
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thiocyanopalladation/carbocyclization transformation identified through enzymatic screening: stereocontrolled tandem C–SCN and C–C bond formation†
G.
Malik‡
,
R. A.
Swyka‡
,
V. K.
Tiwari
,
X.
Fei
,
G. A.
Applegate
and
D. B.
Berkowitz
 *
*
Department of Chemistry, University of Nebraska, Lincoln, NE 68588, USA. E-mail: dberkowitz1@unl.edu
First published on 3rd October 2017
Abstract
Herein we describe a formal thiocyanopalladation/carbocyclization transformation and its parametrization and optimization using a new elevated temperature plate-based version of our visual colorimetric enzymatic screening method for reaction discovery. The carbocyclization step leads to C–SCN bond formation in tandem with C–C bond construction and is highly stereoselective, showing nearly absolute 1,2-anti-stereoinduction (5 examples) for substrates bearing allylic substitution, and nearly absolute 1,3-syn-stereoinduction (16 examples) for substrates bearing propargylic substitution. Based upon these high levels of stereoinduction, the dependence of the 1,2-stereoinduction upon cyclization substrate geometry, and the generally high preference for the transoid vinyl thiocyanate alkene geometry, a mechanistic model is proposed, involving (i) Pd(II)-enyne coordination, (ii) thiocyanopalladation, (iii) migratory insertion and (iv) β-elimination. Examples of transition metal-mediated C–SCN bond formation that proceed smoothly on unactivated substrates and allow for preservation of the SCN moiety are lacking. Yet, the thiocyanate functionality is of great value for biophysical chemistry (vibrational Stark effect) and medicinal chemistry (S,N-heterocycle construction). The title transformation accommodates C-, O-, N- and S-bridged substrates (6 examples), thereby providing the corresponding carbocyclic or heterocyclic scaffolds. The reaction is also shown to be compatible with a significant range of substituents, varying in steric and electronic demand, including a wide range of substituted aromatics, fused bicyclic and heterocyclic systems, and even biaryl systems. Combination of this new transformation with asymmetric allylation and Grubbs ring-closing metathesis provides for a streamlined enantio- and diastereoselective entry into the oxabicyclo[3.2.1]octyl core of the natural products massarilactone and annuionone A, as also evidenced by low temperature X-ray crystal structure determination. Utilizing this bicyclic scaffold, we demonstrate the versatility of the thiocyanate moiety for structural diversification post-cyclization. Thus, the bridging vinyl thiocyanate moiety is smoothly elaborated into a range of derivative functionalities utilizing transformations that cleave the S–CN bond, add the elements of RS-CN across a π-system and exploit the SCN moiety as a cycloaddition partner (7 diverse examples). Among the new functionalities thereby generated are thiotetrazole and sulfonyl tetrazole heterocycles that serve as carboxylate and phosphate surrogates, respectively, highlighting the potential of this approach for future applications in medicinal chemistry or chemical biology.
Introduction
Described herein are our efforts to develop new chemistry for the introduction of the thiocyano functionality into natural product core structures with control of stereochemistry. While important strides have been made in developing transition metal (TM)-mediated bond constructions with simple thiolates,1 TM-based C–S bond formation still lags well behind the corresponding C–C, C–N and C–O-bond formation chemistry. In particular, there are few such reaction manifolds for installing valuable C–SCN bonds.2,3 Two issues loom large here, the longstanding documentation of TM-catalyst poisoning by thiol species1a,4 especially thiocyanate anion,5 and the lability of the SCN functionality.6 The title transformation emerged from a wider search for transformations of synthetic utility employing our in situ enzymatic screening (ISES) approach to reaction discovery.The ISES approach is part of a recent expansion of research in both the synthetic organic and process chemistry communities that explores the interplay of enzymatic chemistry and traditional synthetic chemistry. While there is an important spectrum of activity at this biocatalysis/synthesis interface,7–12 we have been particularly focused on the use of enzymes to screen potential catalytic organic and organometallic combinations across chemical transformations of interest with attention to both throughput13 and information content.14,15 In ISES, enzymes report directly to the experimentalist upon the course of a matrix of test reactions of synthetic organic interest, without the need to draw aliquots, quench or chromatograph samples.16,17 Particularly useful in these endeavors has been the development of visual colorimetric ISES;18 as depicted in Fig. 1A. Note that in this system, successful turnover of an alkyl carbonate substrate by a transition metal catalyst, for example, leads to diffusion of the alcohol-presumably following spontaneous decarboxylation of the alkyl carbonate leaving group-into the adjacent aqueous layer where two ‘reporting enzymes’ are present. In the aqueous ‘reporting layer’, alcohol oxidase oxidizes the alcohol to the corresponding aldehyde, thereby generating an H2O2 equivalent that is itself captured by a second reporting enzyme, peroxidase, and reduced to H2O with the assistance of an ABTS dye cofactor. This reporting chemistry is reminiscent of ELISA technology, with the redox active dye generating two equivalents of the ABTS radical cation per substrate turnover event. This chromophore is both intense (ε404–414 ∼ 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for two equivalents) and absorbs in the visible, leading to the appearance of green color in the enzymatic reporting layer, easily to visible the naked eye.
000 M−1 cm−1 for two equivalents) and absorbs in the visible, leading to the appearance of green color in the enzymatic reporting layer, easily to visible the naked eye.
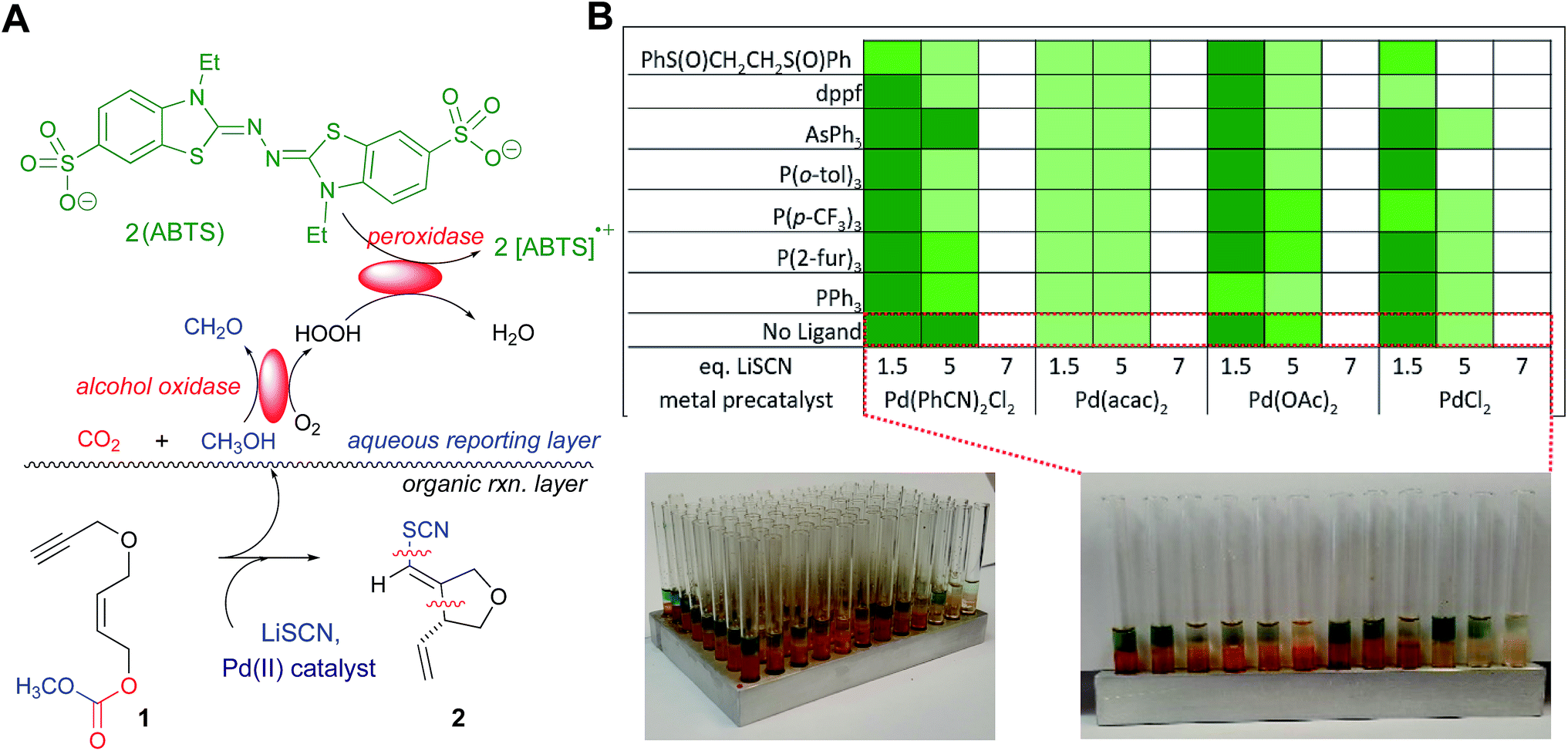 | ||
| Fig. 1 A new elevated temperature format for the plate-based colorimetric enzymatic screen and its use in parametrizing and optimizing the new thiocyanopalladation/carbocyclization transformation. (A) Schematic of the colorimetric, enzyme-based screen for this new bond construction. Note: the green color is due to ABTS radical cation formed from the alcohol oxidase/peroxidase reporting enzyme couple, with intensity of the signal related to the efficiency of organometallic reaction screened. (B) Table illustrating the reaction parameters being probed here – nature of the Pd(II) catalyst, LiSCN loading, and ligand effects. Relative intensity of green shading indicates relative reaction progress after 15 min. (C) Images of the colorimetric enzymatic screen – entire 96-well aluminum plate after heating to 70 °C (sand bath). (D) Close-up view of the first row (white paper backing for clarity; dotted lines show how this row maps onto the schematic) highlighting the effect of LiSCN loading across the array of Pd(II) catalysts screened. | ||
Results and discussion
Utilizing enzymatic screening as a validation/parametrization tool
Utilizing the ISES platform illustrated in Fig. 1A, a broad-based search for (pseudo)halo-carbocyclization chemistry was previously conducted across a matrix of 64 transition metal (TM) complexes × 6 (pseudo)halides × 3 substrates.18b This type of transformation is of particular interest to our group as it results in the installation of (pseudo)halovinyl moiety in the product, in keeping with our longstanding interest in developing halovinyl functionalities for mechanism-based enzyme inhibitors.19 Of the 1152 combinations screened, perhaps the most interesting and unprecedented discovery was the formal transition metal-mediated thiocyano-carbocyclization through which 5-exo-trig ether 1 yields furanoid system 2 upon treatment with LiSCN/PdCl2(PhCN)2. To our knowledge, this single hit in our laboratory is the only example of such a TM-mediated carbocyclization in which C–SCN and C–C bonds are formed concomitantly. Indeed, the only precedents for TM-mediated C–SCN bond formation of which we are aware require special activation conditions. For example, there are a couple of recent examples of additions across π-systems with TMSSCN and a highly activated electrophilic hypervalent iodine–CF3 species.2 And there is a report of a sequence involving TM-mediated arene iodination, followed by non-metal-mediated substitution with thiocyanate.3In this article, we describe the use of a new elevated temperature plate-based variant of colorimetric, enzymatic screening that allows us validate and parametrize this new transformation, and we report on the scope, stereochemical course and application of this chemistry to natural product core synthesis. Finally, by combining stereochemical probe substrates-varying alkene geometry and utilizing strategically placed resident stereocenters-we put forth a mechanistic postulate for this thiocyanopalladation/carbocyclization, consistent with the results of these studies.
Under the conditions of the initial screen, an excess of LiSCN was employed and Pd(II) catalyst-loading was at 10 mol% in an organic layer composed of THF/1,1,2-trichloroethane (TCE) at room temperature over ∼20 min. Building on this single lead data point, we began our studies here with a rapid colorimetric screen across T (35–70 °C in 5 °C increments) and Pd-loading (2.5–10 mol%; see ESI† for details). This led to the conclusion that further screens should be at 60 °C and that 2.5 mol% Pd(II) would suffice. Given these observations, we set out to develop a thermal version of this visual colorimetric ISES. Accordingly, an aluminum 96 well tray (rapidly fabricated in any machine shop) was utilized as it was found that this platform could be conveniently heated in a sand bath. After some experimentation, it was established that the most efficient protocol here involved layering all of the organic/organometallic transformation samples to be screened into the respective wells/tubes in the tray, heating the 8 × 12 array to 60 °C for 15 min, cooling and then layering the aqueous reporting layers (100 μL each) thereupon, using a multichannel pipeter containing 8 arms/pipet tips. Following a 10 min enzymatic development period, the reporting well intensities were catalogued and color-coded accordingly (see Fig. 1C, and the ESI† for more details).
As highlighted in Fig. 1B, four palladium sources were screened: PdCl2(PhCN)2, Pd(acac)2, Pd(OAc)2, and PdCl2. It was found that in addition to PdCl2(PhCN)2, Pd(OAc)2, and PdCl2 also support this chemistry, but Pd(acac)2 appears to but an unacceptable Pd(II) source for this transformation. Most importantly, a significant effect of LiSCN loading on reaction efficiency was clearly visible across all viable Pd(II) catalysts screened. As can be seen in Fig. 1B and C, lower loadings of thiocyanate promote the reaction more effectively, perhaps owing to reduced catalyst poisoning. A preliminary ligand screen was also conducted. The results indicated that whereas sulfoxide ligands appear to inhibit the title transformation, mono- and bidentate phosphine ligands are at least compatible with the cyclization, opening up the possibility of more extensive ligand exploration in the future.
Overall then, this elevated temperature plate-based enzymatic screen led to the conclusion that the title transformation proceeds well at 2.5 mol% Pd(II) with 1.5 eq. of LiSCN in THF (or THF/TCE in the screening) with heating to ∼60 °C (see ESI† for details). In this way, this initial screening-based optimization set the key parameters for studies described here in which we examine the scope of this new transformation with particular attention to functional group tolerance, nature of the bridging functionality and stereochemical course.
We are pleased to report that the Pd(II)-mediated thiocyanocarbocyclization tolerates sulfur-, nitrogen- and carboxylate ester functionalities in the bridging position, and proceeds in very good yield, in general (Table 1). In most cases, high selectivity in favor of the transoid alkene is observed. Thus, the four C- and N-bridged systems, 4, 6, 8, 10 all proceed with transoid selectivities >10:1, and as high as 30:1, in the case of the bridging trifluoroacetamide (6). Only for the bridging chalcogenides is this selectivity more modest, with the ether substrate 1 delivering geometric isomers 2t and 2c in a 4:1 ratio and thioether substrate 11 showing complete erosion of this selectivity. The general trend toward transoid products in these Pd(II)-mediated processes is consistent with initial delivery of the elements of SCN and Pd(II) across the alkyne in anti-fashion (vide infra). This anti-addition aligns with observations in other types metal-mediated enyne cyclizations reported by Lu and Ma,20 Cook21 and by our group.18b
|
|
|||
|---|---|---|---|
| Substrate | Product | X | Yield (t:c)a |
|
a Ratio determined by crude 1H NMR.
b Based on recovered starting material.
c See ESI materials for compound structure.
d dr 1:1.5 ratio in the major transoid product.
|
|||
| 1 | 2 | O | 81% (4:1) |
| 3 | 4 | NTs | 85% (11:1) |
| 5 | 6 | NCOCF3 | 87% (30:1) |
| 7 | 8 | C(COOEt)2 | 60b% (24:1) |
| 9 | 10 | CH(COOEt) | 80% (13:1)d |
| 11 | 12 | S | 85% (1:1.5) |
| 13 | SO2 | Elimination byproduct 14 | |
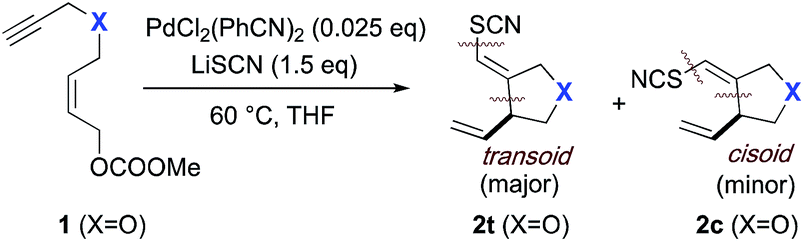
The mixtures of transoid/cisoid products observed for substrates 1 and 11 may be indicative of either a combination of external and internal (i.e. from the metal center) thiocyanate delivery, in these cases, or could be the result of a rapid pre-equilibration in the thiocyanometallation step, prior to migratory insertion (vide infra). No cyclization product was observed with sulfone-bridged substrate 13. Instead a conjugated sulfonyl butadiene species 14 was isolated [see ESI† for details of the formation of byproduct 14 = 1-(2′-thiocyanatoallylsulfonyl)buta-1E,3-diene; formula: C8H9NO2S2] presumably via 1,4-elimination of the carbonate facilitated by the enhanced C–H acidity of α to the sulfonyl functionality.
