Soc. Rev., 2015, 44, 8576 RSC.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS PubMed.
- Y. K. Lee, E.-J. Choi, T. J. Webster, S.-H. Kim and D. Khang, Int. J. Nanomed., 2015, 10, 97 Search PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772 Search PubMed.
- C. D. Oja, S. C. Semple, A. Chonn and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1996, 1281, 31 CrossRef.
- S. C. Semple, A. Chonn and P. R. Cullis, Adv. Drug Delivery Rev., 1998, 32, 3 CrossRef CAS.
- Y. Yan, K. T. Gause, M. M. J. Kamphuis, C.-S. Ang, N. M. O'Brien-Simpson, J. C. Lenzo, E. C. Reynolds, E. C. Nice and F. Caruso, ACS Nano, 2013, 7, 10960 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39 CrossRef CAS PubMed.
- V. Escamilla-Rivera, M. Uribe-Ramirez, S. Gonzalez-Pozos, O. Lozano, S. Lucas and A. de Vizcaya-Ruiz, Toxicol. Lett., 2016, 240, 172 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 118, 14017 CrossRef CAS PubMed.
- Q. Dai, Y. Yan, C.-S. Ang, K. Kempe, M. M. J. Kamphuis, S. J. Dodds and F. Caruso, ACS Nano, 2015, 9, 2876 CrossRef CAS PubMed.
- R. Lin, Y. Li, T. MacDonald, H. Wu, J. Provenzale, X. Peng, J. Huang, L. Wang, A. Y. Wang, J. Yang and H. Mao, Colloids Surf., B, 2017, 150, 261 CrossRef CAS PubMed.
- A. Krais, L. Wortmann, L. Hermanns, N. Feliu, M. Vahter, S. Stucky, S. Mathur and B. Fadeel, Nanomedicine, 2014, 10, 1421 CrossRef CAS PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503 CrossRef CAS PubMed.
- B. A. Kairdolf, X. Qian and S. Nie, Anal. Chem., 2017, 89, 1015 CrossRef CAS PubMed.
- C. Corbo, R. Molinaro, A. Parodi, N. E. T. Furman, F. Salvatore and E. Tasciotti, Nanomedicine, 2016, 11, 81 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28 CrossRef CAS PubMed.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283 CrossRef CAS.
- H. J. Chung, H. Lee, K. H. Bae, Y. Lee, J. Park, S.-W. Cho, J. Y. Hwang, H. Park, R. Langer, D. Anderson and T. G. Park, ACS Nano, 2011, 5, 4329 CrossRef CAS PubMed.
- K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164 CrossRef CAS PubMed.
- B. C. Mei, K. Susumu, I. L. Medintz, J. B. Belehanty, T. J. Mountziaris and H. Mattoussi, J. Mater. Chem., 2008, 18, 4949 RSC.
- Z. Yang, J. A. Galloway and H. Yu, Langmuir, 1999, 15, 8405 CrossRef CAS.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163 CrossRef CAS.
- L. Li, B. Yan, L. Zhang, Y. Tian and H. Zeng, Chem. Commun., 2015, 51, 15780 RSC.
- S. Schottler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailander and F. R. Murm, Nat. Nanotechnol., 2016, 11, 372 CrossRef PubMed.
- H.-J. Sung, A. Luk, N. S. Murthy, E. Liu, M. Jois, A. Joy, J. Bushman, P. V. Moghe and J. Kohn, Soft Mater., 2010, 6, 5196 RSC.
- M. Mahmoudi, V. Serpooshan and S. Laurent, Nanoscale, 2011, 3, 3007 RSC.
- H. Schellekens, W. E. Hennink and V. Brinks, Pharm. Res., 2013, 30, 1729 CrossRef CAS PubMed.
- Z. Wu, W. Tong, W. Jiang, X. Liu, Y. Wang and H. Chen, Colloids Surf., B, 2012, 96, 37 CrossRef CAS PubMed.
- S. Robinson and P. A. Williams, Langmuir, 2002, 18, 8743 CrossRef CAS.
- J. Pieracci, J. V. Crivello and G. Belfort, J. Membr. Sci., 1999, 156, 223 CrossRef CAS.
- Z.-M. Liu, Z.-K. Xu, J.-Q. Wang, J. Wu and J.-J. Fu, Eur. Polym. J., 2004, 40, 2077 CrossRef CAS.
- Z.-M. Liu, Z.-K. Xu, L.-S. Wan, J. Wu and M. Ulbricht, J. Membr. Sci., 2005, 249, 21 CrossRef CAS.
- J. P. Fouassier and J. Lalevee, Polymers, 2014, 6, 2588 CrossRef.
- B. Gupta, C. Plummer, I. Bisson, P. Frey and J. Hilborn, Biomaterials, 2002, 22, 863 CrossRef.
- J. Jiang, L. Zhu, L. Zhu, H. Zhang, B. Zhu and Y. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 12895 CAS.
- (a) Y. Li, R. Lin, L. Wang, J. Huang, H. Wu, G. Cheng, Z. Zhou, T. MacDonald, L. Yang and H. Mao, J. Mater. Chem. B, 2015, 3, 3591 RSC; (b) H. Chen, J. Yeh, L. Wang, X. Wu, Z. Cao, Y. A. Wang, M. Zhang, L. Yang and H. Mao, Biomaterials, 2010, 31, 5397 CrossRef CAS PubMed.
- (a) F. M. Kievit, O. Veiseh, N. Bhattarai, C. Fang, J. W. Gunn, D. Lee, R. G. Ellenbogen, J. M. Olson and M. Zhang, Adv. Funct. Mater., 2009, 19, 2244 CrossRef CAS PubMed; (b) O. Veiseh, F. M. Kievit, J. W. Gunn, B. D. Ratner and M. Zhang, Biomaterials, 2009, 30, 649 CrossRef CAS PubMed.
- Y. Zhang, S. Wen, L. Zhao, D. Li, C. Liu, W. Jiang, X. Gao, W. Gu, N. Ma, J. Zhao, X. Shi and Q. Zhao, Nanoscale, 2016, 8, 5567 RSC.
- J. Lin, S. Wang, P. Huang, Z. Wang, S. Chen, G. Niu, W. Li, J. He, D. Cui, G. Lu, X. Chen and Z. Nie, ACS Nano, 2013, 7, 5320 CrossRef CAS PubMed.
- P. Huang, J. Lin, W. Li, P. Rong, Z. Wang, S. Wang, X. Wang, X. Sun, M. Aronova, G. Niu, R. D. Leapman, Z. Nie and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 13958 CrossRef CAS PubMed.
- R. Z. Xiao, Z. W. Zeng, G. L. Zhou, J. J. Wang, F. Z. Li and A. M. Wang, Int. J. Nanomed., 2010, 5, 1057 CAS.
- G. L. Kenausis, J. Voros, D. L. Elbert, N. Huang, R. Hofer, L. Ruiz-Taylor, M. Textor, J. A. Hubbell and N. D. Spencer, J. Phys. Chem. B, 2000, 104, 3298 CrossRef CAS.
- X. Shi, Y. Wang, D. Li, L. Yuan, F. Zhou, Y. Wang, B. Song, Z. Wu, H. Chen and J. L. Brash, Langmuir, 2012, 28, 17011 CrossRef CAS PubMed.
- Q. Yang, C. Kaul and M. Ulbricht, Langmuir, 2010, 26, 5746 CrossRef CAS PubMed.
- C. Lemarchand, R. Gref and P. Couvreur, Eur. J. Pharm. Biopharm., 2004, 58, 327 CrossRef CAS PubMed.
- K. Park, S. W. Hong, W. Hur, M.-Y. Lee, J.-A. Yang, S. W. Kim, S. K. Yoon and S. K. Hahn, Biomaterials, 2011, 32, 4951 CrossRef CAS PubMed.
- K. Alhareth, C. Vauthier, F. Bourasset, C. Gueutin, G. Ponchel and F. Moussa, Eur. J. Pharm. Biopharm., 2012, 81, 453 CrossRef CAS PubMed.
- P. Xu, G. K. Quick and Y. Yeo, Biomaterials, 2009, 30, 5834 CrossRef CAS PubMed.
- J. S. Rodrigues, N. S. Santos-Magalhaes, L. C. B. B. Coelho, P. Couvreur, G. Ponchel and R. Gref, J. Controlled Release, 2003, 92, 103 CrossRef CAS PubMed.
- D. Labarre, C. Vauthier, C. Chauvierre, B. Petri, R. Muller and M. M. Chehimi, Biomaterials, 2005, 26, 5075 CrossRef CAS PubMed.
- R. E. Marchant, S. Yuan and G. Szakalas-Gratzl, J. Biomater. Sci., Polym. Ed., 1994, 6, 549 CrossRef CAS PubMed.
- (a) Z. G. Estephan, P. S. Schlenoff and J. B. Schlenoff, Langmuir, 2011, 27, 6794 CrossRef CAS PubMed; (b) K. P. Garcia, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516 CrossRef CAS PubMed.
- W. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. G. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530 CrossRef CAS PubMed.
- V. V. Breus, C. D. Heyes, K. Tron and G. U. Nienhaus, ACS Nano, 2009, 3, 2573 CrossRef CAS PubMed.
- A. K. Murthy, R. J. Stover, W. G. Hardin, R. Schramm, G. D. Nie, S. Gourisankar, T. M. Truskett, K. V. Sokolov and K. P. Johnston, J. Am. Chem. Soc., 2013, 135, 7799 CrossRef CAS PubMed.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978 CrossRef CAS PubMed.
- R. D. Vinluan III, J. Liu, C. Zhou, M. Yu, S. Yang, A. Kumar, S. Sun, A. Dean, X. Sun and J. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 11829 Search PubMed.
