
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The α-tertiary amine motif drives remarkable selectivity for Pd-catalyzed carbonylation of β-methylene C–H bonds†
Kirsten F.
Hogg‡
 ,
Aaron
Trowbridge‡
,
Andrea
Alvarez-Pérez
and
Matthew J.
Gaunt
*
,
Aaron
Trowbridge‡
,
Andrea
Alvarez-Pérez
and
Matthew J.
Gaunt
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK. E-mail: mjg32@cam.ac.uk
First published on 9th October 2017
Abstract
The selective C–H carbonylation of methylene bonds in the presence of traditionally more reactive methyl C–H and C(sp2)–H bonds in α-tertiary amines is reported. The exceptional selectivity is driven by the bulky α-tertiary amine motif, which we hypothesise orientates the activating C–H bond proximal to Pd in order to avoid an unfavourable steric clash with a second α-tertiary amine on the Pd centre, promoting preferential cyclopalladation at the methylene position. The reaction tolerates a range of structurally interesting and synthetically versatile functional groups, delivering the corresponding β-lactam products in good to excellent yields.
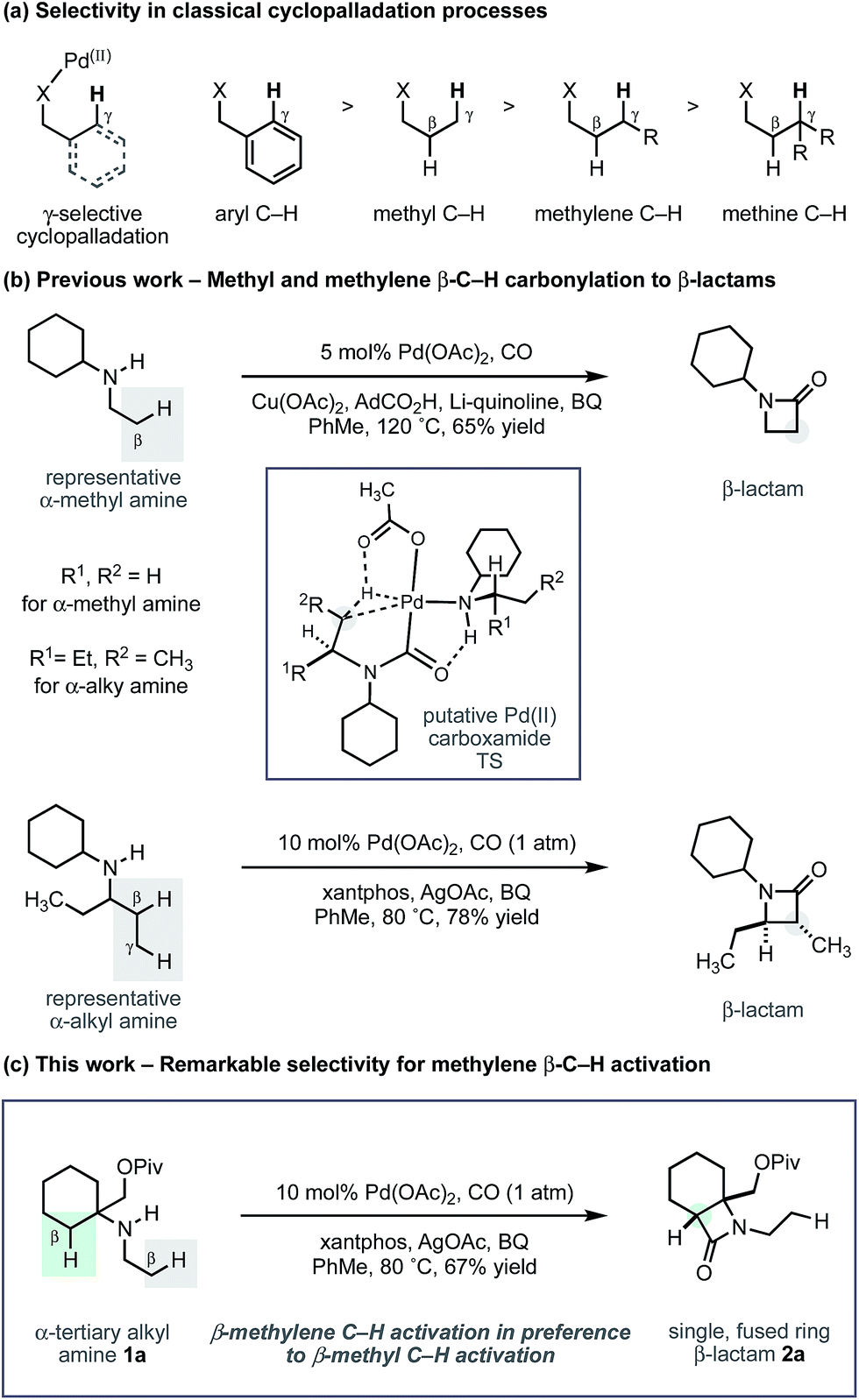
Methods that enable the catalytic functionalization of unreactive aliphatic C–H bonds have great potential in streamlining the synthesis of complex molecules such as natural products or medicinal agents.1 However, these molecules contain many types of C–H bond, each with a subtly different reactivity that is often influenced by an intricate interplay of factors including steric, inductive and conductive effects, and sometimes innate strain.2 As a result, catalytic processes that target certain C–H bonds are an important goal for chemical synthesis, and one that continues to inspire intense research effort.3
Arguably, the most common strategy employed for selective C–H activation involves the use of palladium(II) catalysts, directed to a specific position by a resident polar functional group.4 Known as cyclopalladation, this activation mode most commonly targets the γ-C–H bond with respect to the directing group, to form a 5-membered ring intermediate from which further reaction takes place to install the new functionality. In most cases, the directing motifs needed to facilitate the C–H activation are bespoke auxiliaries or tailored protecting groups that need to be added to (and removed from) an intrinsic functionality of the parent molecule.5 While the use of auxiliaries has enabled many types of C–H activations, by contrast, the number of related transformations directed by functional groups that are native to aliphatic molecules (carboxylic acids, amines, hydroxyl groups) is more limited, despite the emergence of some important recent examples.6
Recently, we reported a new activation mode for C–H carbonylation of unprotected aliphatic secondary amines to form tertiary β-lactams.7 In contrast to other methods, the C–H activation step takes place at the β-C–H bond to the directing nitrogen functionality.8 This change in selectivity is brought about because the reaction follows a pathway that is distinct from classical cyclopalladation-mediated reactions. Rather than C–H activation preceding the CO insertion step, the new pathway uses an amine bound palladium(II) carboxylate to first engage CO to form a carbamoyl–Pd(II) complex. By virtue of CO already being inserted between the amine and the Pd(II) centre, C–H activation via a 5-membered ring transition state now takes place at the β-C–H bond with respect to the resident amine motif. We have shown, firstly, that a wide range of aliphatic amines displaying α-branched methyl groups undergo β-C–H carbonylation to the corresponding β-lactams.7 Secondly, we found that in the absence of suitably disposed methyl groups, the C–H carbonylation was able to target the β-methylene C–H bond under slightly modified conditions to form trans-disubstituted β-lactams.9 The functional group tolerance exhibited by both of these C–H carbonylation processes is particularly notable and gives rise to a range of versatile and diverse β-lactam products.
During the course of our studies to further explore this carbonylation platform, we discovered a remarkable feature inherent to this C–H activation mode. α-Tertiary amines (ATAs) displaying both a β-methyl C–H bond and β-methylene C–H bond undergo exclusive carbonylation at the traditionally less reactive and more hindered methylene position.10 Central to the success of this selective C–H carbonylation is the presence of a fully substituted carbon atom on one side of the amine linkage, which steers the reaction to the C–H bond adjacent to this bulky structural feature (Scheme 1c). Herein, we report the development of a general C–H carbonylation exploiting this selectivity-inducing parameter. The ATA motif is widespread among natural products and pharmaceuticals displaying unique physiochemical properties (Scheme 2).11 However, due to the limited number of methods available in accessing these compounds, we believe that the direct functionalization of ATAs would provide convenient access to a range of molecular scaffolds that would be attractive to practitioners of synthetic and medicinal chemistry.
 | ||
| Scheme 1 Overview of C–H carbonylation of aliphatic amines. | ||
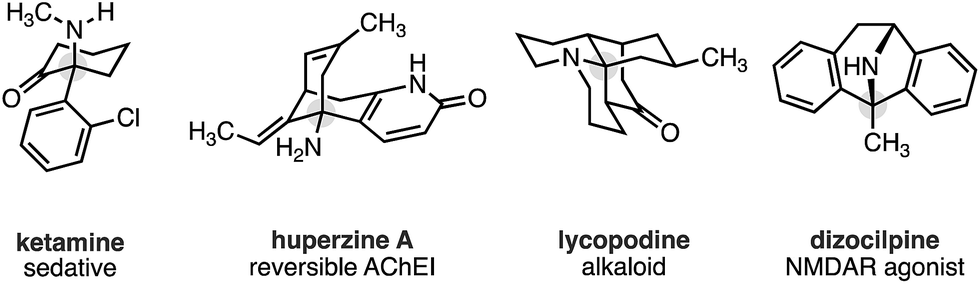 | ||
| Scheme 2 Pharmaceuticals and alkaloids containing the ATA motif. | ||
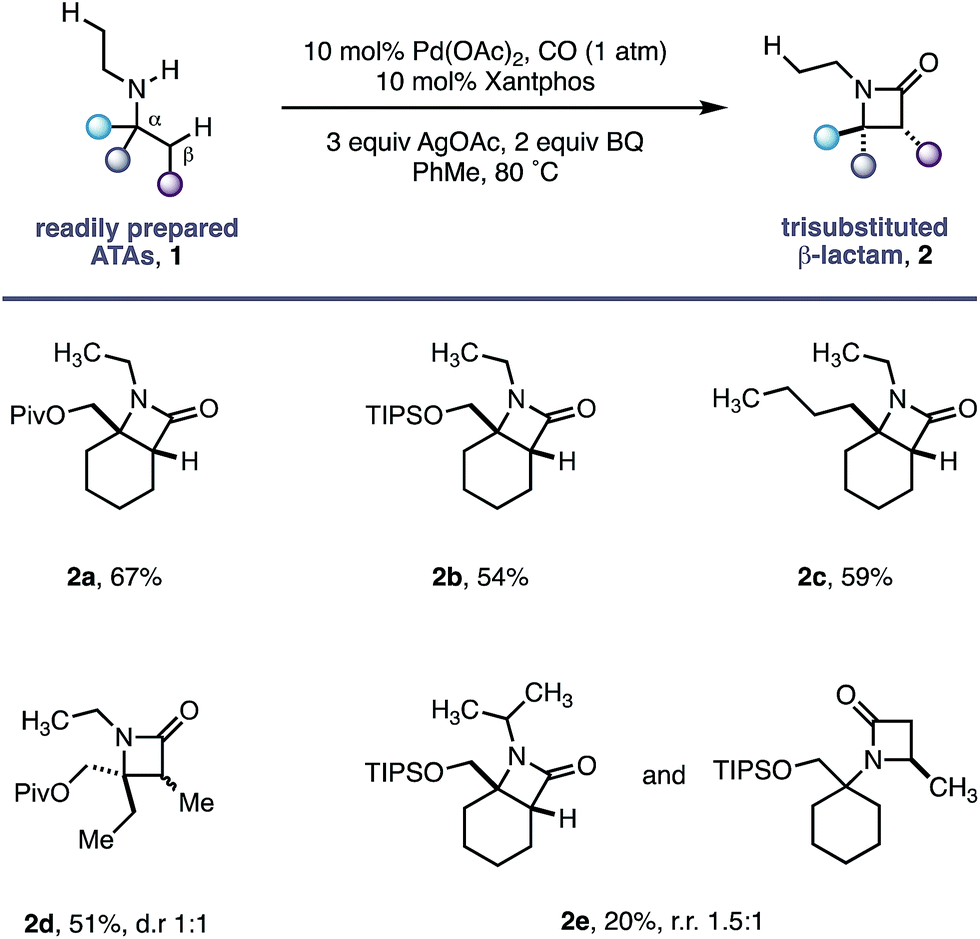
Using the conditions developed for methylene C–H carbonylation,9 using xantphos as a ligand,12 we first assessed substrates displaying a variety of substituents in the α-position on the reacting side of the amine linkage; the secondary amines also contained a β-methyl C–H bond (in the form of an N-ethyl group) on the other side of the free (NH) motif (Table 1). Substrates containing protected α-hydroxymethylene substituents proved effective under the reaction conditions, delivering the fused bicyclic β-lactams (2a and 2b) resulting from selective methylene C–H carbonylation in good yields.13 Moreover, an α-n-butyl chain was also sufficient to deliver the corresponding bicyclic β-lactam 2c in 59% yield, remarkably without any activation of the exocyclic α-alkyl substituent, which contains a competitive methylene β-C–H bond. Exclusive methylene C–H activation also occurred on the corresponding acyclic substrate 1d, further expanding the utility of the methodology. The corresponding N-isopropyl substrate 1e, for which there is a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio of methyl to methylene C–H bonds, afforded a 1.5:1 mixture of β-lactams in favour of the methylene C–H activated product, exemplifying the remarkable selectivity inherent to this C–H activation process.
:4 ratio of methyl to methylene C–H bonds, afforded a 1.5:1 mixture of β-lactams in favour of the methylene C–H activated product, exemplifying the remarkable selectivity inherent to this C–H activation process.
|
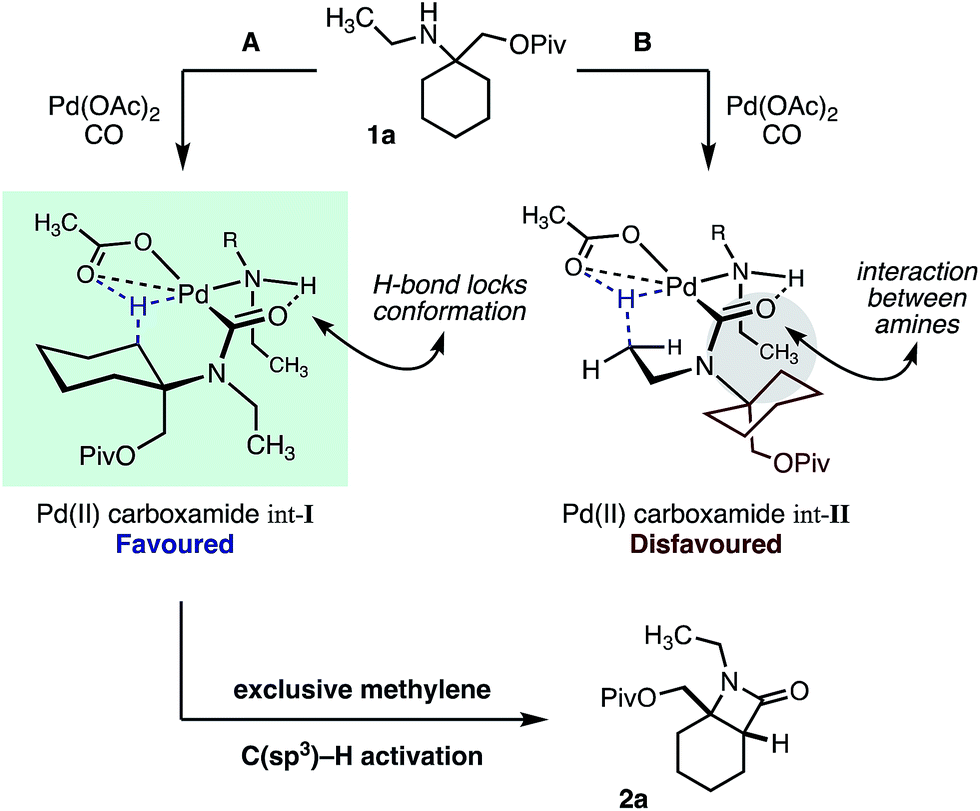
We hypothesize that the selectivity of this methylene C–H carbonylation process arises from the unique Pd(II)–carboxamide intermediate (pathways A and B, Scheme 3). Based on our previous work,7 we propose that a key hydrogen-bond between the carboxamide carbonyl and ligated amine locks the relative conformation of these two substituents, in turn generating two potentially reactive carboxamide intermediates (int-I and int-II). We believe that the large α-tertiary amine substituent generates an unfavorable steric clash with the ligated amine (int-II), resulting in preferential activation of the highlighted methylene C–H bond (pathway A).14 While this model holds for the majority of the substrates, we believe that the large isopropyl amine substituent in amine 1e may result in poorer steric differentiation between the two Pd–carboxamide transition states, leading to the formation of both methylene and methyl activated products (Table 1).
 | ||
| Scheme 3 Mechanistic hypothesis for C–H activation of ATAs. | ||
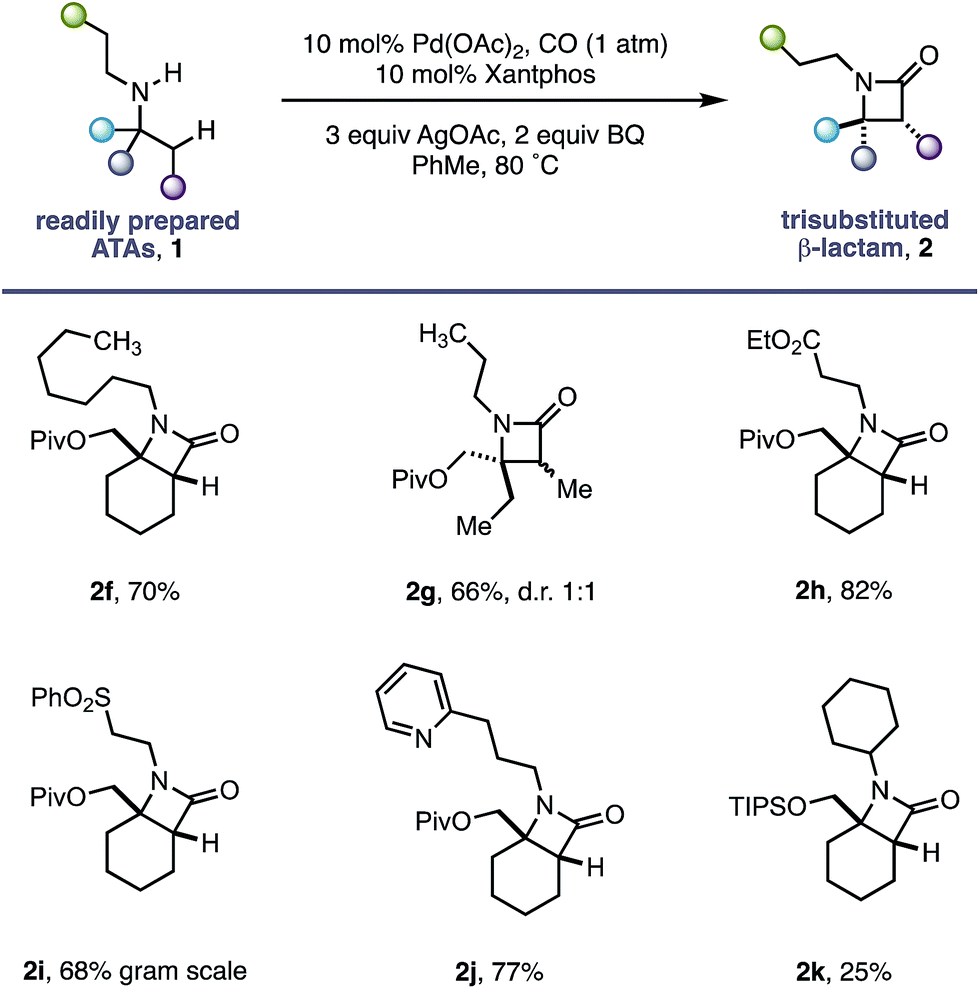
Having successfully demonstrated that a fully substituted centre in the α-position to the amine is sufficient to induce exclusive β-methylene C–H activation, we next explored how substituents on the non-reacting side of the amine affected the carbonylation process (Table 2). The competing classical 5-membered cyclopalladation was not observed in n-propyl-containing amine 2g or n-heptyl amine 2f, affording the corresponding bicyclic β-lactams in a 66% and 70% yield respectively. β-Amino ester 2h and sulfone 2i derivatives bearing acidic α-hydrogens, which have previously been shown to promote C–H activation,9 were tolerated in good yield and on gram scale.
|
The use of Lewis basic heteroaromatics, such as pyridyl motifs, to direct C(sp3)–H activation is well established.5c,15 An amine displaying 2-pyridyl substituent 1j was tolerated in good yield, with no competitive C–H activation on the propyl chain. Impressively, bis-cyclohexyl substrate 1k, bearing two very similar sets of methylene C–H bonds, afforded a single β-lactam 2k with activation occurring exclusively in the α-position to the quaternary carbon centre.
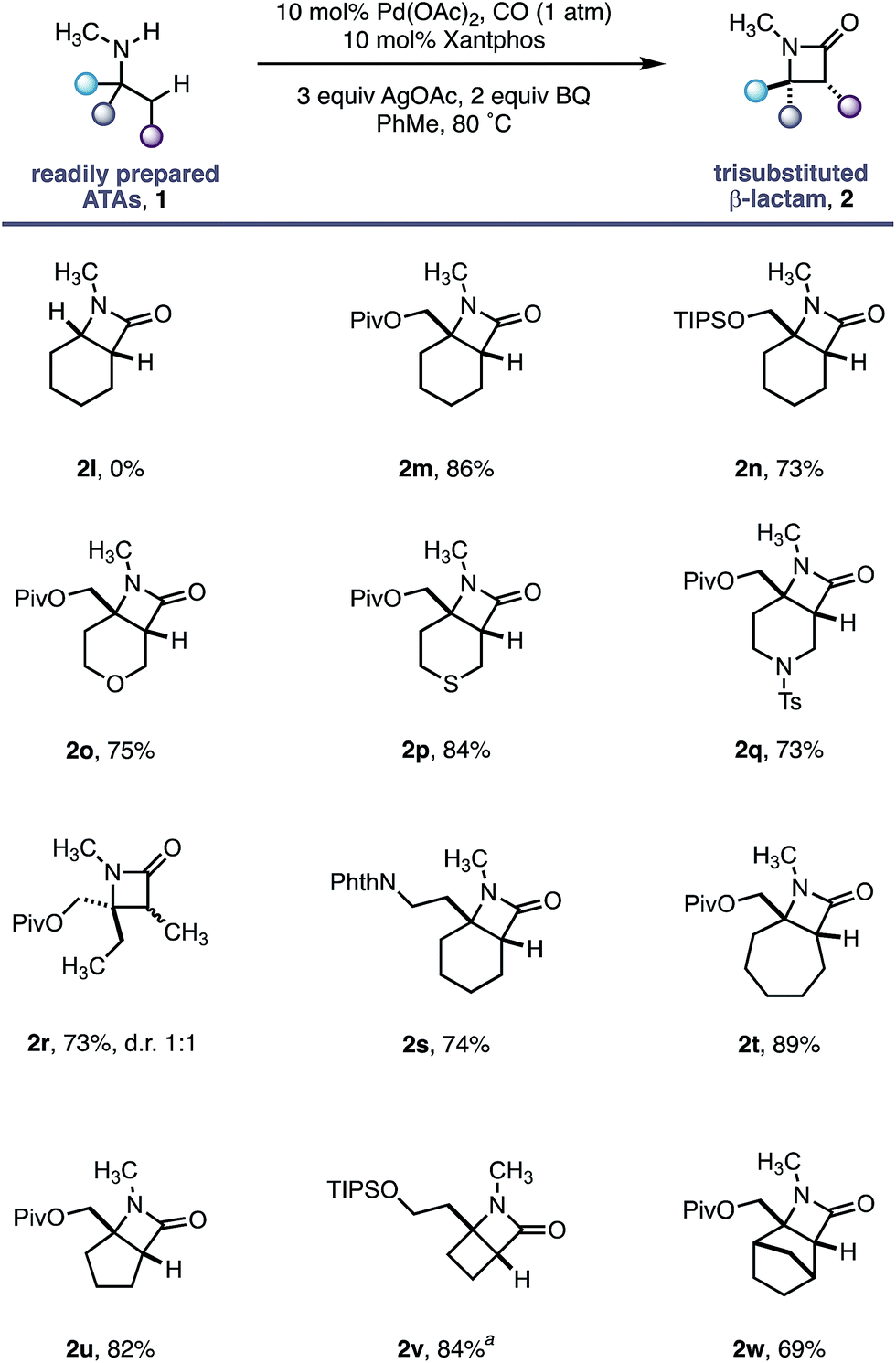
Having established the robustness of this methodology towards a range of functional groups, we turned our attention to substrates containing N-methyl amines (Table 3). Despite the ubiquity of N-Me amines in biologically active molecules and pharmaceutical agents, their deleterious reactivity with many electrophilic transition metal catalysts has rendered them challenging substrates for C–H activation.16 The facile oxidation of N-methylamines to the corresponding imine followed by nucleophilic capture has been exploited in numerous transformations.17 Due to the high pharmaceutical utility of N-methylamines,18 we sought to test the limits of our C–H activation methodology by investigating this important class of amine substrate. By virtue of our geometrically locked Pd–carboxamide intermediate, we reasoned that the N-methyl group would be placed in a remote position relative to the reactive palladium centre, thereby enabling a selective process.
| a Reaction with 10 mol% Pd(OPiv)2 and 3 equiv. AgOPiv. |
|---|
|
As a control experiment, N-methylcyclohexylamine 1l, lacking the important fully substituted α-tertiary centre, was subjected to our optimized conditions; none of the desired β-lactam product was observed and the starting amine decomposed. In line with our hypothesis, α-tertiary amino-alcohol derivatives 1m and 1n delivered the corresponding β-lactams (2m–n) in good yield without any demethylation. Piperidine and tetrahydropyran motifs are common among pharmaceutical agents but their functionalization at C3 and C4 positions can present a significant challenge;19 our methodology delivered the bicyclic β-lactam products 2o and 2q in good yields, allowing for further derivatization of the C3 position. The reaction also proved to be tolerant of a thioether moiety, known to deactivate transition metal catalysts, delivering the β lactam 2p in a good 84% yield. Moreover, the reaction proved extremely versatile across a range of ring sizes (2t to 2w) in good yield. Pleasingly, cyclobutylamine 1v was readily transformed into highly strained 4,4-fused β-lactam 2v, permitting access to functionalized hydrogenated variants of the ‘Dewar-pyridone’ scaffold.20
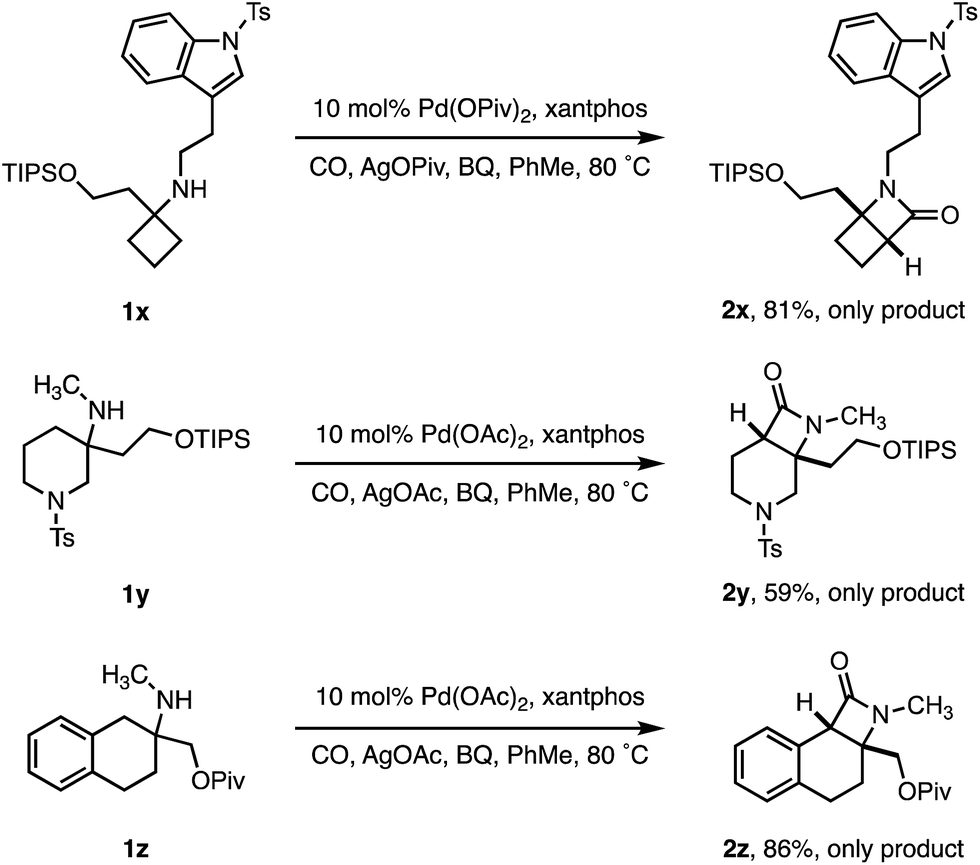
To test the limits of the positional selectivity of the ATA carbonylation among many potentially reactive C–H bonds, we prepared a range of functional amines that could lead to a number of different lactam products (Scheme 4). Indole rings are considered a “privileged” scaffold in medicinal chemistry,21 however, they often undergo facile C(sp2)–H activation.22 We were pleased to observe that tryptamine analogue 1x bearing a cyclobutane ring was readily transformed into the 4,4-fused β-lactam 2x in good yield without any competing C(sp2)–H activation. 3-Methylamino piperidine 1y, containing two different ring C–H bond environments, afforded complete selectivity for the C4 position in useful yield (2y). Similarly, the 2-aminotetralin substrate 1z proved to activate selectively at the benzylic position in good yield, revealing a class of tricyclic β-lactam scaffolds (2z).
 | ||
| Scheme 4 Selectivity of ATA methylene C–H carbonylation. | ||
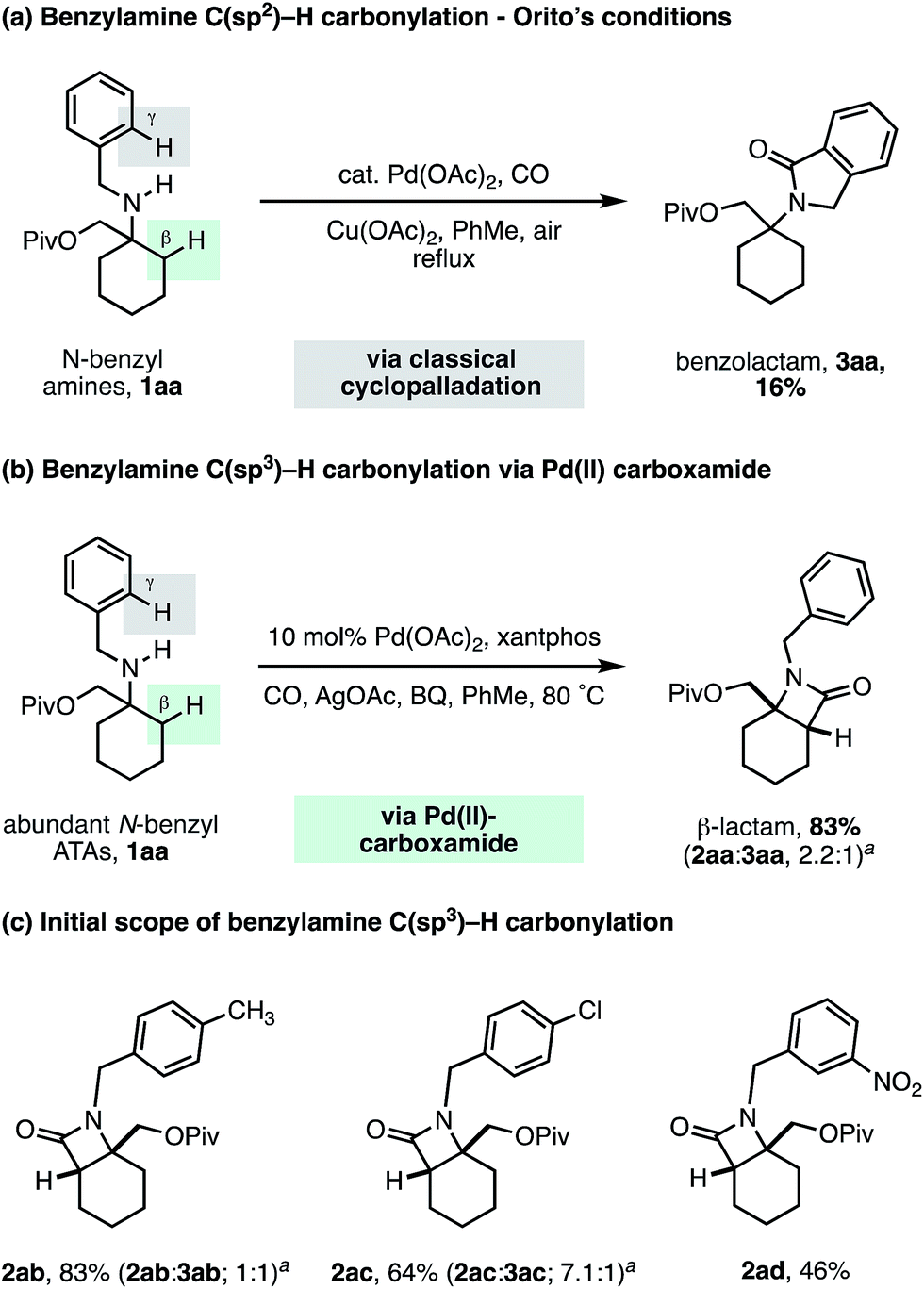
To challenge the capacity of the selective C–H carbonylation process, we next designed a substrate that would place a β-methylene C–H bond in competition with a C(sp2)–H bond on the ortho position of a benzylamine motif. The cyclopalladation of benzylamines is, arguably, one of the most facile and well understood C–H activation processes, with near exclusive C(sp2)–H activation control.23 Orito and coworkers have shown that alkyl-benzyl substituted secondary amines undergo selective C(sp2)–H carbonylation to benzolactams, with no trace of reaction at the C(sp3)–H bond (Scheme 5a).24 To benchmark the reactivity of our alkyl-benzyl amines, we applied Orito's conditions to N-benzyl amine derivative 1aa and found that benzolactam 3aa resulting from C(sp2)–H activation was produced as the sole product (Scheme 5a).
 | ||
| Scheme 5 N-Benzyl ATA substrate scope for selective methylene C–H carbonylation. aRatio of β-lactam 2 to γ-benzolactam 3. | ||
Upon switching to our optimized C–H carbonylation conditions, we were delighted to see that a mixture of β-lactam 2aa and benzolactam 3aa was formed in a good 83% yield with a 2.2:1 ratio in favor of the C(sp3)–H activation product 2aa (Scheme 5b). Encouragingly, we found that changing the electronic properties of the aromatic ring had a significant impact on the product distribution (Scheme 5c). Electron withdrawing substituents favored C(sp3)–H activation, with m-NO2Ph affording exclusively the β-lactam product 2ad, with no C–H activation observed on the aromatic ring. These results suggest that classical C(sp2)–H activation to the benzolactam occurs via an electrophilic cyclopalladation pathway. To the best of our knowledge, this is the first example of a palladium catalyzed C–H activation that is selective for a β-methylene C–H bond in the presence of a γ-C(sp2)–H bond on an aromatic ring.
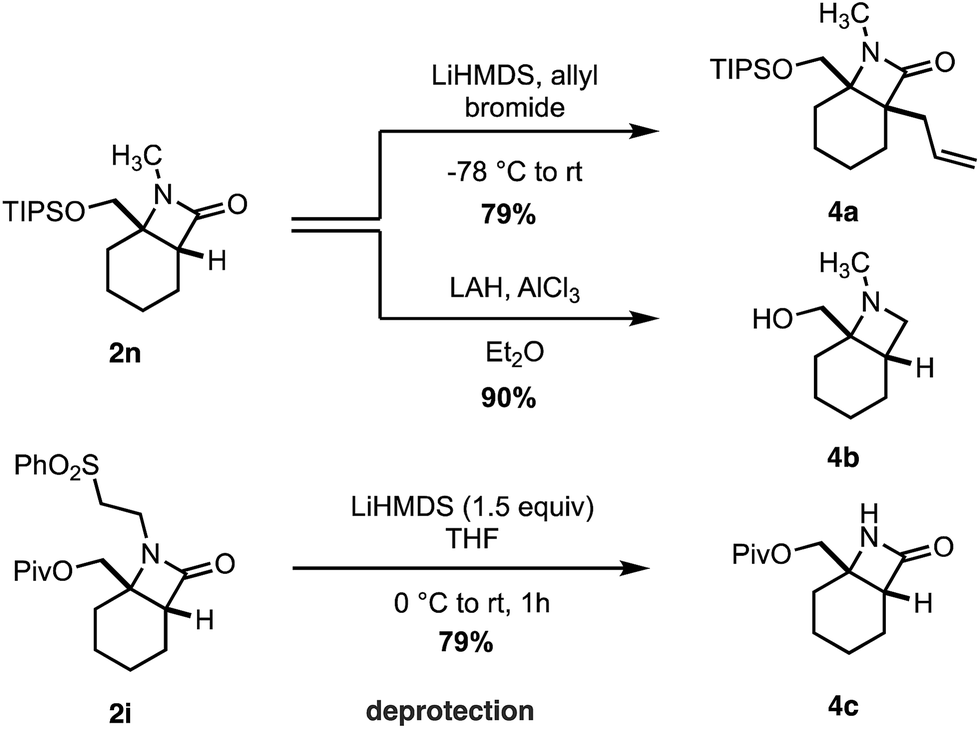
Finally, we transformed the β-lactam products into a range of useful chemical building blocks (Scheme 6). Alkylation to form β-lactams displaying vicinal fully substituted stereocenters proceeded in good yield (4a). Reduction of 2n to the corresponding azetidinyl alcohol 4b, a useful class of scaffold in the design of pharmaceutical agents, occurred in an excellent 90% yield. Importantly, the free (NH)lactam 4c could be obtained in good yield under mild conditions from the corresponding sulfonyl β-lactam 2h, offering a simple lactam deprotection protocol.
 | ||
| Scheme 6 Transformation of β-lactam products. | ||
In conclusion, we have developed a remarkable aliphatic amine C–H carbonylation reaction that is capable of selectively activating β-methylene C–H bonds in the presence of traditionally more reactive C(sp3) and C(sp2)–H bonds. The presence of a fully substituted carbon atom in the α-position to the amine appears to control this unprecedented selectivity. Using this methodology, a range of highly functionalized β-lactam building blocks have been synthesized in good yields, which can further be derivatised in order to access novel heterocyclic scaffolds that we believe we be useful to a range of synthetic and medicinal applications. Computational studies to explore the origin of this unique selectivity in further detail are currently ongoing within our group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to EPSRC (EP/100548X/1), ERC (ERC-STG-259711) and the Royal Society (Wolfson Award) for supporting this research (M. J. G.). We gratefully acknowledge the European Research Council and the UK Engineering and Physical Sciences Research Council (EPSRC) (K. F. H.) and the Herchel Smith Foundation (A. T.) for funding. Mass spectrometry data were acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University.References
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (d) D. Y.-K. Chen and S. W. Youn, Chem. –Eur. J., 2012, 18, 9452 CrossRef CAS PubMed.
- (a) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed; (b) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (c) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed.
- J. F. Hartwig, Acc. Chem. Res., 2017, 50, 549 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- For examples of C–H activation using auxiliaries see: oximes: (a) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS PubMed; oxazolines: (b) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed; picolinamides: (c) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; methyl hydroxamic acids: (d) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed; N-aryl carboxamides: (e) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS PubMed; hydroxyl derivatives: (f) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS PubMed; sulfonamides: (g) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146 CrossRef CAS PubMed.
- C–H activation using native functionality: pyridines: (a) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541 CrossRef CAS PubMed; alcohols: (b) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS PubMed; carboxylic acids: (c) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506 CrossRef CAS PubMed; amines: (d) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS PubMed; (e) J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009 CrossRef CAS PubMed; primary amines using transient directing groups: (f) X. Yu, M. C. Young, C. Wang, D. W. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef PubMed; (g) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS PubMed; (h) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CAS.
- D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851 CrossRef CAS PubMed.
- (a) Z. Huang, C. Wang and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 5299 CrossRef CAS PubMed; (b) N. Gulia and O. Daugulis, Angew. Chem., Int. Ed., 2017, 56, 3630 CrossRef CAS PubMed; (c) D. Dailler, R. Rocaboy and O. Baudoin, Angew. Chem., Int. Ed., 2017, 56, 7218 CrossRef CAS PubMed.
- J. R. Cabrera-Pardo, A. Trowbridge, M. Nappi, K. Ozaki and M. J. Gaunt, Angew. Chem., Int. Ed., 2017, 56, 11958 CrossRef CAS PubMed.
- (a) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114 CrossRef CAS PubMed; for an example of ligand controlled selective methylene C–H activation, see: (b) D. Katayev, M. Nakanishi, T. Bürgi and E. P. Kündig, Chem. Sci., 2012, 3, 1422 RSC.
- A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491 RSC.
- We hypothesize that the role of xantphos (or its mono-oxide) is most likely to stabilize the Pd0 species formed at the end of the catalytic cycle prior to its oxidation to the active PdII species required for the reaction. The reaction also works in the absence of xantphos, but the yield of the reaction is significantly lower; see ref. 9.
- Upon subjection of amine 1a to lower catalyst and ligand loadings of 5 mol% the resulting lactam 2a was obtained in 43% yield, as determined by 1H NMR assay.
- We cannot rule out the possibility of classical angle compression (Thorpe-Ingold effect) influencing the selectivity of the C–H activation.
- Selected examples of pyridine-directed C–H activation: (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) X. Zhao, E. Dimitrijeviic and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466 CrossRef CAS PubMed; (c) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS PubMed; (d) Y. Zhang, J. Feng and C.-J. Li, J. Am. Chem. Soc., 2008, 130, 2900 CrossRef CAS PubMed; (e) W.-Y. Yu, W. N. Sit, K.-M. Lai, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2008, 130, 3304 CrossRef CAS PubMed.
- (a) S. Rosseaux, S. I. Gorelsky, B. K. W. Chung and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 10692 CrossRef PubMed; (b) D.-H. Wang, X.-S. Hao, D.-F. Wu and J.-Q. Yu, Org. Lett., 2006, 8, 3387 CrossRef CAS PubMed.
- (a) S. Murahashi, N. Komiya, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS PubMed; (b) Z. Li and C. J. Li, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS PubMed; (c) Z. Li and C. J. Li, J. Am. Chem. Soc., 2005, 127, 3672 CrossRef CAS PubMed; (d) A. Catino, J. Nichols, B. Nettles and M. Doyle, J. Am. Chem. Soc., 2006, 128, 5648 CrossRef CAS PubMed.
- Amines: Synthesis, Properties, and Applications, ed. Lawrence S. A., Cambridge University, Cambridge, 2006 Search PubMed.
- C3 functionalisation: (a) D. P. Affron and J. A. Bull, Eur. J. Org. Chem., 2016, 139 CrossRef CAS PubMed; (b) J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387 CrossRef CAS PubMed; (c) S. Ye, W. Yang, T. Coon, D. Fanning, T. Neubert, D. Stamos and J.-Q. Yu, Chem. –Eur. J., 2016, 22, 4748 CrossRef CAS PubMed; C4 functionalisation: (d) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220 CrossRef CAS PubMed.
- H. Hongo, K. Iwasa, C. Kabuto, H. Matsuzaki and H. Nakano, J. Chem. Soc., Perkin Trans. 1, 1997, 1747 RSC.
- (a) M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421 CrossRef CAS PubMed; (b) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159 CrossRef CAS PubMed.
- (a) E. Beck and M. J. Gaunt, Topic in Current Chemistry, in C–H activation, J.-Q. Yu and Z. Shi, Springer-Verlag, Heidelberg, 2010, vol. 292, pp. 87–121 Search PubMed; (b) N. Lebrasseur and I. Larrosa, Adv. Heterocycl. Chem., 2012, 105, 309 CrossRef CAS; (c) N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926 CrossRef CAS PubMed; (d) S. Peciado, L. Mendive-Tapia, F. Albericio and R. Lavilla, J. Org. Chem., 2013, 78, 8129 CrossRef PubMed; (e) L. Mendive-Tapia, S. Peciado, J. García, R. Ramón, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- For mechanistic studies, see: (a) A. D. Ryabov, I. K. Sakodinskaya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 12, 2629 RSC; (b) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed; for stoichiometric examples, see: (c) J. Albert, R. M. Ceder, M. Gomez, J. Granell and J. Sales, Organometallics, 1992, 11, 1536 CrossRef CAS; (d) Y. Fuchita, H. Tsuchiya and A. Miyafuji, Inorg. Chim. Acta, 1995, 233, 91 CrossRef CAS; (e) K. Selvakumar and S. Vancheesan, Polyhedron, 1997, 16, 2405 CrossRef CAS; for catalytic examples, see: (f) A. Lazareva and O. Daugulis, Org. Lett., 2006, 8, 5211 CrossRef CAS PubMed; (g) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed; (h) M. Miura, C.-G. Feng, S. Ma and J.-Q. Yu, Org. Lett., 2013, 15, 5258 CrossRef CAS PubMed; (i) A. Rodríguez, J. Albert, X. Ariza, J. Garcia, J. Granell, J. Farràs, A. La Mela and E. Nicolás, J. Org. Chem., 2014, 79, 9578 CrossRef PubMed; (j) P. W. Tan, M. Haughey and D. J. Dixon, Chem. Commun., 2015, 51, 4406 RSC; (k) H. Li, G.-X. Cai and Z.-J. Shi, Dalton Trans., 2010, 39, 10442 RSC.
- (a) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342 CrossRef CAS PubMed; (b) K. Orito, M. Miyazawa, T. Nakamura, A. Horibata, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita, T. Yamazaki and M. Tokuda, J. Org. Chem., 2006, 71, 5951 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and kinetic details. CCDC 1570476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03876c |
| ‡ These authors contributed equally, and are listed alphabetically. |
| This journal is © The Royal Society of Chemistry 2017 |