
Continuous silicon oxycarbide fiber mats with tin nanoparticles as a high capacity anode for lithium-ion batteries†
Aura
Tolosa
 ab,
Mathias
Widmaier
bc,
Benjamin
Krüner
ab,
John M.
Griffin
d and
Volker
Presser
*ab
ab,
Mathias
Widmaier
bc,
Benjamin
Krüner
ab,
John M.
Griffin
d and
Volker
Presser
*ab
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
cRobert Bosch GmbH, Robert-Bosch-Campus 1, 71272 Renningen, Germany
dDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK
First published on 23rd October 2017
Abstract
Continuous fiber mats are attractive electrodes for lithium-ion batteries, because they allow operation at high charge/discharge rates in addition to being free of polymer binders and conductive additives. In this work, we synthesize and characterize continuous Sn/SiOC fibers (diameter ca. 0.95 μm), as a Li-ion battery anode. Our synthesis employs electrospinning of a low-cost silicone resin, using tin acetate in a dual role both as a polymer crosslinker and as a tin precursor (6–22 mass%). The hybrid electrodes present very high initial reversible capacities (840–994 mA h g−1) at 35 mA g−1, and retain 280–310 mA h g−1 at 350 mA g−1. After 100 cycles at 70 mA g−1, the hybrid fibers maintained 400–509 mA h g−1. Adding low amounts of Sn is beneficial not just for the crosslinking of the polymer precursor, but also to decrease the presence of electrochemically inactive silicon carbide domains within the SiOC fibers. Also, the metallic tin clusters contribute to a higher Li+ insertion in the first cycles. However, high amounts of Sn decrease the electrochemical performance stability. In SiOC fibers synthesized at high temperatures (1200 °C), the Cfree phase has a significant influence on the stability of the system, by compensating for the volume expansion from the alloying systems (Sn and SiO2), and improving the conductivity of the hybrid system. Therefore, a high amount of carbon and a high graphitization degree are crucial for a high conductivity and a stable electrochemical performance.
1. Introduction
To meet the growing requirements for mobile electrochemical energy storage,1 it is important to advance present-day electrode materials both in regard to charge/discharge rates (power) and storage capacity (energy).2 Li-ion batteries (LIBs) present high energy density, being today the main choice for consumer electronics and electrical or hybrid vehicles.3 To further improve the capacity of LIBs, significant efforts have been dedicated to replacing graphite anodes (theoretical capacity ≈ 372 mA h g−1) with other elements from group IV (Si, Sn, and Ge; cf.ref. 4) which undergo alloying reactions with lithium leading to high capacity values (4200, 1627, and 993 mA h g−1 theoretical capacity of Si, Ge, and Sn, respectively).5 Such high capacities are accomplished not without complications; for example, in the case of silicon, a complete lithiation (Li22Si5 alloy) corresponds to a volumetric expansion of about 400%,6 leading to poor cycling and huge capacity fading. To improve the specific capacity and cycle life of anode materials, nanoparticles of Si, Sn, or Ge, have been synthesized in a carbon matrix5,6 to compensate for the volumetric changes (volume expansion of graphite ≈ 10%)5 and to improve the electrical conductivity of the system. These Si/C composite materials present a maximum theoretical capacity of 712 mA h g−1, corresponding to 11.7 mass% Si, at which the volumetric expansion of Si can be compensated for by the presence of pores, without decreasing the volumetric capacity.6Since the early work of Wilson and Dahn,7 polymer-derived SiOC materials have been investigated as an alternative to Si/C composites due to the lower cost of synthesis, high capacity values (between 500 and 900 mA h g−1), and reduced capacity fading.6,8 Polymer-derived SiOC synthesized at 1000–1800 °C is predominately amorphous and consists of Si–O–C nanodomains engulfed by carbon.9 The latter is called free carbon (Cfree), that is carbon beyond the stoichiometric amount necessary to saturate all the Si valences.9 The Si–O–C nanodomains consist of amorphous silica, and SiCxO4−x tetrahedral units (with 1 ≤ x ≤ 4).9,10 The microstructure and composition of SiOC depend on the precursor material and the pyrolysis temperature. Above 1000 °C, carbon segregates and forms turbostratic graphite which encapsulates silica domains with local precipitation of SiC by local carbothermal reduction.9 Riedel et al. have demonstrated the effect of the composition and microstructure of SiOC powder electrodes on the electrochemical performance as anodes for LIBs.11–13 Using a polysiloxane as a precursor (polyramic RD-684a), samples pyrolyzed at 900 °C presented the highest initial reversible capacity (738 mA h g−1), but suffered from strong capacity fading.13 At higher synthesis temperature, the stability of the system is improved, with an optimum for samples pyrolyzed at 1100 °C for 3 h with a lower initial reversible capacity (532 mA h g−1).13 Adding divinylbenzene to the polysiloxane precursor leads to an increase in the carbon content of the SiOC.8 It was found that SiOC behaves as a hybrid material, consisting of disordered Si–O–C nanodomains having a high first Li+ insertion capacity of 1300° mA h g−1, and Cfree having an estimated capacity of 350° mA h g−1 (ref. 8). Among them, the material with the highest reversible capacity and best cyclability contained 51 mass% Si–O–C nanodomains and 49 mass% Cfree.8
The incorporation of commercially available or in situ synthesized Si nanoparticles into SiOC has also been investigated,14,15 finding that the SiOC network helps to improve the conductivity of the system and buffers the volume change.15 Compared to pure SiOC synthesized at 1100 °C, incorporating amorphous or crystalline Si leads to higher initial capacities, namely 704 mA h g−1 or 905 mA h g−1, respectively.14 After 30 cycles, the capacity of crystalline Si/SiOC fades below the capacity of pure SiOC, while the amorphous Si/SiOC maintained 85% of the capacity.14 Sn nanoparticles have also been incorporated into low- and high-content carbon SiOC by mixing polysilsesquioxanes with tin(II) acetate, followed by pyrolysis at 1000 °C.16 Adding 25 mass% of Sn increases the initial reversible capacity compared to the high- and low-carbon SiOC samples, by 9% and 140%, respectively.16 The increase in capacity is related to the formation of Li22Sn5 and Li7Sn2, and the increase in rate capability and stability is attributed to the electrically conductive Cfree phase.16 All these studies on polymer-derived SiOC have employed polymer-bound powder electrodes.
Continuous fiber electrodes, being free-standing and polymer binder free, are attractive because the continuous path for electron transport enables operation at much higher charge/discharge rates.17 Also, for hybrid fibers containing carbon and redox-active materials, no additional conductive additives are needed.18 The absence of inactive components (conductive additives and polymer binders) increases the overall gravimetric energy density of the electrode. So far, only continuous fibers have been fabricated by Zhang et al. using nanoparticles dispersed in a polyacrylonitrile (PAN) derived carbon matrix to obtain free-standing electrodes.19–21 The dispersed nanoparticles have been commercially available Si,19–21 Ge,19 and Sn,19,22 or synthetized SiOC and SiO2 particles.21,23 The main issue of this synthesis process is that inorganic nanoparticles show a very high surface energy and tend to agglomerate in an organic medium.24 Therefore, the resulting material is inhomogeneous, affecting the material properties. To obtain homogeneous materials without the need for surface active agents, it is better to employ approaches leading to the in situ formation of nanodomains.24
In our past work, we have demonstrated electrospinning of SiOC electrospun fibers obtained from low-cost silicone resins.17 For this synthesis, one always needs a crosslinker, such as zirconium acetylacetonate or zinc acetylacetonate.17,25 However, parts of these crosslinkers remain in the system forming (undesired/unnecessary) metal compounds.26 We now have explored the direct use of a Sn-containing crosslinker to enable enhanced electrochemical performance of the resulting SiOC–Sn-compound. According to the Ellingham diagrams,27 the Gibbs free energy of SiO2 between 900 and 1300 °C is lower than SnO2; therefore, SiO2 acts as a reducing agent for SnO2, leading to the formation of metallic Sn domains. In this study, we evaluate for the first time polymer-derived SiOC–Sn fibers as anode materials for LIBs. The process variables correspond to the pyrolysis temperature (1000–1200 °C) and Sn-containing crosslinker amount (5–20 mass%). The effect of the process variables on the material properties and electrochemical performance is evaluated.
2. Experimental description
2.1. Materials synthesis
The solvents trichloromethane (TCM, stabilized, GPR Rectapur) and N,N-dimethylformamide (DMF; anhydrous, 99.8%) were purchased from VWR Chemicals and Sigma Aldrich, respectively. The polymers, silicone resin namely H44 (polymethylphenyl-silsesquioxane) and polyvinylpyrrolidone (PVP, Mw ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 g mol−1), were purchased from Wacker Chemie and Sigma Aldrich, respectively. Moisture sensitive tin(II) acetate (SnAc) was purchased from Sigma Aldrich and stored in a moisture free nitrogen atmosphere in a glovebox (H2O, O2 < 0.1 ppm). All chemicals were used as-received.
300000 g mol−1), were purchased from Wacker Chemie and Sigma Aldrich, respectively. Moisture sensitive tin(II) acetate (SnAc) was purchased from Sigma Aldrich and stored in a moisture free nitrogen atmosphere in a glovebox (H2O, O2 < 0.1 ppm). All chemicals were used as-received.
The synthesis of SiOC fibers is based on the approach followed in past studies,17 but modified using PVP to improve rheological behavior for electrospinning.25 30 mass% polymer (75:25 mass ratio of H44:PVP) solutions were prepared in a 1:1 volumetric ratio of TCM:DMF. After the polymers were completely dissolved, the SnAc was added and stirred for 1 h before electrospinning. SnAc was added in different amounts: 2.5, 4.5, and 9.0 mass%. These amounts correspond to stoichiometric ratios of 0.05, 0.1, and 0.2 mol of Sn per 1 mol of Si.
Electrospinning was carried out with a MECC Co. NF-103V nanofiber system. The spinning dope (5 mL) was pumped at 1 mL h−1 through a spinneret (inner diameter: 510 μm) which was inductively charged at 28 kV. The fibers were collected on a stationary grounded target at 21 cm from the spinneret. The spinneret head moved parallel to the collector with a travel distance of 10 cm at a travel velocity of 2 cm s−1. The collected material was then heated at 100 °C for 24 h to stabilize the fiber shape. The pyrolysis of the crosslinked silicone resin was achieved by heating the material at 5 °C min−1 to 200 °C for 2 h and later to 1000 °C or 1200° under argon in a graphite heater (Thermal Technologies, Model 1100C) for 2 h. After the heat treatment, non-woven SiOC fiber mats were cooled to room temperature at a rate of 20 °C min−1. The Sn/SiOC fiber mats were named according to the added Sn content (0.05 Sn, 0.1 Sn, or 0.2 Sn) and the pyrolysis temperature (−1000 or −1200).
SiOC was also produced without a Sn precursor, as a monolith using the same procedure and thermal treatment used for the spinning dope preparation, but without the addition of SnAc. Also, a monolith containing 0.05 Sn was prepared for comparison of the electrochemical performance. These monoliths with and without Sn were named M-0.05 Sn and M-SiOC, respectively, and according to the pyrolysis temperature (−1000 or −1200).
2.2. Materials characterization
Transmission electron microscopy (TEM) was done with a JEOL JEM-2100F system operating at 200 kV in vacuum. Ground fiber mats were dispersed and sonicated in ethanol, and drop cast onto a copper grid with a carbon film. Energy dispersive X-ray spectroscopy (EDX) point analysis was performed using a Thermo Scientific MC100021 detector attached to the TEM chamber. The spectra were acquired at 200 kV and 2 min for point measurements.
To characterize the chemical composition of the system, Fourier-transform infrared spectroscopy (FT-IR), and nuclear magnetic resonance spectroscopy (NMR) were used. FT-IR was performed with a diamond total attenuated reflectance crystal (ATR) using a Bruker Tensor 27 system. Raman spectra were measured using a Renishaw InVia Raman system, and a green laser (532 nm excitation wavelength) with 0.5 mW power on the sample, using a spectral resolution of ca. 1.2 cm−1 and a 50× objective (numerical aperture: 0.9). X-ray diffraction (XRD) was conducted employing a D8 Advance diffractometer (Bruker AXS) with a copper X-ray source (Cu-Kα, 40 kV, 40 mA) in point focus (0.5 mm) and a Goebel mirror. A VANTEC-500 (Bruker AXS) 2D detector (∼25° per step; measurement time, 1000 s per step) was employed. The samples were ground and placed on a Si wafer. Solid-state magic-angle spinning (MAS) NMR measurements were performed on Bruker Avance III spectrometers operating at magnetic field strengths of 9.4 T (13C) and 16.4 T (29Si). The MAS frequency for all experiments was 12.5 kHz. All experiments were performed with direct polarization using a 90°-acquire pulse sequence. A recycle interval of 15 s was used for all experiments. Chemical shifts were referenced to the CH3 resonance of L-alanine at 20.5 ppm for 13C and kaolinite (Al2Si2O5(OH)4) at −91.2 ppm for 29Si.
Thermogravimetric measurements combined with a mass spectrometer (TGA-MS) were performed for the as-spun samples, using a STA 449 F3 Jupiter and QMS 403C Aëolos from Netzsch. The samples were heated to 1200 °C at 5 °C min−1 under N2. Analysis of the tin content was performed by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The samples (30 mg) were placed in microwave vessels, and mixed with 6 mL of HNO3, 4 mL of H2SO4, and 3 mL of HF. The vessels were maintained at 150 °C with a pressure of 300 kPa for several days until a homogenous liquid solution was observed. After cooling, the samples were placed in 50 mL polyethylene flasks and filled to the calibration mark for inductively coupled plasma atomic emission spectroscopy (ICP-AES) using a Horiba Jobin Yvon Ultima 2 system.
:1 by mass (EC/DMC)) from BASF as the electrolyte.
The sheet resistance of the fiber mats was measured with a custom-built spring-loaded four-point probe with blunt gold contacts (tip diameter: 1.5 mm, tip distance: 3 mm). Cyclic voltammetry (CV) was performed using a VSP300 potentiostat/galvanostat (Bio-Logic) and for galvanostatic cycling with potential limitation (GCPL) an Astrol BatSmall battery analyzer was used. For rate capability measurements, the cells were subjected to GCPL between +0.01 V and +3.0 V vs. Li/Li+ at different current rates. Based on a material capacity of 700 mA h g−1, different specific currents were applied, starting at 35 mA g−1 (0.05C) for 5 cycles, followed by GCPL at 70 mA g−1 (0.1C) for 20 cycles, and 140 mA g−1 (0.2C), 350 mA g−1 (0.5C), 700 mA g−1 (1C) and 1400 mA g−1 (2C), for 10 cycles each. Further GCPL was performed at 70 mA g−1 for 40 cycles to complete 100 cycles. To evaluate the electrochemical performance in the first 5 cycles, the cells were tested by CV in the potential range between +0.01 V and +3.0 V vs. Li/Li+ at a scan rate of 0.1 mV s−1. From the GCPL data, the specific capacity (Csp) was calculated according to eqn (1), by integrating the current between the starting time (t0) and end time (t) of the delithiation step, normalized to the total electrode mass (m). The electrode mass corresponds to the mass of the Sn/SiOC fiber mats, which are free of any polymer binder or any conductive additives.
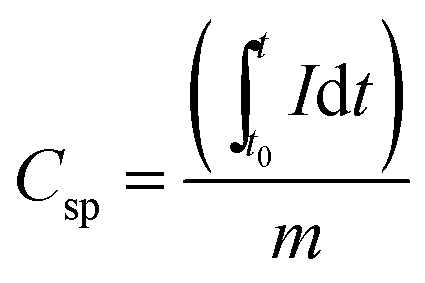 | (1) |
Polymer-bound electrodes were prepared for comparison. The produced monoliths (M-SiOC and M-0.05 Sn) were ground with an agate mortar in ethanol until obtaining a homogeneous slurry, to which 10 mass% polytetrafluoroethylene (PTFE, 60 mass% solution in water, Sigma Aldrich) and 5 mass% carbon black Super C65 (Imerys Graphite & Carbon) were added. The obtained paste was rolled until 126 ± 7 μm thick electrodes were obtained (MTI HR01 rolling machine, MTI).
3. Results and discussion
3.1. Polysilsesquioxane modified fibers
To electrospin SiOC fibers derived from polysilsesquioxanes, one must employ a crosslinker to maintain the fiber shape during pyrolysis. In our previous work on electrospun SiOC fiber mats for electrochemical applications, we had used zinc acetylacetonate as a crosslinker.17 After pyrolysis, low amounts (5–10 mass%) of zinc are present in the fibers. For this reason, we have replaced zinc acetylacetonate with a Sn-containing crosslinker (SnAc) to not only benefit from the crosslinker activity during synthesis, but also to form electroactive Sn after pyrolysis to enhance the energy storage performance.H44 is a commercially available polysilsesquioxane with the chemical formula described in eqn (2), where –OR corresponds to the functional groups where further crosslinking takes place.
| [(C6H5)0.62(CH3)0.31(OR)0.07SiO1.5]n where OR = –OH and –OC2H5 | (2) |
In the absence of metal alkoxides as crosslinkers or polymer modifiers, the condensation reactions described in eqn (3) and (4) take place. In the presence of transition metal alkoxides, the metal alkoxide reacts following the reaction described in eqn (5), exemplified for SnAc.16
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH + HO–Si → Si–O–Si + H2O Si–OH + HO–Si → Si–O–Si + H2O | (3) |
| Si–OH + RO–Si → Si–O–Si + R–OH | (4) |
| Si–OH + Sn(CH3COO)2 → CH3COOSn–O–Si + CH3COOH | (5) |
Based on the reaction described in eqn (5) and the number of crosslinking groups of the polymer, the minimum amount of Sn added was fixed to occupy most of the crosslinking positions, which was 0.05 mol of Sn per 1 mol of Si. Based on this proportion, an excess of Sn was added (0.1 mol and 0.2 mol of Sn per 1 mol of Si). After electrospinning, the samples are named according to the Sn amount (0.05 Sn, 0.1 Sn, or 0.2 Sn), and after pyrolysis according to the tin content and the pyrolysis temperature (for example: 0.05 Sn-1200).
The as-prepared spinning dopes were electrospun and ultrafine fibers with a smooth and uniform morphology were obtained, as confirmed by scanning electron micrographs (Fig. 1A and B). The as-spun fibers had an average diameter of 1.64 ± 0.30 μm (Fig. 1C). During electrospinning, the solvent evaporates, leaving behind the solid fibers containing H44, PVP and SnAc.
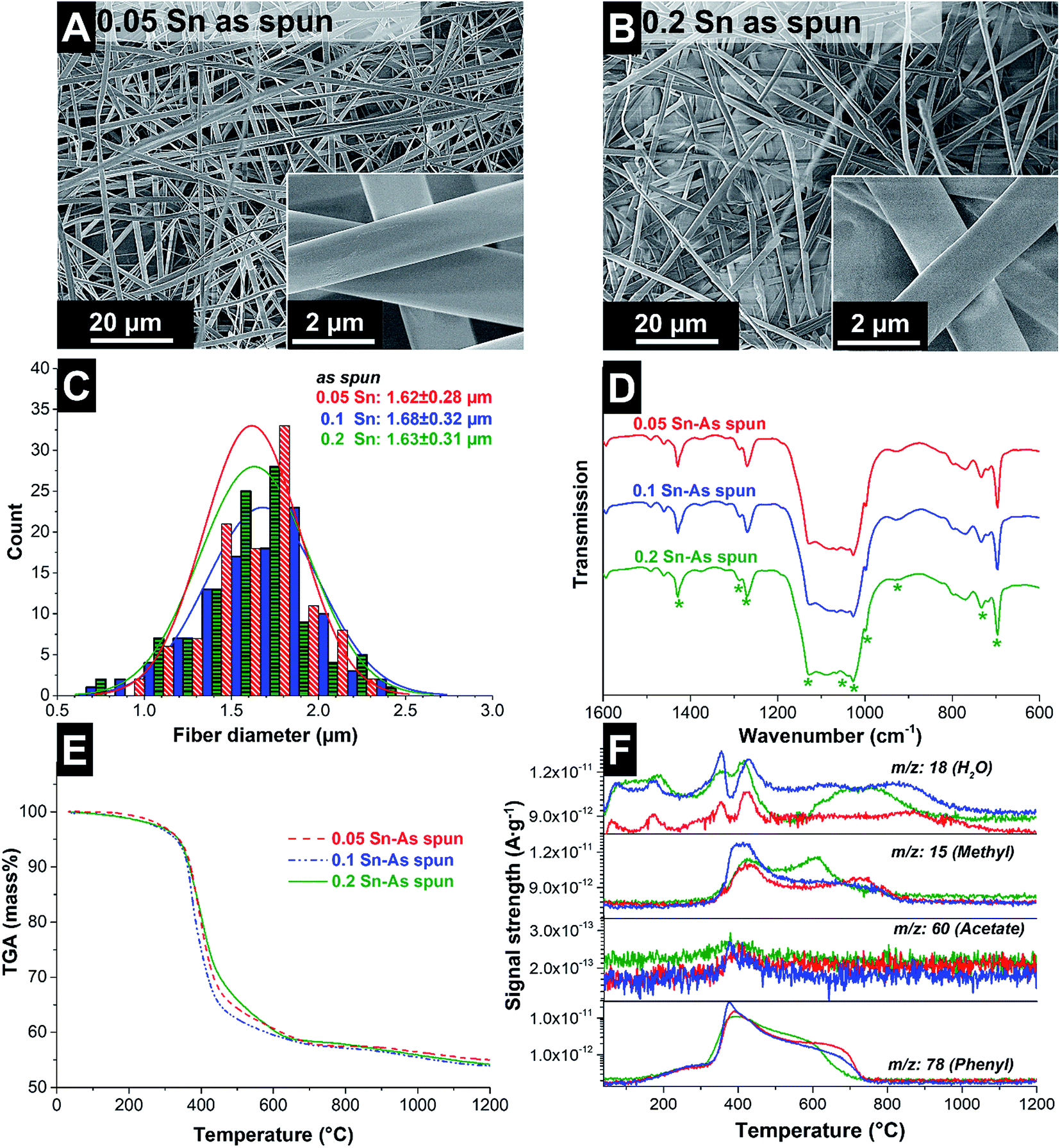 | ||
| Fig. 1 SEM micrographs of the as-spun silicon resin fibers containing 0.05 Sn (A) and 0.2 Sn (B). Fiber diameter distribution (C) and Fourier transform infrared spectra (D) of the as-spun fibers. Thermogravimetric analysis (E) and mass spectra of the most important moieties (F), during heat treatment under an inert atmosphere of the as-spun fibers. | ||
The reactions in the as-spun fibers during heating at 100 °C were characterized by FT-IR (Fig. 1D). The infrared spectra show absorption bands of the H44 polymer: the Si–O stretching vibration of the Si–O–Si ladder-type structure appeared between 1040 and 1027 cm−1 (ref. 17), the band at 1128 cm−1 is related to phenolic groups,30 and the band at 1428 cm−1 comes from the C–H vibration modes of the methyl groups.30 The absorbance of the bridging bond Si–C in the Si–CH3 was observed at 733 cm−1 (ref. 31). The absorption bands corresponding to acetate ligands are either very weak or not observed. This can be related to the complete reaction of Sn in the Si–O–Sn and Sn–O–Sn networks, forming volatile acetic acid. Absorption bands at 695 cm−1 and 928 cm−1 correspond to the stretching modes of Sn–O and Si–O–Si, respectively.16 Also, several signals of the PVP can be observed: the small peak at 1017 cm−1 corresponds to the C–C polymer backbone and the band at 930 cm−1 is the breathing mode of the ring.26 The bands at 1270–1288 cm−1 emerge from the C–N bonds in the polymer.32 Comparing the infrared spectra, no significant differences are observed when increasing the SnAc content. All spectra are similar and confirm the formation of Sn–O bonds, either as a part of the Si–O–Si network, or forming separated Sn–O–Sn domains.
3.2. Sn/SiOC fibers
After the fibers were completely crosslinked, the fibers were treated under Ar at either 1000 °C or 1200 °C to form SiOC/Sn fibers. To understand the effect of the crosslinker during thermal treatment, thermogravimetric analysis coupled with mass spectrometry (TGA-MS) was carried out in an inert atmosphere (Fig. 1E and F). At 250 °C, a mass loss of 4% is observed as water is being desorbed. At 300–600 °C, we see a drastic mass decrease due to redistribution reactions and the decomposition of the polysilsesquioxane. In this temperature range, the initial exchange reactions between the bonds present in the crosslinked resin take place, forming volatile organosilicon species.33 Later, the decomposition of the network occurs by bond-cleavage34 and methyl and phenyl groups diffuse through the bulk material. During the diffusion of the aliphatic and aromatic groups, some condense to form aromatic clusters which will later be seeds for carbon domains, while other leave as volatile compounds. Above 600 °C, hydrogen gas is produced by the aromatic/turbostratic carbon domains, leading to a continuous mass loss of 5%.34 After pyrolysis is completed, a total mass loss of 45% was obtained compared to the initial mass. The Sn–O–Sn domains initially form SnO, which above 600 °C is not favored in the presence of C and Si according to the Ellingham diagrams.27 SnO is reduced to metallic tin, together with the release of hydrogen and CO upon ceramization, and it does not form either silicides or carbides.35After pyrolysis, the fibers present a smooth surface with some beads present on the fiber surface and between the fibers as observed from the scanning electron micrographs (Fig. 2). The material characterization data for the samples treated at 1000 °C are presented in the ESI (Fig. S1 and S3†). The amorphous fibers present an average diameter of 0.96 ± 0.25 μm after pyrolysis (Fig. 3A). There are also some crystalline domains inside the fibers, as observed from the transmission electron micrographs (Fig. 2). Also, the beads located outside the fibers present a crystalline structure engulfed by an amorphous matrix. EDX analysis was carried during TEM analysis (ESI, Fig. S2 and Table S1†), and we determined that the clusters inside and outside the fibers correspond to Sn covered by a SiOC shell. The size and distribution of Sn beads depend on the pyrolysis temperature and the used amount of SnAc, reaching a diameter size up to 0.8–1.0 μm for the sample 0.2 Sn-1000, as observed from the scanning electron micrographs (ESI, Fig. S1†). Fibers synthesized at 1200 °C show a smaller number of beads (Fig. 2) and a larger number of Sn clusters inside the fibers; also, the beads present are smaller (≤200 nm) compared to the samples synthesized at 1000 °C.
 | ||
| Fig. 2 SEM and TEM micrographs (inset) of the SiOC fibers synthesized at 1200 °C, containing different amounts of Sn: 0.05 (A), 0.1 (B), and 0.2 (C). | ||
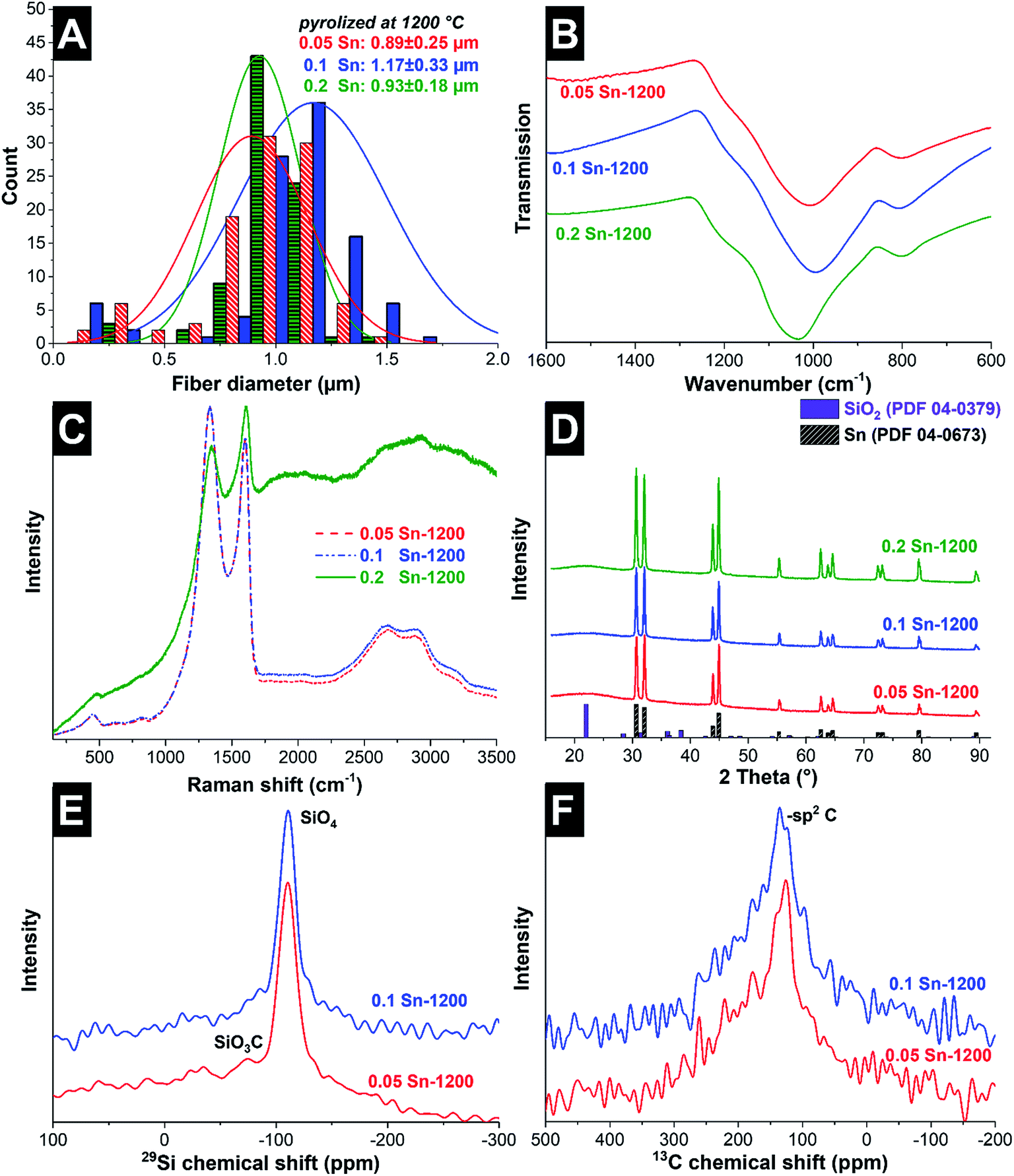 | ||
| Fig. 3 Material characterization of fibers synthesized at 1200 °C. Fiber diameter distribution (A), Fourier transform infrared spectra (B), Raman spectra (C), X-ray diffraction pattern and literature values for diffraction peak positions (D), solid-state 29Si NMR (E), and 13C NMR (F). | ||
During pyrolysis and above 600 °C, metallic tin melts36 and the liquid tin forms droplets due to surface tension.36 This diffusion limited process takes place mainly during the holding time at 1000–1200 °C. Therefore, the process of tin droplet formation depends on the diffusion kinetics of tin in the formed SiOC. The lower diffusion kinetics of metallic tin in the denser structure of SiOC synthesized at high temperature restrict the formation of droplets with large sizes.
The chemical composition was characterized by EDX, confirming the presence of mainly SiOC (Table 1). The tin content measured by EDX is not equal to the initially added amount and presents a high standard deviation due to the segregation of Sn as beads. The tin content, determined with ICP-AES (Table 1), varied in the range of 6–24 mass% and is not statistically different for samples pyrolyzed at different temperatures. Based on the EDX and ICP-AES results, an estimate of the molar ratio Sn/Si was calculated (Table 1). The values confirm that the molar ratio remains unchanged compared to the initial state, meaning that although Sn droplets are formed the initial tin content is maintained. From the EDX results, an estimate of the SiOC composition (Cfree and the Si–O–C domains) can be obtained. The molar amount of O is for every sample at least 2 times the amount of Si, implying a composition of SiO2 as the most likely compound to be formed. For a more accurate characterization of the Si–O–C domains, results from 29Si MAS NMR are presented later. It can be observed that the Cfree content significantly decreases with increasing the tin content: from 3.5–3.7 mol Cfree to 2.0–2.4 mol Cfree per 1 mol of Si for the 0.05 Sn and 0.2 Sn samples, respectively.
| Sample | SiOC composition | Sn/Si molar ratio | EDX results (mass%) | ICP-AES Sn mass% | |||
|---|---|---|---|---|---|---|---|
| C | O | Si | Sn | ||||
| M-SiOC-1000 | 4.6 Cfree + SiO2 | — | 47 ± 2 | 28 ± 1 | 24 ± 3 | — | — |
| 0.05 Sn-1000 | 3.5 Cfree + SiO2 | 0.06 | 36 ± 2 | 28 ± 2 | 24 ± 3 | 11 ± 4 | 6.2 ± 0.1 |
| 0.1 Sn-1000 | 3.0 Cfree + SiO2 | 0.10 | 32 ± 1 | 33 ± 2 | 25 ± 2 | 9 ± 2 | 10.4 ± 0.2 |
| 0.2 Sn-1000 | 2.4 Cfree + SiO2 | 0.24 | 24 ± 2 | 38 ± 2 | 24 ± 3 | 14 ± 6 | 22.1 ± 0.1 |
| M-SiOC-1200 | 4.1 Cfree + SiO1.35C0.32 | — | 52 ± 8 | 21 ± 5 | 26 ± 2 | — | — |
| 0.05 Sn-1200 | 3.7 Cfree + SiO2 | 0.06 | 41 ± 1 | 32 ± 2 | 26 ± 2 | 1 ± 1 | 6.4 ± 0.1 |
| 0.1 Sn-1200 | 3.2 Cfree + SiO2 | 0.10 | 36 ± 1 | 32 ± 1 | 26 ± 1 | 5 ± 1 | 10.7 ± 0.1 |
| 0.2 Sn-1200 | 2.0 Cfree + SiO2 | 0.22 | 23 ± 3 | 37 ± 4 | 27 ± 5 | 13 ± 10 | 21.9 ± 0.1 |
The Cfree phase in the fibers was characterized by Raman spectroscopy (Fig. 3C). All fibers present characteristic peaks for partially graphitized carbon, with corresponding D- and G-modes at 1333–1350 cm−1 and 1602–1609 cm−1, respectively, and higher order and combination modes at 2500–3000 cm−1. The D-mode is related to the breathing modes of six-atom rings and is activated when a defect is present.37 The G-mode is associated with the bond stretching of pairs of sp2-hybridized carbon atoms in rings and chains.38 The position of the G-mode (1602–1609 cm−1) correlates with the coexistence of amorphous carbon (1550 cm−1) and sp2-hybridized carbon chains, which for pure graphite is located at 1581 cm−1 (ref. 38). For further analysis, peak deconvolution was performed by using three Voigt profile peaks in the range of 500–2000 cm−1 (ESI, Fig. S4†). The data of the peak position, integral ID/IG ratio, and full-width at half-maximum (FWHM) are presented in Table 2. Samples treated at 1000 °C present different spectra according to their tin content. Both the D- and G-modes shift to higher wavelengths when increasing the tin content, indicative of a higher presence of amorphous carbon and trans-polyacetylene (between 1200 cm−1 and 1260 cm−1). Also, by increasing the tin content, the ID/IG ratio reduces, which for graphite domains smaller than 2 nm implies that less hexagonal carbon rings (also, defects from the hexagonal carbon rings) are present.38 For the samples synthesized at high temperatures, no effect on the carbon structure is observed as a function of the tin content. All samples present similar Raman spectra, which compared to the samples synthesized at lower temperatures present a sharper G-mode peak and a lower amorphous signal, meaning a higher graphitization degree. The FT-IR spectra of the pyrolyzed samples (Fig. 3B) present mainly one peak centered between 994 and 1033 cm−1, corresponding to the vibration mode of the bond Si–O–Si, and one smaller peak between 801 and 809 cm−1, corresponding to Si–C.31 No bands related to Sn–O–Sn bonds are observed, aligning with the absence of SnO2.
| Sample | Coherence length (nm) | D-mode (cm−1) | G-mode (cm−1) | I D/IG | FWHM (cm−1) | |
|---|---|---|---|---|---|---|
| D-mode | G-mode | |||||
| 0.05 Sn-1000 | 132 | 1335 | 1602 | 2.01 | 200.0 ± 0.6 | 71.4 ± 1.0 |
| 0.1 Sn-1000 | 105 | 1340 | 1603 | 2.03 | 206.6 ± 0.7 | 72.1 ± 0.8 |
| 0.2 Sn-1000 | 101 | 1350 | 1607 | 1.73 | 203.1 ± 1.0 | 71.9 ± 2.0 |
| 0.05 Sn-1200 | 122 | 1335 | 1603 | 1.58 | 180.9 ± 0.5 | 65.4 ± 1.0 |
| 0.1 Sn-1200 | 102 | 1333 | 1604 | 1.67 | 178.3 ± 0.5 | 63.2 ± 0.8 |
| 0.2 Sn-1200 | 138 | 1339 | 1609 | 1.56 | 178.1 ± 0.8 | 65.8 ± 1.4 |
For additional analysis of the crystal structure, XRD patterns of the samples were recorded (Fig. 3D). In all samples, broad signals coming from tetragonal SiO2P41212 (PDF 04-0379; lattice parameters: a = 0.497 nm and c = 0.693 nm) are detected together with sharp peaks corresponding to tetragonal Sn I41/amd (PDF 04-0673; lattice parameters: a = 0.583 nm and c = 3.182 nm). By applying the Scherrer equation, the average coherence length of the metallic Sn in the fibers is about 101–138 nm (Table 2).
To understand the effect of Sn and the pyrolysis temperature on the resulting SiOC network, 29Si MAS and 13C MAS NMR spectra were collected (Fig. 3E and F). The 29Si MAS NMR spectra indicate that in the synthesized samples, the increase in the tin content leads to a decrease in the presence of SiCxO4−x tetrahedral units (with 1 ≤ x ≤ 4); consequently, mainly SiO4 units are obtained. This effect is also observed for higher thermal treatment temperatures. For all samples, the 29Si MAS NMR spectra exhibit an intense peak in the range of −105 ppm to −110 ppm which corresponds to SiO4 units. For 0.05 Sn-1000, a second peak is observed at −71 ppm, corresponding to SiO3C units (ESI, Fig. S3E†).39 The intensity of this peak decreases with increasing the heat treatment temperature and is barely visible for the sample 0.05 Sn-1200 in Fig. 3E. We did not observe any peaks corresponding to SiO2C2 (−34 ppm), SiOC3 (7 ppm), or SiC4 (−11 ppm) in any sample. 13C MAS NMR presents mainly a broad peak around 130 ppm for all samples, which is consistent with sp2-hybridized carbon. The broad character of the peak is related to large anisotropic susceptibility effects which are not fully averaged by MAS.39 As reported by Widgeon et al. for high- and low-carbon SiOC synthesized at 1100 °C, low-carbon SiOC (Cfree = 5.7 mass%) presents more SiO4 units (44% of the Si units), while the high-carbon SiOC (Cfree = 41.3 mass%) presented fewer SiO4 units (32% of the Si units) and a larger number of SiC4 units (17% of the Si units).16,39 The larger number of SiO4 units (above 70%) and the absence of SiC4 units in our system are explained by the reduction of SnO by C and Si, leading to an oxidation of C and Si. The oxidation of carbon to volatile species is supported by the decrease in the carbon content when increasing the tin content, as supported by the EDX results (Table 1), while the silicon content remains constant. For comparison, the elemental composition of monoliths without Sn (M-SiOC) is presented in Table 1, corroborating that the presence of Sn modifies the composition of the SiOC material.
We can conclude that our SiOC material consists of a cellular network of low order graphitic sp2-hybridized carbon, where nanodomains of mainly SiO4 units are sequestered within cells created by the graphitic sheets, and this SiOC material is engulfing larger domains of metallic Sn.
3.3. Electrochemical performance
The fabrication of this hybrid fiber material was motivated by enabling several electrochemical advantages: (i) multiple sites for Li+ insertion in both systems SiOC (Cfree + Si–O–C nanodomains) and metallic Sn, (ii) benefit from the elastic properties of SiOC to mitigate the volume changes of Sn,16 (iii) interfiber space is beneficial to buffer the large volume expansion of the material, and (iv) high conductivity due to the continuous carbon phase in the SiOC fibers. The fiber mats were tested as free-standing electrodes without the addition of any polymer binder or any type of conductive additive. The electrochemical performance was evaluated in 1 M LiPF6 in EC/DMC, between 3 and 0.01 V vs. Li/Li+. Cyclic voltammograms (CVs) were recorded at 0.1 mV s−1, and are presented in Fig. 4 and ESI, Fig. S5.† During the first cycle, all samples showed a cathodic peak below 1 V vs. Li/Li+ from the reductive decomposition of the electrolyte and the formation of a solid electrolyte interphase (SEI). Therefore, initially the system was cycled at low rates (C/20) to promote SEI formation. The latter is a dynamic process in alloy anodes, breaking and reforming in each cycle due to the high volume changes of the electrode.40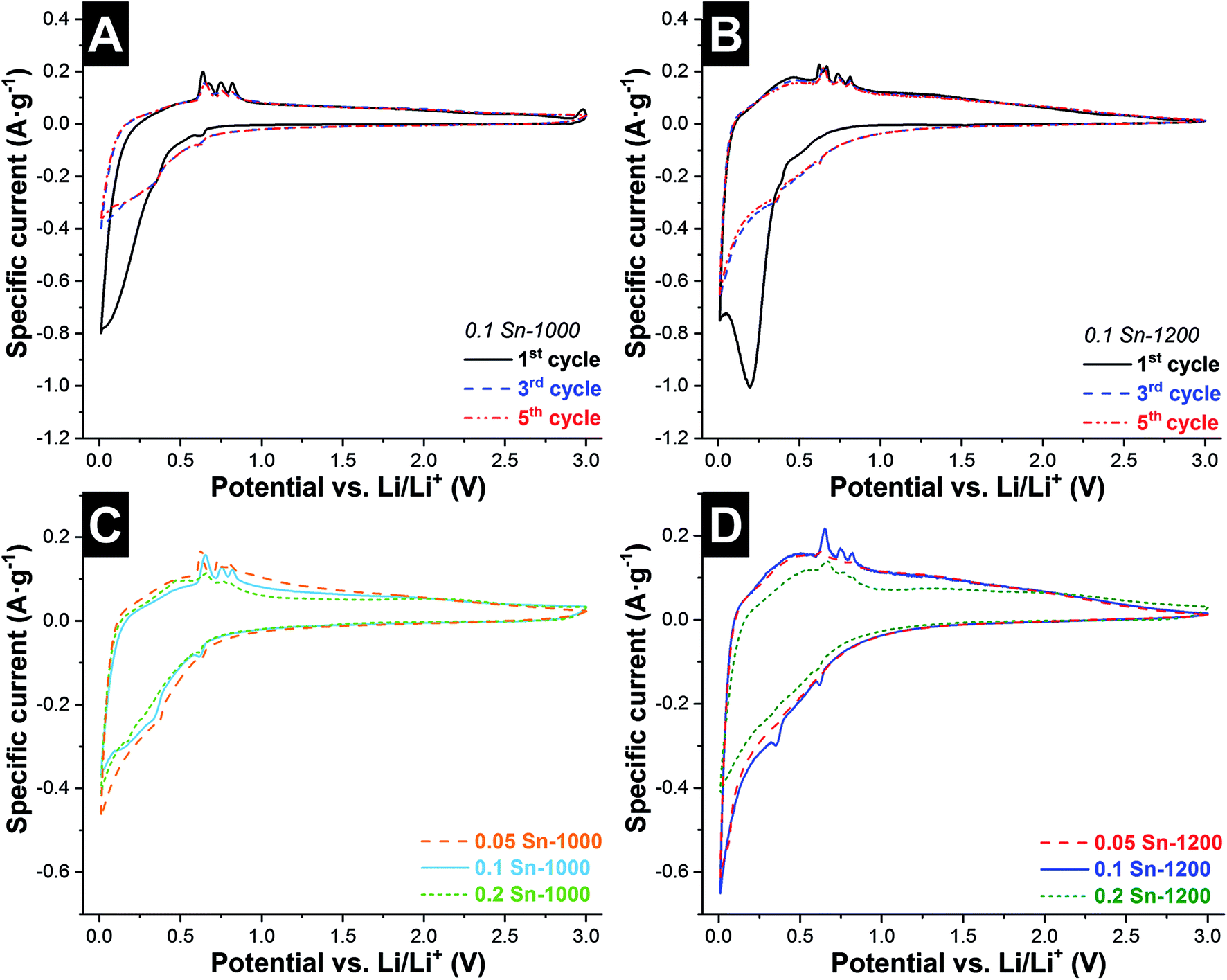 | ||
| Fig. 4 Electrochemical characterization in a half-cell configuration of SiOC/Sn hybrid fibers by cyclic voltammetry at 0.1 mV s−1. First 5 cycles for the samples containing 0.1 Sn synthesized at 1000 °C (A) and 1200 °C (B). 5th cycle of the samples synthesized at 1000 °C (C) and 1200 °C (D). | ||
During the first cycle of the 0.1 Sn-1000 sample, further cathodic peaks at +0.62 V and +0.34 V vs. Li/Li+ are observed, corresponding to LiSn and Li22Sn5, respectively, followed by a broad and large peak at lower voltages, related to the Li+ insertion in the SiOC matrix. All these peaks are blurred by SEI formation reactions during the first cycle, but become clearer in the subsequent cycles. During oxidation, several anodic peaks between +0.63 V and +0.82 V vs. Li/Li+ are observed. During the first cycle, 4 peaks are identified (+0.64, +0.67, +0.75, and +0.82 V vs. Li/Li+), while only 3 peaks occur in the subsequent cycles (+0.65, +0.75 and +0.82 V vs. Li/Li+). The numerous peaks indicate that dealloying in Sn occurs via multiple reactions and stages (Li7Sn2, Li13Sn5, Li5Sn2, Li7Sn3, Li2Sn5, and LiSn),41 and not as the reverse path described by the cathodic peaks during Li+ insertion. The broad peak observed during the oxidation is related to the Li+ extraction of carbon,36 and SiO2 nanodomains. Further anodic peaks are only observed during the first cycle at almost +3 V vs. Li/Li+, which could be related to the reabsorption of hydrogen formed during the reduction step.
Comparing the first cycle of the samples pyrolyzed at 1000 °C, the amplitude of the anodic peaks (between +0.63 V and +0.82 V vs. Li/Li+) increases proportionally to the tin content (ESI, Fig. S5A and B†). The intensity of these peaks in the next cycles decreases proportionally to the tin content. In the 5th cycle (Fig. 4C), all CVs present similar shapes, but some differences in the peak position and intensity are observed. During lithiation, the first cathodic peak is shifted from +0.63 V to +0.61 V vs. Li/Li+, while the intensity of a second peak at +0.36 V vs. Li/Li+ decreases when increasing the tin content. During Li+ extraction, the peaks located at +0.63 V, +0.73 V, and +0.80 V vs. Li/Li+ shift to higher voltages for the sample 0.05 Sn-1000. The increase of the voltage difference, for reduction and oxidation, together with the decrease in the intensity of the peaks, indicates increasing overpotential due to electron transport limitations. As observed for hybrid Sn/C particles, during the first cycles a high capacity fading is observed related to the partial particle pulverization of the Sn particles.36 The large size of the Sn particles in our system and the segregation of some of them due to Sn melting during pyrolysis contribute to a large capacity fading in the first cycles.
Compared to samples synthesized at higher temperatures (Fig. 4B and D), two main differences are observed. For the samples synthesized at 1200 °C, the peaks related to tin redox-reactions are less intense, but are observed at similar voltages, and the CVs present a higher area, which indicates a generally higher capacity. Also, after the 5th cycle, these samples present similar shapes (Fig. 4D). There exist mainly two differences between the materials synthesized at 1000 °C and 1200 °C: the degree of carbon ordering and the particle size of the Sn beads. An increase in the conductivity of the SiOC matrix leads to a higher Li+ insertion into the Si–O–C tetrahedral units and a more homogenous current and voltage distribution throughout the electrode during charging. The effect of the pyrolysis temperature on the conductivity is corroborated by the sheet resistance values, 0.71–0.98 Ω cm and 0.28–0.32 Ω cm for samples synthesized at 1000 °C and 1200 °C, respectively (Table 3). The decrease in the intensity of the Sn alloy peaks is related to the lower formation of long-range ordered structures.41 For the samples synthesized at 1000 °C, lithium is intercalated into Sn in two different mechanisms, the well dispersed nanoparticles in the SiOC fibers and the large segregated Sn sub-micrometer-sized particles outside the fibers. For the samples synthetized at 1200 °C, Sn is mainly present as nanoparticles well dispersed inside the SiOC fibers, which explains the lower but more stable intensity of the Sn alloy peaks.
| Sample | Capacity at 35 mA g−1 (mA h g−1) | Reversible capacity after 100 cycles (mA h g−1) | Sheet resistance (Ω cm) | |||
|---|---|---|---|---|---|---|
| 1st lithiation | 1st delithiation | 5th lithiation | 5th delithiation | |||
| 0.05 Sn-1000 | 1097 | 789 | 725 | 607 | 221 | 0.71 ± 0.10 |
| 0.1 Sn-1000 | 1143 | 881 | 750 | 744 | 281 | 0.90 ± 0.15 |
| 0.2 Sn-1000 | 1093 | 821 | 632 | 613 | 277 | 0.98 ± 0.13 |
| 0.05 Sn-1200 | 1075 | 840 | 804 | 801 | 509 | 0.28 ± 0.05 |
| 0.1 Sn-1200 | 1145 | 939 | 854 | 842 | 457 | 0.31 ± 0.05 |
| 0.2 Sn-1200 | 1178 | 994 | 806 | 791 | 400 | 0.32 ± 0.03 |
For a quantitative characterization of Li+ insertion in the hybrid material, the capacity during lithiation and delithiation was characterized by galvanostatic cycling between 3 and 0.01 V vs. Li/Li+. The voltage profile for the first cycle (35 mA g−1, 0.05C) is presented in Fig. 5A and B and key data are reported in Table 3. During the discharging step, we see a rapid decrease in the voltage before achieving +1 V vs. Li/Li+, at which reduction reactions start to take place, leading later to a voltage plateau at 0.5–0.01 V vs. Li/Li+. The initial insertion capacity was similar for all samples, namely 1075–1178 mA h g−1. The SEI formation and the irreversible Li+ insertion into the nanocomponents42 lead to a low coulombic efficiency of 0.75 and 0.82 for the samples treated at 1000 °C and 1200 °C, respectively. This could be influenced by the carbon ordering, since ordered carbons are characterized by a higher initial charge efficiency.43 It was observed that the coulombic efficiency for the first cycle increases with increasing the tin content and the pyrolysis temperature. However the coulombic efficiency stabilizes faster achieving values close to 1, for low tin content, and also the capacity fading is lower for low tin content, which is related to the partial particle pulverization of the large Sn particles.36 In the case of the samples treated at high temperatures, the initial lithiation/delithiation capacity is proportional to the tin content, while for the sample at 1000 °C, the highest capacity was obtained for the sample 0.1 Sn-1000. A fast decay in the reversible capacity is observed in the next 5 cycles, but with a decrease of the irreversible capacity. After 5 cycles, the samples synthesized at high temperatures maintain a higher delithiation capacity between 842 and 791 mA h g−1.
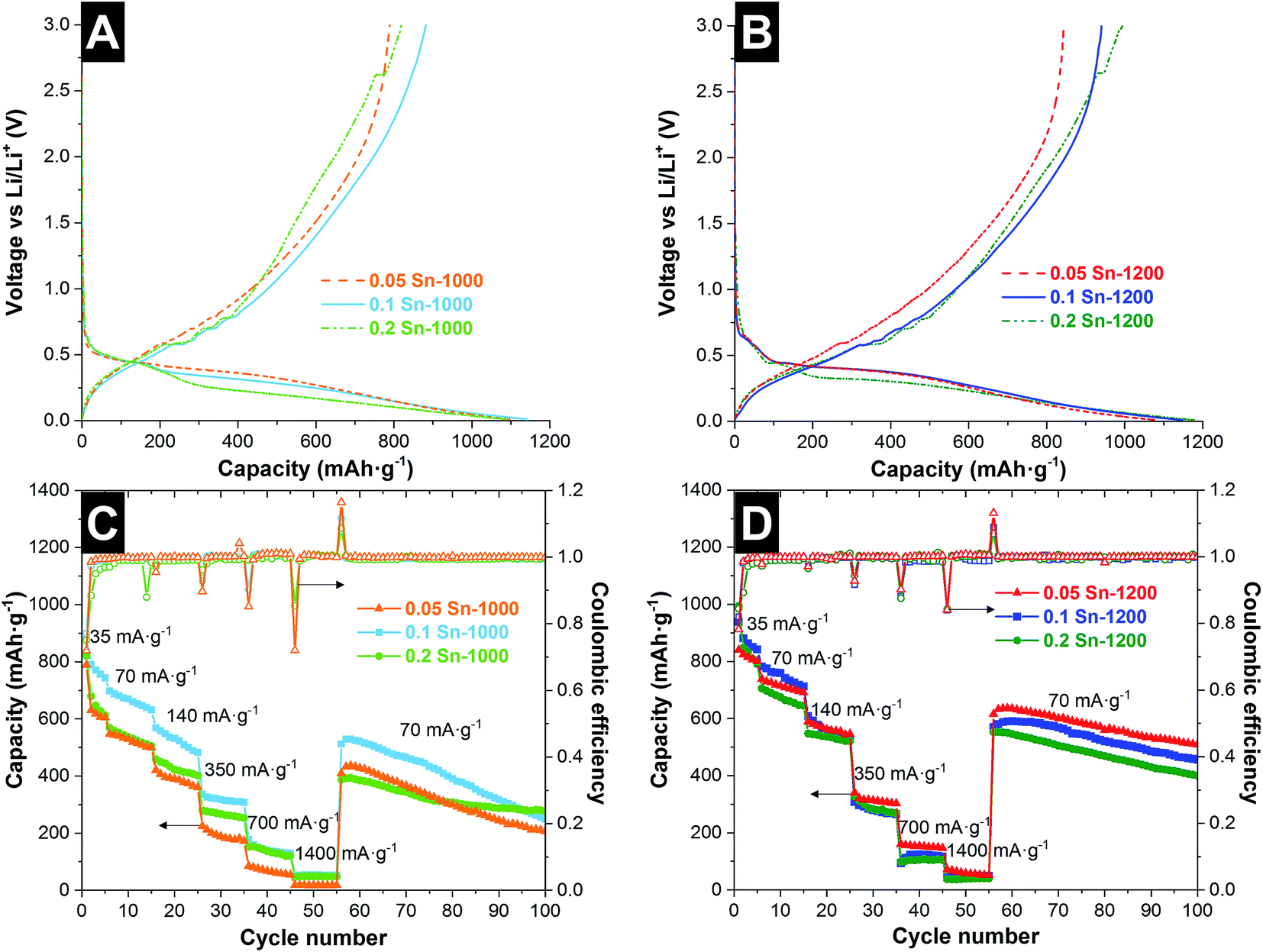 | ||
| Fig. 5 First cycle of galvanostatic lithiation/delithiation at 35 mA g−1 from 3.0 to 0.01 V vs. Li/Li+, for SiOC/Sn hybrid fibers synthesized at 1000 °C (A) and 1200 °C (B). Rate capability performance of samples synthesized at 1000 °C (C) and 1200 °C (D). | ||
At higher rates (140 mA g−1), the samples treated at 1200 °C maintain similar capacity values around 550 mA h g−1. For samples synthesized at lower temperatures, larger differences are observed. The sample 0.1 Sn-1000 presented a higher capacity (525 mA h g−1) while the other samples retained 380–430 mA h g−1. At higher rates, the capacity values for all samples tend to be similar; only the sample 0.05 Sn-1000 presented consistently a lower capacity. For the samples treated at low temperature, after the fast decay in the first cycles, the sample which presents a more stable rate capability performance was 0.2 Sn-1000, which after 100 cycles also maintained the highest capacity of 277 mA h g−1. Samples with a lower amount of Sn presented a higher capacity fading. For the samples synthesized at higher temperatures, higher capacity retention is observed, maintaining between 400 and 509 mA h g−1 after 100 cycles. In this case, the samples present a more similar rate capability behavior and stability.
To explain the effect of the heat treatment and tin content on the observed electrochemical performance, we must consider the theoretical Li+ insertion capacity of the present phases and how the Li-storage takes place in silicon oxycarbides. The theoretical capacity of graphite, SiO2, and Sn corresponds to 372, 1784, and 993 mA h g−1, respectively.5,40 Therefore, our samples have a maximum theoretical capacity of 1150–1300 mA h g−1 when applying a simplified model for the SiOC composition (ESI, Table S2†). There is still an open debate in the literature on the active sites for the Li storage in silicon oxycarbides; however, there are some features which are often reported: SiC is not electrochemically active and both Si–O–C nanodomains and the Cfree phase contribute to the Li+ insertion.8 The proposed sites for reversible lithium storage in SiOC include the Si–O–C tetrahedral units, micropores, and the free carbon phase.8 Without free carbon, lithium storage in the Si–O–C domains cannot occur since a certain amount of carbon is needed to ensure sufficient electronic conductivity.44 Additionally, the electronic conductivity of the free carbon phase can homogenize the voltage and current distribution throughout the electrode in order to avoid local electrode degradation spots.45 While in some aspects, the presence of Cfree is beneficial, there are some aspects in which it is not. The Cfree phase facilitates the formation of Li–O bonds, leading to irreversible lithium uptake in the presence of carbon in the SiOC.16,44 Also, while a higher graphitization degree of carbon is beneficial for a higher conductivity, higher ordered carbons can host less Li+ than less ordered carbons.13
In our system, all three phases contribute to the overall Li+ insertion/extraction. A graphical representation of the phases is presented in the ESI, Fig. S6A.† A higher tin content yields a higher initial reversible capacity; however, this is accompanied by a fast capacity fading after the first 5 cycles. Also, this increase in the reversible capacity with the tin content was only observed for the samples synthesized at 1200 °C, with a more conductive Cfree phase (Table 3). The Cfree phase presents the lowest volumetric expansion (≈10%; ref. 5) and demonstrates to have a crucial role in the stability and rate capability of the system. Therefore, we found that a high and stable performance depends on the electrical conductivity and the volumetric expansion during Li+ insertion/extraction. During Li+ insertion/extraction, the large particle size and the high volumetric expansion of the metallic tin (≈260%)40 may pulverize the particles; therefore, the contribution of the metallic tin to the electrochemical performance decreases during the first cycles. It has been already reported that for C-rich SiOC, the carbon phase accommodates the high volume expansion during alloying with Li+, preventing electrode failure.16 Cracking and pulverization of the active particles and the surrounding matrix lead to the disconnection of some alloy particles from the conductive carbon or current collector, and this isolation of the particles leads to an incomplete delithiation reaction.40 In our case, the fiber morphology is maintained (ESI, Fig. S6B and C†) and the electrodes continue to be free-standing after 100 cycles; possibly, the interfiber space helps to compensate for the volume expansion. However, the Sn beads have disappeared after 100 cycles, and some particles unconnected to the fiber network are observed, which could be related to the pulverization of the Sn beads. To corroborate this failure mechanism during the cyclic test, post-mortem TEM and EDX analysis was performed (ESI, Fig. S6 and S7†). The TEM images are characterized by two different regions: a dense SiOC material and a less dense region with the presence of Sn nanoparticles. The post-mortem SiOC material presents more holes compared to pristine SiOC, and some Sn nanoparticles were observed dispersed outside of the SiOC particles. According to the elemental analysis, the less dense region consists mainly of C, O, F, and P engulfing the Sn nanoparticles, as observed from the TEM images. As can be seen, some of the Sn nanoparticles are seemingly separated from the SiOC matrix during cyclic testing.
The electrochemical performance of the samples 0.05 Sn-1200 and 0.2 Sn-1200 was compared with the literature and Table 4 includes data of the pyrolysis temperature and tin content. The electrochemical data presented in Table 4 correspond mostly to high-carbon SiOC materials for a fair comparison, since low-carbon SiOC is well known for presenting a lower electrochemical performance.8 Our materials present a higher first cycle reversible capacity. This is attributed to several factors: the complete electrode mass contributes to the lithium insertion (no polymer-binder or additional conductive additive), the presence of metallic tin, and the high conductivity of the continuous fiber network. The improved conductivity is indicated by the higher capacity retention at higher rates, despite the higher mass loading of our electrodes. Also, compared to the literature, the effect of pyrolysis on the electrochemical performance of SiOC systems differs. It has been reported that a lower pyrolysis temperature (900 °C) leads to higher capacity values.13 For high-carbon SiOC, a higher pyrolysis temperature leads to an increase in SiC4 units, which are not electrochemically active,8 decreasing the capacity of the system. In our systems, the presence of Sn promoted the increase of SiO4 units and avoided the formation of C containing units, and therefore a heat treatment at higher temperatures helped to obtain a more conductive material, leading to higher capacity values and a higher stability.
| Sample | Electrode | 1st cycle Crev (mA h g−1) | Capacity at high rate (V vs. Li/Li+) | Reference |
|---|---|---|---|---|
| Sn (6 mass%)/SiOC-1200 °C fibers | Free standing (8.6 mg cm−2) | 840 (am: 840) | 303 (am: 303) at 350 mA g−1 | This work; 1 M LiPF6 in EC/DM |
| Sn (22 mass%)/SiOC-1200 °C fibers | Free standing (8.3 mg cm−2) | 994 (am: 994) | 268 (am: 268) at 350 mA g−1 | This work; 1 M LiPF6 in EC/DM |
| SiOC-1100 °C microsized particles | DBD with CA and PVdF (3.3 mg cm−2) | 452 (am: 532) | 274 (am: 323) at 372 mA g−1 | Ref. 13; 1 M LiPF6 in EC/DMC |
| Sn (25 mass%)/SiOC-1000 °C microsized particles | DBD with CA and PVdF (4.5 mg cm−2) | 553 (am: 651) | 230 (am: 270) at 372 mA g−1 | Ref. 16; 1 M LiPF6 in EC/DMC |
| SiOC-1000 °C microsized particles | DBD with CA and PVdF (3–5 mg cm−2) | 483 (am: 568) | 246 (am: 290) at 372 mA g−1 | Ref. 8; 1 M LiPF6 in EC/DMC |
| SiOC-1300 °C microsized particles | DBD with CA and PVdF (3–5 mg cm−2) | 250 (am: 293) | 68 (am: 80) at 372 mA g−1 | Ref. 8; 1 M LiPF6 in EC/DMC |
| (Low carbon) SiOC-1300 °C microsized particles | DBD with CA and PVdF (3–5 mg cm−2) | 619 (am: 728) | 43 (am: 50) at 372 mA g−1 | Ref. 8; 1 M LiPF6 in EC/DMC |
3.4. Comparison to Sn-free SiOC
The chemical characterization results in Section 3.2 have demonstrated that Sn modifies the composition of SiOC. To corroborate the effect of Sn on the electrochemical performance, SiOC monoliths with (M-0.05 Sn) and without Sn (M-SiOC) were prepared and tested. The monoliths were tested as polymer-bound electrodes by admixing carbon black (C65) as the conductive additive. The results are presented in the ESI, Fig. S8.†Comparing the polymer-bound electrodes and the fiber electrodes, a drastic difference is observed in the rate capability performance which is related to the much higher sheet resistivity of the polymer-bound electrodes compared to the fiber mat electrodes (ESI, Table S3†). Comparing the monoliths, when adding 0.05 Sn the first reversible capacity increases from 635 to 681 mA h g−1, and from 677 to 741 mA h g−1 for the samples synthesized at 1000 and 1200 °C, respectively. The monoliths produced at higher temperature present a more stable performance, but in all cases lower than the fiber mat electrodes. The lower stability of the polymer-bound electrodes is related to the higher particle size (5–10 μm) and lower conductivity of the non-continuous network, compared to the fiber mats.
4. Conclusions
We synthesized continuous SiOC fiber mat electrodes free of polymer binders and conductive additives, for the use as an anode material for Li-ion batteries. The synthesis is based on the electrospinning of a low-cost silicone resin, using tin acetate as a polymer crosslinker. We found that adding a low amount of tin acetate (0.05 mol Sn per 1 mol Si) is necessary for a complete crosslinking, maintaining the fiber shape during thermal treatment. After crosslinking and during pyrolysis between 1000 and 1200 °C, a hybrid material is formed, containing a carbon-rich SiOC matrix and metallic tin clusters. During pyrolysis, SnO is reduced to metallic tin, causing the oxidation of carbon and silicon, promoting a higher presence of SiO4 units in the SiOC matrix and reducing the carbon content. This effect depends on the amount of added tin, with low amounts of tin (0.05 mol Sn per 1 mol Si) avoiding the formation of electrochemically inactive SiC4 units. Higher tin amounts decrease the conductivity of the fibers by removing the conductive carbon phase. Since the presence of Sn promotes the existence of SiO4 units, pyrolysis can be performed at higher temperatures (1200 °C) without forming inactive SiC4 and benefiting the formation of a more graphitic and conductive Cfree phase. It was also found that pyrolysis at 1000 °C and high amounts of tin (0.1–0.2 mol per 1 mol Si) lead to a higher segregation of tin into beads outside the fiber shape. This single precursor approach has several advantages for producing continuous Sn/SiOC composite fibers: (i) by an in situ synthesis a uniform distribution of Sn particles in the SiOC matrix is obtained, (ii) the continuous matrix ensures the conductivity of the system and helps to constrain the volume expansion, and (iii) the material properties can be tailored according to the desired electrochemical properties.The hybrid materials synthesized at 1200 °C present very high initial reversible capacities (840–994 mA h g−1) at 35 mA g−1, and retain 280–310 mA h g−1 at 350 mA g−1 specific current. After 100 cycles at 70 mA g−1, the hybrid fibers maintained between 400 and 509 mA h g−1. This capacity values are related to tin alloying and Li+ insertion reactions into the Si–O–C tetrahedral units, the presence of micropores, and the Cfree phase. Without tin segregation outside the fibers and uniform distribution of tin in the SiOC fiber matrix (pyrolysis 1200 °C), a higher amount of Sn is beneficial for a higher reversible capacity in the first cycles. However, the higher volumetric expansion during lithiation of metallic tin possibly explains a higher capacity fading and a lower performance stability. At high rates, all systems have similar capacity values, meaning that tin is not significantly contributing to the Li+ insertion capacity, but mainly the C-rich SiOC. The Cfree phase has a significant influence on the stability of the system. The Cfree phase is present mainly as sp2-hybridized layers, and according to the electrochemical performance of the different samples, it was found that a higher amount of carbon and a higher graphitization degree are crucial for a higher conductivity and a stable electrochemical performance. Adding low amounts of Sn is beneficial not just for the crosslinking of the polymer precursor, but also to decrease the presence of SiC4 electrochemically inactive species in the SiOC, and form metallic tin clusters which contribute to a higher Li+ insertion in the first cycles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the German Federal Ministry for Economic Affairs and Energy (BMWi) in support of the HyBaCap project (award number 03ET6113C). The authors thank Prof. Eduard Arzt for his continuing support, Robert Drumm for his support in TGA-MS measurements and Andrea Jung for her support with ICP-AES measurements (all at INM). We also thank Dr Soumyadip Choudhury, Nicolas Jäckel, Juhan Lee, and Dr Eunho Lim for discussions and brainstorming (all at INM).References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. Proietti Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- R. Dash and S. Pannala, Sci. Rep., 2016, 6, 27449 CrossRef CAS PubMed.
- A. M. Wilson and J. R. Dahn, J. Electrochem. Soc., 1995, 142, 326–332 CrossRef CAS.
- V. S. Pradeep, M. Graczyk-Zajac, R. Riedel and G. D. Soraru, Electrochim. Acta, 2014, 119, 78–85 CrossRef CAS.
- P. Colombo, G. Mera, R. Riedel and G. D. Sorarù, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CAS.
- G. Mera, A. Navrotsky, S. Sen, H.-J. Kleebe and R. Riedel, J. Mater. Chem. A, 2013, 1, 3826–3836 CAS.
- M. Graczyk-Zajac, L. Toma, C. Fasel and R. Riedel, Solid State Ionics, 2012, 225, 522–526 CrossRef CAS.
- P. Dibandjo, M. Graczyk-Zajac, R. Riedel, V. S. Pradeep and G. D. Soraru, J. Eur. Ceram. Soc., 2012, 32, 2495–2503 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, J. Power Sources, 2013, 244, 450–455 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac, S. Lauterbach, H.-J. Kleebe and R. Riedel, J. Power Sources, 2014, 269, 164–172 CrossRef CAS.
- M. Zhu, J. Yang, Z. Yu, H. Chen and F. Pan, J. Mater. Chem. A, 2017, 5, 7026–7034 CAS.
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H.-J. Kleebe and R. Riedel, Adv. Funct. Mater., 2014, 24, 4097–4104 CrossRef CAS.
- A. Tolosa, B. Krüner, N. Jäckel, M. Aslan, C. Vakifahmetoglu and V. Presser, J. Power Sources, 2016, 313, 178–188 CrossRef CAS.
- A. Tolosa, B. Krüner, S. Fleischmann, N. Jäckel, M. Zeiger, M. Aslan, I. Grobelsek and V. Presser, J. Mater. Chem. A, 2016, 4, 16003–16016 CAS.
- S. Li, C. Chen, K. Fu, L. Xue, C. Zhao, S. Zhang, Y. Hu, L. Zhou and X. Zhang, Solid State Ionics, 2014, 254, 17–26 CrossRef CAS.
- Y. Li, B. Guo, L. Ji, Z. Lin, G. Xu, Y. Liang, S. Zhang, O. Toprakci, Y. Hu, M. Alcoutlabi and X. Zhang, Carbon, 2013, 51, 185–194 CrossRef CAS.
- Y. Li, Y. Hu, Y. Lu, S. Zhang, G. Xu, K. Fu, S. Li, C. Chen, L. Zhou, X. Xia and X. Zhang, J. Power Sources, 2014, 254, 33–38 CrossRef CAS.
- X. Xia, X. Wang, H. Zhou, X. Niu, L. Xue, X. Zhang and Q. Wei, Electrochim. Acta, 2014, 121, 345–351 CrossRef CAS.
- M. Dirican, O. Yildiz, Y. Lu, X. Fang, H. Jiang, H. Kizil and X. Zhang, Electrochim. Acta, 2015, 169, 52–60 CrossRef CAS.
- G. Kickelbick, in Hybrid Materials, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 1–48, DOI:10.1002/9783527610495.ch1.
- A. Guo, M. Roso, M. Modesti, J. Liu and P. Colombo, J. Appl. Polym. Sci., 2014, 131, 39836 Search PubMed.
- J. S. Atchison, M. Zeiger, A. Tolosa, L. M. Funke, N. Jäckel and V. Presser, RSC Adv., 2015, 5, 35683–35692 RSC.
- H. J. T. Ellingham, J. Soc. Chem. Ind., London, Trans. Commun., 1944, 63, 125–133 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 2004 Search PubMed.
- T. Oh and C. Choi, J. Korean Phys. Soc., 2010, 56(4), 1150–1155 CrossRef CAS.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. A. Vyvyan, Introduction to Spectroscopy, Cengage Learning, 2008 Search PubMed.
- V. Belot, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 613–623 CrossRef CAS.
- J. Cordelair and P. Greil, J. Eur. Ceram. Soc., 2000, 20, 1947–1957 CrossRef CAS.
- E. Ionescu, C. Terzioglu, C. Linck, J. Kaspar, A. Navrotsky and R. Riedel, J. Am. Ceram. Soc., 2013, 96, 1899–1903 CrossRef CAS.
- W.-M. Zhang, J.-S. Hu, Y.-G. Guo, S.-F. Zheng, L.-S. Zhong, W.-G. Song and L.-J. Wan, Adv. Mater., 2008, 20, 1160–1165 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- S. J. Widgeon, S. Sen, G. Mera, E. Ionescu, R. Riedel and A. Navrotsky, Chem. Mater., 2010, 22, 6221–6228 CrossRef CAS.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- M. Winter and J. O. Besenhard, Electrochim. Acta, 1999, 45, 31–50 CrossRef CAS.
- Y.-S. Hu, R. Demir-Cakan, M.-M. Titirici, J.-O. Müller, R. Schlögl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 1645–1649 CrossRef CAS PubMed.
- Y. Nishi, Electrochem. Soc. Interface, 2016, 25, 71–74 CrossRef.
- M. Graczyk-Zajac, L. Reinold, J. Kaspar, P. Sasikumar, G.-D. Soraru and R. Riedel, Nanomaterials, 2015, 5, 233 CrossRef CAS PubMed.
- M. Widmaier, N. Jäckel, M. Zeiger, M. Abuzarli, C. Engel, L. Bommer and V. Presser, Electrochim. Acta, 2017, 247, 1006–1018 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00431a |
| This journal is © The Royal Society of Chemistry 2018 |