- N. Zhan, G. Palui, M. Safi, X. Ji and H. Mattoussi, J. Am. Chem. Soc., 2013, 135, 13786 CrossRef CAS PubMed.
- K. Boeneman Gemmill, J. R. Deschamps, J. B. Delehanty, K. Susumu, M. H. Stewart, R. H. Glaven, G. P. Anderson, E. R. Goldman, A. L. Huston and I. L. Medintz, Bioconjugate Chem., 2013, 24, 269 CrossRef CAS PubMed.
- W. Zhou, J. Shao, Q. Jin, Q. Wei, J. Tang and J. Ji, Chem. Commun., 2010, 46, 1479 RSC.
- H. Wei, O. T. Bruns, O. Chen and M. G. Bawendi, Integr. Biol., 2013, 5, 108 RSC.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473 CrossRef CAS PubMed.
- S. Chen, L. Liu and S. Jiang, Langmuir, 2006, 22, 2418 CrossRef CAS PubMed.
- (a) S.-H. Ye, C. A. Johnson Jr., J. R. Woolley, H. Nurata, L. J. Gamble, K. Ishihara and W. R. Wagner, Colloids Surf., B, 2010, 79, 357 CrossRef CAS PubMed; (b) X. Lin, K. Fukazawa and K. Ishihara, ACS Appl. Mater. Interfaces, 2015, 7, 17489 CrossRef CAS PubMed.
- L. Zhang, Z. Cao, T. Bai, L. Carr, J.-R. Ella-Menye, C. Irvin, B. D. Ratner and S. Jiang, Nat. Biotechnol., 2013, 31, 553 CrossRef CAS PubMed.
- H. S. Sundaram, X. Han, A. K. Nowinski, J.-R. Ella-Menye, C. Wimbish, P. Marek, K. Senecal and S. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 6664 CAS.
- L. Zhang, H. Xue, C. Gao, L. Carr, J. Wang, B. Chu and S. Jiang, Biomaterials, 2010, 31, 6582 CrossRef CAS PubMed.
- H. S. Sundaram, X. Han, A. K. Nowinski, N. D. Brault, Y. Li, J.-R. Ella-Menye, K. A. Amoaka, K. E. Cook, P. Marek, K. Senecal and S. Jiang, Adv. Mater. Interfaces, 2014, 7, 1400071 CrossRef PubMed.
- T. Ye, Y. Song and Q. Zheng, Colloid Polym. Sci., 2015, 293, 797 CAS.
- Z. Dong, J. Mao, M. Yang, D. Wang, S. Bo and X. Ji, Langmuir, 2011, 27, 15282 CrossRef CAS PubMed.
- Q. Shao and S. Jiang, Adv. Mater., 2015, 27, 15 CrossRef CAS PubMed.
- H.-S. Han, J. D. Martin, J. Lee, D. K. Harris, D. Fukumura, R. K. Jain and M. G. Bawendi, Angew. Chem., Int. Ed., 2013, 52, 1414 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana, D. Dehaini, P. Nguyen, P. Angsantikul, C. H. Wen, A. V. Kroll, C. Carpenter, M. Ramesh, V. Qu, S. H. Patel, J. Zhu, W. Shi, F. M. Hofman, T. C. Chen, W. Gao, K. Zhang, S. Chien and L. Zhang, Nature, 2015, 526, 118 CrossRef CAS PubMed.
- Q. Fu, P. Lv, Z. Chen, D. Ni, L. Zhang, H. Yue, Z. Yue, W. Wei and G. Ma, Nanoscale, 2015, 7, 4020 RSC.
- M. Xuan, J. Shao, L. Dai, J. Li and Q. He, ACS Appl. Mater. Interfaces, 2016, 8, 9610 CAS.
- L. Kaneti, T. Bronshtein, N. M. Dayan, I. Kovregina, N. L. Khait, Y. Lupu-Haber, M. Fliman, B. W. Schoen, G. Kaneti and M. Machluf, Nano Lett., 2016, 16, 1574 CrossRef CAS PubMed.
- H. Sun, J. Su, Q. Meng, Q. Yin, L. Chen, W. Gu, P. Zhang, Z. Zhang, H. Yu, S. Wang and Y. Li, Adv. Mater., 2016, 28, 9581 CrossRef CAS PubMed.
- V. Gujrati, S. Kim, S.-H. Kim, J. J. Min, H. E. Choy, S. C. Kim and S. Jon, ACS Nano, 2014, 8, 1525 CrossRef CAS PubMed.
- (a) A. V. Kroll, R. H. Fang and L. Zhang, Bioconjugate Chem., 2017, 28, 23 CrossRef CAS PubMed; (b) R. H. Fang, Y. Jiang, J. C. Fang and L. Zhang, Biomaterials, 2017, 128, 69 CrossRef CAS PubMed.
- Based on the search on clinicaltrials.gov in September 2017 using terms centered around “nano”.
| This journal is © The Royal Society of Chemistry 2018 |