
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of catabolite control protein A from Staphylococcus aureus as a target of silver ions†
Xiangwen
Liao
 ab,
Fang
Yang
a,
Runming
Wang
cd,
Xiaojun
He
a,
Hongyan
Li
c,
Richard Y. T.
Kao
d,
Wei
Xia
*a and
Hongzhe
Sun
*ac
ab,
Fang
Yang
a,
Runming
Wang
cd,
Xiaojun
He
a,
Hongyan
Li
c,
Richard Y. T.
Kao
d,
Wei
Xia
*a and
Hongzhe
Sun
*ac
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou, 510275, China. E-mail: xiawei5@mail.sysu.edu.cn
bHunan Provincial Key Laboratory for Ethnic Dong Medicine Research, Hunan University of Medicine, Huaihua, 418000, China
cDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: hsun@hku.hk
dDepartment of Microbiology, State Key Laboratory for Emerging Infectious Diseases, The University of Hong Kong, Hong Kong, P. R. China
First published on 2nd October 2017
Abstract
Staphylococcus aureus is one of the most common pathogenic bacteria that causes human infectious diseases. The emergence of antibiotic-resistant strains of S. aureus promotes the development of new anti-bacterial strategies. Silver ions (Ag+) have attracted profound attention due to their broad-spectrum antimicrobial activities. Although the antibacterial properties of silver have been well known for many centuries, its mechanism of action remains unclear and its protein targets are rarely reported. Herein, we identify the catabolite control protein A (CcpA) of S. aureus as a putative target for Ag+. CcpA binds 2 molar equivalents of Ag+via its two cysteine residues (Cys216 and Cys242). Importantly, Ag+ binding induces CcpA oligomerization and abolishes its DNA binding capability, which further attenuates S. aureus growth and suppresses α-hemolysin toxicity. This study extends our understanding of the bactericidal effects of silver.
Introduction
The main carbon catabolite repression (CCR) system is an important global control system of various bacteria, which allows the bacteria to adapt quickly to a preferred carbon source first. This is usually achieved by the repression of genes whose products are involved in the catabolism of alternative, less preferred carbon sources. In Gram-positive bacteria, a highly conserved regulator, catabolite control protein A (CcpA), exerts the important catabolite repression function.1 CcpA is usually activated by its co-regulator via the formation of a complex which recognizes the catabolite-responsive element (cre) sequences and regulates downstream gene expression.2Staphylococcus aureus (S. aureus), a worldwide spread human pathogen, is the leading cause of hospital- and community-acquired infections. The pathogen causes a series of human diseases ranging from minor skin infections to life-threatening sepsis.3 In particular, the emergence of drug-resistant strains of the bacteria, such as methicillin-resistant and vancomycin-resistant S. aureus, poses a huge threat to public health worldwide.4,5 Intriguingly, S. aureus CcpA (SaCcpA) is not only involved in the regulation of carbon metabolism but also affects antibiotic resistance, biofilm formation, toxin expression and even the infectivity of this bacterium, implying its critical role as an important global regulator for bacterial metabolism as well as virulence.6–9 Recently, small molecule inhibitors targeting the S. aureus virulence regulators, SarA or MgrA, are reported to be efficacious in animal models, indicating that targeting these regulator proteins might be a promising anti-bacterial strategy.10,11 Given the important role that CcpA played in S. aureus virulence, this transcription factor could be a feasible anti-bacterial drug target.12 Chemical inhibition of CcpA binding to the cre DNA region could potentially diminish S. aureus virulence.
Silver ions (Ag+) have been used as antibacterial agents for centuries. It is suggested that Ag+ could bind to the thiol group (–SH) of bacterial enzymes and subsequently cause enzyme deactivation.13 However, up to now, few Ag+ protein targets have been identified and characterized. Herein, we demonstrate that SaCcpA serves as a potential target for Ag+ in S. aureus. Ag+ binds specifically to the two cysteines of SaCcpA and abolishes its cre-binding property, which further abrogates S. aureus α-hemolysin secretion and biofilm formation.
Results and discussion
It is reported that silver nanoparticles (AgNPs) could block bacterial sugar metabolism in order to be bactericidal.14 Furthermore, recent studies demonstrated that bacterial strains with a TCA cycle genes knockout were less sensitive to Ag+ treatment.15 All of these data imply that Ag+ targets the bacterial central metabolism pathway. CcpA is an important regulator that coordinates central metabolism in Gram-positive bacteria.1,16 We therefore investigate the possible effect of Ag+ on CcpA physiological function.Sequence alignments of 20 CcpA family proteins from different Gram-positive bacteria species revealed that SaCcpA contains two cysteine residues (Cys216 and Cys242), which are almost absent in other species (Fig. S1†). Given that Ag+ is highly thiophilic, we postulated that Ag+ could bind to SaCcpA. To test this hypothesis, purified SaCcpA was incubated with 3 molar equivalents of Ag+ followed by the removal of excess amounts of Ag+ with a desalting column. By using a BCA assay and inductively coupled plasma mass spectrometry (ICP-MS), the stoichiometry of Ag+ binding to CcpA (monomer) was determined to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that each CcpA monomer binds 2 molar equivalents of Ag+ (Fig. S2†). Subsequently, we examined whether the two cysteine residues are involved in the Ag+ binding. We measured free thiol amounts of the CcpA protein after premixing with different molar ratios of Ag+ by Ellman’s assay. As expected, the free thiols of CcpA decreased with increasing pre-mixed Ag+ concentrations until the Ag+/CcpA molar ratio reached 2:1, confirming that the cysteines participate in Ag+ binding (Fig. S3†). The two cysteines were then individually mutated to serine. Both the ICP-MS measurement and Ellman’s assay showed that the CcpAC216S and CcpAC242S mutants could bind one Ag+ per monomer, while the double Cys mutant CcpAC2S had no Ag+ binding capability, indicating that both cysteines are responsible for Ag+ binding (Fig. S2 and S3†). In line with the results, isothermal titration calorimetry (ITC) data showed that wild-type (WT) CcpA binds 1.94 ± 0.02 molar equivalents of Ag+ with an apparent dissociation constant (Kd) of 0.74 ± 0.03 μM. The single Cys mutant CcpAC242S binds 1.08 ± 0.03 molar equivalents of Ag+ with a much lower affinity (Kd = 7.81 ± 0.61 μM), while the double Cys mutant CcpA2CS had no detectable binding to Ag+ (Fig. 1 and Table S2†).
:1, indicating that each CcpA monomer binds 2 molar equivalents of Ag+ (Fig. S2†). Subsequently, we examined whether the two cysteine residues are involved in the Ag+ binding. We measured free thiol amounts of the CcpA protein after premixing with different molar ratios of Ag+ by Ellman’s assay. As expected, the free thiols of CcpA decreased with increasing pre-mixed Ag+ concentrations until the Ag+/CcpA molar ratio reached 2:1, confirming that the cysteines participate in Ag+ binding (Fig. S3†). The two cysteines were then individually mutated to serine. Both the ICP-MS measurement and Ellman’s assay showed that the CcpAC216S and CcpAC242S mutants could bind one Ag+ per monomer, while the double Cys mutant CcpAC2S had no Ag+ binding capability, indicating that both cysteines are responsible for Ag+ binding (Fig. S2 and S3†). In line with the results, isothermal titration calorimetry (ITC) data showed that wild-type (WT) CcpA binds 1.94 ± 0.02 molar equivalents of Ag+ with an apparent dissociation constant (Kd) of 0.74 ± 0.03 μM. The single Cys mutant CcpAC242S binds 1.08 ± 0.03 molar equivalents of Ag+ with a much lower affinity (Kd = 7.81 ± 0.61 μM), while the double Cys mutant CcpA2CS had no detectable binding to Ag+ (Fig. 1 and Table S2†).
 | ||
| Fig. 1 Isothermal titration calorimetry results of Ag+ binding to CcpA, CcpAC242S and CcpA2CS in 50 mM Tris-HNO3 and 150 mM NaNO3 buffer at pH 7.4. The titrations were carried out at 25 °C. The data were fit to a one-set-of-sites binding model using the Origin software. | ||
As a global transcription factor, CcpA binds to a couple of gene promoter regions (cre sequence), such as the pckA (encoding phosphoenol-pyruvate carboxykinase) and hla (encoding α-hemolysin) promoters. To examine the effect of Ag+ binding on the CcpA’s function, we investigated whether Ag+ affected the CcpA-DNA binding properties in vitro. An electrophoretic mobility shift assay (EMSA) was applied to 35 nM pckA DNA probe (covers the cre sequence of the pckA gene) and a negative control proC probe with increasing concentration of CcpA (0–700 nM monomer concentration). As expected, the significant shift of DNA was only observed for pckA but not proC (Fig. S4a and c†). The results are consistent with a previous report that SaCcpA does not essentially require the association with phosphorylated HPr for efficient DNA binding.8 However, the addition of gradient amounts of Ag+ obviously disrupted the complex formation (Fig. 2a). The double-mutant CcpA2CS binds pckA DNA in a similar way to the WT CcpA (Fig. S4b†). However, the CcpA2CS mutant would not dissociate from the DNA probe even in the presence of Ag+ (Fig. 2b). A similar phenomenon was observed for the hla probe (Fig. S5†). CcpA binding to the cre region was enhanced by phosphorylated HPr (HPr-P).8 However, the EMSA assay demonstrates that Ag+ prevented CcpA-DNA binding even in an excess amount of Hpr-P (Fig. S6a†). Native polyacrylamide gel electrophoresis (PAGE) further confirmed that Ag+ binding also disrupted the DNA-CcpA-(Hpr-P) ternary complex (Fig. S6b†). All these results demonstrate that Ag+ binding completely abolishes CcpA-DNA binding in vitro.
 | ||
| Fig. 2 Electrophoretic mobility shift assay (EMSA) of the CcpA binding to the catabolite responsive elements (cre) of the pckA gene. Approximately 35 nM pckA promoter (189 base pairs) was incubated with 500 nM purified CcpA (a) or CcpA2CS (b) in the presence of 0, 0.4, 0.8, 1.2, 2 and 3 molar equivalents of Ag+. | ||
For further confirmation, we measured the DNA binding capabilities of the WT CcpA and CcpA2CS mutant by BioLayer Interferometry (BLI). A Biotin-labeled pckA DNA probe was immobilized on a streptavidin sensor to enable kinetic analysis of the CcpA binding to the DNA probe. As shown in Fig. 3a, WT CcpA binds strongly to the pckA probe with a Kd value of 16.1 ± 0.47 nM. While in the presence of Ag+, the binding of WT CcpA to the pckA probe is undetectable (Fig. 3b). In contrast, the binding affinities of CcpA2CS to the pckA probe are nearly identical in the absence and presence of Ag+, with Kd values of 15.1 ± 0.28 nM and 20.7 ± 0.51 nM, respectively (Fig. 3c and d and Table S3†). Collectively, these data demonstrate that Ag+ binds to the two Cys residues of CcpA, and Ag+ binding disrupts its DNA binding capability.
 | ||
| Fig. 3 The DNA binding capabilities of CcpA (a), CcpA with Ag+ (b), CcpA2CS (c) and CcpA2CS with Ag+ (d) were measured by BioLayer Interferometry (BLI). Biotinylated pckA (300 nM) was captured on pre-immobilized streptavidin Dip and Read sensor heads for 3 min. Association occurred from 0 to 180 s and dissociation was monitored thereafter for up to 360 s. The Kd values are presented as the mean ± s.e.m. derived from a global fitting of all binding curves. | ||
Previous studies demonstrated that the binding of non-physiological metal ions to proteins usually caused protein aggregation and dysfunction.17,18 To further investigate the mechanism of the Ag+-induced loss of DNA-binding capability of CcpA, we examined the oligomerization states of CcpA before and after Ag+ binding using size-exclusion chromatography (SEC). In the absence of Ag+, WT CcpA eluted at 9.6 ml with a molecular weight (Mw) of 64.9 kDa, corresponding to a dimeric form (Fig. 4a). With increasing amounts of pre-incubated Ag+, the intensities of the dimeric peak of CcpA decreased, whereas a new peak appeared at 8.7 ml with a Mw of 159.7 kDa, indicative of a tetrameric form of CcpA. This result implied that CcpA forms a dimer of dimers after Ag+ binding. A similar phenomenon was observed for the single Cys mutants of CcpA, CcpAC216S and CcpAC242S, which also formed a tetramer after incubation with Ag+ (Fig. S7†). In contrast, the majority of the double mutant CcpA2CS eluted at exactly the same volume as WT CcpA, even in the presence of 2 molar equivalents of Ag+, owing to the loss of the Ag+ binding capability of the protein (Fig. 4b). In line with the SEC results, native PAGE shows that Ag+ binding slowed down the migration rates of WT CcpA, and single mutants CcpAC216S and CcpAC242S in the native PAGE, which is indicative of the formation of a higher molecular weight oligomer upon Ag+ binding. However, Ag+ had no effect on the migration rate of the CcpA double mutant, CcpA2CS (Fig. S8†). Taken together, the binding of Ag+ to CcpA induces its tetramerization, which is possibly attributable to the loss of DNA binding capability.
 | ||
| Fig. 4 The effects of Ag+ binding on the oligomeric states of CcpA (a) and CcpA2CS (b). Size-exclusion chromatography analysis of CcpA incubated with 0, 0.6, 1.2 and 2.0 molar equivalents of Ag+ and CcpA2CS incubated with 0 and 2.0 molar equivalents of Ag+. | ||
Next, we investigated whether CcpA binds Ag+in vivo using the cellular thermal shift assay (CETSA), a method based on the change in protein thermal stability upon ligand binding for studies of the target engagement of drug candidates in a cellular condition.19,20 As shown in Fig. 5a, supplementations of 10 μM Ag+ to the bacterial culture resulted in the apparent aggregation temperature (Tagg) of the intracellular WT CcpA shifting from 49.5 °C to 45.4 °C, indicating that Ag+ binds to CcpA in vivo and such a binding destabilizes the protein. A similar result was obtained when using purified CcpA protein (Fig. S9a†). Since Ag+ binds to the two Cys residues of CcpA, it is plausible to hypothesize that Ag+ would not change the thermal stability of the double-cysteine mutant CcpA2CS due to the loss of Ag+ binding sites. To verify this, a CcpA gene mutant of S. aureus, the Newman strain, was constructed, in which the WT CcpA gene was replaced by a double cysteine mutant gene CcpA2CS (denoted as S. aureus ccpA::ccpA2CS) and a similar CETSA was performed with the mutant strain. As expected, Ag+ treatment did not alter the intracellular CcpA thermal stability in the CcpA mutant strain (Fig. 5b). Similarly, the purified CcpA double mutant CcpA2CS protein had the same thermal denaturation curves in the absence and presence of Ag+ (Fig. S9b†). Collectively, we demonstrated that Ag+ binds to CcpA intracellularly via its two Cys residues.
 | ||
| Fig. 5 Cellular thermal shift assay (CETSA) of wild-type S. aureus (a) and the S. aureus ccpA::ccpA2CS mutant (b) with or without Ag+ treatment. The soluble fractions of the intracellular CcpA or CcpA2CS protein were quantified by a western-blot. The band intensities at different temperatures are normalized to that at 36.6 °C. All experiments were performed in triplicate. | ||
CcpA is the major gene regulator of central metabolism in S. aureus and the CcpA gene knockout was found to retard bacterial growth.9 Since Ag+ binds to CcpA and abolishes its function in S. aureus, this prompted us to investigate whether Ag+ affects S. aureus growth. We examined the bacterial growth of both the WT S. aureus and ccpA::ccpA2CS mutant strains upon supplementation of 30 μM Ag+ into the cultures at the exponential-growth phase (OD600 = 0.6). As shown in Fig. 6a, both the WT and ccpA::ccpA2CS mutant of S. aureus display nearly identical growth curves in the absence of Ag+. Both cultures exhibit typical S-shaped growth curves and enter a stationary-growth phase after around 250 min, indicating that ccpA::ccpA2CS did not affect S. aureus growth significantly. In contrast, the growth rates of both the WT and ccpA::ccpA2CS mutant of S. aureus were remarkably inhibited after the addition of Ag+ until 300 min. However, a clearly different behavior was observed for the WT and mutant cultures after 300 min, with the WT culture displaying a slower growth, yielding cell density which significantly lagged behind that of the ccpA::ccpA2CS mutant culture. After 700 min, the mutant culture with the addition of Ag+ reached almost the same OD600 value as the control group, whereas the WT culture with the addition of Ag+ only reached approximately 60% of the OD600 value of the control group. The results indicated that the S. aureus ccpA::ccpA2CS mutant is less sensitive to Ag+ than the WT. In line with this, the IC50 values of Ag+ for the WT and ccpA::ccpA2CS mutant of S. aureus were calculated to be 79.8 ± 1.1 μM and 94.1 ± 0.8 μM, respectively (Fig. S10†).
 | ||
| Fig. 6 (a) The inhibition effect of Ag+ on the bacterial growth of the wild-type S. aureus and ccpA::ccpA2CS mutant strains. The OD600 was recorded at 20 min intervals, and 30 μM Ag+ was added when OD600 reached 0.6. The secreted α-hemolysin in the supernatant was normalized to OD600. (b) Rabbit erythrocyte lysis activities of S. aureus and the S. aureus ccpA::ccpA2CS mutant with or without 20 μM Ag+. Ag+ was added at the beginning of the bacterial culture. All experiments were performed in triplicate. Results are shown as mean ± sd. The haemolytic activities of the wild-type strain without Ag+ treatment are used as a control and the mean value is set at 100%. The activities in other groups are normalized to the control. The statistical difference is determined by the two-tailed Student’s t-test. | ||
Besides regulation of carbon catabolite repression, CcpA also exerts a critical role in S. aureus virulence factor secretion and biofilm formation.6,7,9 It is reasonable to postulate that Ag+ would interfere with these physiological processes of S. aureus by targeting CcpA. Therefore, the effect of Ag+ on the virulence factor secretion and biofilm formation in both the WT and ccpA::ccpA2CS mutant S. aureus strains was investigated. The promoter region of the hla gene (encoding α-hemolysin) in S. aureus contains the cre sequence that could be recognized by CcpA. Previous studies demonstrated that the hla transcription level was markedly down-regulated in the ccpA knockout S. aureus strain.21 Indeed, secreted α-hemolysin and rabbit erythrocyte lysis activity of a S. aureus mutant strain with the CcpA gene knockout (denoted as S. aureus ΔccpA) is almost undetectable. On the other hand, the S. aureus ccpA::ccpA2CS mutant exhibited a significant decrease on secreted α-hemolysin and retained approximately 50% erythrocyte lysis activity compared to the WT strain, implying that the two Cys residues have a potential role in the regulation of α-hemolysin expression (Fig. S11†). Upon supplementation of 20 μM Ag+, a western-blot showed a significant decrease in secreted α-hemolysin in WT S. aureus. While in the ccpA::ccpA2CS mutant strain, Ag+ caused no obvious change in the secreted α-hemolysin. Consistently, the rabbit erythrocyte lysis activity of WT S. aureus decreased dramatically by 60% after Ag+ treatment. In contrast, the treatment of Ag+ led to much smaller decrease on the lysis activity of the ccpA::ccpA2CS mutant strain, which still exhibited 85% activity compared to the control group (Fig. 6b and c). The results are consistent with qPCR data, which demonstrated that attenuation of the transcription level of CcpA regulated genes (pckA and hla) was much higher in the WT strain than that in the ccpA::ccpA2CS mutant strain (Fig. S12†). Similarly, bacterial biofilm formation is inhibited by Ag+ to a lesser extent in the ccpA::ccpA2CS mutant than in the WT strain (Fig. S13†). It is noteworthy that regulation of α-hemolysin expression and biofilm formation in S. aureus are complicated. For example, α-hemolysin expression is affected by multiple regulatory networks, including the global regulators SarA and MgrA.22,23 It is possible that these regulatory networks were also perturbed upon Ag+ treatment, which could at least partially explain the decrease of erythrocyte lysis activity observed in the ccpA::ccpA2CS mutant strain upon Ag+ treatment. Nevertheless, the evident discrepancy observed between the ccpA::ccpA2CS mutant and WT S. aureus upon Ag+ treatment confirms that CcpA indeed serves as one of the targets of Ag+in vivo.
All of the results demonstrated that the WT S. aureus strain was more sensitive to Ag+ treatment than the ccpA::ccpA2CS mutant strain due to Ag+ binding to the two cysteine residues. To further confirm this, the severity of a S. aureus infection was compared between the WT and ccpA::ccpA2CS mutant in a murine model. The details are described in the ESI.† In brief, groups of female BALB/c mice were inoculated with S. aureus WT or ccpA::ccpA2CS mutant strains to develop abscesses on the skin. Twice-daily treatment of AgNO3 with different concentrations (20 μg ml−1 and 100 μg ml−1) was applied onto the abscesses. The skin abscesses were excised 64 h post-infection, homogenized and serially diluted for CFU quantification. As shown in Fig. 7, the control groups infected with the WT or ccpA::ccpA2CS mutant had similar viable bacterial counts, with logCFU mean values of 8.4 and 8.1 respectively, indicating that double Cys mutation does not significantly perturb S. aureus viability in a murine model. A low dosage of AgNO3 treatment (20 μg ml−1) did not change the bacterial loads in both the WT and ccpA::ccpA2CS mutant infected groups. However, a significant difference was observed when the two infected groups were treated with a high dosage of AgNO3 (100 μg ml−1). In the WT S. aureus infected group, the viable bacterial counts dropped significantly compared to the control group. In contrast, the bacteria counts were almost the same in the ccpA::ccpA2CS mutant infected groups upon a high dosage Ag+ treatment, confirming that WT S. aureus is more sensitive to Ag+ than the ccpA::ccpA2CS mutant. Intriguingly, a significant difference on the dermonecrosis of the skin abscess was observed for the mice infected with two different bacterial strains (Fig. S14†). The mice infected with wild-type S. aureus had much more severe dermonecrosis than those infected with the ccpA::ccpA2CS mutant strain. Given that α-hemolysin is the major contributor to necrotic lesions,24 the observation is consistent with the results that the ccpA::ccpA2CS mutant had a lower α-hemolysin level than the wild-type strain.
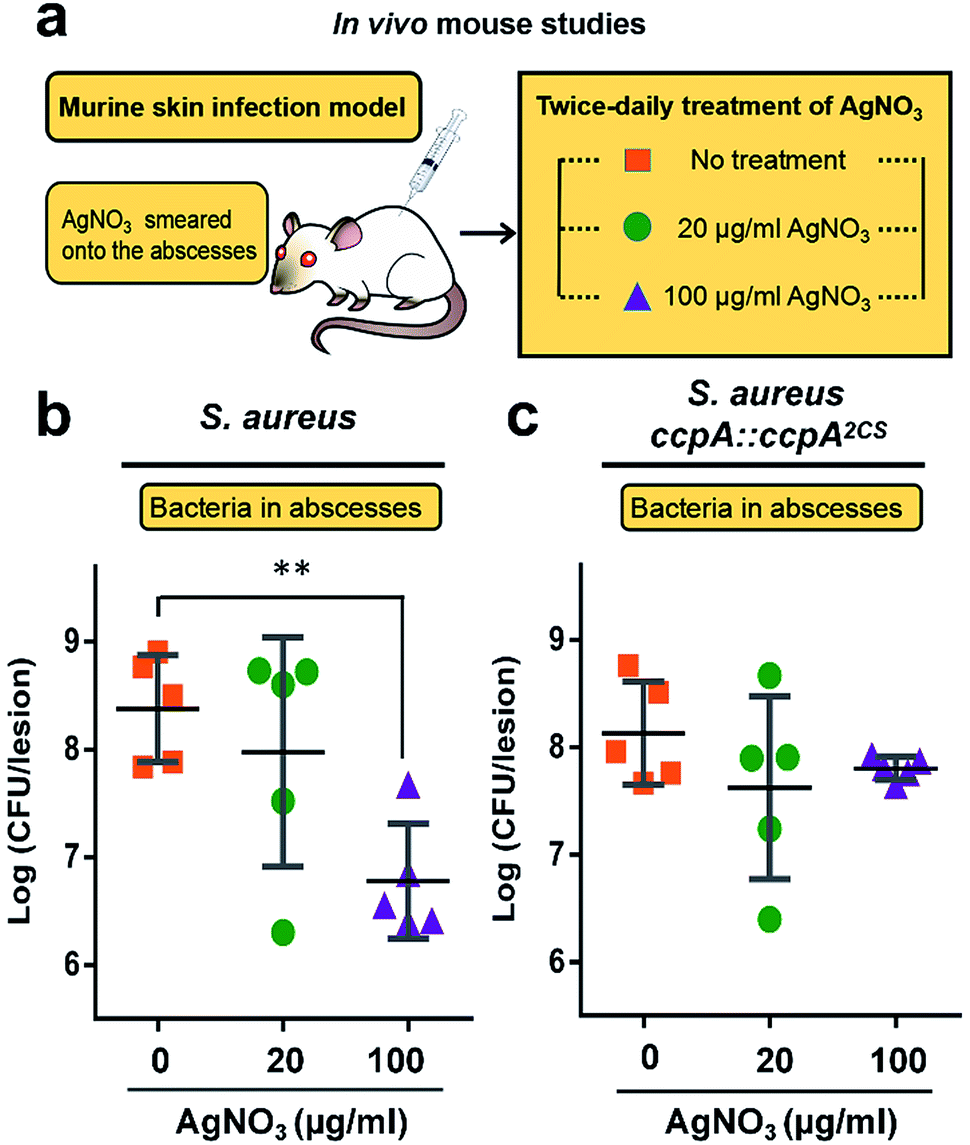 | ||
| Fig. 7 Murine skin infection model (a) to investigate the sensitivity of S. aureus strains to Ag+ treatment. The bacterial load of local abscesses induced by wild-type S. aureus (b) or the ccpA::ccpA2CS mutant (c) was enumerated in control or AgNO3 treatment groups (20 μg ml−1 and 100 μg ml−1). The logCFU values are presented as the mean ± sd. The statistical difference is determined by the Mann–Whitney U test. | ||
Conclusions
CcpA is one of the important global DNA regulators of Gram-positive bacteria. Recent transcriptome and proteome analyses revealed that CcpA has broad effects on gene expression in S. aureus, even in the absence of glucose.25 Particularly, a CcpA gene knockout abrogates biofilm formation and virulence factor expression in S. aureus, which remarkably decreases bacterial pathogenesis. We show clearly that Ag+ binds to CcpA via the two Cys residues both in vitro and in vivo, leading to the disruption of protein functions, thus attenuating bacterial growth, bacterial toxin expression and biofilm formation. Importantly, we demonstrated that WT S. aureus was more sensitive to Ag+ treatment than the ccpA::ccpA2CS mutant in a murine skin infection model. The results herein confirm SaCcpA as an intracellular target for Ag+. It should be noted that metal-based drugs are usually multi-targeted.26 Although it is commonly believed that the antimicrobial activity of silver is due to its interaction with thiol groups in enzymes and proteins, other cellular components are likely to be involved.27–29 Therefore, identification of Ag-binding proteins at a proteome-wide scale may allow extensive exploration of silver targets to advance our understanding on the bactericidal effects of silver.30–32 However, the physiological function of the two cysteine residues in SaCcpA remains unclear. It has been reported previously that several DNA regulators of S. aureus use the cysteine-based oxidation sensing pathway for regulatory functions.33–35 Whether the two cysteines in SaCcpA are also involved in oxidative sensing may warrant further studies.Live subject statement
The animal studies strictly followed the recommendations in “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health. The protocols were approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR) and the University of Hong Kong (Permit no. 4008-16).Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21501200 and 21671203), the Science and Technology Program of Guangzhou, China (Grant No. 201707010038), the RGC of Hong Kong (17305415 and 17333616), the Fundamental Research Funds for the Central Universities and a starting fund from Sun Yat-sen University, and the University of Hong Kong (for the e-SRT on Integrative Biology and a fellowship for RMW).Notes and references
- G. Seidel, M. Diel, N. Fuchsbauer and W. Hillen, FEBS J., 2005, 272, 2566–2577 CrossRef CAS PubMed.
- J. Deutscher, Curr. Opin. Microbiol., 2008, 11, 87–93 CrossRef CAS PubMed.
- F. D. Lowy, N. Engl. J. Med., 1998, 339, 520–532 CrossRef CAS PubMed.
- R. M. Klevens, M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey, S. K. Fridkin and A. Surveillance Investigators, J Am Med Assoc, 2007, 298, 1763–1771 CrossRef CAS PubMed.
- L. B. Rice, Am. J. Infect. Control, 2006, 34, S11–S19 CrossRef PubMed.
- K. Seidl, M. Bischoff and B. Berger-Bachi, Infect. Immun., 2008, 76, 5093–5099 CrossRef CAS PubMed.
- K. Seidl, C. Goerke, C. Wolz, D. Mack, B. Berger-Bachi and M. Bischoff, Infect. Immun., 2008, 76, 2044–2050 CrossRef CAS PubMed.
- C. Li, F. Sun, H. Cho, V. Yelavarthi, C. Sohn, C. He, O. Schneewind and T. Bae, J. Bacteriol., 2010, 192, 3883–3892 CrossRef CAS PubMed.
- K. Seidl, M. Stucki, M. Ruegg, C. Goerke, C. Wolz, L. Harris, B. Berger-Bachi and M. Bischoff, Antimicrob. Agents Chemother., 2006, 50, 1183–1194 CrossRef CAS PubMed.
- F. Sun, L. Zhou, B.-C. C. Zhao, X. Deng, H. Cho, C. Yi, X. Jian, C.-X. X. Song, C.-H. H. Luan, T. Bae, Z. Li and C. He, Chem. Biol., 2011, 18, 1032–1041 CrossRef CAS PubMed.
- R. Arya, R. Ravikumar, R. S. Santhosh and S. A. Princy, Front. Microbiol., 2015, 6, 416 Search PubMed.
- B. Görke and J. Stülke, Nat. Rev. Microbiol., 2008, 6, 613–624 CrossRef PubMed.
- S. Y. Liau, D. C. Read, W. J. Pugh, J. R. Furr and A. D. Russell, Lett. Appl. Microbiol., 1997, 25, 279–283 CAS.
- R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289–1306 CrossRef CAS PubMed.
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190ra81 Search PubMed.
- S. Tobisch, D. Zuhlke, J. Bernhardt, J. Stulke and M. Hecker, J. Bacteriol., 1999, 181, 6996–7004 CAS.
- W. Xia, H. Y. Li and H. Sun, Chem. Commun., 2014, 50, 1611–1614 RSC.
- S. J. Cun and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4943–4948 CrossRef CAS PubMed.
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundback, P. Nordlund and D. M. Molina, Nat. Protoc., 2014, 9, 2100–2122 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. H. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef CAS PubMed.
- J. Leiba, T. Hartmann, M. E. Cluzel, M. Cohen-Gonsaud, F. Delolme, M. Bischoff and V. Molle, J. Biol. Chem., 2012, 287, 43607–43619 CrossRef CAS PubMed.
- A. L. Cheung and A. C. Manna, Infect. Immun., 2005, 73, 4391–4394 CrossRef CAS PubMed.
- S. Ingavale, W. van Wamel, T. T. Luong, C. Y. Lee and A. L. Cheung, Infect. Immun., 2005, 73, 1423–1431 CrossRef CAS PubMed.
- A. D. Kennedy, J. Bubeck Wardenburg, D. J. Gardner, D. Long, A. R. Whitney, K. R. Braughton, O. Schneewind and F. R. DeLeo, J. Infect. Dis., 2010, 202, 1050–1058 CrossRef PubMed.
- K. Seidl, S. Muller, P. Francois, C. Kriebitzsch, J. Schrenzel, S. Engelmann, M. Bischoff and B. Berger-Bachi, BMC Microbiol., 2009, 9, 95 CrossRef PubMed.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- I. Romero-Canelon and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4187–4188 CrossRef CAS PubMed.
- L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y. Lai, G. Jiang and H. Sun, Angew. Chem., 2013, 52, 4916–4920 CrossRef CAS PubMed.
- R. F. S. Lee, S. Theiner, A. Meibom, G. Koellensperger, B. K. Keppler and P. J. Dyson, Metallomics, 2017, 9, 365–381 RSC.
- Y. Wang, L. Hu, F. Xu, Q. Quan, Y. T. Lai, W. Xia, Y. Yang, Y. Y. Chang, X. Yang, Z. Chai, J. Wang, I. K. Chu, H. Li and H. Sun, Chem. Sci., 2017, 8, 4626–4633 RSC.
- P. R. Chen, T. Bae, W. A. Williams, E. M. Duguid, P. A. Rice, O. Schneewind and C. He, Nat. Chem. Biol., 2006, 2, 591–595 CrossRef CAS PubMed.
- P. R. Chen, S. Nishida, C. B. Poor, A. Cheng, T. Bae, L. Kuechenmeister, P. M. Dunman, D. Missiakas and C. He, Mol. Microbiol., 2009, 71, 198–211 CrossRef CAS PubMed.
- Q. J. Ji, L. Zhang, F. Sun, X. Deng, H. H. Liang, T. O. Bae and C. He, J. Biol. Chem., 2012, 287, 21102–21109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures and tables. See DOI: 10.1039/c7sc02251d |
| This journal is © The Royal Society of Chemistry 2017 |