Structure elucidation
Alkene geometry was unambiguously established by a combination of X-ray crystallography and chemical shift correlation using 1H and 13C NMR spectroscopy (Fig. 2). The crystal structure of the NTs-bridged cyclized product 9 was solved and confirms the transoid alkene geometry in the predominant isomer. This result allowed us to align olefin geometry with the chemical shift trends seen in the NMR signatures, particularly of the vinyl thiocyanate functionality. Specifically, the cisoid cyclization products display upfield quaternary vinylic carbon signals (120–122 ppm) relative to those of their transoid counterparts (150–155 ppm). And the transoid cyclization products have upfield-shifted vinyl thiocyanate methine protons (5.8–5.9 ppm) relative to the corresponding methane signals in the cisoid isomers (6.2–6.3 ppm). The crystal structure of 4 also reveals the likely origin of this upfield shift; namely for the vinyl thiocyanate methane H appears to lie within the shielding cone of the π-system from the neighboring terminal vinyl group. The distances between this methine hydrogen and these vinylic carbons are 2.77 and 3.30 Å, respectively, within the range expected for π-shielding effects.22 | ||
| Fig. 2 Assignment of alkene geometry using a combination of X-ray crystallography and correlated 1H and 13C chemical shift patterns. Note the clear positioning of the vinyl thiocyanate methine proton in the shielding region above the π-system of the vinyl group in the transoid isomer, resulting in an ∼0.4 ppm upfield shift, relative to the cisoid isomer. | ||
Stereocontrol and mechanism
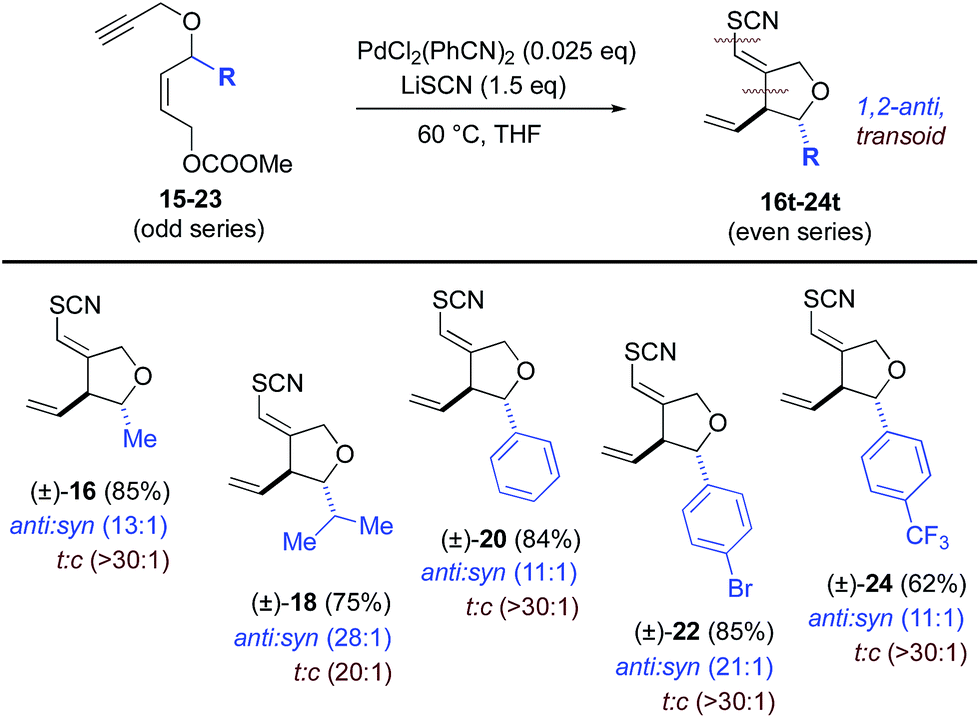
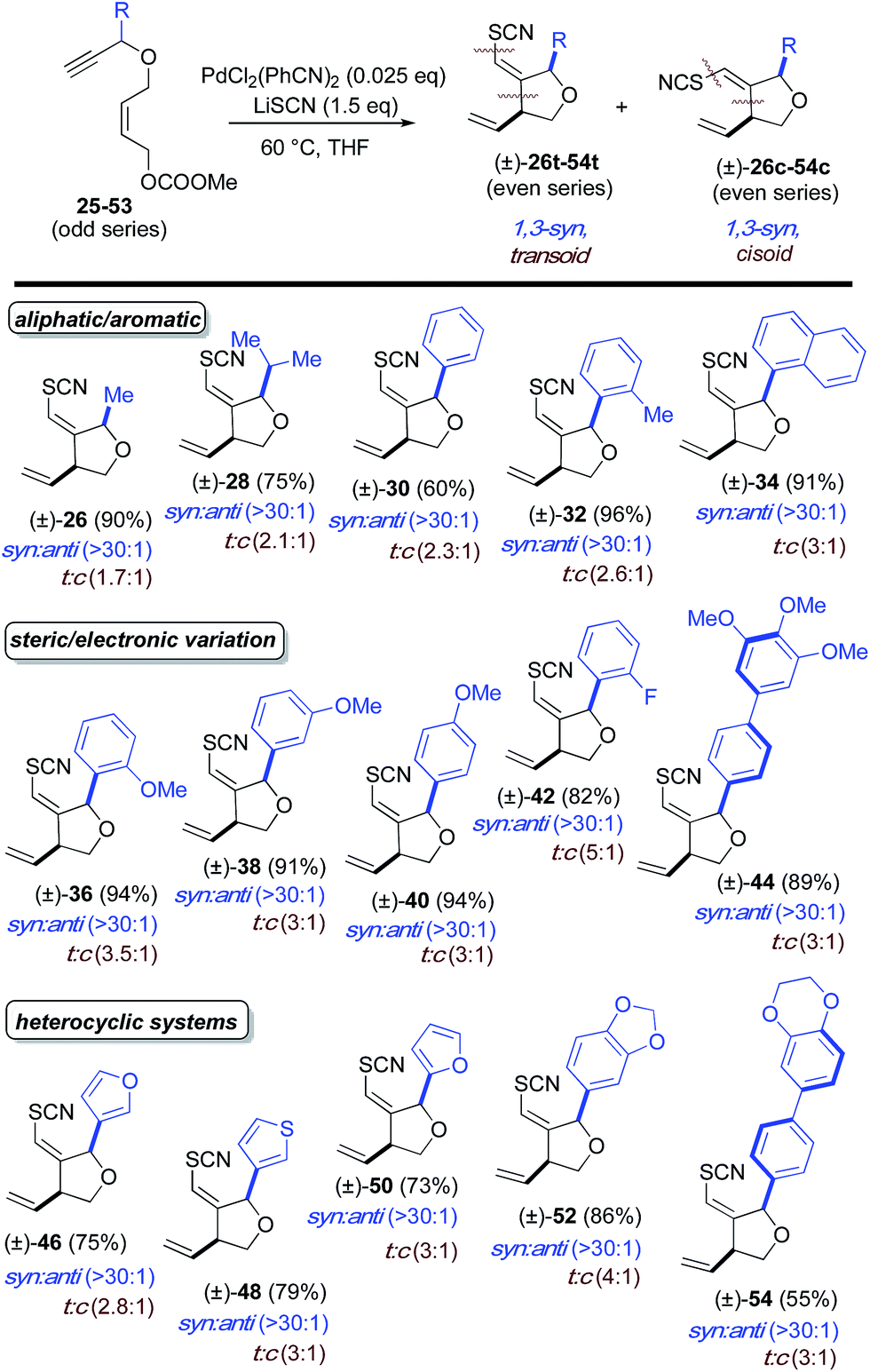
Next, stereochemical course was examined, specifically, the use of extant stereocenters to set relative stereochemistry in the thiocyanopalladation/carbocyclization product. The effects of substituents at both the allylic (Table 2) and propargylic (Table 3) positions upon induction of stereochemistry at the newly formed center were systematically examined. In the former allyl-substituted systems, the 1,2-anti product predominates in ratios greater than 10:1 for all examples tested. In the latter, propargyl-substituted systems, nearly absolute syn-1,3-stereocontrol is observed for all entries, in both geometric isomers of the product. For both substitution patterns, 2D NOESY NMR experiments were conducted to establish these relative stereochemistries. Functional groups accommodated by the title transformation include F (42), Br (22), OMe (36–40), CF3 (24), OCH2O (52), N-Ts (4), NCOCF3 (6), CO2Et (8, 10) and SR (12) (see Tables 1–3). Simple alkyl substituents, such as Me (16, 26) and the sterically demanding i-Pr (18, 28) are well tolerated at both the allylic and propargylic positions. A positional survey was conducted with the methoxy substituent in the propargylic series, with o-, m- and p-OMe (36–40) substrates all giving highly efficient and stereocontrolled cyclization. Heterocycles examined include 2- (50) and 3-furyl (46), 3-thiophenyl (48) and 1,4-dioxane (54) systems. Fused systems are also tolerated, including 1-naphthyl (34) and benzodioxane (54), and the interesting, more highly extended, biaryl systems 44 and 54 also undergo efficient cyclization.
|
a Reaction conditions: 2.5 mol% of PdCl2(PhCN)2 and 1.5 eq. of LiSCN. 5 mol% of PdCl2(PhCN)2 were employed for substrates 15, 17, 19. Ratios were determined by crude 1H NMR. Ratios >30:1 assume an NMR detection limit of approximately 3%; anti:syn ratios were determined by crude NMR for the major alkene formed.
|
|---|
|
|
a Reaction conditions: 2.5 mol% of PdCl2(PhCN)2 and 1.5 eq. of LiSCN. Ratios were determined by crude 1H NMR. Ratios >30:1 assume an NMR detection limit of approximately 3%; anti:syn ratios were determined by crude NMR for the major alkene formed.
|
|---|
|
Selectivity toward the transoid alkene is seen throughout. Most notably, the allyl-substituted systems proceed with a t:c ratio greater than 20:1 while a more modest transoid preference is seen in the propargyl-substituted systems, ranging form 1.7:1 to 5:1. No reaction was observed for R = tBu in the propargylic positon, possibly due to steric encumbrance of thiocyanate approach anti to Pd(II), although initial metal-coordination could also be retarded in this system. From a mechanistic point of view the p-bromophenyl example (22) is intriguing. As will be seen, the mechanism proposed here employs a Pd(II) species throughout. The absence of products resulting from oxidative addition into the C(sp2)-Br bond in 22 is consistent with this mechanism. Moreover, the ability to carry aryl bromide functionality through this new thiocyanocarbocyclization opens up opportunities for additional elaboration of the cyclization products via cross-coupling reactions.
Scheme 1 depicts the mechanism that we currently favor for this thiocyanopalladation/carbocyclization transformation; one that appears to be consistent with the high levels of 1,2- and 1,3-stereoinduction observed. For clarity, the mechanism is illustrated separately for substrates bearing allylic substitution in panel A and for substrates bearing propargylic substitution in panel B. As would be expected for an enyne cyclization, initial coordination of palladium to the 1,6-enyne is proposed, followed by anti-thiocyanopalladation of the triple bond to give vinylpalladium intermediates II and VI, respectively. External nucleophilic delivery of thiocyanate to a Pd(II)-activated alkyne is in accordance with preponderance of transoid products seen across most test substrates. That said, note that Zhang et al. have reported internal halide delivery from a metal center in the case of rhodium-catalyzed cycloisomerizations. These reactions generally give cisoid halovinyl products.23 As has already been noted, such competing mechanisms may be operative for the S-bridged substrate 11.
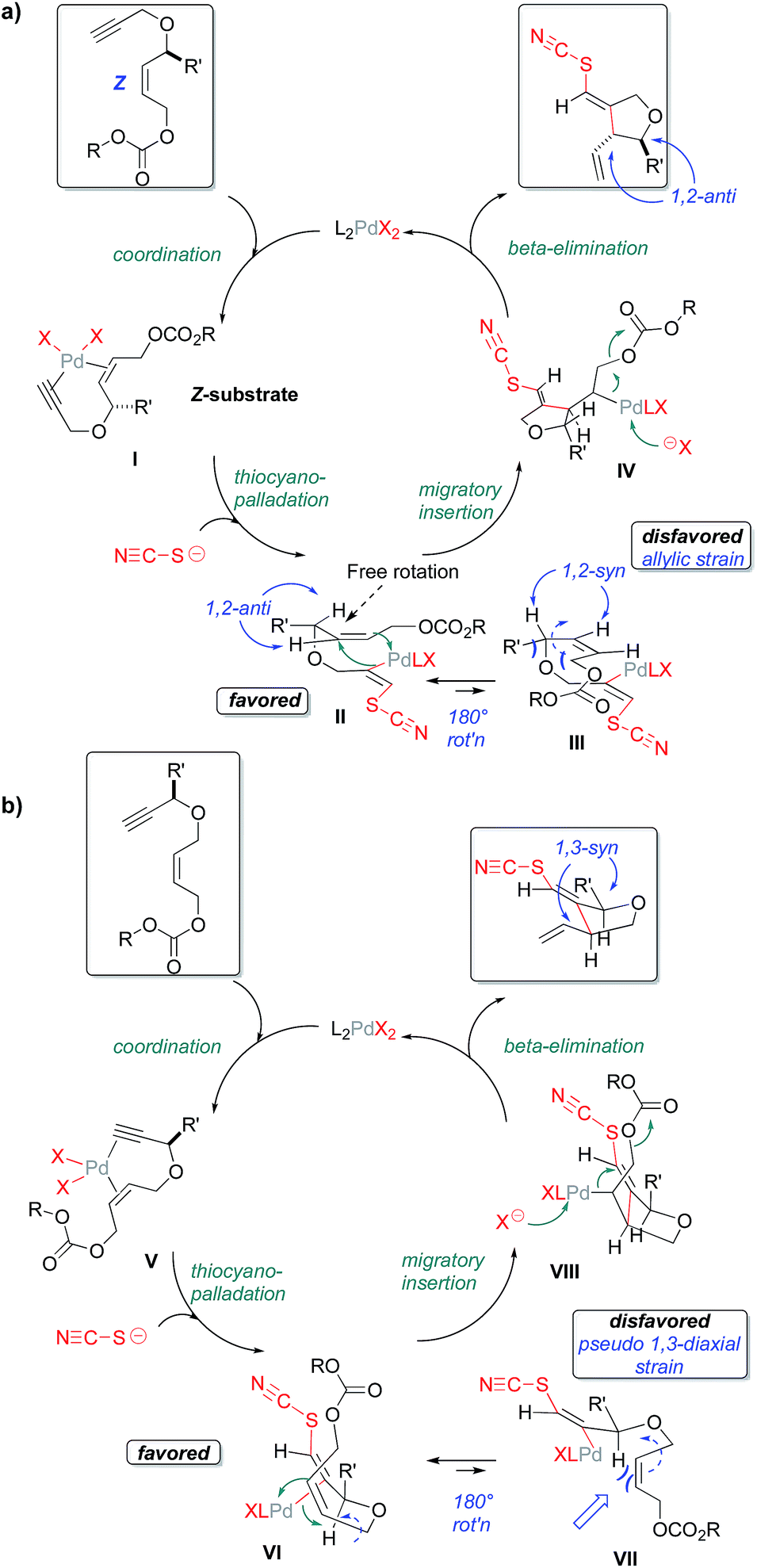 | ||
| Scheme 1 Proposed mechanism for the thiocyanopalladation/carbocyclization. | ||
As an independent test of this mechanism, an enyne substrate bearing an E-configured allylic carbonate was prepared (Scheme 2) and subjected to the cyclization. In this case, a mixture of anti- and syn-1,2 products (syn:anti 1.2:1) is observed. This is to be expected, as the E-alkene geometry opens up a possible migratory insertion transition state leading to the 1,2-syn-configured oxacycle, as well as that leading to the previously seen 1,2-anti-configured oxacycle. This is because the E-olefin geometry in the cyclization substrate relieves the allylic strain inherent in the 1,2-syn-leading transition state for the Z-configured substrate (Scheme 2). The fact that the Z- and E-configured allylic carbonates give such different product distributions also argues against mechanisms proceeding through rapidly equilibrating π-allyl intermediates.
 | ||
| Scheme 2 Starting alkene geometry strongly influences relative stereochemistry. | ||
Stereocontrolled entry into NP core structures
We next set out to exploit the syn-1,3-selectivity observed for propargyl-substituted substrates to develop a streamlined entry into a functionalizable scaffold that maps onto the bridged, bicyclic core of the natural products(NPs) annuionone A24 and massarilactone G25 (Table 4 and Scheme 3). The plan here was to carry a terminal ‘spectator alkene’ into the new cyclization reaction, thereby positioning this alkene for a post-cyclization ring closing metathesis (RCM). There is interest in the annuionones as these NPs display allelopathic properties,26 whereas massarilactones have shown neuraminidase inhibition activity27 and some anti-cancer activity, in vitro.28 This approach was motivated by the work of Waldmann29 and Stockwell,30 both of whom have argued for the ‘privileged’ nature of NP core structures when generating unnatural compound libraries for chemical biology.|
|
||||
|---|---|---|---|---|
| Entry | R | Conditions | Yield | eea |
| a Sign translates to the optical rotation of the major enantiomer obtained. | ||||
| 1 | TMS | (R)-BINOL/Ti(OiPr)4 (10 mol%) tri-n-butylallylstannane, DCM, −20 °C | 25% | +27% |
| 2 | TMS | (−)-DIP-OMe, allylmagnesium chloride, ether, −100 °C | 85% | −64% |
| 3 | TMS | (−)-DIP-Cl, allylmagnesium chloride, ether, −78 °C | 91% | −90% |
| 4 | TMS | Leighton reagent A, DCM, 0 °C | 61% | +65% |
| 5 | TMS | Leighton reagent B, DCM, 0 °C | 35% | +88% |
| 6 | TBS | Leighton reagent B, DCM, 0 °C | 20% | +92% |
| 7 | TBS | (−)-DIP-Cl, allylmagnesium chloride, ether, −78 °C | 77% | −90% |
 | ||
| Scheme 3 Rapid stereoselective access to oxabicyclo[3.2.1]octyl natural product cores. The new cyclization reaction affords core structures outfitted with a versatile thiocyanate functionality as a bioorthogonal probe in and of itself or as a template for subsequent functionalization. | ||
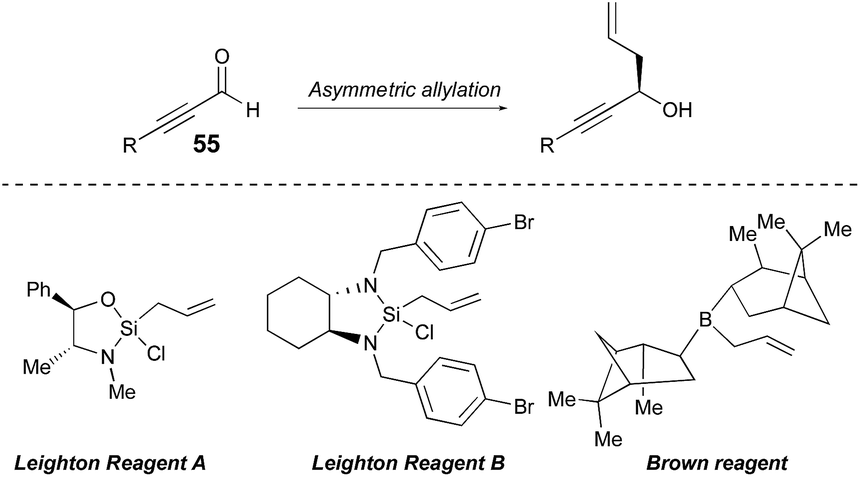
Setting the absolute stereochemistry via asymmetric allylation
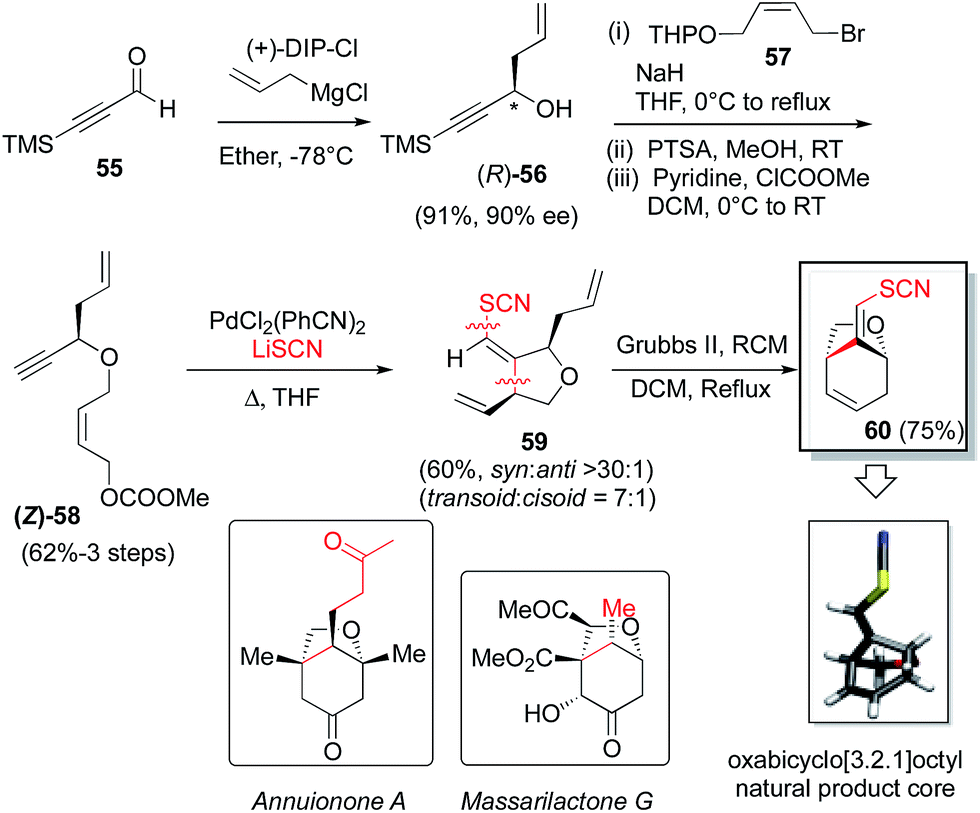
In this endeavor it was envisioned that the absolute stereochemistry could be set via asymmetric allylation of silyl propiolaldehyde (55). There was some precedent for such an approach, albeit with modest success, utilizing either Soderquist's chiral B-allyl-borabicyclo[3.3.2]decane reagent31 (76%, 77% ee)32 or the Duthaler–Hafner allyl titanium TADDOLate reagent33 (62%, 85% ee).32 Accordingly, a range of alternative, asymmetric allylation chemistries were examined here with variation of the metal, the chiral element, the silyl protecting group and the temperature (Table 4). Absolute configuration was determined using reported optical rotation where possible and ultimately confirmed via X-ray crystallography upon the eventual NP core structure.Generation of the Keck-type allyltitanium species via trans-metallation from the allyl stannane in situ with allyl stannane gave low conversions and low ee as well (entry 1).34 Preparation of allyl diisopinocampheylborane Brown reagent from (−)-DIP-OMe and (−)-DIP-Cl and allylmagnesium chloride showed promise (entries 2 and 3). We then turned our attention to the first and second generation allylsilane reagents developed by Leighton from pseudoephedrine35 and trans-1,2-diaminocyclohexane,36 respectively. These reagents presumably act via ‘strain-release Lewis acidity’ as first articulated by Denmark.37 In the event, we were able to achieve 88% ee and 92% ee by pairing the TMS- and TBS-protected propionaldehyde substrates, respectively, with Leighton reagent B (entries 5 and 6). However, very modest yields were observed in both cases (≤35%) and moving to TBS-protected propiolaldehyde with DIP-Cl and allyl magnesium chloride did not improve the ee. Therefore, we chose to retain TMS protection. This is also synthetically expedient, as this silyl ether is cleaved in the course of the subsequent allyl bromide displacement step, presumably due to attack of the released bromide, thereby obviating an extra deprotection step. Thus, utilizing the optimal conditions found, and adjusting for the desired absolute stereochemistry, then, the Brown reagent38 was generated in situ from (+)-DIP-Cl and allylmagnesium chloride following the protocol of Brimble39 and provided the properly configured allylic alcohol 5632 in 91% yield and 90% ee.
Next, deprotonation of alcohol 56, followed by allylic bromide displacement, O-THP-deprotection and carbonate installation gave substrate 58. Thiocyanopalladation/carbocyclization then furnished the desired syn-1,3-tetrahydrofuranoid system, 59 which, upon Grubbs II-catalyzed ring-closing metathesis, provided the oxabicyclo[3.2.1]octyl NP core 60. The absolute stereochemistry of 60 was confirmed by X-ray crystallography utilizing anomalous dispersion with sulfur serving as the requisite heavy atom (see ESI† for details).
Tailoring chemistry – opportunities for DOS
The new reaction modality reported here for introduction of the vinyl thiocyanate functionality into potential protein ligands represents a valuable tool for chemical biology. There has been great interest in exploiting the SCN functional group as a bioorthogonal probe of active site electrostatic environment via vibrational Stark effect studies40 as described by Boxer41 and others42 [for an interesting recent example combining the GFP fluorophore with a Stark effect IR probe, see ref. 43]. A particularly elegant recent example from Hammes–Schiffer and Benkovic utilizes a surgically embedded SCN reporter group in DHFR to demonstrate how the active site microenvironment subtly changes dielectric as the enzyme proceeds along the reaction coordinate for dihydrofolate reduction.28 The title Pd(II)-mediated transformation should allow practitioners to place SCN IR-reporter groups into ligand scaffolds as well, as has been done for nitrile functional groups.44In addition to being a useful bioorthogonal probe for such vibrational Stark effect studies, the thiocyanate moiety is also an outstanding platform for diversity oriented synthesis (DOS). Thus, as is demonstrated herein, each library member bearing an SCN can be selectively diversified by tapping into the unique reactivity of this underexploited functional group. Briefly, the value of such diversification via SCN-tailoring chemistry is significant given current interest in methods to rapidly access novel chemical space.45–47
In the event, the thiocyanate functionality could be utilized as a vehicle to generate aromatic or aliphatic thioethers by condensation with the appropriate Grignard reagent (Scheme 4). These reactions proceeded with clean S–CN cleavage; no competing conjugate addition was observed. In a particularly noteworthy example, selective magnesiation of 4-chlorobromobenzene, followed by condensation with the vinyl thiocyanate 60 providing an aromatic thioether 61, bearing aryl chloride functionality, itself amenable to further elaboration via cross-coupling chemistry.48 Reaction of 60 with isobutylmagnesium chloride and with methylenedioxyphenylmagnesium bromide, led cleanly to thioethers 62 and 63, respectively, demonstrating the generality of this homologation chemistry. S-trifluoromethylation was also achieved upon treatment of 60 with TMSCF3 and TBAF, providing a facile method for accessing the interesting vinyl trifluoromethyl thioether functionality (64).49 In an alternative approach to ‘cleaving’ the SCN group, one may ‘split’ the SCN moiety through the cyanothiolation of alkynes under Pd catalysis. In this manner, vinyl thiocyanate 60 was successfully added across 1-hexyne50 to regioselectively provide tri-substituted alkene 65. Note that, in our hands, this cyanothiolation reaction shows a strong ligand dependence, as investigated by comparing the reactivity of various Pd2dba3-ligand combinations. It was observed that whereas PPh3, Ph2P(CH2)3PPh2 (DPPP) and Pfur3 all support this chemistry well, AsPh3 gives greatly attenuated reactivity and the P(C6F5)3 ligand fails to give the desired alkyne addition product. Of particular interest from a chemical biology perspective, cycloaddition of 60 with sodium azide51 leads to thiotetrazole motif 66, itself a carboxylate surrogate. Mild sulfur oxidation with dimethyl dioxirane then smoothly generates sulfonyl tetrazole 67, a useful phosphate surrogate and a potential chemical tagging tool.52
 | ||
| Scheme 4 Exploiting the SCN moiety for structural diversification. | ||
Conclusions and future directions
In summary, a new elevated temperature plate-based platform for the colorimetric enzymatic screening method has been developed. This thermal enzymatic screening platform is a useful tool for the identification, validation and parameterization of transformations of interest, as is demonstrated herein for a promising new thiocyanopalladation/carbocyclization transformation uncovered in our laboratory.18b This transformation provides ready access to carbacyclic products with malonate-based substrates. Perhaps, more significantly, this Pd(II)-mediated thiocyanato-carbocyclization chemistry also provides for a streamlined and stereocontrolled entry into heterocyclic systems. Indeed, N, O, and S-bridging functionalities are well tolerated in the carbocyclization step, provided that the sulfur is not fully oxidized to the sulfonyl (elimination side reaction observed). In terms of stereochemical course, the thiocyanopalladation/carbocyclization proceeds with nearly absolute anti-1,2- and syn-1,3-stereoselectivity upon incorporation of substituents at the allylic and the propargylic positions, respectively, in the enyne substrate. A broad array of structurally diverse test substrates highlights the functional group tolerance of the title transformation, its stereochemical fidelity and its utility for rapidly increasing molecular complexity in a controlled manner.A mechanism consistent with the results of all probe substrate experiments is posited. The key observations include: (i) the notable stereochemical outcomes of probe substrates bearing propargylic (Scheme 1a – pseudo-1,3-diequatorial preference) and allylic substitution (Scheme 2b – allylic strain model), (ii) the observed product vinyl thiocyanate geometry (implies predominantly anti-addition of SCN and Pd across the substrate alkyne) and (iii) the formation of a significant level of syn-1,2-product from the cyclization of a transoid-allylic carbonate substrate (relief of allylic strain – Scheme 2). Taking into account all of these observations, the following sequence is proposed for the predominant mechanism: (i) eneyne coordination; (ii) thiocyanometallation via external delivery of thiocyanate to the Pd(II) complexed alkyne – consistent with the predominant anti-configuration of the new NCS–C and C–C bonds formed in the products; (iii) migratory insertion – consistent with the 1,2-transoid alkyne addition geometry generally observed; (iv) β-elimination to the final product with release of the Pd(II)-catalyst for the next cycle.
Finally, owing to the exquisite level of stereoselectivity in the cyclization, one can enter the cyclization manifold with enantioenriched substrate and utilize the title transformation to both construct C–SCN and C–C bonds in tandem and to access a single stereoisomeric product bearing two stereogenic centers and one stereogenic vinyl thiocyanate moiety. In the case at hand, a vinyl thiocyanate-appended oxabicyclo[3.2.1]octenyl system resembling the core of the natural products annuionone A and merrilactone G could be efficiently obtained with the title transformation serving as the key step. By combining the new chemistry with DIP-Cl-mediated asymmetric allylation, pre-cyclization, to set the absolute stereochemistry, and Grubbs-RCM, post-cyclization, to close the bicyclic system, one arrives at a strained, densely functionalized bicyclic system quickly, with a bioorthogonal SCN reporting group in place.
As discussed above, the transformation described here would appear to be a fundamentally new type of TM-mediated carbocyclization. The transformation bears some relation to the palladium-mediated formal ene carbocyclization due to Trost and Lautens53 and other metal-mediated enyne carbocyclizations in which a halide or acetate formally donates electron density into the alkyne to induce cyclization.20a–c,21,23,54–58 Trost also recently described related bond construction that may be viewed as a RuII-mediated internal redox bicycloisomerization.59
The ability to install an SCN functional group onto complex natural product-like ligand scaffolds is important as this will provide chemical biologists with a new tool for probing active site environments. The thiocyanate group provides an ideal spectroscopic window for vibrational Stark effect studies that are very sensitive to local electrostatic fields in the environment surrounding the functional group. This technique has been used to study protein ligand interactions, most notably by the groups of Boxer,41,44,60 Hammes–Schiffer,28,42c Londergan42a,42b and Webb.43,61 The most common approach is to generate an active site S–CN functionality by cyanating a cysteine residue with CN–Br. The complementary experiment, wherein the small molecule ligand carries the infrared probe is much rarer; we are aware of an example in which the nitrile functionality on an inhibitor of human aldose reductase was used to probe key substrate–ligand interactions.44 The example utilized a cyano substituent built into the aldose reductase inhibitor. Similar experiments with thiocyanate-bearing ligands are expected to be forthcoming in the future; the title transformation is expected to promote such studies by providing a streamlined entry into functionally dense ligand arrays bearing the SCN moiety, in short order and with control of stereochemistry. The potential for such ligand-based vibrational Stark effect experiments is high, as the observed SCN IR frequency is highly sensitive to the hydration sphere around the SCN functionality and to the hydrogen-bonding environment of the ligand.
Beyond their utility as an IR- or Raman-probes,40 in medicinal chemistry circles, thiocyanates have served as synthetic intermediates to useful drug-like heterocyclic scaffolds, particularly fused cyclic thioureas62 and substituted benzothiazoles61a,63 such as the 2-amino-benzothiazole-based peptidomimetic oligomers recently described by Hamilton that disrupt amyloid peptide fibrillation.64 In this study, we have highlighted the chemical versatility of the thiocyanate moiety, by generating an array of structural variants from a common NP core structure (Scheme 4),29,30 an approach that clearly holds promise for DOS applications.45,46,65 In surveying the literature, it seems clear that the thiocyanate moiety is an under-utilized functionality for chemical diversification in library development.45,46,65,66 In this regard, the new, versatile and stereoselective thiopalladation/carbocyclization transformation described herein will likely open up new vistas to the chemical biology community for both ligand-centered biophysical studies, and for exploiting the rich chemical potential of the SCN moiety.
Funding
The authors gratefully acknowledge the NSF (CHE-1500076) for support. The authors thank the NIH (SIG-1-510-RR-06307, RR016544) and the NSF (CHE-0091975, MRI-0079750, CHE-0923449) for instrumentation and facilities support.Author contributions
G. M., R. A. S., V. T. K., X. F. and G. A. A. performed the research; all authors analyzed and interpreted the results; D. B. B., G. M. and R. A. S. wrote the manuscript while V. T. K., X. F. and G. A. A. contributed to compiling the ESI.Conflict of interests
The authors declare that they have no competing financial interests.Acknowledgements
The authors wish to thank Victor W. Day (U. Kansas) and Douglas R. Powell (U. Oklahoma) for x-ray crystallographic structure determination. This research was facilitated by the IR/D (Individual Research and Development) program associated with DBB's appointment at the National Science Foundation.Notes and references
- (a) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (b) M. A. Fernandez-Rodriguez and J. F. Hartwig, Chem.–Eur. J., 2010, 16, 2355–2359 CrossRef CAS PubMed; (c) M. A. Fernandez-Rodriguez and J. F. Hartwig, J. Org. Chem., 2009, 74, 1663–1672 CrossRef CAS PubMed; (d) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed; (e) M. A. Fernandez-Rodriguez, Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180–2181 CrossRef CAS PubMed.
- (a) N. Zhu, F. Wang, P. Chen, J. Ye and G. Liu, Org. Lett., 2015, 17, 3580–3583 CrossRef CAS PubMed; (b) Z. Liang, F. Wang, P. Chen and G. Liu, Org. Lett., 2015, 17, 2438–2441 CrossRef CAS PubMed.
- F. Wang, X. Yu, Z. Qi and X. Li, Chem.–Eur. J., 2016, 22, 511–516 CrossRef CAS PubMed.
- (a) N. Gao, S. Zheng, W. Yang and X. Zhao, Org. Lett., 2011, 13, 1514–1516 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed; (c) T. Kondo and T.-a. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed.
- D. V. Sokol'skii and A. Y. Matveichuk, Izv. Akad. Nauk Kaz. SSR, Ser. Khim., 1967, 17, 39–43 Search PubMed.
- F. Ke, Y.-Y. Qu, Z.-Q. Jiang, Z.-K. Li, D. Wu and X.-G. Zhou, Org. Lett., 2011, 13, 454–457 CrossRef CAS PubMed.
- (a) S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS; (b) T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- (a) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed; (b) S. Lutz, Science, 2010, 329, 285–287 CrossRef CAS PubMed; (c) U. Arnold, M. P. Hinderaker, B. L. Nilsson, B. R. Huck, S. H. Gellman and R. T. Raines, J. Am. Chem. Soc., 2002, 124, 8522–8523 CrossRef CAS PubMed.
- (a) Y. Okamoto, V. Kohler and T. R. Ward, J. Am. Chem. Soc., 2016, 138, 5781–5784 CrossRef CAS PubMed; (b) T. K. Hyster, L. Knoerr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- (a) J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS PubMed; (b) M. M. Muller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2009, 48, 922–925 CrossRef CAS PubMed; (c) L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Roethlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas III, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- (a) K. Panigrahi, G. A. Applegate, G. Malik and D. B. Berkowitz, J. Am. Chem. Soc., 2015, 137, 3600–3609 CrossRef CAS PubMed; (b) H. Groeger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef CAS PubMed.
- (a) M. Shevlin, ACS Med. Chem. Lett., 2017, 8, 601–607 CrossRef CAS PubMed; (b) A. Buitrago Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49–53 CrossRef CAS PubMed; (c) J. L. Treece, J. R. Goodell, D. Vander Velde, J. A. Porco Jr and J. Aube, J. Org. Chem., 2010, 75, 2028–2038 CrossRef CAS PubMed; (d) E. Wolf, E. Richmond and J. Moran, Chem. Sci., 2015, 6, 2501–2505 RSC.
- (a) M. Teders, L. Pitzer, S. Buss and F. Glorius, ACS Catal., 2017, 7, 4053–4056 CrossRef CAS; (b) D. S. Mannel, M. S. Ahmed, T. W. Root and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 1690–1698 CrossRef CAS PubMed; (c) W. Bentley Keith, P. Zhang and C. Wolf, Sci. Adv., 2016, 2, e1501162 CAS; (d) K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859–871 CrossRef CAS PubMed; (e) J. R. Cabrera-Pardo, D. I. Chai, S. Liu, M. Mrksich and S. A. Kozmin, Nat. Chem., 2013, 5, 423–427 CrossRef CAS PubMed.
- (a) A. Hamberg, S. Lundgren, E. Wingstrand, C. Moberg and K. Hult, Chem.–Eur. J., 2007, 13, 4334–4341 CrossRef CAS PubMed; (b) A. Hamberg, S. Lundgren, M. Penhoat, C. Moberg and K. Hult, J. Am. Chem. Soc., 2006, 128, 2234–2235 CrossRef CAS PubMed; (c) C. M. Sprout and C. T. Seto, Org. Lett., 2005, 7, 5099–5102 CrossRef CAS PubMed.
- (a) D. B. Berkowitz and G. Maiti, Org. Lett., 2004, 6, 2661–2664 CrossRef CAS PubMed; (b) D. B. Berkowitz, M. Bose and S. Choi, Angew. Chem., Int. Ed., 2002, 41, 1603–1607 CrossRef CAS.
- (a) K. R. Karukurichi, X. Fei, R. A. Swyka, S. Broussy, W. Shen, S. Dey, S. K. Roy and D. B. Berkowitz, Sci. Adv., 2015, 1(6), e1500066 Search PubMed; (b) S. Dey, D. R. Powell, C. Hu and D. B. Berkowitz, Angew. Chem., Int. Ed., 2007, 46, 7010–7014 CrossRef CAS PubMed; (c) S. Dey, K. R. Karukurichi, W. Shen and D. B. Berkowitz, J. Am. Chem. Soc., 2005, 127, 8610–8611 CrossRef CAS PubMed.
- (a) S. K. Ginotra, J. A. Friest and D. B. Berkowitz, Org. Lett., 2012, 14, 968–971 CrossRef CAS PubMed; (b) J. A. Friest, S. Broussy, W. J. Chung and D. B. Berkowitz, Angew. Chem., Int. Ed., 2011, 50, 8895–8899 CrossRef CAS PubMed.
- (a) C. D. McCune, M. L. Beio, J. M. Sturdivant, R. de la Salud-Bea, B. M. Darnell and D. B. Berkowitz, J. Am. Chem. Soc., 2017, 139 DOI:10.1021/jacs.7b04690; (b) K. R. Karukurichi, R. de la Salud-Bea, W. J. Jahng and D. B. Berkowitz, J. Am. Chem. Soc., 2007, 129, 258–259 CrossRef CAS PubMed; (c) D. B. Berkowitz, R. de la Salud-Bea and W.-J. Jahng, Org. Lett., 2004, 6, 1821–1824 CrossRef CAS PubMed.
- (a) For pioneering work on Pd(II)-mediated carbocyclization reactions that inspired these studies, see: W. Xu, A. Kong and X. Lu, J. Org. Chem., 2006, 71, 3854–3858 CrossRef CAS PubMed; (b) L. Zhao, X. Lu and W. Xu, J. Org. Chem., 2005, 70, 4059–4063 CrossRef CAS PubMed; (c) X. Xie, X. Lu, Y. Liu and W. Xu, J. Org. Chem., 2001, 66, 6545–6550 CrossRef CAS PubMed; (d) X. Lu, G. Zhu, Z. Wang, S. Ma, J. Ji and Z. Zhang, Pure Appl. Chem., 1997, 69, 553–558 CrossRef CAS; (e) H. Jiang, S. Ma, G. Zhu and X. Lu, Tetrahedron, 1996, 52, 10945–10954 CrossRef CAS; (f) X. Lu, S. Ma, J. Ji, G. Zhu and H. Jiang, Pure Appl. Chem., 1994, 66, 1501–1508 CrossRef CAS.
- G. R. Cook and R. Hayashi, Org. Lett., 2006, 8, 1045–1048 CrossRef CAS PubMed.
- C. S. Wannere and P. v. R. Schleyer, Org. Lett., 2003, 5, 605–608 CrossRef CAS PubMed.
- X. Tong, D. Li, Z. Zhang and X. Zhang, J. Am. Chem. Soc., 2004, 126, 7601–7607 CrossRef CAS PubMed.
- F. A. Macías, R. M. Varela, A. Torres, R. M. Oliva and J. G. Molinillo, Phytochemistry, 1998, 48, 631–636 CrossRef.
- H. Oh, D. C. Swenson, J. B. Gloer and C. A. Shearer, Tetrahedron Lett., 2001, 42, 975–977 CrossRef CAS.
- (a) F. A. Macías, A. López, R. M. Varela, A. Torres and J. M. G. Molinillo, Phytochemistry, 2004, 65, 3057–3063 CrossRef PubMed; (b) T. Anjum and R. Bajwa, Phytochemistry, 2005, 66, 1919–1921 CrossRef CAS PubMed.
- G. F. Zhang, W. B. Han, J. T. Cui, S. W. Ng, Z. K. Guo, R. X. Tan and H. M. Ge, Planta Med., 2012, 78, 76–78 CrossRef CAS PubMed.
- S. Chen, F. Ren, S. Niu, X. Liu and Y. Che, J. Nat. Prod., 2014, 77, 9–14 CrossRef CAS PubMed.
- (a) R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed; (b) H. Lachance, S. Wetzel, K. Kumar and H. Waldmann, J. Med. Chem., 2012, 55, 5989–6001 CrossRef CAS PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- E. Canales, K. G. Prasad and J. A. Soderquist, J. Am. Chem. Soc., 2005, 127, 11572–11573 CrossRef CAS PubMed.
- S. B. Kamptmann and R. Brückner, Eur. J. Org. Chem., 2013, 2013, 6584–6600 CrossRef CAS.
- A. Hafner, R. O. Duthaler, R. Marti, G. Rihs, P. Rothe-Streit and F. Schwarzenbach, J. Am. Chem. Soc., 1992, 114, 2321–2336 CrossRef CAS.
- G. E. Keck, K. H. Tarbet and L. S. Geraci, J. Am. Chem. Soc., 1993, 115, 8467–8468 CrossRef CAS.
- X. Wang, Q. Meng, A. J. Nation and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 10672–10673 CrossRef CAS PubMed.
- (a) L. M. Suen, M. L. Steigerwald and J. L. Leighton, Chem. Sci., 2013, 4, 2413–2417 RSC; (b) W. A. Chalifoux, S. K. Reznik and J. L. Leighton, Nature, 2012, 487, 86–89 CAS; (c) X. Zhang, K. N. Houk and J. L. Leighton, Angew. Chem., Int. Ed., 2005, 44, 938–941 CrossRef CAS PubMed.
- S. E. Denmark, R. T. Jacobs, G. Dai-Ho and S. Wilson, Organometallics, 1990, 9, 3015–3019 CrossRef CAS.
- (a) H. C. Brown and P. V. Ramachandran, Pure Appl. Chem., 1994, 66, 201–212 CrossRef CAS; (b) U. S. Racherla and H. C. Brown, J. Org. Chem., 1991, 56, 401–404 CrossRef CAS.
- M. A. Brimble, P. Bachu and J. Sperry, Synthesis, 2007, 2007, 2887–2893 CrossRef.
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS PubMed.
- (a) A. T. Fafarman, L. J. Webb, J. I. Chuang and S. G. Boxer, J. Am. Chem. Soc., 2006, 128, 13356–13357 CrossRef CAS PubMed; (b) P. A. Sigala, A. T. Fafarman, P. E. Bogard, S. G. Boxer and D. Herschlag, J. Am. Chem. Soc., 2007, 129, 12104–12105 CrossRef CAS PubMed.
- (a) L. Edelstein, M. A. Stetz, H. A. McMahon and C. H. Londergan, J. Phys. Chem. B, 2010, 114, 4931–4936 CrossRef CAS PubMed; (b) H. A. McMahon, K. N. Alfieri, K. A. A. Clark and C. H. Londergan, J. Phys. Chem. Lett., 2010, 1, 850–855 CrossRef CAS PubMed; (c) J. P. Layfield and S. Hammes-Schiffer, J. Am. Chem. Soc., 2013, 135, 717–725 CrossRef CAS PubMed.
- J. D. Slocum and L. J. Webb, J. Am. Chem. Soc., 2016, 138, 6561–6570 CrossRef CAS PubMed.
- L. Xu, A. E. Cohen and S. G. Boxer, Biochemistry, 2011, 50, 8311–8322 CrossRef CAS PubMed.
- S. L. Schreiber, J. D. Kotz, M. Li, J. Aubé, C. P. Austin, J. C. Reed, H. Rosen, E. L. White, L. A. Sklar, C. W. Lindsley, B. R. Alexander, J. A. Bittker, P. A. Clemons, A. de Souza, M. A. Foley, M. Palmer, A. F. Shamji, M. J. Wawer, O. McManus, M. Wu, B. Zou, H. Yu, J. E. Golden, F. J. Schoenen, A. Simeonov, A. Jadhav, M. R. Jackson, A. B. Pinkerton, T. D. Y. Chung, P. R. Griffin, B. F. Cravatt, P. S. Hodder, W. R. Roush, E. Roberts, D.-H. Chung, C. B. Jonsson, J. W. Noah, W. E. Severson, S. Ananthan, B. Edwards, T. I. Oprea, P. J. Conn, C. R. Hopkins, M. R. Wood, S. R. Stauffer, K. A. Emmitte, L. S. Brady, J. Driscoll, I. Y. Li, C. R. Loomis, R. N. Margolis, E. Michelotti, M. E. Perry, A. Pillai and Y. Yao, Cell, 2015, 161, 1252–1265 CrossRef CAS PubMed.
- (a) T. D. Davis, C. J. Gerry and D. S. Tan, ACS Chem. Biol., 2014, 9, 2535–2544 CrossRef CAS PubMed; (b) D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS PubMed; (c) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef PubMed; (d) S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- B. D. Charette, R. G. MacDonald, S. Wetzel, D. B. Berkowitz and H. Waldmann, Angew. Chem., Int. Ed., 2006, 45, 7766–7770 CrossRef CAS PubMed.
- (a) J. Tang, A. Biafora and L. J. Goossen, Angew. Chem., Int. Ed., 2015, 54, 13130–13133 CrossRef CAS PubMed; (b) T. Iwai, T. Harada, K. Hara and M. Sawamura, Angew. Chem., Int. Ed., 2013, 52, 12322–12326 CrossRef CAS PubMed; (c) R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CrossRef CAS PubMed.
- (a) T. Billard, S. Large and B. R. Langlois, Tetrahedron Lett., 1997, 38, 65–68 CrossRef CAS; (b) M.-N. Bouchu, S. Large, M. Steng, B. Langlois and J.-P. Praly, Carbohydr. Res., 1998, 314, 37–45 CrossRef CAS.
- (a) I. Kamiya, J.-i. Kawakami, S. Yano, A. Nomoto and A. Ogawa, Organometallics, 2006, 25, 3562–3564 CrossRef CAS; (b) M. Pawliczek, L. K. B. Garve and D. B. Werz, Org. Lett., 2015, 17, 1716–1719 CrossRef CAS PubMed.
- S. Bhattacharya and P. K. Vemula, J. Org. Chem., 2005, 70, 9677–9685 CrossRef CAS PubMed.
- S. Otsuki, S. Nishimura, H. Takabatake, K. Nakajima, Y. Takasu, T. Yagura, Y. Sakai, A. Hattori and H. Kakeya, Bioorg. Med. Chem. Lett., 2013, 23, 1608–1611 CrossRef CAS PubMed.
- B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1985, 107, 1781–1783 CrossRef CAS.
- (a) J. Song, Q. Shen, F. Xu and X. Lu, Tetrahedron, 2007, 63, 5148–5153 CrossRef CAS; (b) Q. Zhang and X. Lu, J. Am. Chem. Soc., 2000, 122, 7604–7605 CrossRef CAS; (c) G. Zhu and X. Lu, Organometallics, 1995, 14, 4899–4904 CrossRef CAS.
- (a) X. Tong, Z. Zhang and X. Zhang, J. Am. Chem. Soc., 2003, 125, 6370–6371 CrossRef CAS PubMed; (b) A. Lei, M. He and X. Zhang, J. Am. Chem. Soc., 2002, 124, 8198–8199 CrossRef CAS PubMed; (c) P. Cao, B. Wang and X. Zhang, J. Am. Chem. Soc., 2000, 122, 6490–6491 CrossRef CAS.
- O. Jackowski, J. Wang, X. Xie, T. Ayad, Z. Zhang and V. Ratovelomanana-Vidal, Org. Lett., 2012, 14, 4006–4009 CrossRef CAS PubMed.
- L. L. Welbes, T. W. Lyons, K. A. Cychosz and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 5836–5837 CrossRef CAS PubMed.
- (a) M.-C. P. Yeh, H.-F. Chen, Y.-Y. Huang and Y.-T. Weng, J. Org. Chem., 2015, 80, 10892–10903 CrossRef CAS PubMed; (b) A. Saxena, F. Perez and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 1493–1497 CrossRef CAS PubMed; (c) G. M. R. Canlas and S. R. Gilbertson, Chem. Commun., 2014, 50, 5007–5010 RSC; (d) F. Grillet and K. M. Brummond, J. Org. Chem., 2013, 78, 3737–3754 CrossRef CAS PubMed; (e) S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569–20572 CrossRef CAS PubMed; (f) A. D. Jenkins, A. Herath, M. Song and J. Montgomery, J. Am. Chem. Soc., 2011, 133, 14460–14466 CrossRef CAS PubMed; (g) A. Fürstner, K. Majima, R. Martín, H. Krause, E. Kattnig, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 1992–2004 CrossRef PubMed; (h) M. Chen, Y. Weng, M. Guo, H. Zhang and A. Lei, Angew. Chem., 2008, 120, 2311–2314 CrossRef; (i) K. M. Brummond, H. Chen, B. Mitasev and A. D. Casarez, Org. Lett., 2004, 6, 2161–2163 CrossRef CAS PubMed; (j) A. Lei, J. P. Waldkirch, M. He and X. Zhang, Angew. Chem., Int. Ed., 2002, 41, 4526–4529 CrossRef CAS; (k) A. Lei, M. He, S. Wu and X. Zhang, Angew. Chem., Int. Ed., 2002, 41, 3457–3460 CrossRef CAS.
- B. M. Trost, M. C. Ryan, M. Rao and T. Z. Markovic, J. Am. Chem. Soc., 2014, 136, 17422–17425 CrossRef CAS PubMed.
- L. J. Webb and S. G. Boxer, Biochemistry, 2008, 47, 1588–1598 CrossRef CAS PubMed.
- (a) M. P. Hay, S. Turcotte, J. U. Flanagan, M. Bonnet, D. A. Chan, P. D. Sutphin, P. Nguyen, A. J. Giaccia and W. A. Denny, J. Med. Chem., 2010, 53, 787–797 CrossRef CAS PubMed; (b) A. J. Stafford, D. M. Walker and L. J. Webb, Biochemistry, 2012, 51, 2757–2767 CrossRef CAS PubMed.
- (a) M. Buchholz, U. Heiser, S. Schilling, A. J. Niestroj, K. Zunkel and H.-U. Demuth, J. Med. Chem., 2006, 49, 664–677 CrossRef CAS PubMed; (b) C. A. Parrish, N. D. Adams, K. R. Auger, J. L. Burgess, J. D. Carson, A. M. Chaudhari, R. A. Copeland, M. A. Diamond, C. A. Donatelli, K. J. Duffy, L. F. Faucette, J. T. Finer, W. F. Huffman, E. D. Hugger, J. R. Jackson, S. D. Knight, L. Luo, M. L. Moore, K. A. Newlander, L. H. Ridgers, R. Sakowicz, A. N. Shaw, C.-M. M. Sung, D. Sutton, K. W. Wood, S.-Y. Zhang, M. N. Zimmerman and D. Dhanak, J. Med. Chem., 2007, 50, 4939–4952 CrossRef CAS PubMed.
- Q. Chao, K. G. Sprankle, R. M. Grotzfeld, A. G. Lai, T. A. Carter, A. M. Velasco, R. N. Gunawardane, M. D. Cramer, M. F. Gardner, J. James, P. P. Zarrinkar, H. K. Patel and S. S. Bhagwat, J. Med. Chem., 2009, 52, 7808–7816 CrossRef CAS PubMed.
- H. Peacock, J. Luo, T. Yamashita, J. Luccarelli, S. Thompson and A. D. Hamilton, Chem. Sci., 2016, 7, 6435–6439 RSC.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- H. E. Pelish, N. J. Westwood, Y. Feng, T. Kirchhausen and M. D. Shair, J. Am. Chem. Soc., 2001, 123, 6740–6741 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Enzymatic screening procedures, detailed synthetic procedures and compilations of spectroscopic data, as well as copies of NMR spectra for new compounds. CCDC 1513686 and 1513687. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04083k |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |