
3D hierarchical MnO2 microspheres: a prospective material for high performance supercapacitors and lithium-ion batteries†
Syed
Khalid
,
Chuanbao
Cao
 *,
Muhammad
Naveed
and
Waqar
Younas
*,
Muhammad
Naveed
and
Waqar
Younas
Research Center of Materials Science, Beijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: cbcao@bit.edu.cn; Fax: +86 10 6891 2001; Tel: +86 10 6891 3792
First published on 21st August 2017
Abstract
3D hierarchical MnO2 microspheres with an ultrathin nanosheet structure and high specific surface area (184.32 m2 g−1) are synthesized by a rapid microwave heating method in just 10 minutes. In this work, an ionic electrolyte (EMIMBF4/DMF) based asymmetric supercapacitor device is successfully prepared by using 3D hierarchical MnO2 microspheres as the cathode and activated carbon as the anode material. The (EMIMBF4/DMF) electrolyte enables a significant enhancement in the potential windows of individual electrode materials and the asymmetric device which results in much improved electrochemical performance. The asymmetric device operates successfully within a potential window of 3.0 V and exhibits an outstanding energy density of 105 W h kg−1 at a power density of 1494 W kg−1 with good cycling life stability (20% loss after 6000 cycles) at a much higher current density of 6 A g−1. Moreover, 3D hierarchical MnO2 microspheres also exhibit an outstanding Li ion storage performance with a discharge capacity of 715 mA h g−1 even after 200 cycles at a current density of 300 mA g−1. The discharge capacity retention (78% @ the 2nd cycle) after 200 cycles at 300 mA g−1 is the highest amongst those of all the reported anode materials based on MnO2. High specific capacities and outstanding cyclability further indicate their strong potential as an anode material for lithium-ion batteries. The promising energy storage applications can be ascribed to the high specific surface area, mesoporous structure and ultrathin nanosheet building blocks of MnO2 microspheres.
Introduction
Recently, immense research effort has been focused on developing high performance supercapacitors and lithium-ion batteries to meet the requirements of various emerging energy applications.1–3 Supercapacitors play an important role in the field of energy storage due to their high power density, long cycle life and good safety. But their low energy density is still the area of great concern for their industrial scale implementation. High energy density supercapacitors are essentially required for applications such as space flight technology, hybrid vehicles and so on.4 Similarly lithium ion batteries (LIBs) are also the most reliable power sources owing to their high energy density for portable electronic devices, electrical vehicles and green grids.5,6 But their low specific discharge capacity along with poor cycling stability are the main disadvantages associated with their practical application. So it requires more focused work to overcome the deficiencies associated with both supercapacitors and lithium-ion batteries.The performance of supercapacitors and lithium ion batteries can be enhanced by manipulating the morphological features of the electrode material with improved properties.4,7 The role of electrolytes is also the most significant parameter to enhance the operational voltage window and subsequently the energy density of supercapacitors.8,9 The operational voltage window of aqueous electrolyte based supercapacitors is limited to 1.0 & 2.0 V for their symmetric & asymmetric configurations respectively.10–13 Aqueous electrolytes start decomposing beyond this thermodynamically stable voltage limit.12 Organic and ionic liquid electrolytes are alternative options which can provide a much wider operational potential window. But the utility of organic electrolytes cannot be implemented on a large scale due to their toxic, volatile and flammable nature.9 Ionic liquids are considered as the most desirable and safer option to enhance the operational voltage window (2–4 V) due to their negligible vapor pressure, non-toxicity, relatively high ionic conductivity, and good thermal and chemical stability.9,14,15 Ionic liquids have already been used as electrolytes for various carbon based supercapacitors,16–18 but despite this, energy density is still the area of concern which requires further research in this field. It is well known that the specific capacitance offered by carbonaceous materials is much less than those of metal oxides.10 RuO2 has already delivered excellent performance as an electrode material for ionic liquid electrolyte based supercapacitors.19 But its potential cannot be implemented on a large scale due to its high cost and scarcity.20,21 MnO2 is considered to be a good alternative because of its high theoretical capacitance (1370 F g−1), earth abundance, low cost and environmentally friendly nature.9,22–27
X. Zhang et al. have grown MnO2 nanosheets on a CNT/Ni foam electrode by using an electrodeposition method.9 Further they fabricated an asymmetric supercapacitor device based on this material by using 1-butyl-3-methyl-imidazolium hexafluorophosphate ([Bmim]PF6)/N,N-dimethyl formamide (DMF) as an ionic liquid electrolyte.9 They reported a maximum & minimum energy density of 67.5 & 8.5 W h kg−1 at a current density of 0.4 & 24 A g−1.9 But the capacitance retention is only around 12.5% at higher current density which is not appropriate for its practical application.9 The much lower capacitance retention at higher current density may be due to less penetration of the electrolyte inside the electroactive materials.9 Most importantly the cycling performance of the asymmetric device was also not reported in this paper which is the key parameter of its long term application.9
J. K. Chang et al. have grown a MnO2 granular structure directly on Ni foam using an anodic deposition method.24 They reported the electrochemical performance of the as-synthesized material in only three-electrode measurements by using 1-ethyl-3-methylimidazolium-dicyanamide (EMI-DCA) as an ionic liquid electrolyte.24 The maximum specific capacitance (72 F g−1) was achieved at 5 mV s−1 within the potential window of 2.0 V.24 S. Maiti et al. have prepared MnO2 hollow spheres by using a solvothermal method.28 The cyclic capacitance retention of the asymmetric device was around 11% after 5000 cycles at 2 A g−1 by using 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIMBF4) as an ionic liquid electrolyte.28 The above studies indicate that it requires a rigorous development of both the morphological features of MnO2 and electrolytes to significantly enhance energy density, cycle life, and charge–discharge rates while maintaining the highest degree of safety available. They also indicate that electrode material tailoring can maximize the utilization of the ionic liquid electrolyte. This can be achieved by utilizing the suitable morphological features of MnO2 with a compatible ionic liquid electrolyte.
Manganese dioxide (MnO2) is also a very promising anode material for lithium ion batteries due to its much higher theoretical capacity (1233 mA h g−1) and environmental benignity in comparison to other transition metal oxides. MnO2 nanorods,29 nanowires,30 hollow urchin,31 hollow microspheres,32 nanotubes,33 and nanosheets34 have already been reported as anode materials exhibiting a significant enhancement in lithium-ion storage. A few problems still need to be addressed such as volume expansion, low coulombic efficiency in the 1st cycle and fast capacity decays during cycling.30–32,35 Additionally, the low volumetric energy densities due to the low tap density and complicated fabrication methods of these nanomaterials are also the main hindrances towards their optimum utility.36
Numerous reports also indicated that the electrochemical performance of MnO2 is strongly influenced by a variety of factors including preparation conditions, particle size, morphology, surface states, defects and crystallographic structure.3,37–39 So the major parameters such as high capacitance, cycling stability and rate capability can be enhanced by tailoring its morphological features. From this perspective, 3D hierarchical architectures, assembled from 2D nanosheets, can be a fascinating approach to enhance the lithium storage properties. In particular, 2D nanosheets can significantly shorten the migration distance of lithium ions, and the micro-sized assemblies can obtain a high tap density and guarantee architectural integrity during cycling, resulting in improved lithium storage properties.
Capacitance properties are due to the intercalation/deintercalation of protons or cations in MnO2; 3D hierarchical structures, which possess sufficient gaps to accommodate these ions, are also expected to be useful for supercapacitor application.3,37–39 Such a structure will provide in-depth penetration of the electrolyte due to its mesoporosity, shorter pathways and higher specific surface area (SSA).40 In our previous paper, microwave synthesis of 3D hierarchical MnO2 microspheres (3DHM-MnO2) and their application as an anode material for solid state asymmetric supercapacitor devices were already reported.11 We have already synthesized various porous inorganic nanostructures by using this environmentally benign method.10,11,41–47 In this article, we further explore their electrochemical performance as an electrode material for asymmetric supercapacitor devices (ionic electrolyte based) and lithium ion batteries. The main objective is to fabricate high energy density supercapacitors and lithium-ion batteries.
Herein we report the successful fabrication of an asymmetric supercapacitor (ASC) device based on 3DHM-MnO2 as the cathode and activated carbon as the anode material using 0.5 M 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIMBF4)/N,N-dimethyl formamide (DMF, C3H7NO) as the ionic electrolyte. DMF is used to reduce the viscosity of the ionic liquid which will result in an enhancement of the cycling stability of the device. The asymmetric device operates successfully within a potential window of 3.0 V and exhibits an outstanding energy density of 105 W h kg−1 at a power density of 1494 W kg−1 with good cycling stability (20% loss after 6000 cycles). The obtained energy density and cycling stability are much higher than those of most of the reported asymmetric devices (ionic electrolyte based).9,28 The 3D hierarchical MnO2 microstructure provides deep penetration of the ionic electrolyte (EMIMBF4/DMF) due to its higher SSA, mesoporous nature and higher pore volume.
MnO2 microspheres also exhibit remarkable performance as an anode material for lithium ion batteries. They deliver outstanding Li ion storage performance with a discharge capacity of 715 mA h g−1 even after 200 cycles at a current density of 300 mA g−1. The discharge capacity retention of around 78% after 200 cycles at 300 mA g−1 is the highest amongst those of most of the reported anode materials based on MnO2.29–34 The enhanced electrochemical performance in comparison to the reported literature can be attributed to their mesoporous ultrathin nanosheet building block structure, high specific surface area (184.32 m2 g−1), desirable pore distribution and large pore volume (0.416 cm3 g−1).
Experimental
Synthesis of 3D hierarchical MnO2 microspheres
Material synthesis was carried out according to our previous report.11 More briefly, MnO2 microspheres were prepared via an ultrafast and scalable microwave-assisted method. In a typical synthesis 6 mmol of MnCl2·4H2O and 4 mmol of KMnO4 were dissolved in 100 mL of de-ionized (DI) water under constant magnetic stirring for 60 minutes. The above-prepared mixture was heated at 100 °C under microwave irradiation in a SINEO MAS-II microwave reactor at 700 W for 10 minutes. After that the mixture was allowed to cool down naturally and stand for 24 h. The resulting product was washed three times using DI water and ethanol to eliminate any possible contaminants, and then it was dried at 90 °C in an air environment for 12 h.Materials characterization
The phase analysis of the as-prepared material was performed with the help of X-ray diffractometry (Philips X'Pert Pro MPD) with Cu Kα radiation at 40 kV and 40 mA (2θ ranging from 10° to 80°). The morphological features of the as-synthesized material were analyzed using field emission scanning electron microscopy (FESEMS, Hitachi S-4800) and transmission electron microscopy (TEM, JEM-2100F).The surface elemental states of the as-synthesized material were investigated by X-ray photoelectron spectroscopy (XPS) with a monochromatic Al Kα source. The specific surface area and pore size distribution of the samples were characterized by N2 adsorption analyses at 77 K. A COULTER SA 3100 apparatus was employed to perform N2 adsorption isotherms. The Brunauer–Emmett–Teller (BET) method was used to evaluate the specific surface area. The pore size distribution was calculated by using the Barrett–Joyner–Halenda (BJH) model.
Electrochemical measurements
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0. Cyclic voltammetry (CV) and galvanostatic charge/discharge measurements were conducted by using a CHI660D electrochemical workstation and a CT2001A LAND battery testing setup respectively.
:1.0. Cyclic voltammetry (CV) and galvanostatic charge/discharge measurements were conducted by using a CHI660D electrochemical workstation and a CT2001A LAND battery testing setup respectively.
In the case of the three-electrode configuration, the specific capacitance was calculated from CV curves using eqn (1).10
 | (1) |
In the case of the two-electrode asymmetric configuration, the specific capacitance was calculated from CP curves using eqn (2).48,49
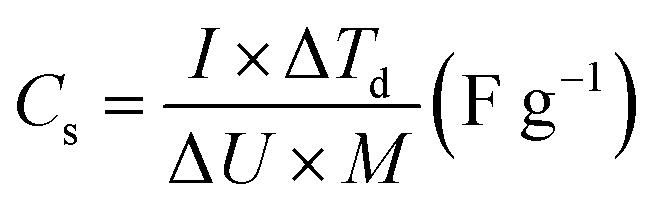 | (2) |
The energy density and power density of the asymmetric device was calculated from the CP curves according to eqn (3) and (4) respectively.48,49
 | (3) |
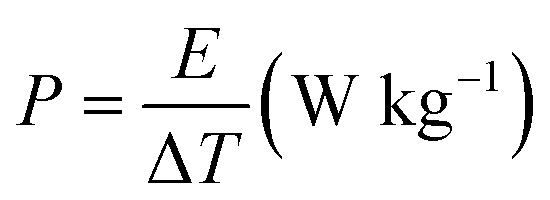 | (4) |
:10:10 wt%, respectively. The above mixture was dissolved in N-methyl-2-pyrrolidone (NMP) to make a slurry. The slurry was homogenously coated on a copper foil using the doctor-blade method. The slurry coated copper foil was dried at 120 °C overnight in a vacuum environment. Prior to its use as an anode for Li-ion batteries, all electrodes were pressed under 10 MPa pressure for 30 seconds. Lithium foil was used as the counter electrode. CR2025 coin cells were used to investigate the electrochemical performance of the anode material. All cells were assembled in an argon-filled glove box. 1 M LiPF6 dissolved in ethyl carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC) (1:1:1 v/v) was used as an electrolyte. Cyclic voltammetry (CV), AC impedance spectroscopy (EIS) and charge–discharge (CD) measurements were carried out on a CHI660D electrochemical workstation and a CT2001A LAND battery testing system respectively.
Results and discussion
Structural and morphological characterizations
The FESEM image (Fig. 1a) reveals a well-defined 3D hierarchical microsphere structure of MnO2. It also indicates the uniform distribution of microspheres with an average diameter of around 250 nm (Fig. 1a). Each microsphere is assembled from numerous ultrathin nanosheets with sufficient void space between them (Fig. 1b). The loosely packed structure with open space is also obvious from the edges of microspheres (Fig. 1b). The lateral size of the nanosheets is much larger than their thickness which results in their bending and curling (Fig. 1b). Numerous pin holes are also obvious on the surface of the nanosheets which indicates their mesoporous nature (Fig. 1b). During synthesis, the aggregation of small nanoparticles is responsible for the formation of these mesopores on the surface of nanosheets. The HRTEM image (Fig. 1c) indicates that the lattice interlayer spacing is around 0.244 nm, which corresponds to the interplanar spacing of the (211) plane of MnO2. The HRTEM result is in accordance with the XRD finding (Fig. 1d), which also indicates that (211) is the major dominant phase of pure MnO2 according to JCPDS card # 44-0141.50 The nanocrystalline nature of MnO2 is also well evident from the broad X-ray diffraction peaks (Fig. 1d). | ||
| Fig. 1 (a) FESEM, (b) TEM, and (c) HRTEM images; (d) XRD and (e) XPS spectra, and (f) the N2 adsorption–desorption isotherm of 3D hierarchical MnO2 microspheres. | ||
Moreover, the XRD pattern (Fig. 1d) also confirms the absence of any other phase or impurities in the as-synthesized product. According to Scherrer's formula (eqn (5)), the average crystallite size of the as-synthesized MnO2 microspheres is calculated to be 8.8 nm.
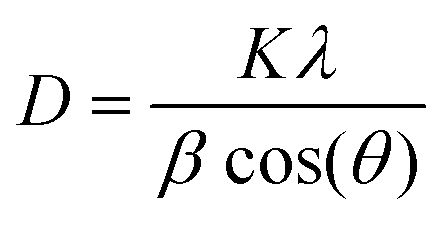 | (5) |
The XPS survey spectrum (Fig. 1e) indicates the high purity of MnO2 with the presence of only C1s (as reference), O and Mn. It also indicates the absence of any detectable impurity in the as-synthesized material. The Mn 2p spectrum (inset of Fig. 1e) shows well-defined valence states at binding energies of 641.95 eV (Mn 2p3/2) and 653.55 eV (Mn 2p1/2). Peak energy separation for Mn2P (inset of Fig. 1e) is 11.6 eV which is the typical characteristic of MnO2.39 Whereas its O 1s spectrum (inset of Fig. 1e) indicates binding energy peaks at 529.55 eV and 531.05 eV, which corresponds to lattice oxygen and surface adsorbed oxygen respectively.51
The BET surface area and porosity of MnO2 microspheres are determined by analyzing their N2 adsorption–desorption isotherm (Fig. 1f). A well-defined hysteresis loop is evident in the relative pressure range of (0.45–1.0) (Fig. 1f). The shape of the isotherm can be ascribed to typical Langmuir type IV, which is indicative of the distinctive mesoporous characteristics of materials.10 The BET surface area of MnO2 microspheres is determined to be 184.32 m2 g−1. The pore size distribution and pore volume were calculated based on the BJH method by using the desorption part of the isotherm. It can be seen that pore size distribution is mainly centered in the mesoporous region (2–50 nm) (inset of Fig. 1f) with an average pore size of 5.158 nm. According to the BJH method, the pore volume of MnO2 microspheres is found to be 0.416 cm3 g−1. MnO2 microspheres possess a much higher specific surface area and pore volume than most of the previous reports.52–56 The high specific surface area is extremely suitable for electrochemical application by providing an enhanced interfacial contact area between the electrolyte and active material. In addition, the large pore volume provides in-depth penetration of the electrolyte at high rates within the pores which also ameliorates their electrochemical performance. So MnO2 microspheres will provide tremendous electrochemical performance owing to their ultrathin nanosheet building blocks, very high specific surface area, narrow pore size distribution and large pore volume.
Electrochemical analysis
To determine the mass balance between the cathode (MnO2) and anode (activated carbon (AC)), the specific capacitances of both electrodes were measured in a three-electrode configuration by using the cyclic voltammetry (CV) method (eqn (1)). The CV curves of 3DHM-MnO2 (Fig. 2a) demonstrate a nearly rectangular shape within the potential window −1.3 to 0.8 V for various scan rates (5 to 100 mV s−1). The slightly distorted rectangular shape is indicative of the pseudocapacitive behavior of 3DHM-MnO2 within a larger potential window (2.1 V).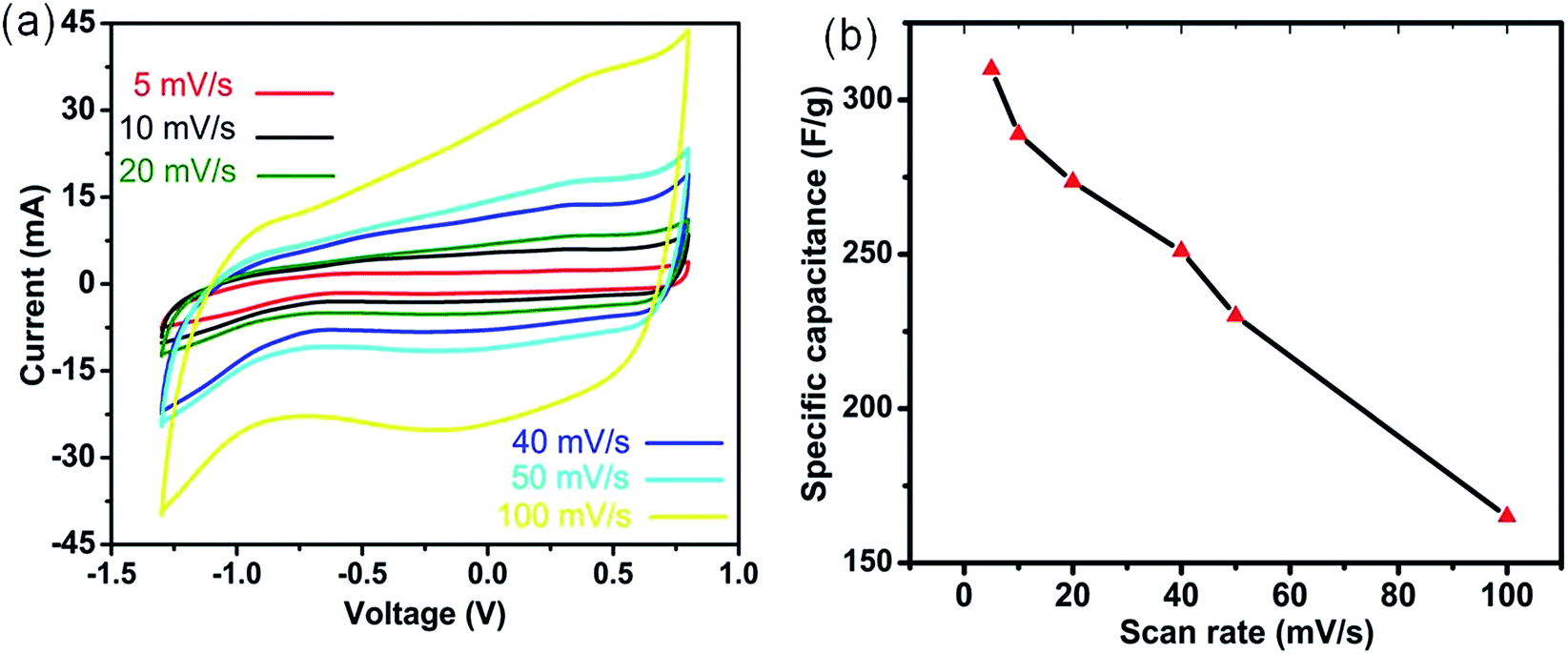 | ||
| Fig. 2 (a) CV curves and (b) specific capacitance of 3DH-MnO2 at various scan rates. | ||
The operational voltage window of MnO2 is double that of its window in aqueous electrolytes.57 The highly symmetric anodic and cathodic sweeps of CV scans (Fig. 2a) are indicative of the better reversibility of the electrode material in (EMIMBF4/DMF) electrolyte. It is also shown that the area under the curve of the CV loop (Fig. 2a) increases with the increase of the scan rate which specifies its high reactivity and great kinetics in EMIMBF4/DMF. The much larger potential window and high rate capability are desirable to enhance the specific energy of the supercapacitor. 3DHM-MnO2 microspheres exhibit a much higher specific capacitance of 310.5 F g−1 at 5 mV s−1 (Fig. 2b).
The electrochemical performance of 3DH-MnO2 microspheres is also investigated in various other ionic electrolytes such as BMIMOtf/DMF and BMIMPF6/DMF. The specific capacitances of 3DH-MnO2 microspheres as a function of the scan rate are also calculated in various ionic electrolytes as shown in Fig. S1.† It can be inferred from Fig. S1† that the specific capacitance of 3DH-MnO2 microspheres is much higher by using EMIMBF4/DMF as the ionic electrolyte. Secondly by using EMIMBF4/DMF as the ionic electrolyte, the capacitance retention at a higher scan rate (100 mV s−1) is much higher (53.2%) than the others (Fig. S1†). So we chose EMIMBF4/DMF as a more compatible optimal ionic electrolyte for enhanced electrochemical performance of 3DH-MnO2 microspheres. The obtained capacitance is much higher than those of the previously reported MnO2 based ionic electrolyte supercapacitors, such as in 1-ethyl-3-methylimidazolium-dicyanamide (72 F g−1 at 5 mV s−1),24 doped butylmethylpyrrolidinium-dicyanamide (125 F g−1 at 5 mV s−1),26 and butylmethylpyrrolidinium-dicyanamide (90 F g−1 at 5 mV s−1).26 The main reason for the higher specific capacitance is the much higher specific surface area (184 m2 g−1) and unique morphological features of 3DHM-MnO2 compared to previous reports.24,26 It is also much higher than its reported double layer capacitance in ionic liquids,58 which also implies that the enhanced specific capacitance is due to the pseudocapacitive behavior of the electrode material. Even at a much higher scan rate (100 mV s−1), a capacitance retention of around 53.2% is still observed (Fig. 2b). 3DHM-MnO2 microspheres exhibit much higher specific capacitance than most of the reported materials for asymmetric devices based on ionic liquid electrolytes.9,28 This can be ascribed to the excellent compatibility of ionic liquid (EMIMBF4/DMF) with the morphological and structural features of MnO2. The low viscosity and resistance of (EMIMBF4/DMF) facilitate much faster kinetics even at high rates. It is also important to understand the charge storage phenomena of MnO2 in (EMIMBF4/DMF). The charge storage mechanism in ionic liquids is totally different from that in aqueous electrolytes.24 In the case of aqueous electrolytes, the charge transfer mechanism is controlled by the reversible uptake of small cations (K+, Na+, Li+ or H+) and electrons.59 The (EMIMBF4/DMF) consists of EMIM+ cations (182 Å3) and BF4− anions (79 Å3), which indicates that the charge insertion/deinsertion is basically governed by anions.28 Because the anionic pore size is more compatible with the average pore size of MnO2 microspheres, it is very difficult for the cations to participate in the charge insertion/deinsertion process in MnO2 due to its much higher pore volume. The EMIM+ cations were just adsorbed on the electrode surface and did not penetrate into the oxide. So the charge storage mechanism of MnO2 in ILs is proposed to be dominated by an anion insertion/deinsertion process that compensated the Mn valence state change (between trivalent and tetravalent), because the cations used (such as imidazolium) were too massive to get involved in the pseudocapacitive reaction. Among the anions studied, BF4− anions, due to their lower pore size, were the most effective one that can reversibly travel into/from the tunnels between the MnO6 octahedral units upon charging/discharging (eqn (6)), contributing to the highest measured pseudocapacitance.59
| MnO2−x(BF−4)2x + 2xe− ↔ MnO2−x + 2x(BF−4) | (6) |
This faradaic redox reaction is the cause of the pseudocapacitive behavior of the Mn oxide electrode in EMIMBF4/DMF ionic liquid electrolyte. The mesopores and high specific surface area of the ultrathin nanosheet building blocks are the main contributing factors towards the enhanced performance even at a much higher scan rate. The enhanced specific capacitance along with good rate capability in EMIMBF4/DMF indicates its potential for high performance asymmetric devices. It also indicates that the EMIMBF4/DMF ionic liquid is highly compatible with 3DHM-MnO2 microspheres to deliver high reversibility and enhanced capacitance. The higher rate capability is due to the much faster penetration of the ionic liquid due to enhanced active sites provided by the mesoporous nanosheet structure of hierarchical microspheres. It is also important to calculate the capacitance value and operational voltage window of activated carbon in (EMIMBF4/DMF). CV curves (Fig. S2a†) demonstrate the well-defined electrical double layer capacitive behavior of AC within the potential window of 2.8 V. The ionic liquid (EMIMBF4/DMF) provides a much larger operational voltage window for AC in comparison to aqueous electrolytes (1.0 V).10 CV curves (Fig. S2a†) also indicate the higher degree of reversibility and rate capability. The calculated specific capacitance of AC at 10 mV s−1 is 227.75 F g−1 (Fig. S2b†).
Prior to fabrication of the asymmetric device the appropriate mass balance between the cathode (3DH-MnO2) and anode (AC) is calculated by balancing the charge storage according to eqn (7). The appropriate mass balance between the cathode and anode at 10 mV s−1 is calculated to be 0.95:1.0.
 | (7) |
All asymmetric devices are fabricated according to the mass balance between the cathode and anode. CP measurements are used to calculate the specific capacitance and cycling stability at various current densities. CP curves (Fig. 3a) indicate the well-defined capacitive behavior of the asymmetric device within the potential window of 3.0 V. CP curves show a non-linear increase and decrease of potential with time which indicates the existence of pseudocapacitive behaviour of the asymmetric device (Fig. 3a). The discharge profile can be divided into two parts, the resistive and the capacitive. The voltage drop at the start of the discharge cycle represents the resistive part (IR drop) which arises due to the equivalent series resistance of the device.
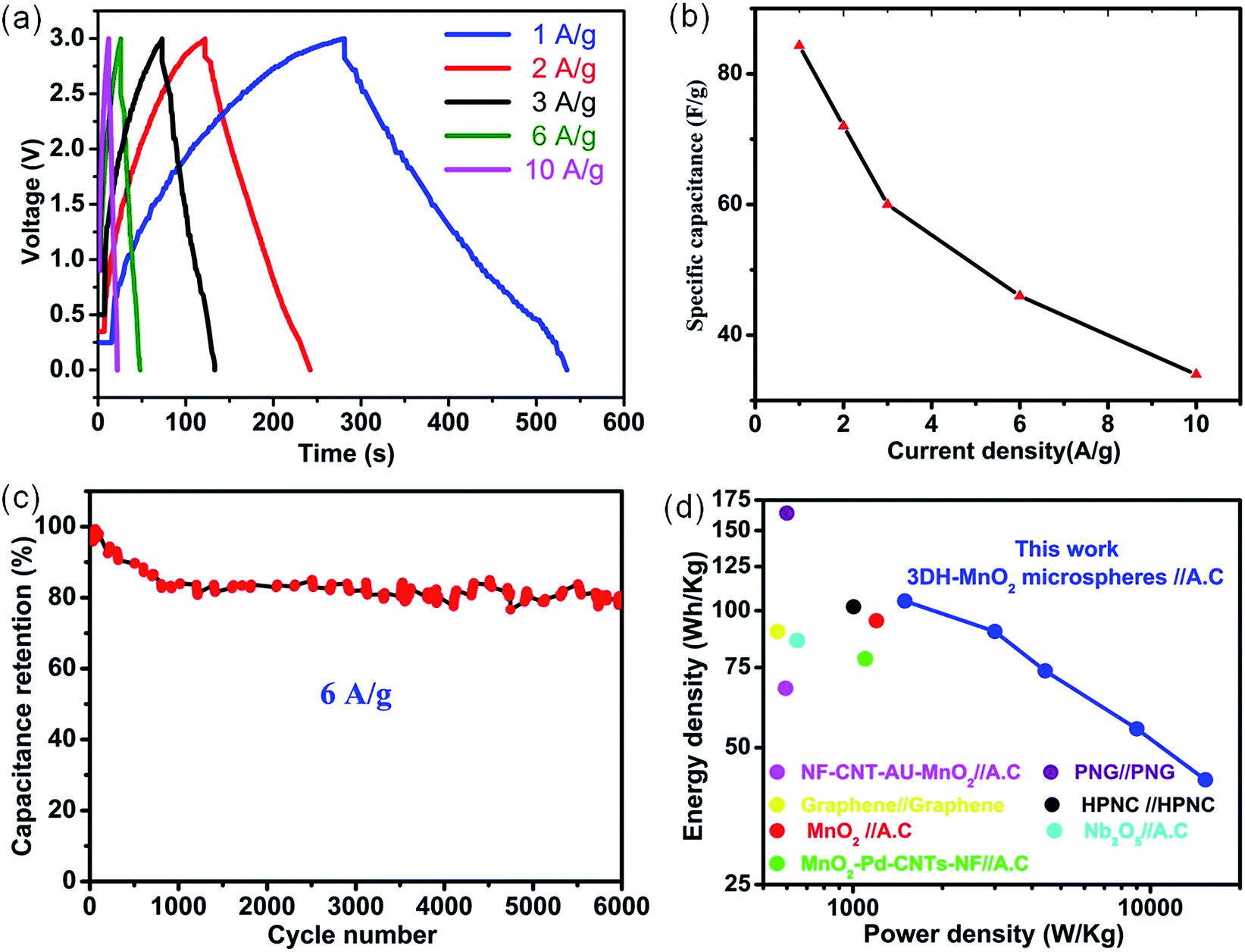 | ||
| Fig. 3 (a) CP curves of the asymmetric devices collected at different current densities. (b) Specific capacitance of the asymmetric device collected from CP curves as a function of current density. (c) Cycling performance of the asymmetric device at the current density of 6 A g−1 for 6000 cycles. (d) Ragone plot of the asymmetric device, the values reported for other devices are added for comparison.8,9,16,28,60–62 | ||
To avoid any error caused by the potential drop (IR drop) during the discharge cycle, both the specific capacitance and energy density of asymmetric devices are calculated by deducting the potential drop contributed by the resistive part during the discharge process at various current densities. CP curves are highly symmetric which indicates the higher degree of reversibility within this potential window. The calculated specific capacitances of ASC devices from CP curves are shown in Fig. 3b. It indicates that the specific capacitance of the asymmetric device based on the total mass of electrode materials is 84.3 F g−1 at 1 A g−1 which is much higher than many previous reports.9,28 The higher specific capacitance is due to the much higher specific surface area in comparison to previous reports. Moreover, the asymmetric device demonstrates a good capacitance retention of 40% at a current density of 10 A g−1 (Fig. 3b). The specific capacitance of the asymmetric device is also calculated by using the CV method (Fig. S3†). It shows a specific capacitance of 91.3 F g−1 at 5 mV s−1 (Fig. S3†), while at a higher scan rate the diffusion of ions occurs mainly in the outer regions of the pores which results in the decrease of specific capacitance (Fig. S3†).63 It is also worthwhile to mention that the specific capacitance values obtained from charge–discharge measurements are almost consistent with those of CV results. The cycling stability is the vital parameter to be determined for long term application of the asymmetric device. The asymmetric device shows (Fig. 3c) remarkable cycling stability with a capacitance retention of 80% after 6000 cycles at 6 A g−1. CD curves for a few cycles at a current density of 6 A g−1 are also presented in Fig. S4.† SEM measurements were carried out to determine the structural changes as a result of the long term cycling test (Fig. S5†). The basic morphology of the MnO2 is overall well preserved (Fig. S5†). This demonstrates that 3DH-MnO2 microspheres possess the essential characteristics for high-performance supercapacitor applications, and meet the requirements of long-term stability. The Ragone plot (Fig. 3d) is used to summarize the electrochemical performance of the ASC device. The ASC device demonstrates an energy density of 105 W h kg−1 and a power density of 1494 W kg−1 at 1 A g−1. Even at a much higher current density of 10 A g−1, the device delivers a much higher power density of 15300 W kg−1 with a sustainable energy density of 42.5 W h kg−1. This also indicates that both energy density and power density are higher than those of many reported devices.8,9,16,28,60–62 A detailed comparison is also made in tabulated form as shown in Table S1.† It can also be inferred from Table S1,† that even at a higher current density of 1 A g−1, the energy density is still higher than the maximum attained energy density of many reported asymmetric devices at low current density.8,9,16,28,60 The significant difference is the better capacity retention and cycling stability even at higher current density in comparison with previous literature. The enhanced asymmetric device performance can be ascribed to the higher compatibility between the (EMIMBF4/DMF) ionic electrolyte and morphological features of 3DHM-MnO2 microspheres. The ultrathin nanosheet building blocks provide a much shorter path for ion transport which is responsible for fast kinetics. The large pore volume (0.416 cm3 g−1) provides facile channels for the transportation of ions. The mesoporous structure and large surface area (184.32 m2 g−1) enhance the contact area between the electrolyte and electrode. The lower viscosity and resistance of the ionic electrolyte are also responsible for the much higher rate capability and capacity retention during cycling operation.
The CV scan was used to investigate the lithium storage phenomena of 3DHM-MnO2 microspheres as an anode material within the potential window (3.0 V vs. Li/Li+). Fig. S6† presents the first three CV cycles at 0.1 mV s−1. It is obvious from the CV pattern that, during the 1st cycle, cathodic peaks appear at 1.42, 1.12 and 0.163 V which correspond to the reduction of manganese oxide to metallic manganese (Mn4+ to Mn0), and the formation of Li2O and the solid electrolyte interphase (SEI) layer. It also indicates that re-oxidation takes place in one step at 1.12 V. After the 1st cycle, the cathodic peak at 1.42 V disappears, whereas the peak at 0.163 shifts to 0.289 V, which is an indication of irreversible textural changes in the electrode.30 But the third cycle is closely similar to the 2nd cycle, which indicates that the lithiation and de-lithiation phenomena are highly reversible after the 1st cycle. The reaction mechanism can be described as follows; during discharge, MnO2 is transformed to Mn, and Li+ is converted to Li2O.
| MnO2 + 4Li → 2Li2O + Mn |
During charging, Mn can facilitate the decomposition of Li2O.
| 2Li2O + Mn → MnO2 + 4Li |
The voltage–capacity curves of the as-prepared 3DHM-MnO2 microspheres tested at 100 mA g−1 are presented in Fig. 4a. Obvious plateaus are observed at 1.0 and 0.4 V in the charge and discharge process respectively which are consistent with the previous reports.32 During the 1st cycle, a discharge capacity of 1345 mA h g−1 is obtained but it reduces to 1190 mA h g−1 in the 2nd cycle. The 1st cycle discharge capacity is much higher than the theoretical capacity of MnO2 (1232 mA h g−1). This can be ascribed to the formation of a SEI layer, interfacial storage, etc. at the electrode surface during the 1st discharge process.64,65 The irreversible capacity loss (20%) during the 1st discharge and charge cycles is also much lower than those of many previously reported MnO2 based electrodes.29,30,32–35 Most importantly this also demonstrates remarkable cycling behavior at 100 mA g−1 with a capacity retention of around 87% @ the 2nd cycle after 50 cycles. It can also be observed that after a few cycles (Fig. 4b), the specific capacity almost remains constant. The discharge cyclic capacity retention is much higher than many previous reports.30–32,35,64 Even at a higher current density of 300 mA g−1, the electrode shows (Fig. S7†) an excellent cyclic response with a capacity retention of around 78% @ the 2nd cycle after 200 cycles. Except for the initial few cycles, the coulombic efficiency of the electrode remains around 99% (Fig. 4b and S7†), which is indicative of highly reversible behavior. The irreversible reaction and formation of the SEI are the main contributing factors towards the irreversible capacity drops in the first few cycles.33 It can also be observed (Fig. 4b and S7†) that the capacity drops initially in the first few cycles and then almost remains stable in the subsequent cycles. This kind of trend can be attributed to morphological features which provide an increased number of vacancies and grain boundaries for lithium ion storage. Moreover in certain regions (Fig. 4b and S7†) the discharge capacity shows an increasing trend which can be attributed to the activation phenomena of ultrathin nanosheets upon cycling.66,67 The rate capability of 3DHM-MnO2 was also evaluated at various current densities (Fig. S8†).
 | ||
| Fig. 4 (a) Charge–discharge potential profiles measured at 100 mA g−1. (b) Charge–discharge capacity and coulombic efficiency as a function of cycle numbers at 100 mA g−1 (plotted from the 1st cycle). | ||
The discharge capacity of the 3DHM-MnO2 electrode at 100 mA g−1 is as high as 1190 mA h g−1 (2nd cycle) and it can be maintained at 990 mA h g−1 after a few cycles. Even at high current densities of 500 & 800 mA g−1, the 3DHM-MnO2 can still deliver discharge capacities of 680 & 590 mA h g−1 respectively (Fig. S8†). The specific capacity restores again to 950 mA h g−1 when the current density is switched back to 100 mA g−1 after 40 cycles at different applied current densities (Fig. S5†). The capacity values remain stable at various current densities for a specific number of cycles which indicates its improved rate capability (Fig. S8†). The structural changes in the electrode material after cycling operation were investigated by using SEM measurements (Fig. S9†). 3DH-MnO2 microspheres exhibit well preserved morphological features as a result of continuous cycling operation (Fig. S9†). This reflects that 3DH-MnO2 microspheres possess the essential characteristics for Li-ion battery applications. EIS was also carried out to understand the reasons for the enhanced lithium ion storage capability of the electrode material. The impedance spectrum consists of a semicircle in the high frequency region and a declining line deviating 90° in the low frequency region. The intercept of the semicircle on the x-axis corresponds to the ohmic electrolyte resistance (Rs) and charge transfer resistance (Rct) respectively. The values of Rs and Rct as indicated in Fig. S10† are 5.28 Ω and 78 Ω respectively. These relatively lower values of Rs and Rct are also responsible for enhanced electrochemical performance. The inclined line in the low frequency region corresponds to the Warburg impedance associated with lithium-ion diffusion.68 The mesoporous porosity, high specific surface area, ultrathin nanosheet structure and low charge transfer resistance are the main contributing factors towards the enhanced lithium storage performance and result in improved capacity retention and cycling stability even at higher current density. The outstanding electrochemical performance as an electrode material for both Li-ion batteries and supercapacitors enhances its potential to be used for industrial scale applications.
Conclusions
3D hierarchical MnO2 microspheres demonstrate excellent electrochemical performance for both supercapacitor and Li-ion battery applications. The remarkable performance could be ascribed to their distinctive morphological features which consist of ultrathin nanosheet building blocks: (i) the high energy density of the asymmetric supercapacitor is due to the increased compatibility between the ionic electrolyte and the mesoporous porosity of MnO2 microspheres, (ii) the large surface-to-volume ratio provides a shorter pathway for both ionic electrolyte and lithium ion transport, (iii) the mesoporous porosity with large pore volume provides facile lithium ion diffusion to the interior of electroactive materials, (iv) the increased number of reactive sites as a result of the much higher specific surface area (184.32 m2 g−1), and (v) the long cycle life is due to the hierarchical structure which provides stress–strain relaxation during the lithiation–delithiation reaction. 3D hierarchical MnO2 microspheres are proved to be a prospective material for high performance ionic electrolyte based supercapacitor and lithium ion battery applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21371023, 50972017) and the Research Fund for the Doctoral Program of Higher Education of China (20101101110026).References
- L. Yuan, X.-H. Lu, X. Xiao, T. Zhai, J. Dai, F. Zhang, B. Hu, X. Wang, L. Gong, J. Chen, C. Hu, Y. Tong, J. Zhou and Z. L. Wang, ACS Nano, 2012, 6, 656–661 CrossRef CAS PubMed.
- L. Yuan, B. Yao, B. Hu, K. Huo, W. Chen and J. Zhou, Energy Environ. Sci., 2013, 6, 470–476 CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 CAS.
- J. Hou, C. Cao, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556–2564 CrossRef CAS PubMed.
- X. Zhang, D. Zhao, Y. Zhao, P. Tang, Y. Shen, C. Xu, H. Li and Y. Xiao, J. Mater. Chem. A, 2013, 1, 3706–3712 CAS.
- S. Khalid, C. Cao, L. Wang and Y. Zhu, Sci. Rep., 2016, 6, 22699 CrossRef CAS PubMed.
- S. Khalid, C. Cao, L. Wang, Y. Zhu and Y. Wu, RSC Adv., 2016, 6, 70292–70302 RSC.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 CAS.
- Y. Zhao, W. Ran, J. He, Y. Huang, Z. Liu, W. Liu, Y. Tang, L. Zhang, D. Gao and F. Gao, Small, 2015, 11, 1310–1319 CrossRef CAS PubMed.
- T. Sato, T. Maruo, S. Marukane and K. Takagi, J. Power Sources, 2004, 138, 253–261 CrossRef CAS.
- K. Hayashi, Y. Nemoto, K. Akuto and Y. Sakurai, J. Power Sources, 2005, 146, 689–692 CrossRef CAS.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- G. A. Tiruye and R. Marcilla, in Applications of Ionic Liquids in Polymer Science and Technology, ed. D. Mecerreyes, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 199–229, DOI:10.1007/978-3-662-44903-5_8.
- A. Balducci, U. Bardi, S. Caporali, M. Mastragostino and F. Soavi, Electrochem. Commun., 2004, 6, 566–570 CrossRef CAS.
- D. Rochefort and A.-L. Pont, Electrochem. Commun., 2006, 8, 1539–1543 CrossRef CAS.
- W. Sugimoto, H. Iwata, Y. Yasunaga, Y. Murakami and Y. Takasu, Angew. Chem., Int. Ed., 2003, 42, 4092–4096 CrossRef CAS PubMed.
- C. Yuan, L. Chen, B. Gao, L. Su and X. Zhang, J. Mater. Chem., 2009, 19, 246–252 RSC.
- R. Liu and S. B. Lee, J. Am. Chem. Soc., 2008, 130, 2942–2943 CrossRef CAS PubMed.
- X. Li and B. Wei, Nano Energy, 2012, 1, 479–487 CrossRef CAS.
- J.-K. Chang, M.-T. Lee, C.-W. Cheng, W.-T. Tsai, M.-J. Deng, Y.-C. Hsieh and I. W. Sun, J. Mater. Chem., 2009, 19, 3732–3738 RSC.
- L. Y. Chen, J. L. Kang, Y. Hou, P. Liu, T. Fujita, A. Hirata and M. W. Chen, J. Mater. Chem. A, 2013, 1, 9202–9207 CAS.
- Y.-S. Li, I. W. Sun, J.-K. Chang, C.-J. Su and M.-T. Lee, J. Mater. Chem., 2012, 22, 6274–6279 RSC.
- C.-H. Yang, I. W. Sun, C.-T. Hsieh, T.-Y. Wu, C.-Y. Su, Y.-S. Li and J.-K. Chang, J. Mater. Chem. A, 2016, 4, 4015–4018 CAS.
- S. Maiti, A. Pramanik and S. Mahanty, RSC Adv., 2015, 5, 41617–41626 RSC.
- X. Fang, X. Lu, X. Guo, Y. Mao, Y.-S. Hu, J. Wang, Z. Wang, F. Wu, H. Liu and L. Chen, Electrochem. Commun., 2010, 12, 1520–1523 CrossRef CAS.
- M.-S. Wu, P.-C. J. Chiang, J.-T. Lee and J.-C. Lin, J. Phys. Chem. B, 2005, 109, 23279–23284 CrossRef CAS PubMed.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404–6410 CrossRef CAS PubMed.
- J. Zhao, Z. Tao, J. Liang and J. Chen, Cryst. Growth Des., 2008, 8, 2799–2805 CAS.
- L. Li, C. Nan, J. Lu, Q. Peng and Y. Li, Chem. Commun., 2012, 48, 6945–6947 RSC.
- M. Kundu, C. C. A. Ng, D. Y. Petrovykh and L. Liu, Chem. Commun., 2013, 49, 8459–8461 RSC.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002–1006 CrossRef CAS PubMed.
- Z. Chen, P.-C. Hsu, J. Lopez, Y. Li, J. W. F. To, N. Liu, C. Wang, S. C. Andrews, J. Liu, Y. Cui and Z. Bao, Nat. Energy, 2016, 1, 15009 CrossRef CAS.
- C. Yuan, L. Hou, L. Yang, D. Li, L. Shen, F. Zhang and X. Zhang, J. Mater. Chem., 2011, 21, 16035–16041 RSC.
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915–2921 CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y.-M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- Y. Zhu, H. Guo, Y. Wu, C. Cao, S. Tao and Z. Wu, J. Mater. Chem. A, 2014, 2, 7904–7911 CAS.
- Y. Zhu, C. Cao, J. Zhang and X. Xu, J. Mater. Chem. A, 2015, 3, 9556–9564 CAS.
- J. Zhang, Y. Zhu, C. Cao and F. K. Butt, RSC Adv., 2015, 5, 58568–58573 RSC.
- Y. Zhu, H. Guo, H. Zhai and C. Cao, ACS Appl. Mater. Interfaces, 2015, 7, 2745–2753 CAS.
- Y. Zhu and C. Cao, Electrochim. Acta, 2015, 176, 141–148 CrossRef CAS.
- S. Khalid, C. Cao, A. Ahmad, L. Wang, M. Tanveer, I. Aslam, M. Tahir, F. Idrees and Y. Zhu, RSC Adv., 2015, 5, 33146–33154 RSC.
- Y. Zhu, C. Cao, S. Tao, W. Chu, Z. Wu and Y. Li, Sci. Rep., 2014, 4, 5787 CrossRef CAS PubMed.
- F. Luan, G. Wang, Y. Ling, X. Lu, H. Wang, Y. Tong, X.-X. Liu and Y. Li, Nanoscale, 2013, 5, 7984–7990 RSC.
- H. Wang, H. Yi, C. Zhu, X. Wang and H. Jin Fan, Nano Energy, 2015, 13, 658–669 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- G. Zhao, J. Li, L. Jiang, H. Dong, X. Wang and W. Hu, Chem. Sci., 2012, 3, 433–437 RSC.
- J. Yue, X. Gu, L. Chen, N. Wang, X. Jiang, H. Xu, J. Yang and Y. Qian, J. Mater. Chem. A, 2014, 2, 17421–17426 CAS.
- P. Pal, S. K. Pahari, A. K. Giri, S. Pal, H. C. Bajaj and A. B. Panda, J. Mater. Chem. A, 2013, 1, 10251–10258 CAS.
- L. Benhaddad, L. Makhloufi, B. Messaoudi, K. Rahmouni and H. Takenouti, ACS Appl. Mater. Interfaces, 2009, 1, 424–432 CAS.
- C. Yu, G. Li, L. Wei, Q. Fan, Q. Shu and J. C. Yu, Catal. Today, 2014, 224, 154–162 CrossRef CAS.
- M. Huang, Y. Zhang, F. Li, L. Zhang, R. S. Ruoff, Z. Wen and Q. Liu, Sci. Rep., 2014, 4, 3878 CrossRef PubMed.
- A. Lewandowski and M. Galiński, J. Phys. Chem. Solids, 2004, 65, 281–286 CrossRef CAS.
- J.-K. Chang, M.-T. Lee, W.-T. Tsai, M.-J. Deng and I. W. Sun, Chem. Mater., 2009, 21, 2688–2695 CrossRef CAS.
- D. Zhao, Y. Zhao, X. Zhang, C. Xu, Y. Peng, H. Li and Z. Yang, Mater. Lett., 2013, 107, 115–118 CrossRef CAS.
- D. Liu, C. Fu, N. Zhang, Y. Li, H. Zhou and Y. Kuang, J. Solid State Electrochem., 2017, 21, 759–766 CrossRef CAS.
- F. Idrees, J. Hou, C. Cao, F. K. Butt, I. Shakir, M. Tahir and F. Idrees, Electrochim. Acta, 2016, 216, 332–338 CrossRef CAS.
- H. Pang, B. Zhang, J. Du, J. Chen, J. Zhang and S. Li, RSC Adv., 2012, 2, 2257–2261 RSC.
- M.-S. Wu and P.-C. J. Chiang, Electrochem. Commun., 2006, 8, 383–388 CrossRef CAS.
- L. Feng, Z. Xuan, H. Zhao, Y. Bai, J. Guo, C.-w. Su and X. Chen, Nanoscale Res. Lett., 2014, 9, 290 CrossRef PubMed.
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215–5220 RSC.
- J. Maier, Faraday Discuss., 2007, 134, 51–66 RSC.
- L. Wang, Z. Nie, C. Cao, Y. Zhu and S. Khalid, J. Mater. Chem. A, 2015, 3, 6402–6407 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00317j |
| This journal is © The Royal Society of Chemistry 2017 |