
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II)-catalyzed olefination via carbonyl reductive cross-coupling†
Wei
Wei‡
 ab,
Xi-Jie
Dai‡
a,
Haining
Wang
a,
Chenchen
Li
a,
Xiaobo
Yang
a and
Chao-Jun
Li
*a
ab,
Xi-Jie
Dai‡
a,
Haining
Wang
a,
Chenchen
Li
a,
Xiaobo
Yang
a and
Chao-Jun
Li
*a
aDepartment of Chemistry, FQRNT Center for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. W., Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, Shandong, China
First published on 9th October 2017
Abstract
Natural availability of carbonyl groups offers reductive carbonyl coupling tremendous synthetic potential for efficient olefin synthesis, yet the catalytic carbonyl cross-coupling remains largely elusive. We report herein such a reaction, mediated by hydrazine under ruthenium(II) catalysis. This method enables facile and selective cross-couplings of two unsymmetrical carbonyl compounds in either an intermolecular or intramolecular fashion. Moreover, this chemistry accommodates a variety of substrates, proceeds under mild reaction conditions with good functional group tolerance, and generates stoichiometric benign byproducts. Importantly, the coexistence of KOtBu and bidentate phosphine dmpe is vital to this transformation.
Efficient construction of carbon–carbon double bonds has long been a central pursuit in the synthetic community. Recent decades have witnessed impressive accomplishments in the field of carbonyl olefination.1–6 Well-known milestones include, among others, the Wittig reaction,4 the Peterson reaction,5 the Julia olefination6 and the Tebbe–Petasis olefination.7 Parallel to these classical olefination methods, the McMurry reaction mediated by low-valent titanium (LVT) reagents enables direct reductive homo-couplings of carbonyl compounds for facile synthesis of olefins (Scheme 1, eqn (a)).8 Mechanistically, the synergy between oxophilic titanium(III)/(IV) and strong metal reductants (e.g. LiAIH4 and alkali metals) is crucial to form metal pinacolates as key intermediates. One problematic scenario pertaining to the original protocol, however, is that the cross-coupling of two unsymmetrical carbonyl compounds generally affords a statistic mixture of coupling products.8 Despite McMurry-type variants (e.g. external ligands and auxiliaries) having been developed to bypass this issue,9 two challenges endure: (1) stoichiometric quantities of metal wastes accompanied by the excessive usage of metal-based reagents, and (2) poor chemoselectivity and functional group tolerance stemmed from the presence of strong metal reductants. As the synthetic community calls for more sustainable and efficient chemical syntheses, carbonyl cross-coupling represents an ideal strategy to access olefins because naturally prevailing carbonyl functionalities are generally regarded as renewable feedstocks.
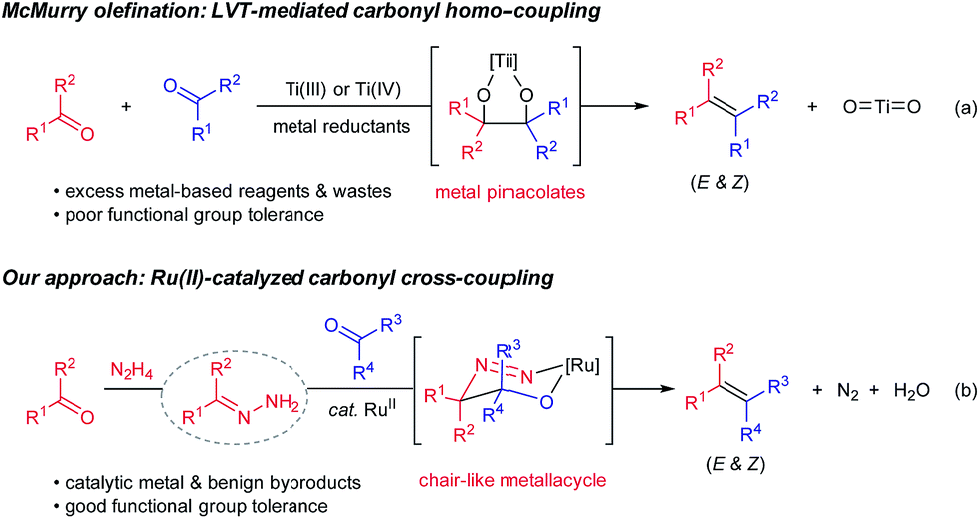 | ||
| Scheme 1 Olefination methods via reductive carbonyl coupling. | ||
Very recently, we have disclosed a ruthenium-catalyzed deoxygenation reaction for the highly selective cleavage of aliphatic primary C–O bonds.10 Building on the similar ruthenium(II) catalysis, we further demonstrated its robustness in catalyzing a series of new carbon–carbon bond forming processes through addition reactions to various carbonyl compounds, imines and activated alkenes.11–13 Variations of these precedents notwithstanding, one of their commonalities is to use aldehydes/ketones as alkyl carbanion equivalents via hydrazone formation. As a continuation of our interests in utilizing such carbanion equivalents for useful synthetic transformations, we describe herein the development of a ruthenium(II)-catalyzed, hydrazine-mediated olefination reaction via carbonyl reductive cross-coupling (Scheme 1, eqn (b)). This catalytic method features good functional group tolerance and generates nitrogen and water as the only environmentally benign byproducts in stoichiometric quantities.14,15
Initially, propionaldehyde 1a and benzophenone 2a were used as carbonyl partners in our investigation. By varying the reaction conditions employed in carbonyl addition,11 we detected the corresponding olefin 3a in 51% yield from the cross-coupling between preformed hydrazone of 1a and 2a after 12 h (Table 1, entry 1). In line with our basicity rationale, a variety of bases were prioritized in the optimization process. In fact, improved yields were consistently observed using stronger ionic bases such as hydroxides and alkoxides, among which KOtBu gave the best result (Table 1, entries 1–5; Table S3, ESI†). In contrast, organic base (e.g. Et3N) and weaker inorganic base (e.g. K2CO3) were significantly inferior (Table 1, entries 6 and 7). Moreover, the amount of base matters significantly for this reaction. Specifically, lower loadings than substoichiometric quantity (i.e. 50 mol%) were associated with yield attenuation (Table S4, ESI†). As expected, the olefination reaction did not proceed in the absence of base (Table 1, entry 8). In addition to the basicity, both ruthenium(II) pre-catalyst and bidentate phosphine ligand 1,2-bis(dimethylphosphino)ethane (dmpe) are essential for the elimination to occur. Likewise, no desired product was obtained without either one of them (Table 1, entries 9 and 10). Subsequent screenings on various ruthenium catalysts (Table S1, ESI†) and phosphine ligands (Table 1, entries 11–14; Table S2, ESI†)16 revealed that the combination of [Ru(p-cymene)Cl2]2 and dmpe provided the biggest catalytic turnover number, and thus the highest yield. Although other reaction parameters such as solvent, additive and temperature play minor roles in the current reaction, the following observations are worthy noting. For example, ether solvents (e.g. THF, 1,4-dioxane, 1,2-dimethoxyethane) favor the olefination over other types of solvents (Table S5, ESI†). Addition of cesium fluoride as an additive slightly increases the reaction yield (Table S6, ESI†).
|
|
|||
|---|---|---|---|
| Entry | L | Base | Yieldb (%) |
| a 1a (0.28 mmol, 1.4 equiv.), N2H4·H2O (0.3 mmol, 1.5 equiv.), THF (0.14 mL), rt, 30 min; 2a (0.20 mmol, 1.0 equiv.), [Ru(p-cymene)Cl2]2 (0.003 mmol, 1.5 mol%), ligand (0.006 mmol, 3.0 mol%), base (0.1 mmol, 50 mol%), additive: CsF (0.15 mmol, 75 mol%), 45 °C, 12 h, under N2. b Yields were determined by crude 1H NMR using mesitylene as an internal standard. c Without [Ru(p-cymene)Cl2]2. | |||
| 1 | dmpe | K3PO4 | 51 |
| 2 | dmpe | KOtBu | 84 |
| 3 | dmpe | NaOtBu | 80 |
| 4 | dmpe | CsOH | 82 |
| 5 | dmpe | KOH | 78 |
| 6 | dmpe | Et3N | 0 |
| 7 | dmpe | K2CO3 | 4 |
| 8 | dmpe | — | 0 |
| 9 | — | KOtBu | 0 |
| 10c | dmpe | KOtBu | 0 |
| 11 | dppb | KOtBu | 11 |
| 12 | dppp | KOtBu | 15 |
| 13 | dppm | KOtBu | 4 |
| 14 | P(p-Tolyl)3 | KOtBu | 4 |

With the standard reaction conditions in hand, we moved on to study the scope of electrophilic carbonyl partners (Table 2). In general, this chemistry covers a broad spectrum of benzophenone and its derivatives, regardless of their electronic nature. Consequently, the corresponding olefins (3a–h) were obtained in moderate to good yields. For unsymmetrical benzophenones, a mixture of stereoisomers (E/Z isomers) was produced (3e–h). Similarly, acetophenones bearing a wide range of aryl substituents were surveyed, with the formation of E/Z isomers (E-isomer predominant) in moderate to good yields (3i–ip). Notably, functional groups that are commonly incompatible with traditional carbonyl olefination approach such as unprotected alcohol, ester and amide groups, were well tolerated in this chemistry and thus amenable to further functionalization (3ij, 3in and 3ip). Intriguingly, aromatic ketones substituted by linear alkyl chains (2–5 carbons) did not hamper the reaction leading to the desired products in good yields (3j–l). Aromatic aldehyde and aliphatic ketones can also serve as coupling partners, including the relatively complex oxo-steroid compound. Asymmetric azines were observed as major products with cyclic aliphatic ketones (3o). However, the complex reaction mixture was resulted in the case of acyclic aliphatic ketone (3n), whereby the corresponding asymmetric azine was obtained as one of the major side products. Low yields were therefore seen in all these substrates at this stage.
| a 1a (0.28 mmol, 1.4 equiv.), N2H4·H2O (0.3 mmol, 1.5 equiv.), THF (0.14 mL), rt, 30 min; 2 (0.20 mmol, 1.0 equiv.), [Ru(p-cymene)Cl2]2 (0.003 mmol, 1.5 mol%), dmpe (0.006 mmol, 3.0 mol%), KOtBu (0.1 mmol, 50 mol%), CsF (0.15 mmol, 75 mol%), 45 °C, 12 h, under N2. b Isolated yields and the ratio of E/Z isomers were determined by crude 1H NMR analysis. c 80 °C. d K3PO4 (0.1 mmol, 50 mol%), 12 h. e Yields were determined by crude 1H NMR using mesitylene as an internal standard. Yields of asymmetric azines were given in the parentheses. |
|---|

|
Subsequently, the scope of nucleophilic carbonyl coupling partners was tested (Table 3). Linear saturated aliphatic aldehydes, irrespective of carbon chain length, all delivered the corresponding olefins in moderate to good yields (4a–f). Although aromatic aldehydes were poorly coupled with aromatic ketones (i.e. The reductive coupling of benzaldehyde with benzophenone leading to product 4g) due to competing alcohol formation, their couplings with other aromatic aldehydes typically proceeded well to yield various stilbenes with excellent stereoselectivity (4h–k). In the latter cases, using K2CO3 as the base at elevated temperature was necessary. Encouragingly, more sterically demanding aromatic ketones (i.e. acetophenone) worked as nucleophilic partner, providing 4l in moderate yield with high stereoselectivity. Finally, the intramolecular olefination was evaluated with 3-(2-benzoylphenyl)propanal 1m and 6-oxo-6-phenylhexanal 1n. The corresponding cyclohexene derivatives 4m and 4n were obtained in 51% and 48% yields, respectively.
| a 1 (0.28 mmol, 1.4 equiv.), N2H4·H2O (0.3 mmol, 1.5 equiv.), THF (0.14 mL), rt, 30 min; 2 (0.20 mmol, 1.0 equiv.), [Ru(p-cymene)Cl2]2 (0.003 mmol, 1.5 mol%), dmpe (0.006 mmol, 3.0 mol%), KOtBu (0.1 mmol, 50 mol%), CsF (0.15 mmol, 75 mol%), 45 °C, 12 h, under N2. b Isolated yields and the ratio of E/Z isomers were determined by crude 1H NMR analysis. c 60 °C, 24 h. d [(C6Me6)RuCl2]2 (0.003 mmol, 1.5 mol%) was used as catalyst. e K2CO3 (0.1 mmol, 50 mol%), 120 °C, 24 h. |
|---|

|
Mechanistically, two scenarios are possible to form olefins. One is metal-assisted decomposition of the corresponding asymmetric azine. The other is base-mediated elimination of the corresponding alcohol. In other words, both asymmetric azines and alcohols might have been generated prior to olefins. To exclude these possibility, two parallel control experiments were conducted with the presynthesized azine 6i and 1,1-diphenylbutan-1-ol 5a under standard conditions, respectively (Scheme 2). However, olefin products (3i and 3a) were not detected in both cases. The above results strongly suggested that olefins were eliminated from a transient intermediate, rather than azines or alcohols, via a E1cB-type mechanism.
 | ||
| Scheme 2 Control experiments for the olefin formation. | ||
Based on all experimental data, the preliminary computational calculations done on the previous carbonyl addition chemistry11 and studies from others,14 a postulated reaction pathway was proposed as shown in Scheme 3. The bidentate phosphine coordinated complex I is initially generated by a ligand dissociation/association between [Ru(p-cymene)Cl2]2 and dmpe, followed by a ligand association with carbonyl-derived hydrazone B and carbonyl C in the presence of KOtBu, giving rise to complex II and III, respectively. Formation of the coordinately intermediate III sets the stage for the intramolecular isomerization. This concerted process yields the key six-membered ring intermediate IV by forming a new carbon–carbon bond between A and C.14 Base-catalyzed decomposition of diimide intermediate IVvia a E1cB-type mechanism produces the desired olefins along with the formation of ruthenium hydroxide species V. To turnover the cycle, V then reacts with hydrazone B to release water and active species II (X = Cl). Alternatively, the chloride anionic ligand on V could have been replaced by B, leaving hydroxide on the active species VI (X = OH) and other ruthenium complexes (III–V).
 | ||
| Scheme 3 Possible reaction mechanism. | ||
Conclusions
In summary, we have developed a ruthenium(II)-catalyzed, hydrazine-mediated olefination method via reductive carbonyl coupling reactions. This chemistry possesses a distinct mechanistic profile and highlights the use of naturally abundant carbonyl functionalities for efficient olefin synthesis. Other striking features include cross-coupling capability, mild reaction conditions, good functional group tolerance and stoichiometric benign byproducts. Taken together, our findings are expected to spur more interest in developing catalytic methods in this field. Further investigations on increasing the reaction scope, synthetic applications and mechanistic details are undergoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Canada Research Chair (Tier 1) foundation, FQRNT (CCVC), NSERC, CFI and McGill University. We also thank the CSC (China Scholarship Council) for the postdoctoral fellowship (W. Wei).References
- (a) S. E. Kelly, Alkene Synthesis, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 1, pp. 729–817 Search PubMed; (b) D. M. Hodgson and L. T. Boulton, in Preparation of Alkenes, ed. J. M. J. Williams, Oxford University Press, Oxford, 1996, p. 81 Search PubMed; (c) R. Dumeunier and I. E. Markó, in Modern Carbonyl Olefination, ed. T. Takeda, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- (a) A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS PubMed; (b) X. Guo, J. Wang and C.-J. Li, J. Am. Chem. Soc., 2009, 131, 15092–15093 CrossRef CAS PubMed; (c) S. Takemoto, E. Shibata, M. Nakajima, Y. Yumoto, M. Shimamoto and H. Matsuzaka, J. Am. Chem. Soc., 2016, 138, 14836–14839 CrossRef CAS PubMed; (d) J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Nature, 2016, 533, 374–379 CrossRef CAS PubMed.
- (a) W. S. Wadsworth and W. D. Emmons Jr, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS; (b) C. R. Johnson, J. R. Shanklin and R. A. Kirchhoff, J. Am. Chem. Soc., 1973, 95, 6462–6463 CrossRef CAS; (c) S. Sano, T. Takehisa, S. Ogawa, K. Yokoyama and Y. Nagao, Chem. Pharm. Bull., 2002, 50, 1300–1302 CrossRef CAS PubMed; (d) Z.-Y. Peng, F.-F. Ma, L.-F. Zhu, X.-M. Xie and Z. Zhang, J. Org. Chem., 2009, 74, 6855–6858 CrossRef CAS PubMed.
- (a) G. Wittig and G. Geissler, Liebigs Ann. Chem., 1953, 580, 44–57 CrossRef CAS; (b) G. Wittig and U. Schollkopf, Ber. Dtsch. Chem. Ges., 1954, 87, 1318–1330 CrossRef; (c) J. Boutagy and R. Thomas, Chem. Rev., 1974, 74, 87–99 CrossRef CAS; (d) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- (a) D. J. Peterson, J. Org. Chem., 1968, 33, 780–784 CrossRef CAS; (b) J. Huang, C. Wu and W. D. Wulff, J. Am. Chem. Soc., 2007, 129, 13366–13367 CrossRef CAS PubMed; (c) D. J. Ager, The Peterson Olefination Reaction, Organic Reactions, ed. L. A. Paquette, 1990, 38, pp. 1–224 Search PubMed.
- (a) M. Julia and J. M. Paris, Tetrahedron Lett., 1973, 14, 4833–4836 CrossRef; (b) P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 2563–2585 RSC; (c) K. Ando, T. Kobayashi and N. Uchida, Org. Lett., 2015, 17, 2554–2557 CrossRef CAS PubMed.
- (a) F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS; (b) I. Beadham and J. Micklefield, Curr. Org. Synth., 2005, 2, 231–250 CrossRef CAS; (c) N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392–6394 CrossRef CAS.
- (a) J. E. McMurry and M. P. Fleming, J. Am. Chem. Soc., 1974, 96, 4708–4709 CrossRef CAS; (b) J. E. McMurry and L. R. Krepski, J. Org. Chem., 1976, 41, 896–897 CrossRef CAS; (c) S. Tyrlik and I. Wolochowicz, Bull. Soc. Chim. Fr., 1973, 2147–2148 CAS; (d) B. E. Kahn and R. D. Rieke, Chem. Rev., 1988, 88, 733–745 CrossRef CAS; (e) J. E. McMurrv, Chem. Rev., 1989, 89, 1513–1524 CrossRef; (f) T. Takeda and A. Tsubouchi, The McMurry Coupling and Related Reactions, Organic Reactions, ed. S. E. Denmark, 2013, 82, pp. 1–470 Search PubMed.
- (a) N. Balu, S. K. Nayak and A. Banerji, J. Am. Chem. Soc., 1996, 118, 5932–5937 CrossRef CAS; (b) C. Villiers and M. Ephritikhine, Chem.–Eur. J., 2001, 7, 3043–3051 CrossRef CAS PubMed; (c) M. H. Chisholm and J. A. Klang, J. Am. Chem. Soc., 1989, 111, 2324–2325 CrossRef CAS; (d) X.-F. Duan, J. Zeng, J.-W. Lü and Z.-B. Zhang, J. Org. Chem., 2006, 71, 9873–9876 CrossRef CAS PubMed; (e) H. R. Diéguez, A. López, V. Domingo, J. F. Arteaga, J. A. Dobado, M. Mar Herrador, J. F. Quílez del Moral and A. F. Barrero, J. Am. Chem. Soc., 2010, 132, 254–259 CrossRef PubMed; (f) L. Zhang, X. Yu, L. Zhang, X. Zhou and Y. Lin, Org. Chem. Front., 2014, 1, 929–935 RSC.
- (a) X.-J. Dai and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5433–5440 CrossRef CAS PubMed ; for an iridium-catalyzed version, see: ; (b) J.-L. Huang, X.-J. Dai and C.-J. Li, Eur. J. Org. Chem., 2013, 6496–6500 CrossRef CAS.
- H. Wang, X.-J. Dai and C.-J. Li, Nat. Chem., 2016, 9, 374–378 CrossRef PubMed.
- N. Chen, X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6260–6263 CrossRef CAS PubMed.
- X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6302–6306 CrossRef CAS PubMed.
- A few examples for the formation of carbon-carbon double bond via the coupling of stoichiometric metalloazines with carbonyl compounds, see: (a) G. M. Arvanitis, J. Schwartz and D. Van Engen, Organometallics, 1986, 5, 2157–2159 CrossRef CAS; (b) J. A. Smegal, I. K. Meier and J. Schwartz, J. Am. Chem. Soc., 1986, 108, 1322–1323 CrossRef CAS; (c) G. M. Arvanitis and J. Schwartz, Organometallics, 1987, 6, 421–423 CrossRef CAS; (d) G. M. Arvanitis, J. Smegal, I. Meier, A. C. C. Wong, J. Schwartz and D. Van Engen, Organometallics, 1989, 8, 2717–2723 CrossRef CAS ; for a pioneering work on using alkylhydrazone anion for olefination with carbonyls via a multistep process mediated by PCl3, see ; (e) J. E. Baldwin, R. M. Adlington, J. C. Bottard, J. N. Kolhe, I. M. Newington and M. W. D. Perry, Tetrahedron, 1986, 42, 4235–4246 CrossRef CAS.
- A recent review on ruthenium-catalyzed transfer hydrogenation for C–C bond formation, see: F. Perez, S. Oda, L. M. Geary and M. J. Krische, Top. Curr. Chem., 2016, 374, 365–387 CrossRef PubMed.
- For the reactions of [RuCl2(η6-arene)]2 with tertiary phosphines, please see. (a) R. A. Zelonka and M. C. Baird, Can. J. Chem., 1972, 50, 3063–3072 CrossRef CAS; (b) M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC; (c) H. Werner and R. Werner, Chem. Ber., 1982, 115, 3766–3780 CrossRef CAS; (d) M. A. Bennett, T.-N. Huang and J. L. Latten, J. Organomet. Chem., 1984, 272, 189–205 CrossRef CAS; (e) M. A. Bennett and J. L. Latten, Aust. J. Chem., 1987, 40, 841–849 CrossRef CAS; (f) P. Pertici, S. Bertozzi, R. Lazzaroni, G. Vitulli and M. A. Bennett, J. Organomet. Chem., 1988, 354, 117–121 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c7sc04207h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |