
Functional peptide-based nanoparticles for photodynamic therapy
Kai
Han
 ,
Zhaoyu
Ma
and
Heyou
Han
*
,
Zhaoyu
Ma
and
Heyou
Han
*
State Key Laboratory of Agricultural Microbiology, College of Science, Bio-Medical Center of Huazhong Agricultural University, Huazhong Agricultural University, Wuhan 430070, China. E-mail: hyhan@mail.hzau.edu.cn; Tel: +86 027 87282043
First published on 16th November 2017
Abstract
Photodynamic therapy as a non-invasive approach has obtained great research attention during the last decade. However, photodynamic therapy still suffers from low tumor selectivity and therapeutic inefficacy due to the unspecific distribution of photosensitizers in normal tissues/cells. To overcome these hurdles, functional peptides have been introduced in photodynamic therapy systems due to their advantages of functional diversity, bioactivity, high biocompatibility and biodegradability. Herein, we review various peptide-based self-assemblies or hybrid nanoparticles that have already been reported to achieve tumor tissue, cell or subcellular organelle targeted photodynamic therapy. The role of tumor microenvironments, cellular/subcellular location, and physical/chemical properties of peptide-based nanoparticles in facilitating the photodynamic therapy efficiency are discussed in-depth. The novel development of peptide-based nanoparticles described here should offer great potential to achieve better photodynamic therapy in tumors.
 Kai Han | Dr Kai Han is a Principal Investigator in the Bio-Medical Center of Huazhong Agricultural University and an associate professor in the College of Science, Huazhong Agricultural University (China). He received his bachelor's degree from Wuhan University in 2010 and his PhD in polymer chemistry and physics from Wuhan University in 2015. His research area focuses on the construction of functional peptide self-assembly. The targeted applications concern tumor microenvironment targeted photodynamic therapy, gene delivery, controlled drug release and tumor-specific imaging. |
 Zhaoyu Ma | Zhaoyu Ma received her bachelor's degree in Chemical Engineering and Technology from Wuhan Institute of Technology (2016). She is currently pursuing her MS degree under the supervision of Prof. Heyou Han at the State Key Laboratory of Agricultural Microbiology, College of Science, Huazhong Agricultural University. Her research area mainly focuses on the construction of functional peptide-based nanoparticles for tumor targeted imaging and phototherapy. |
 Heyou Han | Dr Heyou Han is a professor in the State Key Laboratory of Agricultural Microbiology, College of Science from Huazhong Agricultural University (China). He also serves as the Executive Vice Dean of the Graduate School in Huazhong Agricultural University. He received his PhD in analytic chemistry from the Wuhan University in 2000. From 2000 to 2002, he was a post-doctoral researcher at the University of Science and Technology of China. His research area mainly focuses on developing new analysis methods to detect pathogenic microorganisms including virus and bacteria in agriculture. He is also interested in tumor bioimaging and therapy. |
1. Introduction
Tumor is one of the leading causes of death in the world. Currently, chemotherapy is the major treatment modality along with surgery for tumor treatment, and great efforts in improving chemotherapy have achieved significant improvements in patient survival during the last few decades.1–5 However, numerous tumor-related deaths still occur every year, partly because of the poor selectivity and accumulation of drugs in the tumor, which result in severely systemic side-effects to patients.6,7 As an alternative, photodynamic therapy (PDT) has obtained increasing research attention.8–15 PDT is a local and non-invasive technique for cancer treatment. Usually, PDT requires exposure of cells or tissues to the phototherapeutic agents, also known as photosensitizers. Then irradiation with the appropriate wavelength is provided, usually in the red or near-infrared region, in order to activate the ground state photosensitizer to the excited triplet state. The triplet-state photosensitizer can participate in a one-electron oxidation–reduction reaction with a neighbouring molecule. Consequently, free radical intermediates are generated, which can react with oxygen to form peroxy radicals and various reactive oxygen species (ROS).16,17 Alternatively, the triplet-state photosensitizer can also transfer energy to ground state oxygen, producing singlet oxygen. The highly reactive singlet oxygen can destroy the tumor cells and tissues through damage of various biological molecules including lipids,18 proteins19 and nucleic acids.20During PDT, photosensitizers are always inherently non-toxic in the dark. Meanwhile, the light-induced toxicity can be restricted to a confined area through directing light irradiation. As a result, PDT has its own merits compared to conventional chemotherapy and radiotherapy, including negligible systemic side effects, excellent functional and cosmetic results, repeatability and reduced long-term morbidity.21–26 To date, PDT has already been applied together with surgery and other traditional treatments as a part of synergistic antitumor therapies in the clinic. It can suppress the growth of various tumors such as neck cancers, skin cancer, early and obstructive lung cancer and so on.27–29 Recent studies also confirm that PDT can even activate the immune response of the body to kill tumor cells.30,31 In spite of the great success of PDT, PDT is still considered as an alternative or supporting remedy due to certain limitations. For instance, like most traditional chemotherapy drugs, photosensitizers also have unsatisfactory tumor accumulation efficacy after in vivo injection.32,33 Meanwhile, the free diffusion of photosensitizer inside tumor cells dramatically compromises the therapeutic efficacy of PDT due to the very short half-life (less than 20 ns) and limited action region (below 40 nm) of ROS.34,35 Besides, there is also still a lack of methods to monitor the treatment response.
To solve these dilemmas, various nanomaterials have been developed. Among these materials, peptides that consist of natural amino acids exhibit unique merits such as bioactivity, biocompability, biodegradability and functional diversity.36–41 Currently, various peptide sequences with specific functions have been screened including the tumor homing peptide, cell penetration peptide, nuclear localization peptide sequence and so on. Peptides are extensively used in biomedical applications such as drug delivery, gene delivery, biomarker imaging, and tissue engineering.42–47 Specifically, these peptides can not only self-assemble into nanoparticles but also modify the surface of nanoparticles. These modifications endow the nanoparticles with tumor tissue or subcellular organelle specific location ability, providing a promising method to overcome the limitations of current PDT.48 Here, we have attempted to provide an overview of the present status and prospects of peptide-based nanoparticles for PDT by taking specific illustrations from recently published articles. The examples given in this review do not mean that other pioneering contributions made by a large number of researchers have been neglected. We will highlight how the modification of functional peptides achieves the tumor specific PDT and maximizes the PDT efficacy (Fig. 1). We hope to inspire new ideas in this promising and burgeoning field.
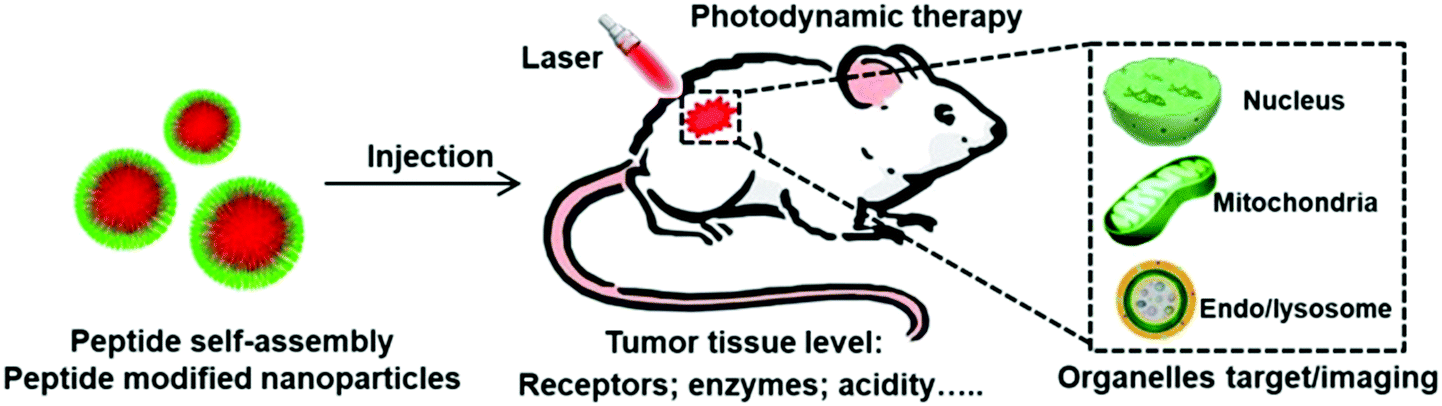 | ||
| Fig. 1 Schematic illustration showing peptide self-assembly and peptide modified nanoparticles for tumor microenvironment responsive photodynamic therapy and subcellular organelle (including endo/lysosome, mitochondria and nucleus) targeted photodynamic therapy and imaging. | ||
2. Tumor extracellular microenvironment-triggered targeted photodynamic therapy
As mentioned above, photosensitizers cannot accumulate in the tumor efficiently, which increases the dosage during treatment. The most widely used strategy in transporting photosensitizer to the tumor is to utilize the leaky vessels in the tumor to realize the passive targeting of nanoparticles. It is believed that the highly heterogeneous tumor tissue is perfused by abnormal and leaky microvasculature. In tumor blood vessels, the rapid and defective angiogenesis results in great gaps between endothelial cells, facilitating selective extravasation of nanoparticles into the tumor. At the same time, the lymphatic drainage in tumor tissue is also impaired, which further allows nanoparticles to release drugs in the tumor region. This phenomenon is also called the “enhanced permeability and retention” (EPR) effect.49,50 Although the passive targeting approach has achieved great success, it still suffers from many inherent limitations: (1) tumor tissue is highly heterogeneous. Consequently, the permeability of vessels may vary even in a single tumor mass. (2) The passive targeting approach requires the nanoparticles to have a long blood circulation time, making the preparation of nanoparticles difficult. (3) The tumor has a high interstitial pressure, limiting the diffusion of nanoparticles from the intravascular region to tumors.51The most promising way to address these challenges is to construct tumor microenvironment activatable nanoparticles to realize prolonged retention in the tumor, active binding on the tumor cell's surface and enhanced internalization by tumor cells. Over the past few decades, it has become recognized that both cumulative gene mutations and a significantly changed tumor microenvironment result in tumor growth and progression.52 The tumor microenvironment differs from the normal tissue environment, which is more complex. Acidosis, some overexpressed receptors, hypoxia, high levels of bioreductive molecules and the specific expression of some proteases are the main biological characteristics in tumor tissue.53–55 These discrepancies between tumor and normal tissues in the microenvironment, integrated with the bioactivity of peptides, provide great potential in tumor targeted PDT. In the following section, we focus on some examples and the underlying design principles for utilizing receptors, enzymes, and acidity as the major targets for tumor targeted PDT.
2.1. Receptor mediated enhanced PDT
Many receptors such as folate receptor, transferrin receptor and sialic acid receptor are overexpressed on the membrane of tumor cells,56–58 when compared with normal tissues. Meanwhile, these receptors will accelerate the cellular internalization of nanoparticles in tumor cells via a ligand/receptor induced endocytic pathway. Clearly, the overexpressed receptors are specific and potential binding targets of interest in tumor-targeted PDT. Among these receptors, integrin αvβ3 has obtained great attention during the last few years. Integrin ανβ3 is strongly up-regulated in many solid tumor cells and endothelial cells of new vasculature in tumor tissues.59 The RGD (arginine-glycine-aspartic acid) peptide is the most commonly used and effective short peptide that can specifically bind to integrin receptors on tumor cells.60 Therefore, the peptide containing the RGD sequence has been an ideal targeting ligand for modification on the surface of nanoparticles. For instance, Li et al. developed a cyclic RGD (cRGD) modified red emissive aggregation induced emission (AIE) photosensitizer-based dot for image-guided PDT via a simple and straightforward one step strategy.61 cRGD could bind to receptors more strongly than that of linear RGD. The specific interaction between integrin ανβ3 and cRGD on AIE dots triggered the ligand–receptor-mediated endocytosis, facilitating cellular internalization of the dots into the targeted tumor cells. Upon light illumination at the appropriate time point, the AIE dots in the tumor site emit red fluorescence and the generated ROS induced tumor cell death.Interestingly, recent studies demonstrated that RGD-based peptide can not only act as a tumor-homing peptide, it can also serve as a tumor-penetrating peptide to increase vascular and tissue permeability using a mechanism dependent on αν integrin and neuropilin-1.62,63 Oupický et al. constructed iRGD-modified hybrid PLGA/lipid nanoparticles for co-delivery of the photosensitizer indocyanine green and the hypoxia-activated prodrug tirapazamine for an improved antitumor therapeutic effect in metastatic breast cancer.64 Both the results of 3D tumor spheroids in vitro and orthotopic breast tumors in vivo revealed that the iRGD modified nanoparticles loaded with ICG and tirapazamine showed significantly improved penetration in the tumor, leading to improved PDT.
2.2. Enzyme responsive PDT
Matrix metalloproteinases (MMPs) are the most widely used enzymes that are overexpressed in tumor tissue. They are important in the remodelling of normal tissue and play critical roles in cancer.65,66 The presence of extracellular and membrane-bound MMPs in tumors can aid the degradation of the extracellular matrix by neoplastic cells, whilst they facilitate tumor motility and direct cell invasion.67,68 Currently, MMPs have long been of interest as pharmaceutical targets, and various MMP responsive peptides including Pro-Leu-Gly-Val-Arg (PLGVR) and Pro-Leu-Arg-Leu-Ala (PLRLA) have been used to develop tumor targeted PDT systems.69,70Zhang et al. developed a photosensitizer–peptide conjugate containing a metalloproteinase-2 (MMP-2)-sensitive sequence for targeted PDT.71 This peptide PpIX-R9GPLGLAGE8 was composed of protoporphyrin IX (PpIX) as the photosensitizer, cationic R9 as the cell-penetrating peptide (CPP) and negatively charged E8 as the masking peptide, using the MMP-2-sensitive Gly-Pro-Leu-Gly-Leu-Ala-Gly (GPLGLAG) peptide sequence as the linker. In normal tissue, the cationic CPP R9 was shielded by the negatively charged E8 peptide via electrostatic attraction. While in tumor tissue, the overexpressed MMP-2 protein cleaved the GPLGLAG sequence between Gly and Leu, leading to the detachment of the masking peptide and the subsequent recovery of the function of CPP. This exposed CPP could accelerate the internalization of the peptide in tumor cells, realizing enhanced PDT. In a similar way, Wu et al. took advantage of zwitterionic stealth peptide EKEKEKEKEKEKEKEKEKEK to shield CPP, using Pro-Leu-Gly-Leu-Ala-Gly (PLGLAG) as the MMP-2 substrate.72 The zwitterionic stealth peptide endowed aminolevulinic acid (ALA) prodrug nanocarriers with a strong antifouling ability. Meanwhile, the shielded cationic CPP can be activated by MMP-2 in the tumor, resulting in an enhanced cellular uptake of ALA and PDT efficacy.
Although the above studies utilize MMP-triggered CPP exposure to realize tumor targeted PDT, they cannot provide information about the irradiation region and time during PDT. Choosing the appropriate and precise therapeutic window is still challenging.73 Recently, a new concept of “stimuli-triggered imaging-guided” therapy has been proposed to realize visible and precise phototherapy. Wilson's group designed a photodynamic molecular beacon, in which the photosensitizer was quenched by the black hole quencher BHQ-3 using GPLGLARK as a linker. The quenched fluorescence of the photosensitizer would be restored in the tumor region due to the existence of MMP-7 enzymes, which depicted the tumor outline and killed tumor cells.74 Zhang et al. used gold nanoparticles to quench the fluorescence of PpIX via fluorescence resonance energy transfer (FRET).75 The overexpressed MMP-2 in the tumor cleaved the PLGVR linker between gold nanoparticles and PpIX, leading to the recovery of fluorescence of PpIX as well as imaging-guided PDT.
Currently, this imaging-guided therapeutic strategy can provide information on where to provide the light irradiation. However, it is largely dependent on the fluorescence recovery of the photosensitizer in the presence of MMPs. It is worth noting that the intensity of the fluorescence imaging signal could also be affected by the local content of biosensors in the tumor region. As a result, the background fluorescence is inevitably always mistaken for weak fluorescence. This false positive fluorescence will cause undesired side effects.76,77 To overcome these limits, Zhang et al. developed a ratiometric fluorescence biosensor TPPP for MMP-2 responsive AIE-guided PDT.78 As shown in Fig. 2, the biosensor TPPP consisted of an AIE molecule tetraphenylethylene (TPE) and PpIX using the PEGylated PLGVR peptide sequence as a linker. The fluorescence of TPE was negligible when the biosensor was self-assembled into nanoparticles. The overexpressed MMP-2 in the tumor region could hydrolyze the PLGVR sequence, resulting in the detachment of TPE and the PEGylated photosensitizer. Then the liberated TPE molecule could emit blue fluorescence, which guided the PDT. Specifically, the therapeutic PpIX also acted as the fluorescence internal reference. Fig. 2F shows that the ratiometric fluorescence ratio between TPE and PpIX could precisely evaluate the MMP-2 expression level at as low as 0.29 mg L−1, regardless of the concentration of the biosensor in the tumor, which avoided the false positive fluorescence.
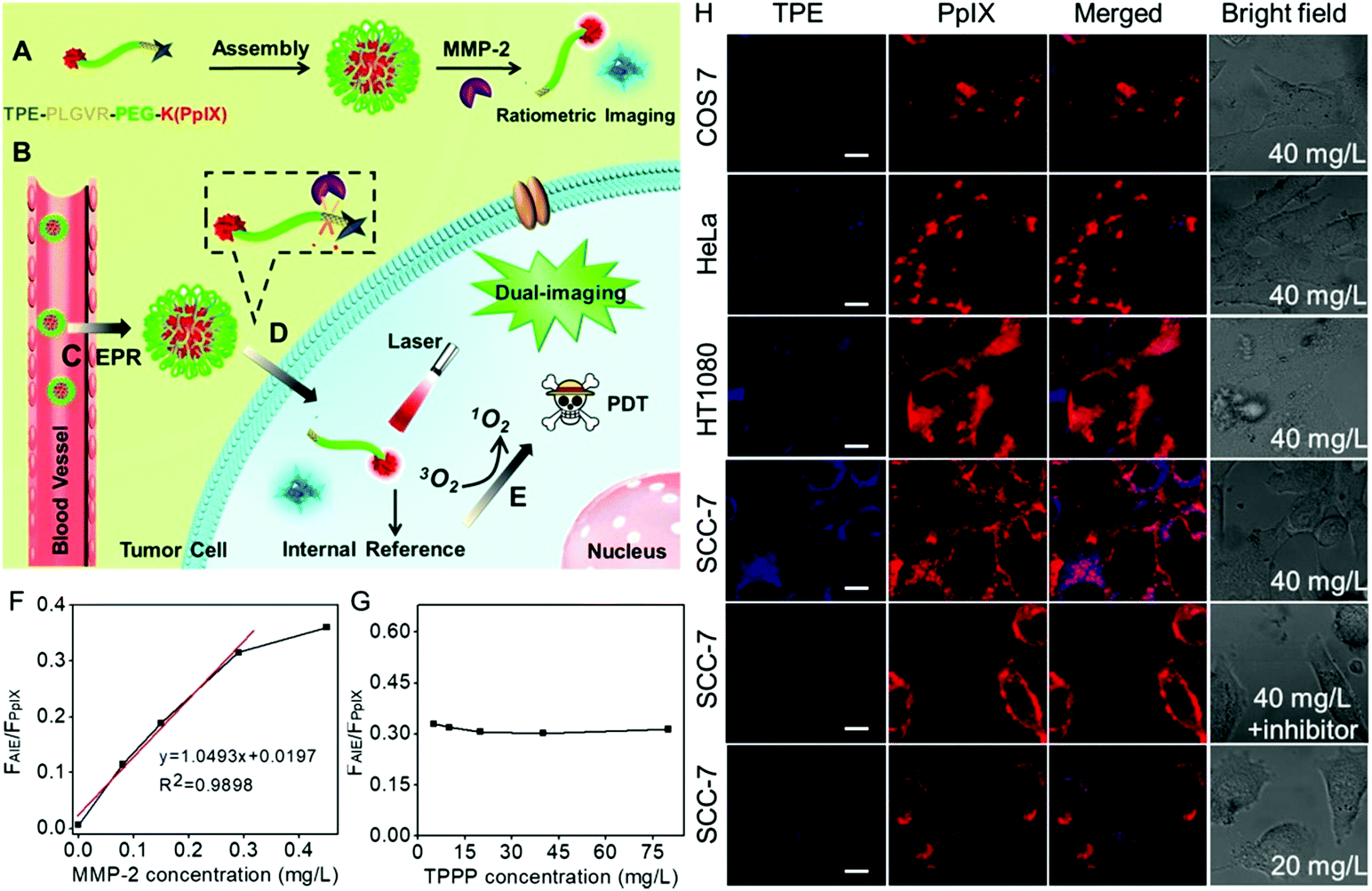 | ||
| Fig. 2 (A) Self-assembly of peptide, (B) intravenous injection of TPPP into mice via vein injection and then (C) TPPP accumulated in the tumor region via enhanced permeability and retention, (D) hydrolysis of TPPP and the fluorescence recovery of TPE, and (E) AIE-guided photodynamic antitumor therapy. (F) Fluorescence intensity ratios of TPE and PpIX when the peptide (80 mg L−1) was incubated with various concentrations of MMP-2 for 8 h. (G) Fluorescence intensity ratios of TPE and PpIX when TPPP at different concentrations was incubated with 0.29 mg L−1 of MMP-2 for 8 h. (H) Fluorescence recovery of TPE when 40 mg L−1 of peptide was incubated with COS7 cells, HeLa cells, HT1080 cells, and SCC-7 cells, SCC-7 cells with MMP-2 inhibitor or decreasing the concentration of peptide to 20 mg L−1 in SCC-7 cells. Reproduced with permission.78 Copyright 2016, American Chemical Society. | ||
In tumor tissue, there are various stromal cells that secrete factors, acting like the soil and fertilizers of tumor cells. Different from the stromal cells in normal tissues, stromal cells in tumor tissue can affect some features of tumor cells and the tumor progression.79 Among various stromal cells, tumor-associated fibroblasts originate from many types of normal cells in response to complex interactions with tumor cells. They can specifically secrete certain proteins including fibroblast-activation protein (FAP) and fibroblast specific protein.80 These bioactive enzymes have been proven to mediate tumor fibrosis, angiogenesis and metastasis and become important biomarkers in tumor tissue.81 Taking advantage of the overexpression of FAP, Zheng et al. developed a FAP responsive photodynamic molecular beacon (FAP-PPB), which comprised a fluorescent photosensitizer, BHQ-3, and a peptide linker (TSGPNQEQK) specific to FAP.82 In tumor tissue, FAP could specifically cleave FAP-PPB. Then the fluorescence of the photosensitizer was restored in the FAP-expressing cells while leaving non-expressing FAP cells undetectable. Moreover, FAP-PPB exhibited FAP-specific photocytotoxicity toward HEK-mFAP cells whereas it was non-cytotoxic to HEK-Vector cells.
2.3. Acidity responsive PDT
Another characteristic feature of the extracellular matrix in most tumors is the mild acidity (ranging from 6.5 to 7.2), when compared with normal tissues.83–85 The decreased pH is due to the elevated glycolysis and plasma membrane proton-pump activity of tumor cells. Consequently, more lactic acid molecules accumulate in tumor cells and then leach out to the extracellular matrix. Meanwhile, most tumors have insufficient blood supply and poor lymphatic drainage, which further contribute to the acidity in the tumor.86 Although this acidic microenvironment provides a growth advantage for tumor cells in vivo, tumor acidity has also been widely used to construct tumor acidity responsive nanoparticles for tumor targeted therapy.87–89One of the typically used strategies is the charge reversal system developed by Wang and co-workers.90,91 Generally, the polylysine-based amphiphilic polymer was prepared and the amino group was modified with acidity-sensitive 2,3-dimethylmaleic anhydride (DMA), generating an amide bond conjoined with a carboxylic acid group. The negatively charged polymer can self-assemble into micelles and exhibits relative stability at a neutral pH value, but rapidly degrades under a slightly acidic environment on exposure to positively charged amino groups. In other words, the polymer can become positively charged in the tumor region and readily internalized by tumor cells, leading to enhanced gene transfection or drug delivery. Inspired by this tumor-acidity-triggered charge-reversal strategy, Gao et al. used DMA to modify lysine residues’ amines in CPP to inactivate the cell penetration function.92 Once accumulated in tumor tissues, the tumor extracellular acidity detached DMA groups and activated the masked CPP peptide, leading to fluorescence/MR dual-mode imaging-guided PDT.
Recently, studies have demonstrated that not only can the surface charge change affect the cellular internalization of nanoparticles, but also the shape of nanoparticles.93,94 It is well known that there are rich hydrogen bonds among peptides, which can drive the formation of various self-assemblies with different shapes. Taking advantage of these properties, Han et al. synthesized a chimeric peptide (PEAK-DMA) for tumor acidity-triggered shape switching for tumor enhanced PDT.95 PEAK-DMA used an alkylated PpIX for PDT and the DMA modified AEAEAKAKAEAEAKAK peptide sequence for self-assembly. The AEAEAKAKAEAEAKAK peptide sequence has two surfaces in neutral solution, i.e., a polar surface that has alternatively charged ionic side chains (Glu and Lys) and a nonpolar surface that has alanines (Ala).96 As shown in Fig. 3, PEAK-DMA could self-assemble into spherical nanoparticles at the normal tissue due to the introduction of acidity sensitive DMA groups, which prevented the electrostatic and hydrogen bond interactions between AEAEAKAKAEAEAKAK. However, the tumor extracellular acidity triggered the detachment of DMA groups. The ionic complementarity between Lys and Glu was recovered, which together with hydrogen bonding, hydrophobicity, and van der Waals interactions, could drive the formation of short rod-like nanoparticles. This acidity triggered sphere-to-rod shape switch improved the specific internalization and retention of the peptide in the tumor, achieving enhanced PDT efficacy both in vitro and in vivo.
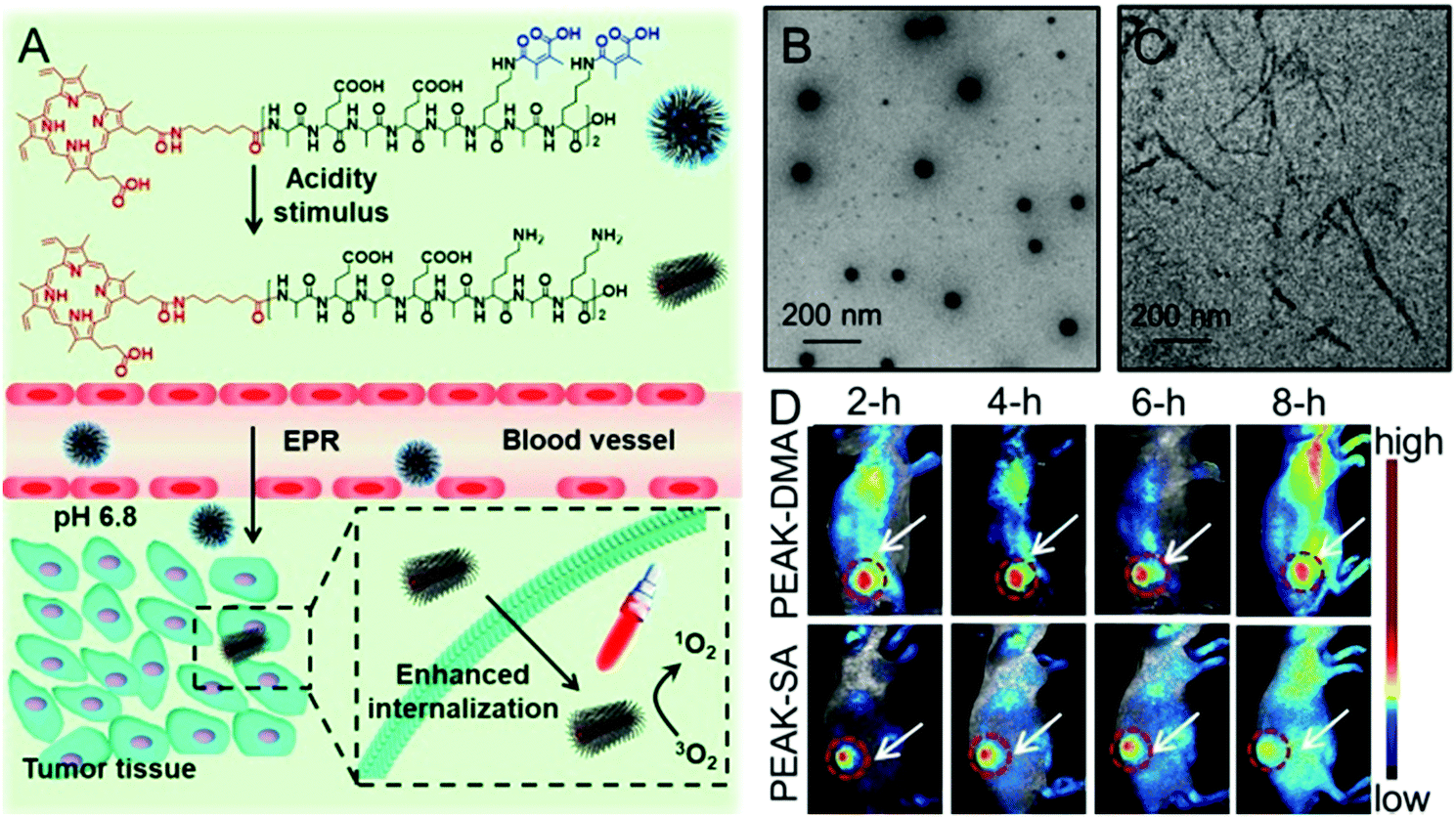 | ||
| Fig. 3 (A) Schematic illustration of acidity-triggered geometrical shape switch for enhanced PDT: intravenous injection of PEAK-DMA, and tumor acidity-triggered sphere-to-rod shape switch. This shape switch enhanced the cellular internalization and enhanced PDT efficacy in vivo. TEM images of PEAK-DMA at (B) pH 7.4 and (C) pH 6.8. (D) In vivo fluorescence images of H22 tumor-bearing mice at preset times after intravenous injection of PEAK-DMA and PEAK-SA. Reproduced with permission.95 Copyright 2017, American Chemical Society. | ||
It is known that the cell membrane is the most important protective barrier in living cells, which provides a stable environment for the efficient intracellular cell metabolism processes. Direct destruction of the tumor cell membrane significantly elevated the PDT efficacy. Liu et al. designed a charge reversible chimeric peptide (C16-K(PpIX)RRK(DMA)K(DMA)-PEG) with cell membrane-targeting property for enhanced PDT.107 The modification of DMA groups prolonged the in vivo circulation of the peptide. While in the tumor region, tumor acidity could hydrolyze DMA. Subsequently, the synergetic effect of the exposed positively charged RRKK peptide and lipophilic palmitic acid realized long-time drug retention of the peptide on the tumor cell membrane, achieving in situ PDT on the cell membrane.
Except for the introduction of pH sensitive DMA groups into the peptide, the acidic environment can also directly lead to the partial protonation of negatively charged residues including Asp or Glu, which may trigger peptide folding and change the peptide conformation including the α-helix, β-sheet and so on.108
For example, pH (low) insertion peptides (pHLIP) are randomly coiled in the physiological environment, while tumor acidity can trigger pHLIP to form a transmembrane α-helix structure. This α-helix structure has been proven to anchor and penetrate into the cell membrane. Taking advantage of this property, Luo et al. developed a pH-driven membrane-anchoring photosensitizer (pHMAPS).109 This α-helix structure of pHLIP in the acidic tumor could assist pHMAPS to anchor on the cancer cell membrane. Then PDT could damage the plasma membrane in situ, leading to improved PDT. Similarly, pHLIP could be also modified on hollow gold nanospheres,110 which could enhance the intracellular delivery of the photosensitizer by the transmembrane ability of pHLIP at mild acidity, leading to tumor specific internalization.
Our group observed that this partial protonation of negatively charged residues of Asp or Glu under tumor acidity can also improve the hydrophobicity of the peptide.111 As shown in Fig. 4, the enhanced hydrophobicity could mediate the morphology switch of peptide PpIX-PEG8-RDEVDGK(TPE)V (denoted as PPDT) from sphere to rod, resulting in selective cellular internalization and enhanced PDT against the tumor. Moreover, PDT further initiated cell apoptosis. The subsequent formation of caspase-3 enzyme cleaved the DEVDG peptide, realizing high signal/noise ratio apoptosis imaging.
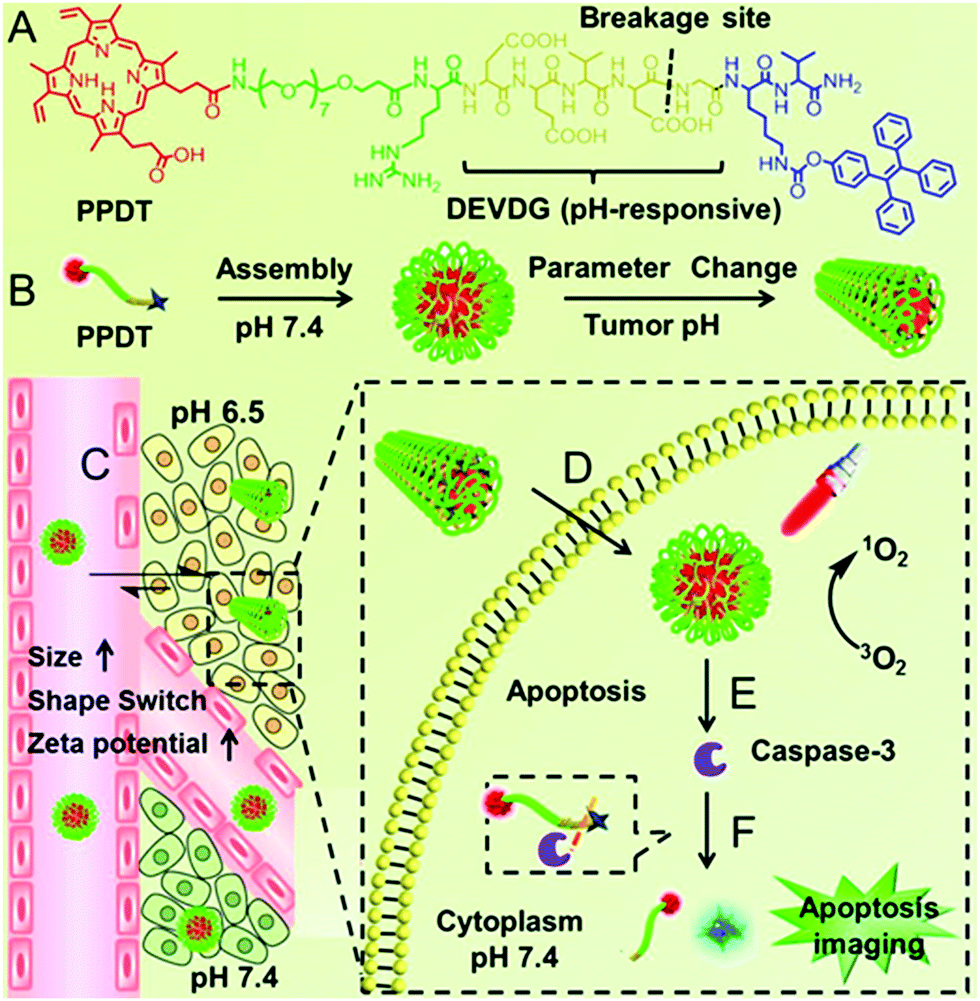 | ||
| Fig. 4 (A) The chemical structure of PPDT and (B) pH responsive self-assembly. (C) I.V. injection of PPDT and in the acidic tumor region, the size enlargement, morphology switching and increased ζ-potential of PPDT mediated prolonged tumor retention and (D) enhanced cellular uptake. (E) PDT could induce cell apoptosis and (F) the generated caspase-3 enzyme cleaved the DEVDG peptide sequence, leading to AIE imaging. Reproduced with permission.111 Copyright 2016, American Chemical Society. | ||
3. Tumor subcellular organelle targeted photodynamic therapy
After the cellular internalization of photosensitizer in the tumor, another great challenge for PDT is the intracellular localization of the photosensitizer. It is well documented that ROS is highly reactive. Both the diffusion distance and half-span of ROS are very short, so the ROS damage to biosubstances is just restricted to the immediate vicinity of ROS generation. However, the generation of ROS for most photosensitizers is in the cytoplasm, which greatly decreases PDT efficacy. It is recognized that cell fate is significantly affected by the intracellular organelles. Therefore, the precise subcellular localization of the photosensitizer will improve PDT efficacy. To date, various peptides capable of localizing on specific subcellular organelles have already been demonstrated, and some examples are shown in Table 1. In this section, we focus on some examples and the underlying design principles using the endo/lysosome, mitochondria and nucleus as the major destinations for enhanced PDT.| Targeted organelles | Peptide sequence | Peptide origin | Ref. |
|---|---|---|---|
| Endosome |
HHHHHHHH
KKALLALALHHLAHLALHLALALKKA GIGAVLKVLTTGLPALISWIKRKRQQ |
LAH4
Melittin |
97 |
| Lysosome | YQRLC | Lysosomal targeting peptide | 100 |
| AGYLLGKINLALAALAKKIL | Transportan 10 | 101 | |
| Mitochondrion | (KLAKLAK)2 | Proapoptotic peptide | 102 |
| (RLARLAR)2 | Proapoptotic peptide | 103 | |
| MALLRGVFIVAAKRTPFGAYGC | Mitochondrial 3-oxoacyl-coenzyme-A | 104 | |
| Nucleus | PKKKKRKV | HIV-1 TAT protein NLS | 105 |
| GLFEAIEGFIENGWEGMIDGWYGC | Influenza derived fusogenic peptide | 106 |
3.1. PDT in endosomes
Many nanoparticles are expected to enter endosomes after cellular internalization. Endosomes are recognized to be acidic (pH ∼ 5) and rich in various enzymes, which are hostile to bioactive substances, especially DNA, RNA or proteins. Currently, the “proton sponge” effect has been widely used to overcome the endosomal trap.112,113 However, the “proton sponge” effect alone is insufficient for rapid endosomal disruption.114 Recently, it has been reported that the generated ROS can disrupt the endosomal membranes via the oxidization of lipid, thereby leading to enhanced cytoplasmic delivery of bioactive substances. This phenomenon is also termed the photochemical internalization (PCI) effect.115 The PCI effect has been successfully applied for the delivery of both low molecular weight anticancer agents (such as camptothecin)116 and high molecular weight biosubstances (such as proteins and nucleic acids)117–119 under various therapies.Although the PCI effect can enhance the endosome escape and in vitro transfection of DNA or RNA, the generated ROS during PCI may also damage these genetic materials. To avoid this damage, quick endosomal escape and short-time light irradiation are required to ensure the transfection efficacy. Recently, Zhang et al. designed a chimeric peptide (Fmoc-ADDA-H8R8-PLGVR-PEG8) to simultaneously transport PpIX and therapeutic DNA to tumor cells.120 As shown in Fig. 5, the chimeric peptide can be selectively taken up by MMP-2 rich tumor cells owing to the hydrolysis of PLGVR peptide sequence, exfoliation of PEG as well as the increase of the surface charges of nanoparticles. Importantly, both the PCI effect of PpIX and the “proton sponge” effect of H8 sequence were employed, which dramatically decreased the light irradiation time and enhanced the endosomal escape of chimeric peptide/PpIX/DNA complexes, leading to improved DNA expression. Meanwhile, the short-time light irradiation led to undetectable changes in cell viability owing to the screened phototoxicity of PpIX, which ensured highly efficient gene transfection. After the gene transfection, the screened phototoxicity of PpIX was subsequently activated by long-time irradiation to achieve high synergistic efficacy of PDT and gene therapy.
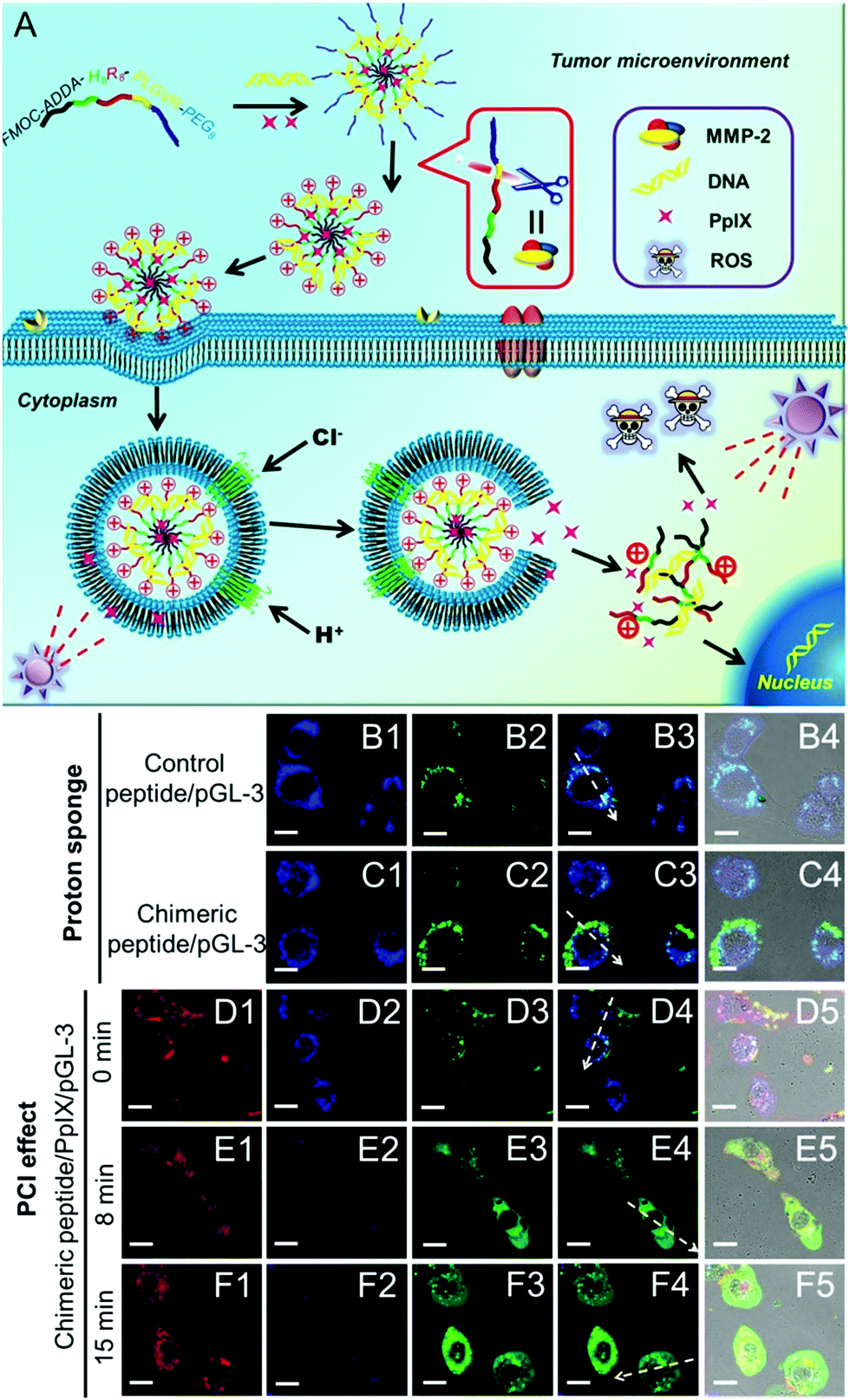 | ||
| Fig. 5 (A) Schematic diagram of the chimeric peptide/PpIX/p53 system. The chimeric peptide can encapsulate PpIX and DNA and undergo PEG detachment under the MMP-2 enzyme. After entering the endosome, the “proton sponge” effect of the H8 sequence and the PCI effect under short-time light irradiation facilitate endosomal escape, leading to optimized gene/photodynamic therapies. Endosome escape behaviors were observed by CLSM: (B1–B4) control peptide/pGL-3 complexes; (C1–C4) chimeric peptide/pGL-3 complexes. (D1–D4, E1–E4, F1–F4) chimeric peptide/PpIX/pGL-3 complexes with 0, 8, and 15 min light irradiation, respectively. Red signal: PpIX. Blue signal: LysoTracker blue; green signal: YOYO-1 labelled chimeric peptide/pGL-3 complexes. Scale bare: 15 μm. Reproduced with permission.120 Copyright 2015, Wiley. | ||
3.2. PDT in lysosomes
Generally, the endosome will gradually get more acidic with the aid of ATPase, and nanoparticles that cannot escape from the endosome are expected to enter the lysosomes.121,122 Lysosomes are single membrane-bound vesicles that have various digestive enzymes. As one of the vital organelles, lysosomes participate in many crucial biological activities including apoptosis and even metabolism.123 Specifically, some enzymes including cathepsin B are overexpressed in lysosomes of the tumor. Integrating photosensitizer and cathepsin B responsive peptide substrate, many theranostic agents have been developed for PDT.124,125 Tian et al. adopted the graphene oxide (GO) sheet to inhibit both fluorescence and 1O2 generation of close proximity-Ce6-GRRGKGGFFFF (Fig. 6).126 After the biosensor was specifically internalized into the lysosome of cancer cells, the GRRGKGGFFFF peptide sequence could be cleaved by cathepsin B. Subsequently, Ce6 was liberated from the GO sheet, inducing the efficient formation of 1O2 to damage the lysosome as well as lysosomal cell death. The lighted Ce6 upon lysosomal destruction provided real-time self-feedback information on therapeutic efficacy. Choi et al. designed a smart dual-targeted theranostic agent in which the photosensitizers were within close proximity to folic acid using RRK peptide as a linker.127 Both near-infrared fluorescence emission and ROS generation of the photosensitizers are quenched by folic acid. When the conjugates specifically bind to the folic acid receptor (first target) and then enter the lysosome, the RRK (Agr-Arg-Lys) peptide linker in the conjugate was cleaved by a cancer-selective cathepsin B enzyme (second target), resulting in the release of the photosensitizers from folic acid. Consequently, the fluorescence and ROS of the photosensitizer was completely recovered inside the target cancer cells.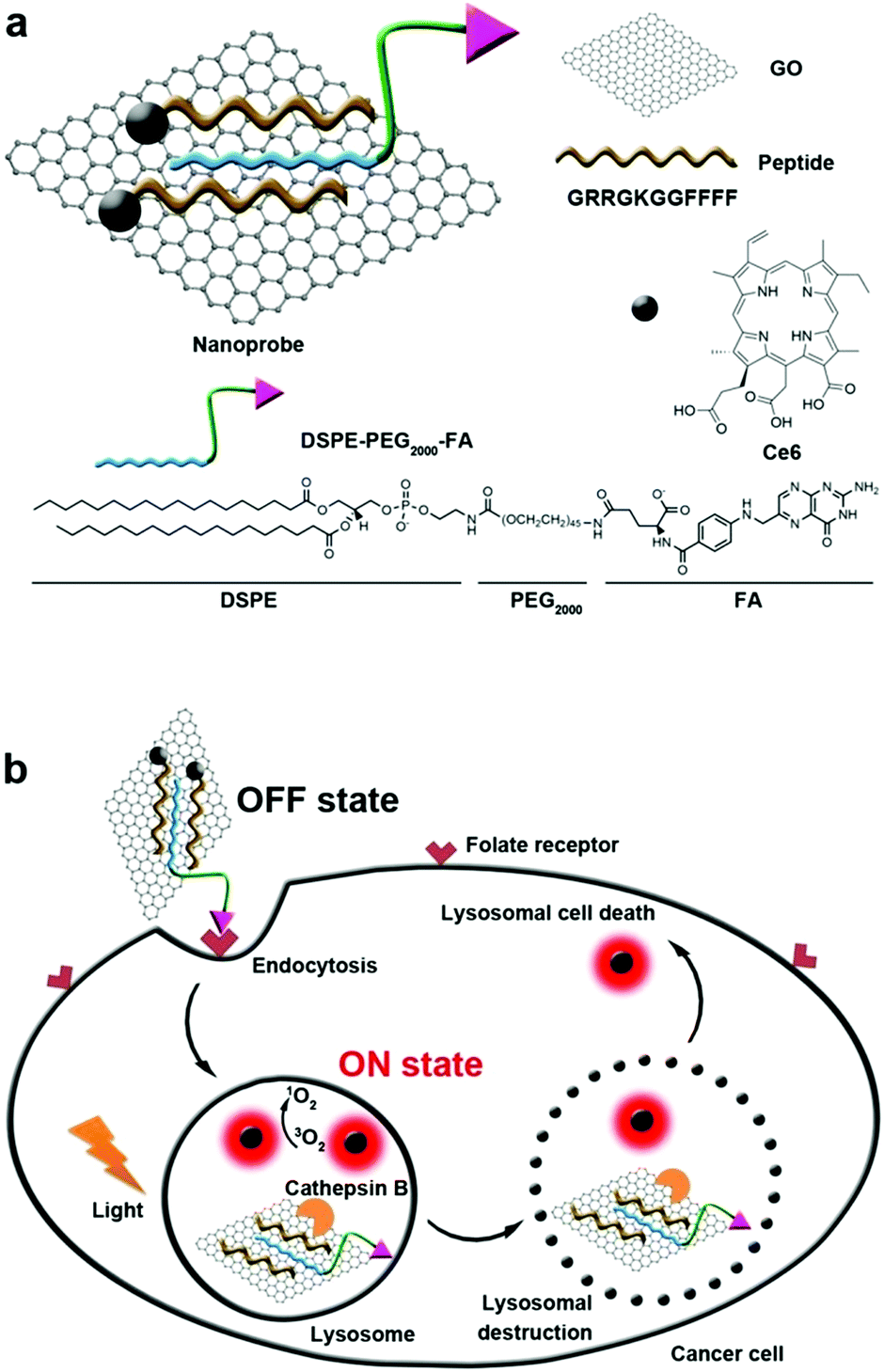 | ||
| Fig. 6 (a) Construction of biosensor. (b) Folate mediated cellular internalization of biosensor and then cathepsin B activated recovery of ROS and fluorescence of photosensitizer. PDT destroyed lysosome and lead to cell death. Reproduced with permission.126 Copyright 2015, American Chemical Society. | ||
Currently, most photosensitizers are hydrophobic and tend to aggregate in aqueous media due to their rigid planar structures. This aggregation will lead to fluorescence quenching and reduced therapeutic efficacy.128,129 A gratifying result is that scientists have discovered a cathepsin B responsive, AIE fluorogen based photosensitizer for PDT. Liu et al. fabricated dicyanovinyl-containing AIE fluorogens, which could react with biological thiol molecules and could generate ROS upon irradiation with light.130 These water soluble AIE fluorogens were non-fluorescent and could not generate ROS with light irradiation. After cRGD selectively recognized tumor cells and cathepsin B specifically cleaved the GFLG (Gly-Phe-Leu-Gly) peptide sequence, the enhanced hydrophobicity led to the aggregation of AIE fluorogens and activated PDT. These activatable photosensitizers did not contain any quencher or energy acceptor, yet they exhibited a high signal-to-noise ratio fluorescence and ROS formation.
3.3. PDT in mitochondria
Mitochondria are the energy centers of cells, which are widely spread in the cytoplasm. Recently, mitochondria-targeted DDSs have rapidly obtained considerable attention and numerous reports have implicated mitochondria as important targets for PDT,131–134 partly because of the fact that drugs can be transported to mitochondria without conquering additional hurdles including the karyotheca; this can significantly simplify the preparation of DDSs. On the other hand, mitochondria play a decisive role in the intrinsic pathway of apoptosis;135,136 mitochondria located photosensitizers have been demonstrated to be more efficient in killing cells than those that localize at other cellular sites.Mitochondria specific localization of drugs is usually driven by the electrochemical potential gradient across the inner mitochondrial membrane.137,138 The most used ligands for mitochondrial location are cationic triphenylphosphine and α-helical pro-apoptosis (KLAKLAK)2. It was reported that (KLAKLAK)2 could target and disrupt mitochondrial membranes and mediate the initiation of the apoptosis process, acting as a bio-drug. Recently, Zhang et al. fabricated a self-delivery system PpIX-PEG-(KLAKLAK)2 (denoted as PPK), achieving mitochondria-targeted photodynamic tumor therapy.139 As shown in Fig. 7A, both PpIX and (KLAKLAK)2 peptide are therapeutic indexes, resulting in high drug loading efficacy. The PCI effect of PpIX with short-time light irradiation disrupted the stability of the cell membrane and improved the cellular internalization of PPK. Moreover, PPK could target mitochondria with the aid of the (KLAKLAK)2 peptide. Importantly, the in situ generation of ROS in mitochondria dramatically decreased the mitochondrial membrane potential and destroyed the intercellular energy center (Fig. 7B–E). As a result, significant cell death was observed during PDT.
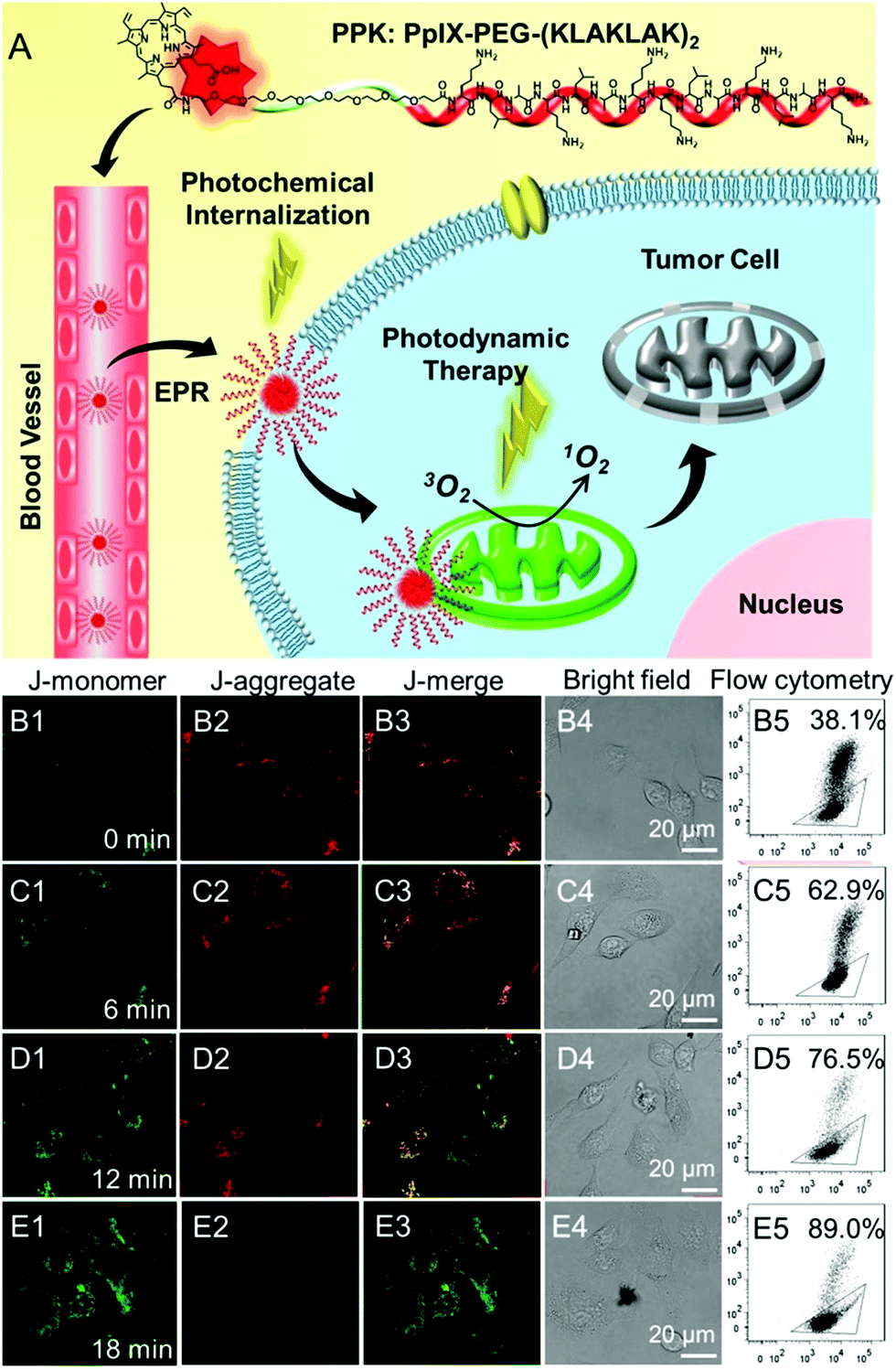 | ||
| Fig. 7 (A) Schematic diagram of the mitochondria-targeted self-delivering process: PPK could passively target tumor cells via EPR effect and then the PCI effect under a short-time light irradiation accelerated cellular internalization. The peptide could realize in situ PDT in mitochondria under long-time light irradiation with the guidance of (KLAKLAK)2. (B–E) CLSM images and the flow cytometric analysis of mitochondrial membrane potential for HeLa cells when the cells were incubated with PPK and then performed with different time irradiations: (B1–B5) 0 min light irradiation; (C1–C5) 6 min light irradiation; (D1–D5) 12 min light irradiation; (E1–E5) 18 min light irradiation. The triangle separated populations with high (inside the triangle) and low membrane potential (outside the triangle). The horizontal axis is green fluorescence (dead cell) while the vertical axis is red fluorescence (normal cell). Reproduced with permission.139 Copyright 2015, Wiley. | ||
As the energy center of cells, mitochondria also manipulate the generation of various biosubstances such as intracellular adenosine 5′-triphophate (ATP) and caspase-3 enzyme.140 Inspired by the fact that active drug efflux mediated by the efflux transporter is mainly ATP-dependent, our group developed an amphiphilic mitochondria-targeted peptide-based DDS to overcome drug resistance.141 As shown in Fig. 8, this amphiphilic peptide could encapsulate doxorubicin (DOX) with high efficacy. It can also achieve in situ PDT in mitochondria due to the introduction of (KLAKLAKK)2. The generated ROS significantly disrupted the mitochondria during PDT, which remarkably decreased the content of intracellular ATP. As a result, the DOX efflux was remarkably inhibited, realizing combined chemo-/photodynamic therapies and suppressing drug resistance. The relationship between the ATP amount and the drug efflux was also studied (Fig. 8C). Interestingly, the amount of intracellular ATP exhibited a linear relationship with the efflux amount of DOX, suggesting ATP content could determine the drug efflux.
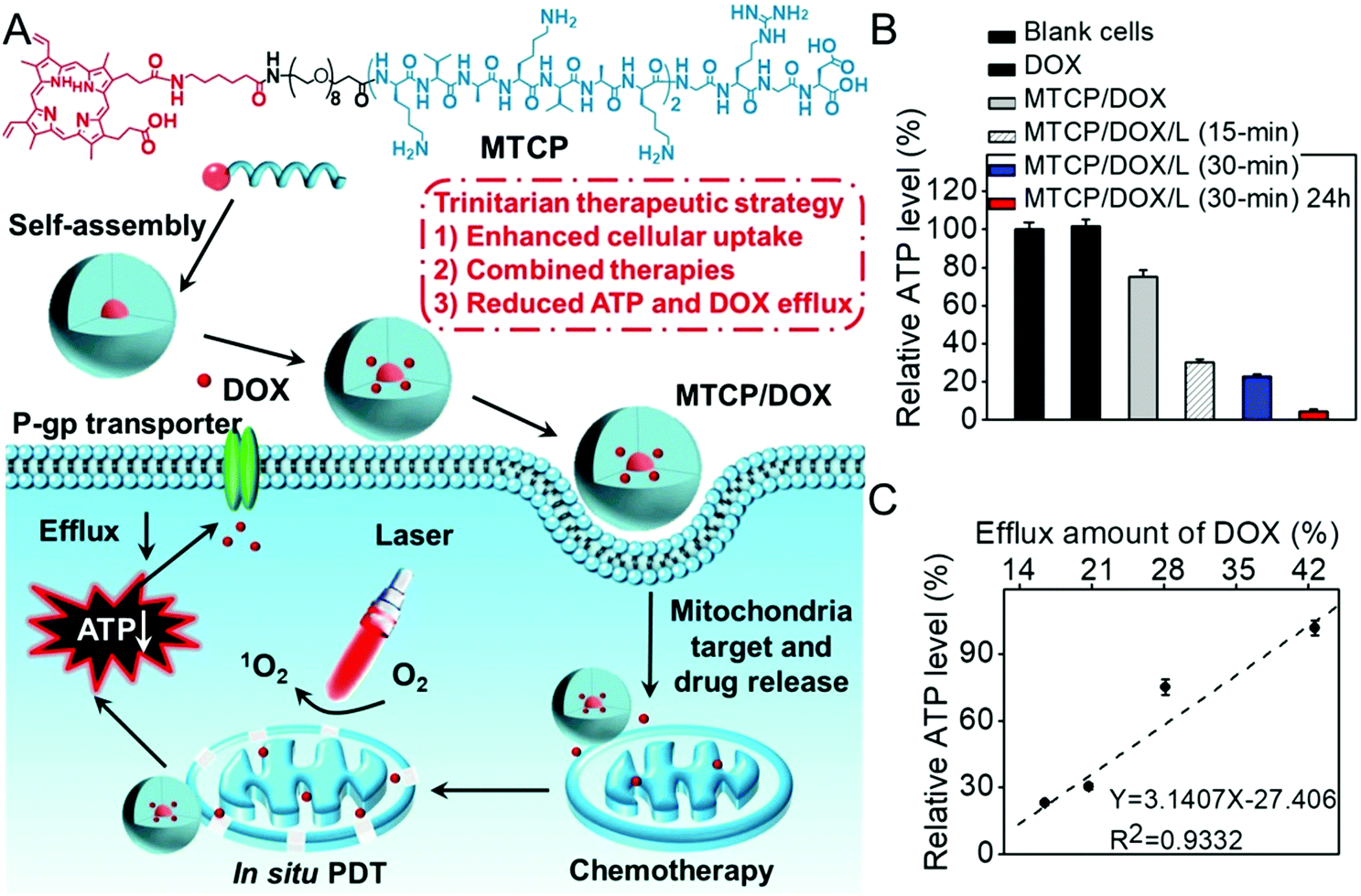 | ||
| Fig. 8 (A) Chemical structure of mitochondria-targeted peptide and schematic illustration of mitochondria-disruption mediated reversal of drug resistance in tumor cells. The peptide can encapsulate DOX and (KLAKLAK)2-mediated mitochondria-targeted accumulation. Then in situ PDT in mitochondria decreased intracellular ATP generation and inhibited DOX efflux. (B) Relative intracellular ATP level after different treatments and (C) the relationship with efflux amount of DOX. Reproduced with permission.141 Copyright 2016, American Chemical Society. | ||
3.4. PDT in nuclei
The nucleus is believed to be the most hypersensitive intracellular organelle, since the genetic DNA and transcription machinery reside at the nucleus.142 The nucleus possesses the double lipid bilayer of the nuclear envelope, which can separate the nucleus from the cytosol. This pair of peripheral membranes is perforated by nuclear pore complexes. The cell nucleus can maintain the integrity of genes by regulating its genetic expression. The final destination of many anticancer drugs is the DNA inside the nucleus. However, the domains of unstructured phenylalanine-glycine repeats exist in the inner nuclear pore complex channel, forming a steric permeability barrier to larger macromolecules. The central channel of the nuclear pore complexes has been reported as being 39 nm in diameter. Thus, nanoparticles larger than 39 nm or molecules larger than approximately 40 kDa require active transport.143,144To date, many peptide sequences have been screened to transport biosubstances into the nucleus.145,146 For example, VQRKRQKLMP is derived from the transcription factor NF-kB, which functions to internalize NF-kB into the nucleus. Blackmore et al. confirmed that VQRKRQKLMP could direct two Ru(II) polypyridyl complexes to the nucleus and showed the first evidence of targeted delivery and localization of a ruthenium complex using a transcription factor sequence.147
The most widely used nuclear localization peptide sequence (NLS) is PKKKRKV undoubtedly.148,149 Our group constructed a chimeric peptide PpIX-Ahx-(PEG8)2-PKKKRKV (denoted as PAPP-DMA) for acidity-triggered tumor/nucleus dual-targeted PDT.150 As shown in Fig. 9, the DMA group could both avoid the nonspecific adsorption of peptide in vivo and disguise the NLS sequence. The mildly acidic environment of the tumor triggers the detachment of the DMA group and then the surface charge of the peptide self-assembly dramatically increased, which enhanced the uptake of the chimeric peptide in the tumor. More importantly, the NLS sequence acted both as the cellular internalization accelerator and the nuclear translocation guider. It guided the nuclear transport of the peptide, leading to in situ PDT in the nucleus, which dramatically simplified the preparation.
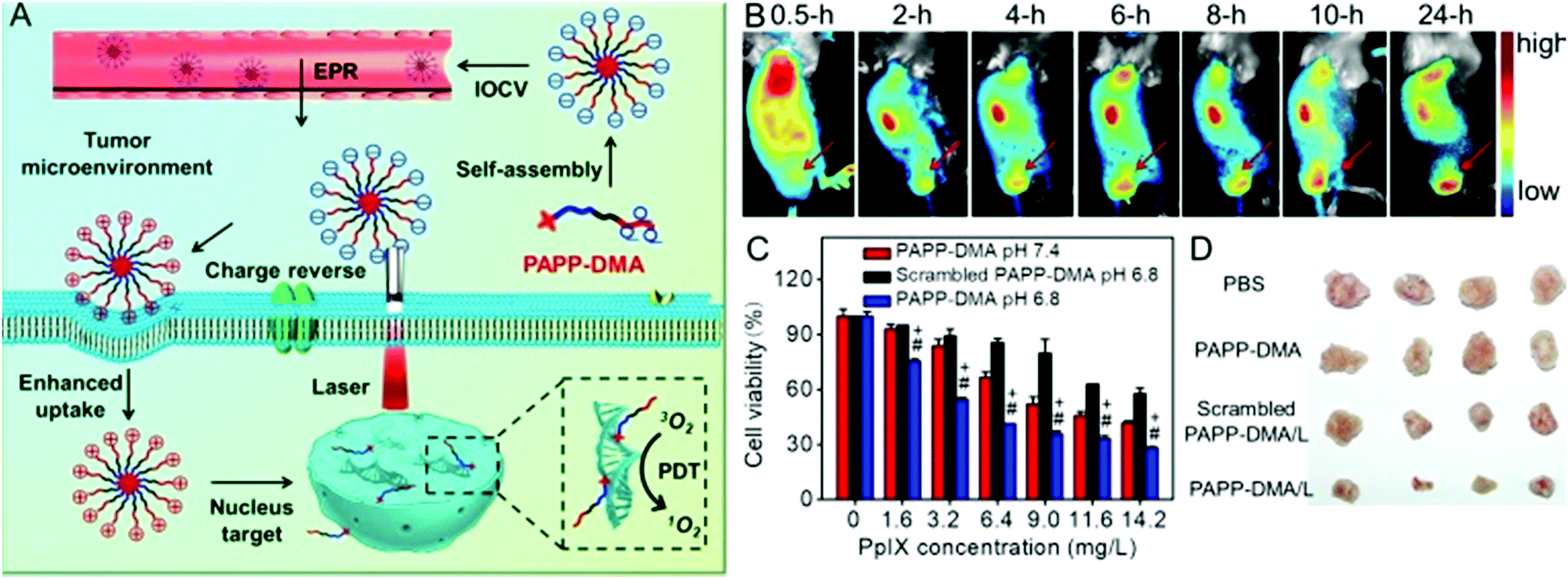 | ||
| Fig. 9 (A) Schematic illustration of tumor and intranuclear delivery of photosensitizer for enhanced PDT: self-assembly of PAPP-DMA was intravenously injected into the vessel. EPR effect mediated the tumor accumulation of PAPP-DMA and then the tumor acidic environment triggered charge reversal, accelerated cellular uptake and the NLS peptide guided in situ PDT in nuclei. (B) In vivo biodistribution images of PAPP-DMA nanoparticles in H22 tumor-bearing mice at preset times after intravenous injection. The red arrow points to tumor tissue. (C) Cytotoxicity in vitro of PAPP-DMA nanoparticles at different pHs (6.8 and 7.4) and scrambled PAPP-DMA at pH 6.8 with 2 min light irradiation. (D) Post-sacrifice tumor images at the 11th day after in vivo injection of various samples. Reproduced with permission.150 Copyright 2016, Wiley. | ||
Except for the typical NLS sequence, more and more studies have demonstrated that the transcriptional activator (TAT) peptide sequence can also guide the nanoparticles to enter the nucleus. Tang et al. designed a nuclear targeted dual-photosensitizer for PDT against multidrug resistant cancer.151 Ce6 was modified on the surface of a core/shell structure of nano-photosensitizer upconversion@TiO2. Meanwhile, the TAT peptide was anchored for nuclear targeting. The introduction of rare earth elements including Er and Tm triggered the generation of multiple ROS (˙OH and 1O2) for the dual-photosensitizer. The nano-sized photosensitizer accompanied with nuclear targeting could generate multiple ROS in the nucleus in situ regardless of P-glycoprotein, achieving enhanced PDT against multidrug resistant cancer. Although the TAT peptide could facilitate the active nuclear entry of nanoparticles, the size of nanoparticles was a critical factor in the translocation. Shi et al. synthesized monodispersed MSNs-TAT with various particle sizes and demonstrated that only MSNs-TAT with a diameter of 50 nm or smaller can efficiently target the nucleus.152 Shi et al. also observed ROS generation upon irradiation right inside the nuclei for destroying DNA instantaneously.153 The in vivo result further revealed that the nuclear-targeted delivery could decrease the photosensitizer dose to a rather low value (2 mg kg−1 per body weight) as well as the irradiation dose to an extremely low value (6 J cm−2).
4. Conclusions and future perspectives
During the last few decades, great progress has been made in developing functional nanoparticles for tumor-targeted PDT. These nanoparticles showed improved performance in addressing the existing challenges of PDT including enhanced tumor accumulation, facilitated cellular internalization and maximized PDT efficacy. In this review, we highlight several examples of peptide-based nanoparticles for PDT. With the aid of a bioactive peptide, these peptide-based nanoparticles can target the tumor extracellular microenvironments or localize in the intracellular organelles. The target of tumor extracellular microenvironments can facilitate cell internalization in the tumor site, while the subcellular organelle localization dramatically improved the therapeutic efficacy. These peptide-based nanoparticles show great potential in minimizing drug dose and side effects.Despite many encouraging outcomes, it should be noted that there still exist many challenges that restrict the extensive clinical application of PDT. First, the penetration of light, even near infrared light, is limited, making PDT treatment in deep tumor difficult. Second, many peptide-based, tumor-targeted PDT systems can achieve success in vitro and in vivo using the mouse as the animal model. However, in vivo studies using the mouse as the model are greatly simpler than the realistic situation. Clinical trials are still rare but desirable. Meanwhile, the inherent biodegradability of the peptide may also compromise the bioactivity in vivo. And the efficacy of the peptide is sometimes not so efficient, especially compared with antibodies or enzymes. Third, after clinical PDT, patients are always required to stay in the dark for as long as one month in order to avoid potential phototoxicity. This process also restricts the further application of PDT. The screening of a more efficient tumor penetration or target peptide can decrease photosensitizer dosage and overcome this problem to some extent. On the other hand, developing tumor microenvironment responsive, peptide-based photodynamic systems that can completely quench the ROS generation should also have great potential to solve this problem. Finally, the long-term safety should be systemically investigated. In any case, this review clearly illustrates the potential and strength of peptide-based biomaterials and the development of functional peptide-based nanoparticles can provide new opportunities for more efficient tumor targeted PDT.
Abbreviations
| ALA | Aminolevulinic acid |
| AIE | Aggregation-induced-emission |
| BHQ-3 | Black hole quencher 3 |
| ATP | Adenosine 5′-triphophate |
| CPPs | Cell-penetrating peptides |
| DDS | Drug delivery system |
| DMA | 2,3-Dimethylmaleic anhydride |
| DOX | Doxorubicin |
| EPR | Enhanced permeability and retention |
| FAP | Fibroblast-activation protein |
| FAP-PPB | FAP-triggered photodynamic molecular beacon |
| FRET | Fluorescence resonance energy transfer |
| MMPs | Matrix metalloproteinases |
| PCI | Photochemical internalization |
| PDT | Photodynamic therapy |
| PpIX | Protoporphyrin IX |
| ROS | Reactive oxygen species |
| TAT | Transcriptional activator |
| TPE | Tetraphenylethylene |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51603080, 21375043 and 21778020), the Fundamental Research Funds for the Central Universities (2662015QD026), the National Key Research Development Program of China (2016YFD0500700) and the Sci-tech Innovation Foundation of Huazhong Agricultural University (2662017PY042).References
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- Y. Ping, D. Ding, R. A. N. S. Ramos, H. Mohanram, K. Deepankumar, J. Gao, G. Tang and A. Miserez, ACS Nano, 2017, 11, 4528–4541 CrossRef CAS PubMed.
- R. Xu, G. Zhang, J. Mai, X. Deng, V. Segura-Ibarra, S. Wu, J. Shen, H. Liu, Z. Hu, L. Chen, Y. Huang, E. Koay, Y. Huang, J. Liu, J. E. Ensor, E. Blanco, X. Liu, M. Ferrari and H. Shen, Nat. Biotechnol., 2016, 34, 414–418 CrossRef CAS PubMed.
- X. Liu, A. Situ, Y. Kang, K. R. Villabroza, Y. Liao, C. H. Chang, T. Y. Donahue, A. E. Nel and H. Meng, ACS Nano, 2016, 10, 2702–2715 CrossRef CAS PubMed.
- J. Chen, S. Ratnayaka, A. Alford, V. Kozlovskaya, F. Liu, B. Xue, K. Hoyt and E. Kharlampieva, ACS Nano, 2017, 11, 3135–3146 CrossRef CAS PubMed.
- C. He, X. Duan, N. Guo, C. Chan, C. Poon, R. R. Weichselbaum and W. B. Lin, Nat. Commun., 2016, 7, 12499–12511 CrossRef CAS PubMed.
- A. Kamkaew, F. Chen, Y. Zhan, R. L. Majewski and W. Cai, ACS Nano, 2016, 10, 3918–3935 CrossRef CAS PubMed.
- H. Huang and J. F. Lovell, Adv. Funct. Mater., 2017, 27, 1603524 CrossRef PubMed.
- Y. Yu, J. Geng, E. Y. X. Ong, V. Chellappan and Y. N. Tan, Adv. Healthcare Mater., 2016, 5, 2528–2535 CrossRef CAS PubMed.
- P. Kalluru, R. Vankayala, C. S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2016, 26, 7908–7920 CrossRef CAS.
- J. Ge, Q. Jia, W. Liu, M. Lan, B. Zhou, L. Guo and X. Meng, Adv. Healthcare Mater., 2016, 5, 665–675 CrossRef CAS PubMed.
- N. Zhang, F. F. Zhao, Q. L. Zou, Y. X. Li, G. H. Ma and X. H. Yan, Small, 2016, 12, 5936–5943 CrossRef CAS PubMed.
- K. Liu, R. R. Xing, Q. L. Zou, G. H. Ma, H. Mçhwald and X. H. Yan, Angew. Chem., Int. Ed., 2016, 55, 3036–3039 CrossRef CAS PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- X. Yao, X. Chen, C. He, L. Chen and X. Chen, J. Mater. Chem. B, 2015, 3, 4707–4714 RSC.
- H. Park and K. Na, Biomaterials, 2013, 34, 6992–7000 CrossRef CAS PubMed.
- A. El-Hussein, J. Nanomed. Nanotechnol., 2016, 7, 2–7 Search PubMed.
- F. Berg, J. Wilken, C. A. Helm and S. Block, J. Phys. Chem. B, 2015, 119, 25–32 CrossRef CAS PubMed.
- W. Piao, K. Hanaoka, T. Fujisawa, S. Takeuchi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2017, 139, 13713–13719 CrossRef CAS PubMed.
- J. Wang, Y. Li, R. Mao, Y. Wang, X. Yan and J. Liu, J. Mater. Chem. B, 2017, 5, 5793–5805 RSC.
- S. Y. Li, H. Cheng, B. R. Xie, W. X. Qiu, J. Y. Zeng, C. X. Li, S. S. Wan, L. Zhang, W. L. Liu and X. Z. Zhang, ACS Nano, 2017, 11, 7006–7018 CrossRef CAS PubMed.
- X. Wen, Y. Li and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2017, 19, 140–152 CrossRef CAS PubMed.
- W. Yu, J. Zhu, Y. Wang, J. Wang, W. Fang, K. Xia, J. Shao, M. Wu, B. Liu, C. Liang, C. Ye and H. Tao, Oncotarget, 2017, 8, 39833–39848 Search PubMed.
- K. Ma, R. R. Xing, T. F. Jiao, G. Z. Shen, C. J. Chen, J. B. Li and X. H. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 30759–30767 CAS.
- C. Hur, N. S. Nishioka and G. S. Gazelle, Dig. Dis. Sci., 2003, 48, 1273–1283 CrossRef PubMed.
- N. Yavari, S. Andersson-Engels, U. Segersten and P. U. Malmstrom, Can. J. Urol., 2011, 18, 5778–5786 Search PubMed.
- R. Allison, K. Moghissi, G. Downie and K. Dixon, Photodiagn. Photodyn. Ther., 2011, 8, 231–239 CrossRef PubMed.
- M. Korbelik, J. H. Sun and J. J. Posakony, Photochem. Photobiol., 2001, 73, 403–409 CrossRef CAS PubMed.
- Y. G. Qiang, C. M. N. Yow and Z. Huang, Med. Res. Rev., 2008, 28, 632–644 CrossRef CAS PubMed.
- Y. P. Zeng, S. L. Luo, Z. Y. Yang, J. W. Huang, H. Li, C. Liu, W. D. Wang and R. Li, J. Mater. Chem. B, 2016, 4, 2190–2198 RSC.
- Z. J. Zhou, J. B. Song, L. M. Nie and X. Y. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- J. M. Dąbrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 Search PubMed.
- X. S. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 16040531 Search PubMed.
- S. Menegatti, M. Zakrewsky, S. Kumar, J. S. De Oliveira, J. A. Muraski and S. Mitragotri, Adv. Healthcare Mater., 2016, 5, 602–609 CrossRef CAS PubMed.
- W. Wei, L. Petrone, Y. Tan, H. Cai, J. N. Israelachvili, A. Miserez and J. H. Waite, Adv. Funct. Mater., 2016, 26, 3496–3507 CrossRef CAS PubMed.
- Y. Wang, M. R. Newman, M. Ackun-Farmmer, M. P. Baranello, T. J. Sheu, J. E. Puzas and D. S. Benoit, ACS Nano, 2017, 11, 9445–9458 CrossRef CAS PubMed.
- M. A. Khalily, G. Bakan, B. Kucukoz, A. E. Topal, A. Karatay, H. G. Yaglioglu, A. Dana and M. O. Guler, ACS Nano, 2017, 11, 6881–6892 CrossRef CAS PubMed.
- W. Zhang, K. Müller, E. Kessel, S. Reinhard, D. He, P. M. Klein and E. Wagner, Adv. Healthcare Mater., 2016, 5, 1493–1504 CrossRef CAS PubMed.
- J. J. Hu, D. Xiao and X. Z. Zhang, Small, 2016, 12, 3344–3359 CrossRef CAS PubMed.
- Z. Meng, L. Luan, Z. Kang, S. Feng, Q. Meng and K. Liu, J. Mater. Chem. B, 2017, 5, 74–84 RSC.
- S. Wong, M. S. Shim and Y. J. Kwon, J. Mater. Chem. B, 2014, 2, 595–615 RSC.
- V. Bhagat and M. L. Becker, Biomacromolecules, 2017, 18, 3009–3039 CrossRef CAS PubMed.
- M. Abbas, Q. Zou, S. K. Li and X. H. Yan, Adv. Mater., 2017, 29, 1605021 CrossRef PubMed.
- Q. L. Zou, M. Abbas, L. Y. Zhao, S. K. Li, G. Z. Shen and X. H. Yan, J. Am. Chem. Soc., 2017, 139, 1921–1927 CrossRef CAS PubMed.
- V. Mikhalevich, I. Craciun, M. Kyropoulou, C. G. Palivan and W. Meier, Biomacromolecules, 2017, 18, 3471–3480 CrossRef CAS PubMed.
- C. G. England, H. Im, L. Feng, F. Chen, S. A. Graves, R. Hernandez, H. Orbay, C. Xu, S. Y. Cho, R. J. Nickles, Z. Liu, D. S. Lee and W. Cai, Biomaterials, 2016, 100, 101–109 CrossRef CAS PubMed.
- L. Wang, J. Huang, H. Chen, H. Wu, Y. Xu, Y. Li, H. Yi, Y. A. Wang, L. Yang and H. Mao, ACS Nano, 2017, 11, 4582–4592 CrossRef CAS PubMed.
- J. Z. Du, C. Q. Mao, Y. Y. Yuan, X. Z. Yang and J. Wang, Biotechnol. Adv., 2014, 32, 789–803 CrossRef CAS PubMed.
- J. Liu, Z. Luo, J. Zhang, T. Luo, J. Zhou, X. Zhao and K. Cai, Biomaterials, 2016, 83, 51–65 CrossRef CAS PubMed.
- C. J. Cheng, R. Bahal, I. A. Babar, Z. Pincus, F. Barrera, C. Liu, A. Svoronos, D. T. Braddock, P. M. Glazer, D. M. Engelman, W. M. Saltzman and F. J. Slack, Nature, 2015, 518, 107–110 CrossRef CAS PubMed.
- T. Ji, Y. Zhao, Y. Ding and G. Nie, Adv. Mater., 2013, 25, 3508–3525 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- F. Sonvico, S. Mornet, S. Vasseur, C. Dubernet, D. Jaillard, J. Degrouard, J. Hoebeke, E. Duguet, P. Colombo and P. Couvreur, Bioconjugate Chem., 2005, 16, 1181–1188 CrossRef CAS PubMed.
- X. Wu, M. Yu, B. Lin, H. Xing, J. Han and S. Han, Chem. Sci., 2015, 6, 798–803 RSC.
- T. Zhao, X. W. He, W. Y. Li and Y. K. Zhang, J. Mater. Chem. B, 2015, 3, 2388–2394 RSC.
- B. A. Othman, C. Greenwood, A. F. Abuelela, A. A. Bharath, S. Chen, I. Theodorou, T. Douglas, M. Uchida, M. Ryan, J. S. Merzaban and A. E. Porter, Adv. Healthcare Mater., 2016, 5, 1310–1325 CrossRef CAS PubMed.
- K. Zhang, P. Li, Y. He, X. Bo, X. Li, D. Li, H. Chen and H. Xu, Biomaterials, 2016, 99, 34–46 CrossRef CAS PubMed.
- M. Li, Y. Gao, Y. Y. Yuan, Y. Z. Wu, Z. F. Song, B. Z. Tang, B. Liu and Q. C. Zheng, ACS Nano, 2017, 11, 3922–3932 CrossRef CAS PubMed.
- H. L. Gao, Y. Xiong, S. Zhang, Z. Yang, S. J. Cao and X. G. Jiang, Mol. Pharmaceutics, 2014, 11, 1042–1052 CrossRef CAS PubMed.
- X. D. Xu, J. Wu, Y. L. Liu, M. Yu, L. L. Zhao, X. Zhu, S. Bhasin, Q. Li, E. Ha, J. J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2016, 55, 7091–7094 CrossRef CAS PubMed.
- Y. Z. Wang, Y. Xie, J. Li, Z. H. Peng, Y. Sheinin, J. P. Zhou and D. Oupicky, ACS Nano, 2017, 11, 2227–2238 CrossRef CAS PubMed.
- G. S. Butler and C. M. Overall, Biochemistry, 2009, 48, 10830–10845 CrossRef CAS PubMed.
- A. Mohsen, P. Collery, R. Garnotel, B. Brassart, N. Etique, G. M. Sabry, R. E. Hassan, P. Jeannesson, D. Desmaëlea and H. Morjani, Metallomics, 2017, 9, 1176–1184 RSC.
- C. M. Overall and O. Kleifeld, Nat. Rev. Cancer, 2006, 6, 227–239 CrossRef CAS PubMed.
- Q. J. He, S. R. Guo, Z. Y. Qian and X. Y. Chen, Chem. Soc. Rev., 2015, 44, 6258–6286 RSC.
- A. Llopis-Lorente, B. Lozano-Torres, A. Bernardos, R. Martínez-Máñez and F. Sancenón, J. Mater. Chem. B, 2017, 5, 3069–3083 RSC.
- F. Y. Zhou, B. Feng, T. T. Wang, D. Wang, Q. S. Meng, J. F. Zeng, Z. W. Zhang, S. L. Wang, H. J. Yu and Y. P. Li, Adv. Funct. Mater., 2017, 27, 1606530 CrossRef.
- S. Y. Li, H. Cheng, W. X. Qiu, L. H. Liu, S. Chen, Y. Hu, B. R. Xie, B. Li and X. Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28319–28329 CAS.
- J. N. Wu, H. J. Han, Q. Jin, Z. H. Li, H. Li and J. Ji, ACS Appl. Mater. Interfaces, 2017, 9, 14596–14605 CAS.
- D. M. Wang, B. Liu, Z. W. Quan, C. X. Li, Z. Y. Hou, B. G. Xing and J. Lin, J. Mater. Chem. B, 2017, 5, 2209–2230 RSC.
- G. Zheng, J. Chen, K. Stefflova, M. Jarvi, H. Li and B. C. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8989–8994 CrossRef CAS PubMed.
- K. Han, J. Y. Zhu, S. B. Wang, Z. H. Li, S. X. Cheng and X. Z. Zhang, J. Mater. Chem. B, 2015, 3, 8065–8069 RSC.
- Y. C. Chen, C. C. Zhu, Z. H. Yang, J. J. Chen, Y. F. He, Y. Jiao, W. J. He, L. Qiu, J. J. Cen and Z. J. Guo, Angew. Chem., 2013, 125, 1732–1735 CrossRef.
- A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640–9641 CrossRef CAS PubMed.
- K. Han, S. B. Wang, Q. Lei, J. Y. Zhu and X. Z. Zhang, ACS Nano, 2015, 9, 10268–10277 CrossRef CAS PubMed.
- Y. L. Dai, C. Xu, X. L. Sun and X. Y. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- Z. P. Zhen, W. Tang, M. Wang, S. Y. Zhou, H. Wang, Z. H. Wu, Z. L. Hao, Z. B. Li, L. Liu and J. Xie, Nano Lett., 2017, 17, 862–869 CrossRef CAS PubMed.
- Q. Y. Gong, W. Shi, L. H. Li, X. F. Wu and H. M. Ma, Anal. Chem., 2016, 88, 8309–8314 CrossRef CAS PubMed.
- P. C. Lo, J. Chen, K. Stefflova, M. S. Warren, R. Navab, B. Bandarchi, S. Mullins, M. Tsao, J. D. Cheng and G. Zheng, J. Med. Chem., 2009, 52, 358–368 CrossRef CAS PubMed.
- D. Li, Y. Ma, J. Du, W. Tao, X. Du, X. Yang and J. Wang, Nano Lett., 2017, 17, 2871–2878 CrossRef CAS PubMed.
- S. Shen, H. J. Li, K. G. Chen, Y. C. Wang, X. Z. Yang, Z. X. Lian, J. Z. Du and J. Wang, Nano Lett., 2017, 17, 3822–3829 CrossRef CAS PubMed.
- H. Z. Deng, X. F. Zhao, J. J. Liu, L. D. Deng, J. H. Zhang, J. F. Liu and A. J. Dong, J. Mater. Chem. B, 2015, 3, 9397–9408 RSC.
- J. W. Wojtkowiak, D. Verduzco, K. J. Schramm and R. J. Gillies, Mol. Pharmaceutics, 2011, 8, 2032–2038 CrossRef CAS PubMed.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem. In. Ed., 2010, 49, 3621–3626 CrossRef CAS PubMed.
- T. Feng, X. Ai, G. An, P. Yang and Y. Zhao, ACS Nano, 2016, 10, 4410–4420 CrossRef CAS PubMed.
- F. L. Wang, W. Sun, L. Li, L. J. Li, Y. Y. Liu, Z. R. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 27563–27574 CAS.
- H. J. Li, J. Z. Du, J. Liu, X. J. Du, S. Shen, Y. H. Zhu, X. Y. Wang, X. D. Ye, S. M. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed.
- C. Y. Sun, S. Shen, C. F. Xu, H. J. Li, Y. Liu, Z. T. Cao, X. Z. Yang, J. X. Xia and J. Wang, J. Am. Chem. Soc., 2015, 137, 15217–15224 CrossRef CAS PubMed.
- M. Gao, F. Fan, D. Li, Y. Yu, K. Mao, T. Sun, H. Qian, W. Tao and X. Yang, Biomaterials, 2017, 133, 165–175 CrossRef CAS PubMed.
- S. Barua, J. W. Yoo, P. Kolhar, A. Wakankar, Y. R. Gokarn and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3270–3275 CrossRef CAS PubMed.
- D. Li, Z. M. Tang, Y. Gao, H. L. Sun and S. B. Zhou, Adv. Funct. Mater., 2016, 26, 66–79 CrossRef CAS.
- K. Han, J. Zhang, W. Y. Zhang, S. B. Wang, L. M. Xu, C. Zhang, X. Z. Zhang and H. Y. Han, ACS Nano, 2017, 11, 3178–3188 CrossRef CAS PubMed.
- M. Bibian, J. Mangelschots, J. Gardiner, L. Waddington, M. M. Diaz Acevedo, B. G. De Geest, B. Van Mele, A. Madder, R. Hoogenboom and S. Ballet, J. Mater. Chem. B, 2015, 3, 759–765 RSC.
- S. L. Lo and S. Wang, Biomaterials, 2008, 29, 2408–2414 CrossRef CAS PubMed.
- L. Prongidi-Fix, M. Sugawara, P. Bertani, J. Raya, C. Leborgne, A. Kichler and B. Bechinger, Biochemistry, 2007, 46, 11255–11262 CrossRef PubMed.
- W. Zhang, J. Song, R. Liang, X. Zheng, J. Chen, G. Li, B. Zhang, X. Yan and R. Wang, Bioconjugate Chem., 2013, 24, 1805–1812 CrossRef CAS PubMed.
- C. D. Dekiwadia, A. C. Lawrie and J. V. Fecondo, J. Pept. Sci., 2012, 18, 527–534 CrossRef CAS PubMed.
- L. D. Field, J. B. Delehanty, Y. Chen and I. L. Medintz, Acc. Chem. Res., 2015, 48, 1380–1390 CrossRef CAS PubMed.
- H. M. Ellerby, W. Arap, L. M. Ellerby, R. Kain, R. Andrusiak, G. D. Rio, S. Krajewski, C. R. Lombardo, R. Rao, E. Ruoslahti, D. E. Bredesen and R. Pasqualini, Nat. Med., 1999, 5, 1032–1038 CrossRef CAS PubMed.
- I. Nakase, S. Okumura, S. Katayama, H. Hirose, S. Pujals, H. Yamaguchi, S. Arakawa, S. Shimizu and S. Futaki, Chem. Commun., 2012, 48, 11097–11099 RSC.
- J. Salaklang, B. Steitz, A. Finka, C. P. O'Neil, M. Moniatte, A. J. van der Vlies, T. D. Giorgio, H. Hofmann, J. A. Hubbell and A. Petri-Fink, Angew. Chem., Int. Ed., 2008, 47, 7857–7860 CrossRef CAS PubMed.
- W. Qu, S. Y. Qin, S. Ren, X. J. Jiang, R. X. Zhuo and X. Z. Zhang, Bioconjugate Chem., 2013, 24, 960–967 CrossRef CAS PubMed.
- W. Zhang, K. Müller, E. Kessel, S. Reinhard, D. He, P. M. Klein, M. Höhn, W. Rödl, S. Kempter and E. Wagner, Adv. Healthcare Mater., 2016, 5, 1493–1504 CrossRef CAS PubMed.
- L. H. Liu, W. X. Qiu, Y. H. Zhang, B. Li, C. Zhang, F. Gao, L. Zhang and X. Z. Zhang, Adv. Funct. Mater., 2017, 27, 1700220 CrossRef.
- S. Wang, Z. Teng, P. Huang, D. Liu, Y. Liu, Y. Tian, J. Sun, Y. Li, H. Ju, X. Chen and G. Lu, Small, 2015, 11, 1801–1810 CrossRef CAS PubMed.
- G. F. Luo, W. H. Chen, S. Hong, Q. Cheng, W. X. Qiu and X. Z. Zhang, Adv. Funct. Mater., 2017, 27, 1702122 CrossRef.
- M. Yu, F. Guo, J. Wang, F. Tan and N. Li, ACS Appl. Mater. Interfaces, 2015, 7, 17592–17597 CAS.
- K. Han, W. Y. Zhang, Z. Y. Ma, S. B. Wang, L. M. Xu, J. Liu, X. Z. Zhang and H. Y. Han, ACS Appl. Mater. Interfaces, 2017, 9, 16043–16053 CAS.
- B. Herranz-Blanco, M. A. Shahbazi, A. R. Correia, V. Balasubramanian, T. Kohout, J. Hirvonen and H. A. Santos, Adv. Healthcare Mater., 2016, 5, 1904–1916 CrossRef CAS PubMed.
- L. F. Qi, W. J. Shao and D. L. Shi, J. Mater. Chem. B, 2013, 1, 654–660 RSC.
- Y. Y. Won, R. Sharma and S. F. Konieczny, J. Controlled Release, 2009, 139, 88–93 CrossRef CAS PubMed.
- N. Nishiyama, A. Iriyama, W. Jang, K. Miyata, K. Taka, Y. Inoue, H. Takahashi, Y. Yanag, Y. Tamaki, H. Koyama and K. Kataoka, Nat. Mater., 2005, 4, 934–941 CrossRef CAS PubMed.
- N. Nishiyama, A. Iriyama, W. D. Jang, K. Miyata, K. Itaka and Y. Inoue, Nat. Mater., 2005, 4, 934–941 CrossRef CAS PubMed.
- K. Berg, A. Dietze, O. Kaalhus and A. Høgset, Clin. Cancer Res., 2005, 11, 8476–8485 CrossRef CAS PubMed.
- E. Zeira, A. Manevitch, Z. Manevitch, E. Kedar, M. Gropp and N. Daudi, FASEB J., 2007, 21, 3522–3533 CrossRef PubMed.
- H. Cabral, M. Nakanishi, M. Kumagai, W. D. Jang, N. Nishiyama and K. Kataoka, Pharm. Res., 2009, 26, 82–92 CrossRef CAS PubMed.
- K. Han, Q. Lei, H. Z. Jia, S. B. Wang, W. N. Yin, W. H. Chen, S. X. Cheng and X. Z. Zhang, Adv. Funct. Mater., 2015, 25, 1248–1257 CrossRef CAS.
- K. Han, S. Chen, W. H. Chen, Q. Lei, Y. Liu, R. X. Zhuo and X. Z. Zhang, Biomaterials, 2013, 34, 4680–4689 CrossRef CAS PubMed.
- K. Han, J. Yang, S. Chen, J. X. Chen, C. W. Liu, C. Li, H. Cheng, R. X. Zhuo and X. Z. Zhang, Int. J. Pharm., 2012, 436, 555–563 CrossRef CAS PubMed.
- B. Turk, D. Turk and V. Turk, Biochim. Biophys. Acta, 2000, 1477, 98–111 CrossRef CAS.
- H. Shi, X. He, Y. Yuan, K. Wang and D. Liu, Anal. Chem., 2010, 82, 2213–2220 CrossRef CAS PubMed.
- X. Liang, Y. Yang, L. Wang, X. B. Zhu, X. W. Zeng, X. J. Wu, H. B. Chen, X. D. Zhang and L. Mei, J. Mater. Chem. B, 2015, 3, 9383–9396 RSC.
- J. W. Tian, L. Ding, Q. B. Wang, Y. P. Hu, L. Jia, J. S. Yu and H. X. Ju, Anal. Chem., 2015, 87, 3841–3848 CrossRef CAS PubMed.
- J. Kim, C. H. Tung and Y. Choi, Chem. Commun., 2014, 50, 10600–10603 RSC.
- J. X. Chen, H. Y. Wang, C. Li, K. Han, X. Z. Zhang and R. X. Zhuo, Biomaterials, 2011, 32, 1678–1684 CrossRef CAS PubMed.
- H. Ding, B. D. Sumer, C. W. Kessinger, Y. Dong, G. Huang, D. A. Boothman and J. Gao, J. Controlled Release, 2011, 151, 271–277 CrossRef CAS PubMed.
- Y. Y. Yuan, C. J. Zhang, M. Gao, R. Y. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- Y. Guan, H. G. Lu, W. Li, Y. D. Zheng, Z. Jiang, J. L. Zou and H. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 26731–26739 CAS.
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519 CrossRef CAS PubMed.
- K. Han, Y. Liu, W. N. Yin, S. B. Wang, Q. Xu, R. X. Zhuo and X. Z. Zhang, Adv. Healthcare Mater., 2014, 3, 1765–1768 CrossRef CAS PubMed.
- J. P. Liu, C. Z. Jin, B. Yuan, Y. Chen, X. G. Liu, L. N. Jia and H. Chao, Chem. Commun., 2017, 53, 9878–9881 RSC.
- G. X. Feng, W. Qin, Q. L. Hu, B. Z. Tang and B. Liu, Adv. Healthcare Mater., 2015, 4, 2667–2676 CrossRef CAS PubMed.
- J. S. Xu, F. Zeng, H. Wu, C. M. Yu and S. Z. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 9287–9296 CAS.
- P. M. R. Pereira, S. Silva, M. Bispo, M. Zuzarte, C. Gomes, H. Girão, J. A. S. Cavaleiro, C. A. F. Ribeiro, J. P. C. Tomé and R. Fernandes, Bioconjugate Chem., 2016, 27, 2762–2769 CrossRef CAS PubMed.
- Y. Song, Q. R. Shi, C. Z. Zhu, Y. N. Luo, Q. Lu, H. Li, R. F. Ye, D. Du and Y. H. Lin, Nanoscale, 2017, 9, 15813–15824 RSC.
- K. Han, Q. Lei, S. B. Wang, J. J. Hu, W. X. Qiu, J. Y. Zhu, W. N. Yin, X. Luo and X. Z. Zhang, Adv. Funct. Mater., 2015, 25, 2961–2971 CrossRef CAS.
- H. C. Chen, J. W. Tian, D. Y. Liu, W. J. He and Z. J. Guo, J. Mater. Chem. B, 2017, 5, 972–979 RSC.
- K. Han, J. Y. Zhu, H. Z. Jia, S. B. Wang, S. Y. Li, X. Z. Zhang and H. Y. Han, ACS Appl. Mater. Interfaces, 2016, 8, 25060–25068 CAS.
- P. Xu, E. A. Van Kirk, Y. Zhan, W. J. Murdoch, M. Radosz and Y. Q. Sheng, Angew. Chem., Int. Ed., 2007, 46, 4999–5002 CrossRef CAS PubMed.
- F. Dosio, S. Arpicco, B. Stella and E. Fattal, Adv. Drug Delivery Rev., 2016, 97, 204–236 CrossRef CAS PubMed.
- L. P. Qiu, T. Chen, I. Öçsoy, E. Yasun, C. C. Wu, G. Z. Zhu, M. X. You, D. Han, J. H. Jiang, R. Q. Yu and W. H. Tan, Nano Lett., 2015, 15, 457–463 CrossRef CAS PubMed.
- C. W. Chang, R. M. Couñago, S. J. Williams, M. Bodén and B. Kobe, Traffic, 2013, 14, 1144–1154 CAS.
- S. S. Han, Z. Y. Li, J. Y. Zhu, K. Han, Z. Y. Zeng, W. Hong, W. X. Li, H. Z. Jia, Y. Liu, R. X. Zhuo and X. Z. Zhang, Small, 2015, 11, 2543–2554 CrossRef CAS PubMed.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658–2660 RSC.
- H. Y. Wang, J. X. Chen, Y. X. Sun, J. Z. Deng, C. Li, X. Z. Zhang and R. X. Zhuo, J. Controlled Release, 2011, 155, 26–33 CrossRef CAS PubMed.
- A. Huefner, W. L. Kuan, R. A. Barker and S. Mahajan, Nano Lett., 2013, 13, 2463–2470 CrossRef CAS PubMed.
- K. Han, W. Y. Zhang, J. Zhang, Q. Lei, S. B. Wang, J. W. Liu, X. Z. Zhang and H. Y. Han, Adv. Funct. Mater., 2016, 26, 4351–4361 CrossRef CAS.
- Z. Z. Yu, W. Pan, N. Li and B. Tang, Chem. Sci., 2016, 7, 4237–4244 RSC.
- L. M. Pan, Q. J. He, J. N. Liu, Y. Chen, M. Ma, L. L. Zhang and J. L. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- L. M. Pan, J. N. Liu and J. L. Shi, Adv. Funct. Mater., 2014, 24, 7318–7327 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |